

SUMMARY
LpxC is an essential enzyme in the lipid A biosynthetic pathway in Gram-negative bacteria. Several promising antimicrobial lead compounds targeting LpxC have been reported, though they typically display a large variation in potency against different Gram-negative pathogens. We report that inhibitors with a diacetylene scaffold effectively overcome the resistance caused by sequence variation in the LpxC substrate-binding passage. Compound binding is captured in complex with representative LpxC orthologs, and structural analysis reveals large conformational differences that mostly reflect inherent molecular features of distinct LpxC orthologs, whereas ligand-induced structural adaptations occur at a smaller scale. These observations highlight the need for a molecular understanding of inherent structural features and conformational plasticity of LpxC enzymes for optimizing LpxC inhibitors as broad-spectrum antibiotics against Gram-negative infections.
Keywords: LpxC, Lipid A, Antibiotic, Protein-inhibitor complex
INTRODUCTION
Development of novel antibiotics against multidrug-resistant Gram-negative infections by targeting the UDP-3-O-(acyl)-N-acetylglucosamine deacetylase (LpxC) in the lipid A biosynthetic pathway has been a major research focus for industrial groups and academic laboratories in the last two decades (Barb and Zhou, 2008; Raetz, 1998; Raetz, et al., 2007). Lipid A (endotoxin) is a glucosamine-based saccharolipid that serves as a hydrophobic membrane anchor of lipopolysaccharide (LPS). It is the major lipid component of the outer leaflet of the outer membrane of most Gram-negative bacteria and shields bacterial cells from damage by external agents, such as antibiotics and detergents. Lipid A biosynthesis is an essential pathway in virtually all Gram-negative bacteria, and it starts with the LpxA-catalyzed acylation of UDP-N-acetylglucosamine (UDP-GlcNAc, Figure 1A). With the exception of Gram-negative organisms that synthesize lipid A from the analog UDP-2-acetamido-3-amino-2,3-dideoxy-α-D-glucopyranose (UDP-GlcNAc3N) (Raetz, et al., 2007), the acylation reaction is thermodynamically unfavorable. Therefore, the second reaction in the lipid A pathway, the deacetylation of UDP-3-O-(acyl)-N-acetylglucosamine catalyzed by LpxC, is generally considered as the committed step of lipid A biosynthesis. Following deacetylation, seven additional enzymes are needed to synthesize Kdo2-lipid A, the active component of endotoxin and the LPS substructure sufficient to maintain bacterial viability.
Figure 1. Potent LpxC Inhibitors Block the Committed Step of Lipid A Biosynthesis.
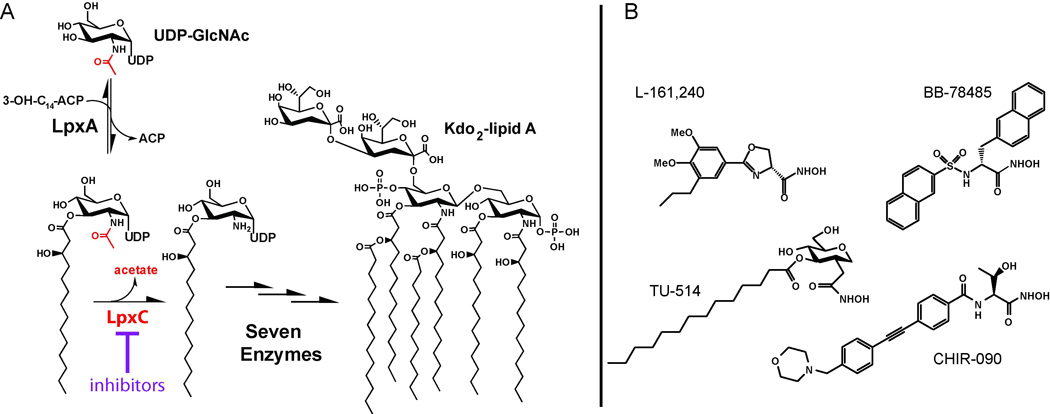
(A) LpxC is an essential enzyme that catalyzes the first irreversible step of the lipid A biosynthetic pathway. (B) Representative LpxC inhibitors.
The lpxC gene is essential and conserved in virtually all Gram-negative organisms. Structural studies of LpxC have revealed a unique protein fold, indicating that highly specific LpxC inhibitors can be developed as novel antibiotics (Barb, et al., 2007; Buetow, et al., 2006; Coggins, et al., 2003; Coggins, et al., 2005; Gennadios and Christianson, 2006; Gennadios, et al., 2006; Hernick, et al., 2005; Mochalkin, et al., 2008; Shin, et al., 2007; Whittington, et al., 2003). Consistent with this notion, several well-characterized LpxC inhibitors (Figure 1B) have been reported to display various degrees of antibiotic activity against Gram-negative bacteria, most notably Escherichia coli (Barb and Zhou, 2008; Raetz, et al., 2007). Very recently, a large number of LpxC inhibitors with vastly different chemical scaffolds have appeared in literature (Kline, et al., 2002; Pirrung, et al., 2003) and in patent applications (Anderson, et al., 2004; Dobler, et al., 2010; Mansoor, et al., 2008; Mansoor, et al., 2010; Moser, et al., 2008; Raju, et al., 2010; Siddiqui, et al., 2007; Takashima, et al., 2010; Yoshinaga, et al., 2008); however, the potency and spectrum of inhibition of these compounds have yet to be systematically investigated.
