

Abstract
MST1 (mammalian STE20-like kinase 1) is a serine/threonine kinase that is cleaved and activated by caspases during apoptosis. Overexpression of MST1 induces apoptotic morphological changes such as chromatin condensation, but the mechanism is not clear. Here we show that MST1 induces apoptotic chromatin condensation through its phosphorylation of histone H2AX at Ser-139. During etoposide-induced apoptosis in Jurkat cells, the cleavage of MST1 directly corresponded with strong H2AX phosphorylation. In vitro kinase assay results showed that MST1 strongly phosphory-lates histone H2AX. Western blot and kinase assay results with a mutant S139A H2AX confirmed that MST1 phos-phorylates H2AX at Ser-139. Direct binding of MST1 and H2AX can be detected when co-expressed in HEK293 cells and was also confirmed by an endogenous immunoprecipitation study. When overexpressed in HeLa cells, both the MST1 full-length protein and the MST1 kinase domain (MST1-NT), but not the kinase-negative mutant (MST1-NT-KN), could induce obvious endogenous histone H2AX phosphorylation. The caspase-3 inhibitor benzyloxycarbonyl-DEVD-fluoromethyl ketone (Z-DEVD-fmk) attenuates phosphorylation of H2AX by MST1 but cannot inhibit MST1-NT-induced histone H2AX phosphorylation, indicating that cleaved MST1 is responsible for H2AX phosphory-lation during apoptosis. Histone H2AX phosphorylation and DNA fragmentation were suppressed in MST1 knockdown Jurkat cells after etoposide treatment. Taken together, our data indicated that H2AX is a substrate of MST1, which functions to induce apoptotic chromatin condensation and DNA fragmentation.
Keywords: Caspase, DNA, Histones, Protein Phosphorylation, Serine Threonine Protein Kinase
Introduction
MST12 (mammalian STE20-like kinase 1) is a ubiquitously expressed serine/threonine kinase. This kinase was originally discovered as a mammalian homologue of the budding yeast gene STE20, which encodes the mitogen-activated protein kinase kinase kinase kinase (MAPKKKK) enzyme of the Saccharomyces cerevisiae MAPK cascade (1, 2). MST1 was shown to activate MKK6, MAPK, MKK7, or SAPK when co-transfected with each of them in mammalian cells (3), suggesting that MST1 might also function as a MAPKKKK to activate these pathways.
MST1 has an N-terminal catalytic domain and a C-terminal regulatory region. Deletion analysis revealed that the C-terminal domain of MST1 serves an inhibitory function (4). MST1 has two caspase cleavage sites situated between its N-terminal kinase domain and its C-terminal regulatory domain (3, 5). During apoptosis induced by a wide variety of stimuli, such as staurosporine, engagement of CD95/Fas, etoposide, UV, okadaic acid, serum starvation, anti-tumor drugs, or arsenite, MST1 is cleaved and activated by a process that involves both phosphorylation and removal of the C-terminal inhibitory domain by caspases (2, 3, 5–11).
Human MST1 contains two functional nuclear export signals in the C-terminal domain, and full-length MST1 is excluded from the nucleus and localized to the cytoplasm. During apoptosis upon caspase-mediated cleavage, the C-terminal domain will be released from the N-terminal kinase domain, resulting in nuclear localization of the MST1 kinase domain (12). The activated MST1 translocates from the cytoplasm to the nucleus and enhances the induction of peripheral chromatin condensation (12, 13). This evidence suggests that MST1 is an important caspase substrate that induces chromatin compaction during apoptosis.
Overexpression of MST1 induces apoptotic morphological changes such as chromatin condensation, and MST1 is required for apoptosis induced by certain genotoxic agents, including UV and staurosporine (10, 11). MST1 is more efficient at inducing chromatin condensation when it is constitutively localized to the nucleus by mutation of its nuclear export signals. Moreover, inhibition of MST1 nuclear translocation by mutation of its cleavage sites reduces its ability to induce chromatin condensation (12). Staurosporine treatment induces chromatin condensation, MST1 cleavage, and nuclear translocation (3, 10). Staurosporine-induced chromatin condensation is partially inhibited by the expression of a kinase-negative mutant of MST1, suggesting that MST1 kinase activity is important in this process (12).
During apoptosis, a commonly observed specific histone modification is the phosphorylation of histone H2B at Ser-14, and MST1 has been identified as the activating kinase (14). However, at least one report showed that MST1 can also induce chromatin condensation independent of histone H2B phosphorylation (13). Histone H2AX phosphorylation was also found to play a role in apoptosis. The appearance of strong H2AX phosphorylation is concurrent with the initiation of DNA fragmentation (15). Our previous studies showed that DNA fragmentation did not occur in H2AX-deficient murine embryonic fibroblasts, indicating an essential role in apoptosis (16). Recently, phosphorylation of histone H2AX at serine 139 (γ-H2AX) was also found to coincide with phosphorylation of H2B at serine 14 (17). This evidence suggested that phosphorylation of H2AX is also an important apoptotic “histone code” (18).
Here we provide evidence that MST1 can phosphorylate H2AX in vitro and ex vivo. MST1-induced chromatin condensation is dependent on MST1 cleavage and kinase activity. Both H2AX phosphorylation and DNA fragmentation were inhibited in MST1 knockdown Jurkat cells after etoposide treatment. Our data therefore indicate that caspase-mediated cleavage of MST1 is responsible for the phosphorylation of H2AX, which is essential for chromatin condensation and DNA fragmentation during apoptosis.