Among the well-characterized compounds, CHIR-090 is the best LpxC inhibitor reported to date, killing both E. coli and Pseudomonas aeruginosa in bacterial disk diffusion assays with an efficacy rivaling that of ciprofloxacin (McClerren, et al., 2005). Surprisingly, CHIR-090 is ~600-fold less effective against LpxC orthologs from the Rhizobium family than against E. coli LpxC (EcLpxC) (Barb, et al., 2007), raising concerns of rapid evolution of antibiotic resistance for CHIR-090-sensitive strains through point mutations (Barb, et al., 2007). Through structural and biochemical studies of the Aquifex aeolicus LpxC (AaLpxC)/CHIR-090 complex, we have revealed the molecular basis of the intrinsic resistance of Rhizobium leguminosarum LpxC (RlLpxC) to CHIR-090 (Barb, et al., 2007) (Figure 2A). These studies showed that CHIR-090 occupies the hydrophobic substrate-binding passage consisting of the Insert II region of Domain II in LpxC. The diphenyl-acetylene group of CHIR-090 penetrates through this hydrophobic substrate-binding passage, with the first phenyl group (proximal to the hydroxamate group) located close to the active site and next to the entrance of the hydrophobic passage, the acetylene group threading through the narrowest part of the passage, and the second phenyl ring (distal to the hydroxamate group) emerging from the passage. The exit of the substrate-binding passage contains a critical glycine residue that is conserved in LpxC orthologs sensitive to CHIR-090 inhibition. In Rhizobium LpxC, however, this critical glycine residue is replaced by a serine residue, which narrows the exit of the substrate-binding passage and decreases its susceptibility to CHIR-090 inhibition by generating van der Waals clashes with the distal phenyl ring of CHIR-090 (Figure 2A). Consistent with this notion, a single Ser-to-Gly mutation that broadens the exit to the substrate-binding passage renders RlLpxC 100-fold more sensitive to CHIR-090 inhibition, whereas an EcLpxC mutant with a narrower passage is more resistant to CHIR-090 inhibition compared to the wild-type enzyme (Barb, et al., 2007). The knowledge that crucial residues in the hydrophobic substrate-binding passage of CHIR-090-resistant LpxC orthologs can cause van der Waals clashes with the distal aromatic ring of CHIR-090 motivated us to evaluate novel inhibitors based on a narrower scaffold for their ability to overcome this resistance mechanism.
Figure 2. Inhibitors Based on the Diacetylene Scaffold Overcome the Resistance Mechanism Represented by RlLpxC and Display Enhanced Antibiotic Activity against E. coli and P. aeruginosa.
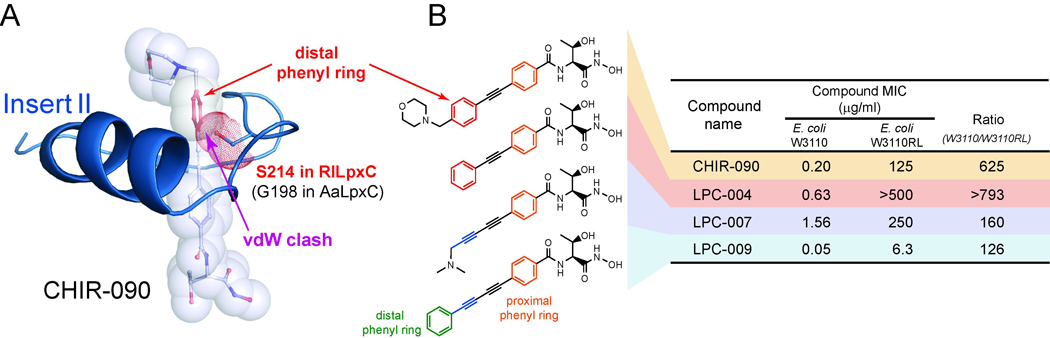
(A) Antibiotic resistance generated by steric clashes between S214 in RlLpxC and the distal ring of CHIR-090. CHIR-090 and the S214 side-chain are shown in stick and transparent-sphere models. The distal phenyl ring of CHIR-090 and the side-chain of S214 in RlLpxC are highlighted in red. The Insert II conformation was generated using homology modeling based on the structure of the AaLpxC/CHIR-090 complex (PDB entry: 2JT2), which contains a Gly (G198) at the corresponding position of S214 in RlLpxC. (B) The MICs of each compound against strains of wild-type E. coli (W3110) and E. coli with its genomic lpxC gene replaced by that of R. leguminosarum (W3110RL). The narrow diacetylene scaffold not only overcomes the CHIR-090 resistance of RlLpxC, but also displays enhanced antibiotic activity against E. coli.
Here, we report the biochemical and structural characterization of compounds based on the linear diacetylene scaffold. These compounds effectively diminish the resistance of RlLpxC. Excitingly, the best compound, LPC-009, also shows a general enhancement of potency (2- to 64-fold) over CHIR-090 against a variety of clinically important Gram-negative pathogens, including E. coli, P. aeruginosa, Salmonella typhimurium, Klebsiella pneumonia, Vibrio cholera, Bordetella bronchiseptica, Burkholderia cepacia, Burkholderia cenocepacia, and Burkholderia dolosa. Furthermore, we have captured the binding mode of LPC-009 in complex with LpxC enzymes from E. coli, A. aeolicus, and P. aeruginosa. Structural comparison of diverse LpxC enzymes in complex with the same LPC-009 inhibitor reveals large, inherent conformational variations of individual LpxC orthologs and unexpected inhibitor flexibility. The binding of distinct inhibitors to the same LpxC enzyme also induces noticeable conformational changes, but at a smaller scale. Thus, understanding the contribution of inherent molecular features of representative LpxC enzymes and their associated conformational dynamics is essential for the design of broad-spectrum LpxC-targeting antibiotics.
RESULTS
Compounds Based on the Diacetylene Scaffold Potently Inhibit a Wide Range of LpxC orthologs
Because point mutations that narrow the LpxC substrate-binding passage can be a source of antibiotic resistance to CHIR-090, we reasoned that compounds with a narrower chemical scaffold—the diacetylene group—would be able to overcome the CHIR-090 resistance represented by RlLpxC. The diacetylene scaffold was among the many chemical structures initially discussed in the international patent WO 2004/062601 A2 (Anderson, et al., 2004), but its effect on the antibiotic profile was not quantified. To facilitate the analysis, we constructed a RlLpxC knock-in strain of E. coli by replacing the genomic lpxC gene with that of R. leguminosarum (W3110RL) (Barb, et al., 2007). Because the W3110RL strain is identical to the wild-type E. coli strain W3110 except for the lpxC gene, any difference in the minimum inhibitory concentration (MIC) values should directly reflect the different KI values of a compound against LpxC enzymes, but not other factors such as membrane permeability. As expected, the MIC values of CHIR-090 are vastly different for these two bacterial strains, and a ~600-fold variation in MIC values correlates quite well with the KI differences measured by enzyme kinetic studies (Barb, et al., 2007). LPC-004, which was identical to CHIR-090 except that the morpholine moiety was removed, was still significantly less effective (~800 fold) for RlLpxC than for EcLpxC (Figure 2B), confirming that the bulky distal phenyl group, but not the morpholine group, is responsible for its decreased activity against RlLpxC. Replacing the phenyl-acetylene group with a (N,N-dimethylamino)methyl-diacetylene group generated a weaker inhibitor (LPC-007) for E. coli, but the ratio of the MIC values for W3110 and W3110RL started to diminish (160-fold). Finally, addition of a phenyl ring to the diacetylene group (LPC-009) further reduced the difference of the MIC values between the two bacterial strains to ~126-fold (Figure 2B), suggesting that the narrow diacetylene group can effectively reduce the intrinsic resistance of RlLpxC to CHIR-090. Excitingly, LPC-009 also appears to be 4-fold more potent than CHIR-090 for inhibiting the growth of E. coli in MIC assays (Figure 2B). In order to establish that the diacetylene-based compound LPC-009 indeed displays a superior antibiotic profile over CHIR-090, we measured the MICs of LPC-009 and CHIR-090 against a panel of Gram-negative human pathogens, including P. aeruginosa, Salmonella typhimurium, Klebsiella pneumonia, Vibrio cholera, Bordetella bronchiseptica, Burkholderia cepacia, Burkholderia cenocepacia, and Burkholderia dolosa. Invariably, LPC-009 was determined to be a more potent compound than CHIR-090, showing a 2- to 64-fold enhancement of antibiotic activity (Table 1).
Table 1.
MIC Values
| MIC (µg/ml) | ||
|---|---|---|
| CHIR-090 | LPC-009 | |
|
Pseudomonas Aeruginosa PAO1 |
1.6 | 0.74 |
|
Salmonella Typhimurium LT2 |
0.16 | 0.024 |
|
Klebsiella Pneumoniae 43816 |
0.64 | 0.10 |
|
Vibrio Cholerae P4 (P27459ΔctxAB::KmR, SmR) |
0.16 | 0.010 |
|
Bordetella bronchiseptica RB50 |
16 | 2.0 |
|
Burkholderia cepacia ATCC 25146 |
>32 | 12 |
|
Burkholderia cenocepacia GIIIa J2315 lineage ET12 |
>32 | 32 |
|
Burkholderia dolosa AU0158 lineage 5LC6 |
8.0 | 0.125 |
To further evaluate the inhibition of EcLpxC by LPC-009, we performed detailed enzymatic assays. A KIapp of 0.55 ± 0.09 nM and a corresponding KI value of 0.18 ± 0.03 nM were calculated based on the assumption of competitive inhibition and a measured KM of 2.5 ± 0.2 µM for EcLpxC (see Experimental Procedures for details). Interestingly, we observed a similar fractional inhibition of product accumulation with or without inhibitor pre-incubation (1 hr) with enzyme prior to initiating the reaction, suggesting that unlike the slow tight-binding inhibitor CHIR-090, LPC-009 does not appear to inhibit EcLpxC in a time-dependent fashion.
Overall Structure of EcLpxC in Complex with LPC-009
To probe the molecular details of the LpxC/LPC-009 interaction, we determined the structure of EcLpxC in complex with LPC-009 at 1.65 Å resolution (crystallographic statistics shown in Table 2). EcLpxC is the most divergent ortholog to structurally characterized LpxC orthologs, such as AaLpxC (34% sequence identity) and P. aeruginosa LpxC (PaLpxC, 57% sequence identity). It is identical to Shigella sonnei LpxC and highly similar to Salmonella typhimurium LpxC and Yersinia pestis LpxC (> 92% sequence identity). Therefore, E. coli LpxC is an excellent model enzyme for understanding the behavior of LpxC enzymes from these clinically important Gram-negative pathogens.