EXPERIMENTAL PROCEDURES
Plasmids
The pCE-MST1-FL, pCE-MST1-NT, and pCE-MST1-NT-KN plasmids were kindly provided by Professor Hiroshi Nishina (Department of Developmental and Regenerative Biology, Medical Research Institute, Tokyo Medical and Dental University) (13). The lentivirus plasmids expressing shMST1 were purchased from Addgene (Huntsville, AL), and the pCDNA4-H2AX, pGE5-GST-H2AX, and pGE5-GST-H2AX-S139A plasmids were constructed as reported previously (16).
Cells
Jurkat cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) in a 37 °C 5% CO2 incubator. HeLa cells were cultured in Eagle's minimum essential medium supplemented with 10% FBS.
Reagents
Etoposide was purchased from Sigma-Aldrich. The JetPEI transfection reagent and caspase-3 inhibitor Z-DEVD-fmk were obtained from Roche Applied Science. Recombinant human histone H2AX and H2B were from Millipore (Billerica, MA). The antibodies against γ-H2AX, Myc-Tag, total JNKs, phosphorylated JNKs, caspase-3, MST1, and β-actin were obtained from Cell Signaling Biotechnology, Inc. (Beverly, MA). Antibodies to detect Myc-HRP and histones H2B and H2AX were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), Upstate Biotech Millipore (Charlottesville, VA), and Abcam Inc. (Cambridge, MA), respectively.
Etoposide Treatment of Cells
Jurkat cells (5 × 106) were seeded in 10-cm dishes and treated for various times with culture medium supplemented with etoposide (50 μm).
Total Cellular Protein or Histone Extraction and Western Blot Analysis
Cellular proteins were extracted by disrupting cells in Nonidet P-40 lysis buffer (50 mm Tris-HCl, pH 7.4, 0.5% Nonidet P-40, 150 mm NaCl, 1 mm EGTA, 1 mm Na3VO4, 1 mm NaF, 1 mg/ml aprotinin, 1 mg/ml leupeptin, 1 mg/ml pepstatin, and 1 mm PMSF). Histones were extracted according the method described previously (19). The protein samples were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked at room temperature for 1 h with 5% nonfat milk in Tris-buffered saline containing Tween 20 (TBST). Primary antibodies were incubated with membranes at 4 °C overnight. Membranes were incubated with the appropriate secondary antibody in TBST for 1 h at room temperature. Proteins were detected by enhanced chemiluminescence (ECL) (Amersham Biosciences).
Immunoprecipitation
For immunoprecipitation, HEK293 cells were transfected with pCDNA4-H2AX and pCE-MST1-FL or pCE-MST1-NT. Cells were harvested and disrupted in lysis buffer as above at 36 h after transfection. Cell lysates were incubated with anti-His (Santa Cruz Biotechnology) at 4 °C overnight and protein A/G-Sepharose beads (Santa Cruz Biotechnology) for an additional 4 h. Duplicate blots were made from the same set of experiments. One blot was probed with anti-Myc-HRP (Santa Cruz Biotechnology) to detect MST1-FL or MST1-NT in the immunoprecipitated complex, and the other blot was probed with anti-His-HRP (Santa Cruz Biotechnology). Proteins were revealed with ECL reagents (Amersham Biosciences). The expression of histone H2AX and MST1-FL or MST1-NT was detected by anti-Xpress and anti-Myc-HRP separately. For endogenous immunoprecipitation, Jurkat cells were harvested at different times after etoposide treatment. Cell lysates were immunoprecipitated with anti-MST1, and the immunoprecipitated complex was probed with antibodies to detect H2AX or γ-H2AX.
Immunofluorescence Staining
To study the function of MST1 in H2AX phosphorylation, HeLa cells transfected with MST1-FL, MST1-NT, or MST1-NT-KN were fixed in 4% paraformaldehyde and permeabilized in 0.5% Triton X-100 for 30 min at 36 h after transfection. Fixed cells were then incubated with Myc-Tag mouse monoclonal or γ-H2AX rabbit polyclonal antibodies (Cell Signaling Biotechnology, Inc.) followed by incubation with red fluorescent Alexa Fluor 568 dye-labeled anti-mouse IgG or green fluorescent Alexa Fluor 488 dye-labeled anti-rabbit (Invitrogen), respectively. For the localization study, HeLa cells transfected with MST1-NT or MST1-NT-KN were fixed and stained with a Myc-Tag mouse monoclonal antibody followed by incubation with red fluorescent Alexa Fluor 568 dye-labeled anti-mouse IgG. Nuclei were stained with Hoechst. To study the influence of caspase-3 inhibition on MST1-mediated histone H2AX phosphorylation, a caspase-3 inhibitor (10 μm) was added to the medium right after transfection. All samples were viewed with a confocal fluorescence microscope system (NIKON C1si confocal spectral imaging system, NIKON Instruments Co., Melville, NY).