Table 2.
Data Collection and Refinement Statistics
| EcLpxC/LPC-009 | AaLpxC/LPC-009 | PaLpxC/LPC-009 | |
|---|---|---|---|
| Space group | P 61 | P 61 | P 212121 |
| Cell dimensions | |||
| a, b, c (Å) | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 |
| α, β, γ (°) | 106.7, 106.7, 52.7 | 65.6, 65.6, 132.3 | 52.6, 73.6, 88.3 |
| Reflections (unique/total) | 41012 / 348669 | 88659 / 662791 | 88611 / 521390 |
| Resolution range (Å) | 26.60-1.65 (1.71-1.65) a | 23.88-1.25 (1.30-1.25) | 22.99-1.28 (1.33-1.28) |
| Completeness (%) | 99.6 (96.6) | 100 (100) | 99.7 (98.0) |
| I/σ | 33.0 (3.3) | 23.0 (3.7) | 20.8 (2.25) |
| R-merge (%) | 7.4 (49.7) | 12.1 (47.1) | 11.5 (45.6) |
| No. of atoms b | |||
| Protein | 2378 | 2230 | 2432 |
| LPC-009 | 27 | 27 | 27 |
| Water | 310 | 455 | 413 |
| Other molecules | 48 | 12 | 103 |
| R-factor (%) | 17.2 | 15.4 | 18.1 |
| R-free (%) | 20.2 | 16.1 | 19.2 |
| Av. B-factor (Å2) c | |||
| Protein | 35.87 | 14.52 | 18.00 |
| LPC-009 | 26.53 | 19.31 | 16.68 |
| Catalytic zinc | 19.15 | 11.55 | 8.75 |
| Water | 45.57 | 31.79 | 34.19 |
| Rmsd from ideal geometry | |||
| Bond lengths (Å) | 0.008 | 0.004 | 0.009 |
| Bond angles (°) | 0.924 | 0.929 | 0.880 |
| Ramachandran plot | |||
| Favored (%) | 97.32 | 97.04 | 98.25 |
| Allowed (%) | 100.00 | 100.00 | 100.00 |
| MolProbity | |||
| All-atom clashscore | 2.51 | 1.57 | 1.69 |
| Clashscore percentile d | 99th | 99th | 98th |
Values in parentheses are for highest-resolution shell.
Non-hydrogen atoms. The deposited structures include riding hydrogen atoms used in refinement.
Hydrogen atoms are exclude.
100th percentile is the best among structures of comparable resolution; 0th percentile is the worst
EcLpxC consists of two domains with a β-α-α-β sandwich fold similar to that of AaLpxC or PaLpxC (Figure 3A) (Barb, et al., 2007; Buetow, et al., 2006; Coggins, et al., 2003; Coggins, et al., 2005; Gennadios and Christianson, 2006; Gennadios, et al., 2006; Hernick, et al., 2005; Mochalkin, et al., 2008; Shin, et al., 2007; Whittington, et al., 2003). Each domain contains a layer of helices packing against a layer of five-stranded β-sheet with the helices “sandwiched” between the two main β-sheets. Additionally, each domain contains a unique insert region (Insert I of Domain I and Insert II of Domain II) that adopts a distinct structure. Insert I forms a small three-stranded β-sheet that partially defines the boundary of the active site, whereas Insert II adopts a β–α–β structure that forms a topologically closed hydrophobic passage to harbor substrate or inhibitors.
Figure 3. Structure of the EcLpxC/LPC-009 Complex.
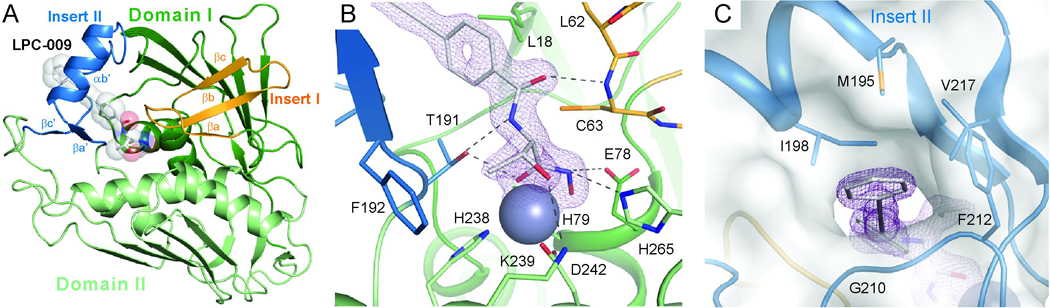
(A) Overall structure of the complex. EcLpxC is shown in the ribbon diagram and LPC-009 in the space-filling model. (B) Interactions between LPC-009 and LpxC in the active site. (C) External view of the hydrophobic substrate-binding passage. Insert I and Insert II are highlighted in orange and blue, respectively. LPC-009 and selected active site residues are shown in the stick model. The active site zinc ion is shown in the space-filling model. Purple mesh represents 2Fo-Fc map density (contoured at 1.6σ) surrounding the LPC-009. The surface plot in panel (C) is generated by PYMOL (DeLano Scientific LLC).
Consistent with our structure-based prediction, the overall binding mode of LPC-009 is similar to that of CHIR-090, with its threonyl-hydroxamate group occupying the active site and its diacetylene group penetrating through the hydrophobic substrate-binding passage (Figure 3A). In the active site, the hydroxamate group chelates the catalytic zinc ion with a square-pyramidal geometry (Figure 3B) defined by its two oxygen atoms and three LpxC residues, H79, H238 and D242. The hydroxamate group is oriented perpendicular to the β-strands of Insert II, and its position is held by hydrogen bonds with the T191 hydroxyl group of Insert II on one side of the active site and the side-chains of the catalytically important residues E78 and H265 on the opposite side. In addition to these hydroxamate-mediated interactions, the threonyl moiety of LPC-009 forms extensive interactions with highly conserved residues in the active site, including (1) van der Waals contacts between its methyl group and the aromatic ring of F192, which lies at the base of the active site and (2) hydrogen bonds between its amide and hydroxyl groups and the side-chains of T191 and K239, respectively. In addition to these highly conserved interactions, a unique hydrogen bond is observed between the carbonyl of the carboxyl-amide group of LPC-009 and the backbone amide of C63 of EcLpxC.
At the entrance of the hydrophobic substrate-binding passage, the proximal phenyl ring of LPC-009 faces the methyl groups of L18, and the narrow diacetylene group of LPC-009 penetrates through and emerges from the hydrophobic passage. Although LPC-009 also contains a distal phenyl ring similar to CHIR-090, it is located further away from the exit and is less likely to lead to the possible emergence of antibiotic resistance through mutation of residues at the exit of the hydrophobic passage that would clash with the inhibitor. Instead, this distal phenyl ring of LPC-009 is held by favorable van der Waals contacts with a hydrophobic “clamp” consisting of residues outside the substrate-binding passage, including I198, M195, F212 and V217 (Figure 3C).
Inherent Conformational Differences of LpxC Orthologs
A number of AaLpxC structures in complex with different small molecules have been reported, revealing an essentially identical enzyme conformation regardless of the bound inhibitor (Barb, et al., 2007; Buetow, et al., 2006; Coggins, et al., 2003; Coggins, et al., 2005; Gennadios and Christianson, 2006; Gennadios, et al., 2006; Hernick, et al., 2005; Shin, et al., 2007; Whittington, et al., 2003). However, a distinct orientation of the Insert II helix has been observed in PaLpxC in complex with BB-78485 (Mochalkin, et al., 2008). Compared to AaLpxC, the PaLpxC Insert II helix is rotated ~16° around a pivot point near the N-terminus of the helix, generating a movement of ~7 Å at the C-terminal end. It was proposed that binding to the bulky naphthalene ring of BB-78485 is responsible for this large conformational difference (Mochalkin, et al., 2008). However, the comparison used different LpxC orthologs bound to distinct inhibitors, and thus it is not clear whether the observed structural variation reflects conformational plasticity in response to inhibitor binding or species-specific differences in the structures of LpxC orthologs, or a combination of both. The discovery of LPC-009 as a potent broad-spectrum inhibitor has allowed us to resolve this ambiguity by determining and comparing high-resolution structures of two additional LpxC orthologs, AaLpxC and PaLpxC, in complex with LPC-009 (crystallographic statistics shown in Table 2). To avoid masking local conformational changes, we superimposed the AaLpxC/LPC-009 and PaLpxC/LPC-009 complexes onto the EcLpxC/LPC-009 complex using only the backbone Cα atoms of the two main β-sheets and four helices that are highly conserved in sequence and structure across LpxC orthologs (Figure 4A).
Figure 4. Species-specific Conformational Differences of LpxC Orthologs.