In Vitro Kinase Assay
For the in vitro kinase assay to detect γ-32P incorporation, the histone H2AX, histone H2B (Millipore, Inc.), GST-H2AX, or GST-H2AX-S139A proteins were each mixed together with active MST1 (Upstate Biotech Millipore), 1 mCi of [γ-32P]ATP, 50 μm unlabeled ATP, and 1× kinase buffer A (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm EGTA, 1 mm DTT, 0.01% Brij 35; Cell Signaling Technology) and incubated at 30 °C for 30 min. The reactive products were separated by SDS-PAGE for autoradiography. To detect histone H2AX phosphorylation by Western blot, the H2AX protein, GST-H2AX, or GST-H2AX-S139A protein was mixed together with active MST1, 0.2 mm ATP, and 1× kinase buffer and incubated at 30 °C for 30 min. The reactive products were separated by 15% SDS-PAGE before Western blot analysis. In addition, the activity of overexpressed MST1 was detected by an immunoprecipitation kinase assay. Overexpressed Myc-MST1-FL, Myc-MST1-NT, or Myc-MST1-NT-KN was immunoprecipitated by anti-Myc tag and mixed together with 0.5 μg of the histone H2AX protein, 0.2 mm ATP, and 1× kinase buffer and incubated at 30 °C for 30 min and then analyzed by SDS-PAGE and autoradiography.
Flow Cytometry Analysis
Etoposide-induced apoptosis of Jurkat cells was examined using the annexin V FITC apoptosis detection kit (MBL Medical & Biological Laboratories, Nagoya, Japan) according to the protocol provided. Briefly, cells were centrifuged, washed once with RPMI 1640 medium containing serum, and incubated with annexin V-conjugated FITC. Apoptosis was analyzed by flow cytometer (FACSCalibur, Becton Dickinson).
DNA Fragmentation
Jurkat cells were treated with etoposide (50 μm) for various times. The cells were disrupted by adding lysis buffer (10 mm Tris-HCl, pH 8.0, 10 mm EDTA, 0.5% Triton X 100) and incubated on ice for 45–90 min followed by centrifugation at 12,000 × g for 45 min at 4 °C. The supernatant fraction containing fragmented DNA was mixed with 5 μl of protease K at 50 °C for 3 h and centrifuged at 12,000 × g for 30 min at 4 °C. The DNA was extracted twice with phenol:chloroform:isopropyl alcohol (25:24:1) and once by chloroform. One-tenth volume of 3 m NaAc and 2 volumes of ethanol were added to the DNA extraction part and kept at −20 °C for 1–2 h or overnight before centrifugation at 12,000 × g for 30 min at 4 °C. The DNA pellet was washed with 70% ethanol and resuspended in Tris-EDTA buffer with 1 mg/ml RNase A. DNA fragments were separated by 2% agarose gel electrophoresis, stained with ethidium bromide, and photographed under UV light.
RESULTS
MST1 Cleavage Coincides with Strong H2AX Phosphorylation and DNA Fragmentation during Apoptosis
To study the function of MST1 and the possible relationship between MST1 and histone H2AX phosphorylation in apoptosis, Jurkat cells were treated with etoposide to induce apoptosis. Results of flow cytometry analysis showed that etoposide effectively induces apoptosis in Jurkat cells (Fig. 1A). MST1 is reportedly cleaved and activated by caspase-3 during apoptosis (3, 10), and our results confirmed that the cleavage of MST1 was highly consistent with caspase-3 cleavage (Fig. 1B). The γ-H2AX protein abundance and DNA fragmentation levels were also detected, and results showed that γ-H2AX is detected shortly (0.5 h) after etoposide stimulation, but strong H2AX phosphorylation appeared only after MST1 was cleaved. Both the strong histone H2AX phosphorylation and DNA fragmentation corresponded with MST1 cleavage (Fig. 1B). H2AX has been found to be essential for DNA fragmentation during ultraviolet A-induced apoptosis (16). To determine the essential role of H2AX in etoposide-induced DNA fragmentation, we treated H2AX+/+ and H2AX−/− murine embryonic fibroblasts with etoposide and found that DNA fragmentation occurs in H2AX+/+ murine embryonic fibroblasts but not in H2AX−/− murine embryonic fibroblasts (Fig. 1C). Strong H2AX phosphorylation and DNA fragmentation corresponded very well in several other cell lines, including HL-60 and HeLa cells (supplemental Fig. 1, A and B). Thus, these results indicate an essential role for H2AX in DNA fragmentation and a strong association between MST1 cleavage and histone H2AX phosphorylation during apoptosis.
FIGURE 1.

MST1 cleavage corresponds with strong histone H2AX phosphorylation and DNA fragmentation during apoptosis. A, Jurkat cells were treated with etoposide (50 μm). Cells were harvested at the indicated times and subjected to flow cytometry after annexin V/propidium iodide staining to assess apoptosis. The means of three independent determinations ± S.D. are shown. Ctrl, control. B and C, Jurkat or H2AX+/+ and H2AX−/− murine embryonic fibroblasts were treated with etoposide (50 μm) and harvested at the indicated times. Lysates were prepared for the detection of MST1 and caspase-3. Histone proteins were extracted for the detection of γ-H2AX and total histone H2AX. DNA was extracted for the DNA ladder assay as described under “Experimental Procedures.”
MST1 Phosphorylates Histone H2AX at Ser-139 in Vitro
To study the possibility that MST1 can directly phosphorylate histone H2AX, an in vitro kinase assay was conducted. Results showed that MST1 strongly phosphorylated histone H2AX in vitro (Fig. 2A, third lane). The known MST1 substrate, histone H2B, was used as a positive control (Fig. 2A, fifth lane). Western blot analysis using an antibody to detect γ-H2AX showed that MST1 phosphorylates H2AX at Ser-139 (Fig. 2B). To further confirm that MST1 phosphorylates H2AX at Ser-139, purified wild-type and mutant GST-H2AX (H2AX-S139A) were used as substrates for an in vitro kinase assay. Results indicated that the phosphorylation of the H2AX-S139A mutant was dramatically decreased (Fig. 2C). Western blotting was also performed after an in vitro kinase assay using purified wild-type or mutant GST-H2AX (H2AX-S139A) as substrate, confirming that MST1 phosphorylates H2AX at Ser-139 (Fig. 2D). Taken together, these data indicate that MST1 phosphorylates histone H2AX in vitro, and a major phosphorylation site is Ser-139.