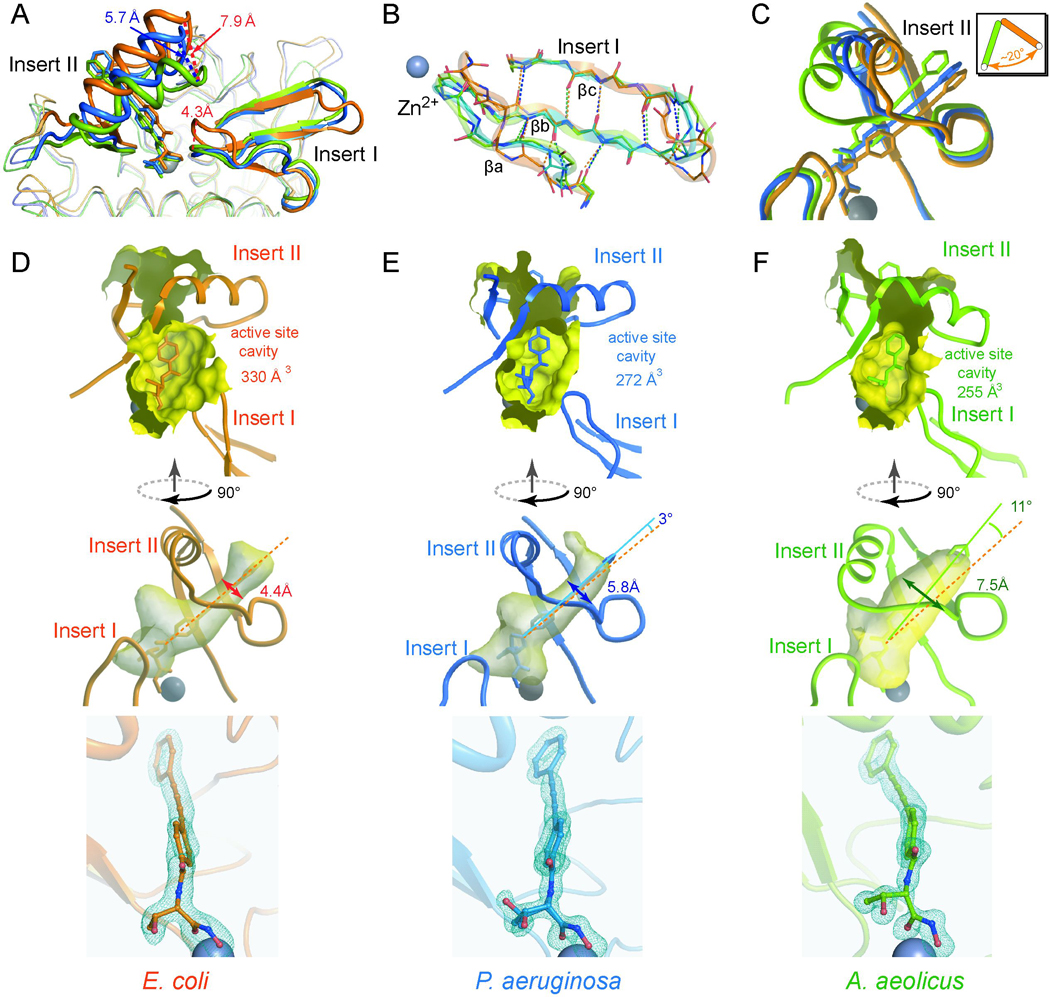
(A) Superimposition of EcLpxC/LPC-009 (orange), PaLpxC/LPC-009 (blue) and AaLpxC/LPC-009 (green). The bound LPC-009 is shown in the stick model, and the catalytic zinc ion in the sphere model. Close-up views around the Insert I and Insert II regions are shown in panels (B) and (C) respectively. The effects of the inherent conformational differences on the size of the active site cavity and the shape and orientation of the hydrophobic substrate-binding passage, and the bound compound are shown for LpxC enzymes from E. coli, P. aeruginosa and A. aeolicus in panels (D), (E), and (F) respectively. Fo-Fc omit map density contoured at 3.4σ (blue mesh) is shown around LPC-009 in the bottom panels, illustrating different curvatures of the diacetylene group of LPC-009 bound to distinct LpxC orthologs. Two conformations of the threonyl group of LPC-009 were used to fit the electron density in the PaLpxC complex, panel (E).
Insert I and Active Site
There are sizeable structural differences in the Insert I region that partially defines the active site boundary (Figure 4B). All three LpxC orthologs have Insert I regions with similar lengths and topologies; however, the three-stranded β-sheets contain β-bulges at different locations. In EcLpxC, the β-bulge is located after the β-turn connecting the second and third β-strands (βb-βc), whereas in PaLpxC and AaLpxC, the β-bulge is part of the first strand (βa). Consequently, the βa-strand in EcLpxC forms the typical inter-strand hydrogen bonds of a flat antiparallel β-sheet, while the βa-strand in PaLpxC and AaLpxC contains an extra residue that loops out of the plane of the β-sheet and is not involved in the main-chain hydrogen bonding network. The EcLpxC loop connecting βa- βb is one residue longer, pushing it away from the active site ~4.3 Å compared to the loop positions of AaLpxC and PaLpxC (Figure 4A,B) and creating a more spacious active site, with a cavity volume of 330 Å 3, whereas the active sites in PaLpxC and AaLpxC are significantly smaller, with cavity volumes of 272 Å 3 and 255 Å 3, respectively (Figure 4D,E,F).
Insert II Substrate-binding Passage
Much to our surprise, there are large differences in the orientation of the Insert II helix in all three LpxC enzymes (EcLpxC, AaLpxC and PaLpxC) despite being bound to the same LPC-009 inhibitor (Figure 4A,C). In AaLpxC, the Insert II helix is positioned closest to the active site, which generates the widest hydrophobic passage of all three LpxC orthologs with a diameter of ~7.5 Å. In contrast, the Insert II helix in both EcLpxC and PaLpxC is rotated ~20° around the N-terminal end of the helix (Figure 4C), moving away from the active site and resulting in a narrower substrate passage with a diameter of 4.4 Å for EcLpxC and 5.8 Å for PaLpxC (Figure 4D,E). The largest difference in the Insert II helix orientation occurs between EcLpxC and AaLpxC, with the position of the C-terminal end of the helix deviating by nearly 7.9 Å. This is followed by ~5.7 Å difference between PaLpxC and AaLpxC, and ~2 Å between EcLpxC and PaLpxC (Figure 4A).
In response to the movement of the Insert II helix, LPC-009 also rotates around the threonyl-hydroxamate head group in a counter clockwise fashion at an angle of ~11° in AaLpxC and ~3° in PaLpxC compared to its position in EcLpxC (Figure 4D,E,F). Interestingly, in contrast to its apparently “rigid” chemical structure, the diacetylene group is slightly curved, a distortion that has only been previously reported in chemically constrained diacetylene compounds (Baldwin, et al., 1998). The curvature of the diphenyl-diacetylene group is most prominent in the AaLpxC complex, followed by the EcLpxC and PaLpxC complexes. This unexpected observation suggests that there is not a huge energetic penalty for distorting the diacetylene group to allow for adaptation to the different curvatures of the hydrophobic passage in individual LpxC orthologs.
It is important to note that the conformational difference between AaLpxC/LPC-009 and PaLpxC/LPC-009 is on a similar scale as the previously reported structural discrepancy between AaLpxC/TU-514 and PaLpxC/BB-78485 (Mochalkin, et al., 2008), suggesting that the structural divergence in these latter complexes is largely driven by an inherent, species-specific conformational variation, rather than by ligand-binding.
Ligand-induced Conformations of LpxC
Our observation of LpxC conformational differences across orthologs does not rule out the possibility that the enzyme is able to respond structurally to the binding of distinct inhibitors. To evaluate the contribution of conformational plasticity to inhibitor binding, we compared structures of the same LpxC ortholog in complex with different inhibitors. Interestingly, superimposition of AaLpxC/LPC-009 with existing AaLpxC structures shows very limited conformational variation. On the other hand, comparison of the PaLpxC/LPC-009 and PaLpxC/BB-78485 (PDB entry: 2VES) structures reveals noticeable conformational changes in the active site and hydrophobic substrate-binding passage (Figure 5). These differences are particularly pronounced in the hydrophobic passage—to accommodate the two bulky and rigid naphthalene groups of BB-78485, the Insert II helix and its supporting loop (β1-β2 loop) are pushed ~2.0–2.4 Å away from the active site compared to the corresponding structural elements in the PaLpxC/LPC-009 complex. This positions the Insert II helix closer to the loop immediately following it, which expands the active site and closes off the hydrophobic passage in the PaLpxC/BB-78485 complex (Figure 5).
Figure 5. Inhibitor-induced Structural Plasticity.
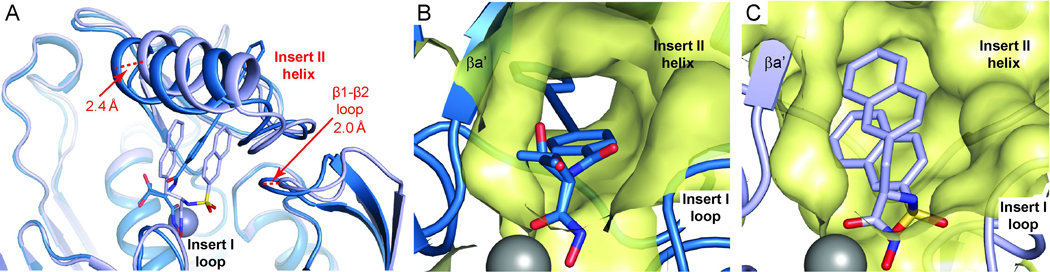
(A) Binding of LPC-009 and BB-78485 to PaLpxC causes noticeable conformational changes in the Insert I and Insert II regions that generate an open substrate-binding passage accommodating LPC-009 (panel B), but a closed conformation for BB-78485 (panel C). Complexes of PaLpxC/LPC-009 and PaLpxC/BB-78485 are shown in sky blue and pale blue, respectively. Surfaces of the hydrophobic substrate-binding passage are colored in yellow. Two conformations of the threonyl group of LPC-009 were observed in the PaLpxC complex.
DISCUSSION
The rise of multidrug-resistant microbes and their dire impact on human health highlight the pressing need for novel antibiotics with a distinct mode of action to combat Gram-negative infections. While LpxC is an attractive and validated novel drug target for these efforts, development of lead compounds has been hampered by a lack of broad-spectrum antibiotic activity against a wide range of Gram-negative bacteria. Here, we detail a successful strategy for designing a broad-spectrum LpxC inhibitor—identifying key residues that confer inhibitor resistance in representative LpxC orthologs and searching for compounds to circumvent these stumbling blocks. The diacetylene-based LPC-009 inhibitor presents an important first step in this process, as our combined kinetic and structural studies show that LPC-009 inhibits a wide range of orthologs with promising efficacy. Furthermore, LPC-009 has made it possible to solve the structure of three LpxC orthologs in complex with a common inhibitor, including the first structure of the EcLpxC enzyme, which provides an excellent model for several clinically-important pathogens.
In addition to providing the basis for further inhibitor optimization, the crystal structures of EcLpxC, AaLpxC and PaLpxC in complex with LPC-009 allow us to probe the intrinsic conformational variability across LpxC orthologs, while avoiding the contributions from inhibitor-specific conformational changes. Contrary to the prevailing hypothesis, our study shows that distinct LpxC orthologs naturally possess large conformational differences in the Insert II region, which generate distinct shapes and angles of the hydrophobic substrate-binding passage. It is important to note that the natural substrate of LpxC is not always the same—while EcLpxC and PaLpxC utilize acylated UDP-GlcNAc as a substrate, AaLpxC uses acylated UDP-GlcNAc3N. The difference of ester versus amide bonds in these substrates may lead to unique geometrical constraints that may affect their binding to LpxC (Supplementary Figure S1); indeed, different angles between the acyl chain and UDP moiety have been observed for R-3-hydroxylauroyl UDP-GlcNAc3N and R-3-hydroxymyrisoyl UDP-GlcNAc in the product complexes of LpxA enzymes from Leptospira and E. coli, respectively (Robins, et al., 2009; Williams and Raetz, 2007). It seems likely that the hydrophobic passages of LpxC orthologs are optimized for processing these distinct substrates, which would partially explain the inherent conformational differences in the LPC-009 complexes.