FIGURE 2.

MST1 phosphorylates H2AX at Ser-139 in vitro. A, histones H2AX and H2B were used as substrates for active MST1. Reactive products were resolved by SDS-PAGE followed by autoradiography or Coomassie blue staining. auto-p, autophosphorylation. B, the H2AX protein was used as a substrate for an in vitro kinase assay. Reactive products were subjected to SDS-PAGE and Western blot to detect histone H2AX and γ-H2AX. C, equal molar amounts of GST-H2AX or GST-H2AX-S139A were used as substrates for MST1 in an in vitro kinase assay. Reactive products were resolved by SDS-PAGE followed by autoradiography or Coomassie Blue staining. D, the same in vitro kinase assay as shown in C was conducted, and reactive products were subjected to SDS-PAGE and Western blot to detect histone H2AX, γ-H2AX, and MST1.
MST1 Interacts with Histone H2AX
To confirm whether MST1 interacts with histone H2AX ex vivo, plasmids containing full-length MST1 (pCE-Myc-MST1-FL), MST1 kinase domain (pCE-Myc-MST1-NT), or MST1 C-terminal regulatory domain (pCE-Myc-MST1-CT) and histone H2AX (pCDNA4-H2AX) were co-transfected into HEK293 cells. Histone H2AX was immunoprecipitated with anti-His, and MST1 was detected using a Myc-HRP antibody. The results showed that both MST1-FL and MST1-NT could interact with histone H2AX, but MST1-CT could not (Fig. 3A). To determine whether MST1 can bind with and phosphorylate endogenous histone H2AX, Jurkat cells were treated with etoposide and harvested at different time points. MST1 was immunoprecipitated with anti-MST1, and H2AX and γ-H2AX were detected with the appropriate antibody (Fig. 3B). This result confirmed that MST1 could bind and phosphorylate histone H2AX ex vivo.
FIGURE 3.

MST1 interacts with H2AX ex vivo. A, HEK293 cells were co-transfected with pCE-MST1-FL, pCE-MST1-NT, or pCE-MST1-CT and pCDNA4-H2AX. At 36 h after transfection, cells were harvested, and the overexpressed H2AX was immunoprecipitated (IP) with anti-His. The expression level of MST1-FL or MST1-NT or MST1-CT was detected with anti-Myc-Tag, and the expression level of H2AX was detected by anti-His. B, Jurkat cells were treated with etoposide and harvested at different times. MST1 was immunoprecipitated with anti-MST1, and H2AX and γ-H2AX were detected with the appropriate antibody.
Overexpression of MST1 Induces Apoptotic H2AX Phos-phorylation in HeLa Cells
To study the function of MST1-mediated H2AX phosphorylation, full-length MST1 (MST1-FL), MST1 kinase domain (MST1-NT), or kinase-negative MST1 (MST1-NT-KN) was each transfected into HeLa cells. Histones were extracted for the detection of γ-H2AX. Results indicated that MST1-FL was cleaved after its overexpression in HeLa cells. Furthermore, both MST1-FL and MST1-NT could induce histone H2AX phosphorylation at Ser-139, and H2AX phosphorylation in the MST1-NT transfected group was strongest (Fig. 4A, second and third lanes). In contrast, the MST1-NT-KN cells could not induce histone H2AX phosphorylation as effectively as MST1-NT, suggesting that MST1-induced histone H2AX phosphorylation is dependent on its kinase activity (Fig. 4B, fourth lane). An immunoprecipitation (IP) kinase assay was conducted to confirm the kinase activity of overexpressed MST1-FL and MST1-NT (Fig. 4B, second and third lanes). Commercially available active MST1 was used as a positive control (Fig. 4B, fifth lane). By using immunofluorescence staining, we confirmed the localization of full-length MST1 and the MST1 kinase domain. These results indicated that the full-length kinase-negative MST1 (MST1-KN) is mainly located in the cytoplasm, whereas the kinase-negative MST1 (MST1-NT-KN) is primarily localized in the nucleus (Fig. 4C). Immunofluorescence staining results also showed that strongly phosphorylated γ-H2AX could be detected in MST1-FL and MST1-NT transfected cells but not in the MST1-NT-KN group (Fig. 4D). Taken together, these results suggested that both full-length MST1 and the MST1 kinase domain can interact with histone H2AX, and either the overexpressed MST1-FL or MST1-NT can induce H2AX phosphorylation ex vivo, which is depen-dent on MST1 kinase activity.
FIGURE 4.

MST1 overexpression induces apoptotic H2AX phosphorylation in HeLa cells. A, HeLa cells were transfected with pCE-MST1-FL, pCE-MST1-NT, or pCE-MST1-NT-KN. At 36 h after transfection, cells were harvested, and lysates and histone proteins were prepared. The expression level of MST1-FL or MST1-NT was detected with anti-Myc-Tag, and the level of γ-H2AX was detected in the acid-extracted histones. B, HeLa cells were transfected with pCE-MST1-WT, pCE-MST1-NT, or pCE-MST1-NT-KN. At 36 h after transfection, cells were harvested, the overexpressed MST1 was immunoprecipitated (IP) with anti-Myc-Tag, and an in vitro kinase assay was conducted as described under “Experimental Procedures.” IB, immunoblot. C, HeLa cells growing on a slide chamber were transfected with pCE-MST1-KN or pCE-MST1-NT-KN. At 36 h after transfection, cells were fixed with paraformaldehyde, stained for Myc-Tag (MST1, red) and DAPI (blue), and observed by confocal immunofluorescence microscopy. D, HeLa cells growing on a slide chamber were transfected with pCE-MST1-FL, pCE-MST1-NT, or pCE-MST1-NT-KN. At 36 h after transfection, cells were fixed with paraformaldehyde, stained for Myc-Tag (MST1, red), γ-H2AX (green), and Hoechst 33342 (blue), and observed by confocal immunofluorescence microscopy. The bar graphs represent the percentage of apoptotic chromatin condensation in transfected cells from three independent experiments (±S.D.). At least 200 cells were counted each time. The asterisk indicates p < 0.05.
MST1-induced H2AX Phosphorylation Is Dependent on MST1 Cleavage
To study the dependence of MST1 cleavage in the induction of H2AX phosphorylation, pCE-Myc-MST1-FL and pCE-Myc-MST1-NT plasmids were each transfected into HeLa cells, and cells were treated or not with the caspase-3 inhibitor, Z-DEVD-fmk. In the MST1-FL transfected cells, Western blot results showed that the caspase-3 inhibitor could completely suppress the cleavage of MST1. Correspondingly, the inhibitor also dramatically suppressed the phosphorylation of H2AX (Fig. 5A). Immunofluorescence staining results also showed that the caspase-3 inhibitor almost completely blocked the translocation of MST1 into the nucleus, and phosphorylation of histone H2AX was markedly suppressed (Fig. 5A). In contrast, the caspase-3 inhibitor was not effective in suppressing MST1-NT-induced H2AX phosphorylation (Fig. 5, A and B). These results indicated that MST1-mediated H2AX phosphorylation is dependent on MST1 cleavage.
FIGURE 5.

MST1-induced histone H2AX phosphorylation is dependent on its cleavage in HeLa cells. A, HeLa cells were transfected with the pCE-MST1-FL or pCE-MST1-NT plasmid. The caspase-3 inhibitor Z-DEVD-fmk (20 μm) was added to the medium immediately after transfection. At 36 h after transfection, cells were harvested, and lysates and histone proteins were prepared. The expression level of MST1-FL or MST1-NT was detected with anti-Myc-Tag, and the level of γ-H2AX expression was detected in the acid-extracted histones. B, HeLa cells growing on a slide chamber were transfected with the pCE-MST1-FL or pCE-MST1-NT plasmid (NT). The caspase-3 inhibitor Z-DEVD-fmk (20 μm) was added to the medium immediately after transfection. At 36 h after transfection, cells were fixed with paraformaldehyde, stained for Myc-Tag (MST1, red), γ-H2AX (green), and Hoechst 33342 (blue), and observed by confocal immunofluorescence microscopy. The bar graphs represent the percentage of apoptotic chromatin condensation in transfected cells from three independent experiments (±S.D.). At least 200 cells were counted each time. The asterisk indicates p < 0.05.
MST1 Is Required for DNA Fragmentation through Phosphorylation of Histone H2AX
Our previous results showed that histone H2AX is essential for DNA fragmentation during ultraviolet A-induced apoptosis (16). To study the potential role of MST1 in H2AX phosphorylation and DNA fragmentation during apoptosis, Jurkat cells were infected with shRNA expressing lentivirus targeting MST1, and stable MST1 knockdown cells were selected by puromycin. The shMock and shMST1 knockdown cells were then treated with etoposide, and histone H2AX phosphorylation and DNA fragmentation were assessed. Results showed that both histone H2AX phosphorylation and DNA fragmentation were suppressed in MST1 knockdown cells (Fig. 6A). These data suggested that MST1 is required for H2AX phosphorylation and DNA fragmentation during apoptosis. Ataxia telangiectasia mutated protein (ATM) is critical for DNA damage-induced H2AX phosphorylation; to test the role of ATM in apoptotic H2AX phosphorylation, we detected ATM and phosphorylated ATM (Ser-1981) in both shMock and shMST1 knockdown Jurkat cells. Results indicated that ATM can be cleaved in both shMock and shMST1 knockdown Jurkat cells (Fig. 6A), which agrees with previous results (20). However, phosphorylated ATM can still be detected, and no obvious difference in phosphorylation is observed between shMock and shMST1 knockdown cells. This suggests a role for MST1 in the decreased H2AX phosphorylation in MST1 knockdown cells. We further found that cleavage of caspase-3 was also inhibited in MST1 knockdown cells. Caspase-3 activity was confirmed using a caspase-3 activity assay kit. The data showed that caspase-3 activity was also decreased, which corresponded with the decreased caspase-3 cleavage (Fig. 6B). The inhibition of apoptosis in MST1 knockdown cells was also confirmed by flow cytometry analysis (Fig. 6C). Because MST1 has been suggested to be involved in the activation of JNKs, we determined the phosphorylation level of JNKs in MST1 knockdown cells. Results indicated that the phosphorylation of JNKs was only slightly suppressed. Thus, these results indicate that MST1 plays an important role in apoptosis, including its phosphorylation of histone H2AX, which is associated with caspase cleavage and DNA fragmentation.
FIGURE 6.

MST1 is required for histone H2AX phosphorylation and DNA fragmentation during apoptosis. A, mock and MST1 knockdown Jurkat transfectant cells (shMock and shMST1) were treated with 50 μm etoposide for 6 h. Each cell type was divided into three groups: one was used to extract histones to detect γ-H2AX and H2AX; one was prepared as a cell lysate to detect MST1, caspase-3, phosphorylated JNKs and total JNKs, and phosphorylated ATM and total ATM; and the other was used for the DNA ladder assay. B, mock and MST1 knockdown transfectant Jurkat cells were treated as in A. Cell lysates were prepared, and 50 μg were used for the caspase-3 activity assay. Ctrl, control; Etp, etoposide. C, mock and MST1 knockdown Jurkat cells (shMock and shMST1) were treated with 50 μm etoposide for 6 h. Cells were harvested and subjected to flow cytometry analysis after annexin V/propidium iodide staining to assess apoptosis. The percentage of early apoptosis is shown in the bar graphs. The means of three independent determinations ±S.D. are shown, and the asterisk indicates p < 0.05.
DISCUSSION
Apoptosis is an evolutionarily conserved process, which is essential for morphogenesis, development, differentiation, and homeostasis in multicellular organisms. Typical apoptosis has several characteristic morphological features, such as cell rounding, membrane blebbing, cytoskeletal disassembly, chromatin condensation, the formation of apoptotic bodies, and DNA fragmentation (21–25). Cascade activation of caspases plays a crucial role in the process of apoptosis. To date, almost 400 substrates for the apoptosis-associated caspases have been reported (26). Some of the substrates, such as poly(ADP-ribose) polymerase (PARP) (27), gelsolin (28), and inhibitor of caspase-activated DNase (ICAD) (29), are actually the executors of apoptotic death.
Several kinases have also been identified as substrates of caspases, including MST1 (3, 7), PAK2 (p21-activated kinase 2) (30), and PKC (31–33). Caspase-mediated cleavage occurs within a linker region between their catalytic domain and inhibitory domain, thereby separating the catalytic and inhibitory domains (34), which results in the activation of the kinase. Notably, ectopic expression of the catalytic fragment of these kinases is sufficient to cause apoptosis, whereas the expression of the catalytically inactive or cleavage-resistant mutant forms of the protein markedly attenuates or delays cell death that is triggered by genotoxic agents. The mechanism explaining the process by which the cleaved kinases participate in process of apoptosis is not well studied.
MST1 is a ubiquitously expressed serine/threonine kinase and has been reported to be a substrate of caspase-3. Caspase-mediated cleavage markedly increases the activity of the MST kinase by 10-fold and also causes translocation of the cleaved kinase domain to the nucleus (4, 7). Overexpression of the MST1 kinase domain can induce apoptotic chromatin condensation (3, 13). The only substrate that has been reported to be phosphorylated by cleaved MST1 is histone H2B (14). In HL-60 cells, after apoptotic stimulation (UV or etoposide), histone H2B can be phosphorylated at Ser-14, which is a unique histone modification associated with apoptotic chromatin. In the present study, strong histone H2B phosphorylation could be detected in HL-60 cells after etoposide treatment. However, obvious histone H2B phosphorylation in apoptotic Jurkat cells could not be detected (supplemental Fig. 2), indicating the existence of other MST1 substrates besides histone H2B.
Histone H2AX is a variant of the H2A protein family that differs from the other H2A histones by the presence of a unique COOH-terminal tail that contains a highly conserved SQEY motif with a serine residue at position 139 (35). Serine 139 becomes rapidly phosphorylated after the induction of DNA double strand breaks induced by ionizing radiation (36) or replication stress caused by UV or hydroxyurea (37, 38). The phosphorylated histone H2AX, referred to as γ-H2AX, is required for the recruitment of DNA repair proteins, such as Brca1, 53bp1, and Nbs1-Mre11-Rad50, to the site of DNA damage (39–41). Members of the phosphatidylinositol 3-kinase family, including ATM, ataxia telangiectasia and Rad3-related protein (ATR), and DNA-dependent protein kinase, reportedly are involved in the responses of mammalian cells to double strand breaks (42, 43).
Besides its association with double strand breaks, γ-H2AX has been found to play a role in apoptosis. Its appearance is concurrent with the initiation of DNA fragmentation, suggesting that γ-H2AX formation is an early chromatin modification following initiation of DNA fragmentation during apoptosis (15). The appearance of a high γ-H2AX level during apoptosis is referred to as apoptosis-associated γ-H2AX. An immunofluorescence study showed that the apoptosis-associated-H2AX is distinguished from the γ-H2AX activated during double strand break by: 1) its greater intensity; 2) the prevention of its appearance by a caspase inhibitor; and 3) the concurrent activation of caspase-3 in the same cells (44). Our previous work showed that γ-H2AX is essential for DNA fragmentation during ultraviolet A-induced apoptosis. Although we demonstrated that JNKs were important for H2AX phosphorylation, we found that in apoptotic Jurkat cells, JNK activation appears to match with the early H2AX phosphorylation but is decreasing while the H2AX phosphorylation is still increasing. Thus, other kinases that mediate strong H2AX phosphorylation during apoptosis need to be identified.
Because histone H2AX phosphorylation could be attenuated by a caspase inhibitor, kinases that can be activated by caspase cleavage are potential candidates for mediating histone H2AX phosphorylation during apoptosis. In a screening of possible kinases that might be responsible for the phos-phorylation of histone H2AX, we observed that MST1 strongly phosphorylated histone H2AX (Fig. 2A), and Ser-139 was identified as a major phosphorylation site (Fig. 2, B–D). During etoposide-induced apoptosis in Jurkat cells, we found that the cleavage of MST1 matches the appearance of strong histone H2AX phosphorylation (Fig. 1B). MST1 could bind and phosphorylate H2AX (Fig. 3B). In HeLa cells, the overexpression of MST1 induced the phosphorylation of endogenous histone H2AX. MST1-induced histone H2AX phos-phorylation was dependent on its cleavage and kinase activity (Figs. 4, A and D, and 5, A and B). Furthermore, in MST1 knockdown Jurkat cells, H2AX phosphorylation, caspase-3 cleavage, and DNA fragmentation were inhibited after etoposide treatment, indicating that MST1 is responsible for the strong H2AX phosphorylation, which is essential for DNA fragmentation (Fig. 6, A and B).
Based on these results, we suggest the following model (Fig. 7). (i) With apoptotic stimuli, such as etoposide, the cleavage of caspase-3 mediates the cleavage of MST1 and promotes its activation; (ii) the cleaved MST1 N-terminal kinase domain then translocates into the nucleus and mediates the phos-phorylation of histone H2AX; and (iii) phosphorylated histone H2AX then recruits caspase-activated DNase to mediate DNA fragmentation.
FIGURE 7.
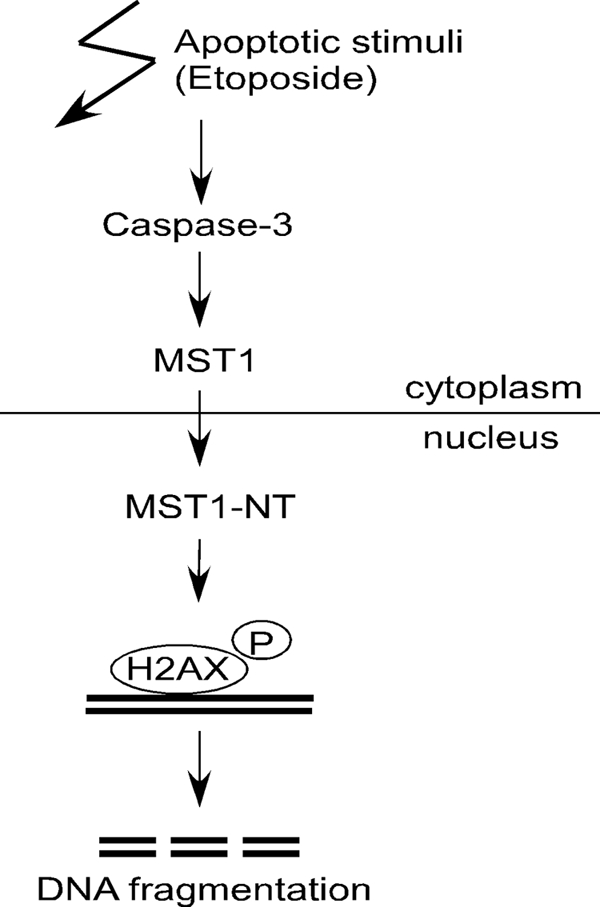
Model of the proposed function of MST1 in histone H2AX phosphorylation and DNA fragmentation during apoptosis. Once caspase-3 is activated during etoposide-induced apoptosis, MST1 will be cleaved and activated. The cleaved N-terminal kinase domain translocates into the nucleus and mediates the phosphorylation (P) of histone H2AX, and the phosphorylated H2AX then recruits caspase-activated DNase to mediate DNA fragmentation.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants CA077646, CA027502, CA111536, CA120388, R37CA081064, and ES017548 (to Z. D.) and by The Hormel Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
- MST1
- mammalian STE20-like kinase 1
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- ATM
- ataxia telangiectasia-mutated.
REFERENCES
- 1.Creasy C. L., Chernoff J. (1995) Gene 167, 303–306 [DOI] [PubMed] [Google Scholar]
- 2.Taylor L. K., Wang H. C., Erikson R. L. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10099–10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graves J. D., Gotoh Y., Draves K. E., Ambrose D., Han D. K., Wright M., Chernoff J., Clark E. A., Krebs E. G. (1998) EMBO J. 17, 2224–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creasy C. L., Ambrose D. M., Chernoff J. (1996) J. Biol. Chem. 271, 21049–21053 [DOI] [PubMed] [Google Scholar]
- 5.Graves J. D., Draves K. E., Gotoh Y., Krebs E. G., Clark E. A. (2001) J. Biol. Chem. 276, 14909–14915 [DOI] [PubMed] [Google Scholar]
- 6.Kakeya H., Onose R., Osada H. (1998) Cancer Res. 58, 4888–4894 [PubMed] [Google Scholar]
- 7.Lee K. K., Murakawa M., Nishida E., Tsubuki S., Kawashima S., Sakamaki K., Yonehara S. (1998) Oncogene 16, 3029–3037 [DOI] [PubMed] [Google Scholar]
- 8.Lu M. L., Sato M., Cao B., Richie J. P. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 8977–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reszka A. A., Halasy-Nagy J. M., Masarachia P. J., Rodan G. A. (1999) J. Biol. Chem. 274, 34967–34973 [DOI] [PubMed] [Google Scholar]
- 10.Watabe M., Kakeya H., Onose R., Osada H. (2000) J. Biol. Chem. 275, 8766–8771 [DOI] [PubMed] [Google Scholar]
- 11.Watabe M., Kakeya H., Osada H. (1999) Oncogene 18, 5211–5220 [DOI] [PubMed] [Google Scholar]
- 12.Ura S., Masuyama N., Graves J. D., Gotoh Y. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 10148–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ura S., Nishina H., Gotoh Y., Katada T. (2007) Mol. Cell Biol. 27, 5514–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung W. L., Ajiro K., Samejima K., Kloc M., Cheung P., Mizzen C. A., Beeser A., Etkin L. D., Chernoff J., Earnshaw W. C., Allis C. D. (2003) Cell 113, 507–517 [DOI] [PubMed] [Google Scholar]
- 15.Rogakou E. P., Nieves-Neira W., Boon C., Pommier Y., Bonner W. M. (2000) J. Biol. Chem. 275, 9390–9395 [DOI] [PubMed] [Google Scholar]
- 16.Lu C., Zhu F., Cho Y. Y., Tang F., Zykova T., Ma W. Y., Bode A. M., Dong Z. (2006) Mol. Cell 23, 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solier S., Pommier Y. (2009) Cell Cycle 8, 1853–1859 [DOI] [PubMed] [Google Scholar]
- 18.Jenuwein T., Allis C. D. (2001) Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 19.Shechter D., Dormann H. L., Allis C. D., Hake S. B. (2007) Nat. Protoc. 2, 1445–1457 [DOI] [PubMed] [Google Scholar]
- 20.Smith G. C., d'Adda di Fagagna F., Lakin N. D., Jackson S. P. (1999) Mol. Cell Biol. 19, 6076–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan J., Shaham S., Ledoux S., Ellis H. M., Horvitz H. R. (1993) Cell 75, 641–652 [DOI] [PubMed] [Google Scholar]
- 22.Wyllie A. H. (1980) Nature 284, 555–556 [DOI] [PubMed] [Google Scholar]
- 23.Vaux D. L., Korsmeyer S. J. (1999) Cell 96, 245–254 [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G., Galluzzi L., Vandenabeele P., Abrams J., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., El-Deiry W. S., Golstein P., Green D. R., Hengartner M., Knight R. A., Kumar S., Lipton S. A., Malorni W., Nuñez G., Peter M. E., Tschopp J., Yuan J., Piacentini M., Zhivotovsky B., Melino G. (2009) Cell Death Differ. 16, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerr J. F., Wyllie A. H., Currie A. R. (1972) Br. J. Cancer 26, 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lüthi A. U., Martin S. J. (2007) Cell Death Differ. 14, 641–650 [DOI] [PubMed] [Google Scholar]
- 27.Lazebnik Y. A., Kaufmann S. H., Desnoyers S., Poirier G. G., Earnshaw W. C. (1994) Nature 371, 346–347 [DOI] [PubMed] [Google Scholar]
- 28.Kothakota S., Azuma T., Reinhard C., Klippel A., Tang J., Chu K., McGarry T. J., Kirschner M. W., Koths K., Kwiatkowski D. J., Williams L. T. (1997) Science 278, 294–298 [DOI] [PubMed] [Google Scholar]
- 29.Wolf B. B., Schuler M., Echeverri F., Green D. R. (1999) J. Biol. Chem. 274, 30651–30656 [DOI] [PubMed] [Google Scholar]
- 30.Rudel T., Bokoch G. M. (1997) Science 276, 1571–1574 [DOI] [PubMed] [Google Scholar]
- 31.Emoto Y., Manome Y., Meinhardt G., Kisaki H., Kharbanda S., Robertson M., Ghayur T., Wong W. W., Kamen R., Weichselbaum R. (1995) EMBO J. 14, 6148–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeVries T. A., Neville M. C., Reyland M. E. (2002) EMBO J. 21, 6050–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghayur T., Hugunin M., Talanian R. V., Ratnofsky S., Quinlan C., Emoto Y., Pandey P., Datta R., Huang Y., Kharbanda S., Allen H., Kamen R., Wong W., Kufe D. (1996) J. Exp. Med. 184, 2399–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dix M. M., Simon G. M., Cravatt B. F. (2008) Cell 134, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redon C., Pilch D., Rogakou E., Sedelnikova O., Newrock K., Bonner W. (2002) Curr. Opin. Genet. Dev. 12, 162–169 [DOI] [PubMed] [Google Scholar]
- 36.Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M. (1998) J. Biol. Chem. 273, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 37.Hanasoge S., Ljungman M. (2007) Carcinogenesis 28, 2298–2304 [DOI] [PubMed] [Google Scholar]
- 38.Ward I. M., Chen J. (2001) J. Biol. Chem. 276, 47759–47762 [DOI] [PubMed] [Google Scholar]
- 39.Sedelnikova O. A., Pilch D. R., Redon C., Bonner W. M. (2003) Cancer Biol. Ther. 2, 233–235 [DOI] [PubMed] [Google Scholar]
- 40.Celeste A., Fernandez-Capetillo O., Kruhlak M. J., Pilch D. R., Staudt D. W., Lee A., Bonner R. F., Bonner W. M., Nussenzweig A. (2003) Nat Cell Biol. 5, 675–679 [DOI] [PubMed] [Google Scholar]
- 41.Ward I. M., Minn K., Jorda K. G., Chen J. (2003) J. Biol. Chem. 278, 19579–19582 [DOI] [PubMed] [Google Scholar]
- 42.Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J. (2001) J. Biol. Chem. 276, 42462–42467 [DOI] [PubMed] [Google Scholar]
- 43.Park E. J., Chan D. W., Park J. H., Oettinger M. A., Kwon J. (2003) Nucleic Acids Res. 31, 6819–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang X., Okafuji M., Traganos F., Luther E., Holden E., Darzynkiewicz Z. (2004) Cytometry A 58, 99–110 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.