The unexpected observation of large, intrinsic structural variation among LpxC orthologs helps explain the inadequacies of some of the earlier inhibitors. For example, the unique Insert I conformation of EcLpxC, wherein the βa- βb loop is pushed away form the active site to generate an enlarged cavity, provides a molecular explanation for the potent inhibition of EcLpxC by BB-78485—the bulky naphthalene groups of this compound require a more spacious active site cavity to effectively compete with substrate for binding to the enzyme (Figure 4D). Consistently, BB-78485 does not bind AaLpxC even at mM concentrations, and its antibiotic activity toward an E. coli strain with the native lpxC gene replaced by PaLpxC is at least 20-fold higher than the wild-type E. coli strain (data not shown).
To accommodate the inherent conformational discrepancy of LpxC orthologs and to achieve a better inhibition profile, an attractive strategy of inhibitor design is to engineer flexibility into the chemical scaffold so that the compound can adapt to the distinct shapes and orientations of the active site and hydrophobic passage in various LpxC enzymes. A prime example of this approach is the substrate analog inhibitor TU-514, which contains a flexible acyl chain occupying the hydrophobic passage and inhibits a wide range of LpxC enzymes (Jackman, et al., 2000). Interestingly, the diacetylene group has been observed to adopt “warped” conformations in chemically-constrained compounds (Baldwin, et al., 1998), suggesting that the diacetylene group may have a greater intrinsic flexibility than expected. Given our structural observation of a distorted diacetylene group in different LpxC enzymes, the success of LPC-009 may indeed benefit from the ability of the diacetylene group to adapt to the different trajectories of the substrate-binding passage in distinct LpxC orthologs.
Despite the similarity between CHIR-090 and LPC-009, LPC-009 does not appear to display time-dependent inhibition, whereas CHIR-090 has been shown as a slow, tight-binding inhibitor for a number of LpxC orthologs. Although the molecular basis for such difference needs further investigation, it is likely that the elasticity of the diacetylene bond significantly lowers the energetic barrier and results in a much faster transition rate from the initial LpxC/LPC-009 encounter complex to the final complex, rendering such transition undetectable on the time scale (seconds to minutes) of enzyme kinetic assays.
A final consideration for inhibitor design is that some LpxC enzymes may be inherently more flexible than others. For example, AaLpxC displays a largely rigid structure despite binding to different small molecules, while PaLpxC shows pronounced ligand-induced conformational changes. A thorough understanding of the structural plasticity of key LpxC enzymes by probing their dynamic properties in solution may enable us to design broad-spectrum antibiotics optimized for the less flexible orthologs, while allowing other LpxC enzymes to adapt dynamically.
SIGNIFICANCE
The essential LpxC enzyme in the lipid A biosynthetic pathway is a promising antibiotic target for developing novel therapeutics against multidrug-resistant Gram-negative pathogens. The majority of LpxC inhibitors discovered so far display a narrow range of antibiotic activity. This study presents a structure-based strategy for lead optimization. We show that compounds based on the diacetylene scaffold potently inhibit a wider range of LpxC enzymes by overcoming the resistance mechanism caused by sequence and conformational heterogeneity in the LpxC substrate-binding passage. The structural revelation of large, inherent conformational variation of distinct LpxC enzymes and the relatively small scale of inhibitor-induced structural plasticity provides a molecular explanation for the limited efficacy of existing compounds and a rationale to exploit more flexible scaffolds for further optimization of LpxC-targeting antibiotics to treat a wide range of Gram-negative infections.
EXPERIMENTAL PROCEDURES
Chemical Synthesis
CHIR-090 and LPC-004 were synthesized according to published procedures (Anderson, et al., 2004). Synthesis of LPC-007 and LPC-009 was started from the acylation of L-threonine methyl ester with sodium 4-ethynylbenzoate. Under modified Sonogashira coupling conditions (Nicolaou, et al., 1982), the resulting 4-ethynylbenzamide was coupled with substituted acetylene to afford diacetylene methyl ester. Finally, the methyl ester was converted into the corresponding hydroxamic acid (LPC-007 and LPC-009) by treatment with hydroxylamine under basic conditions.
Protein Purification
Plasmids encoding wild-type EcLpxC, PaLpxC (residues 1–299) with a C40S mutation, and AaLpxC lacking the eight C-terminal amino acids and containing a C181A mutation (1–274) were prepared following established procedures (Barb, et al., 2007; Mochalkin, et al., 2008). An EcLpxC construct lacking the C-terminal five amino acids (1–300) was prepared by using the QuikChange site-directed mutagenesis kit (Stratagene) using a plasmid containing the full-length EcLpxC as the template. LpxC proteins were overexpressed in BL21(DE3)STAR cells (Invitrogen) grown in LB media and purified using anion-exchange (Q-Sepharose Fast Flow, Amersham) and size exclusion (Sephacryl S-200 HR, Amersham) chromatography. Purified proteins were concentrated and buffer-exchanged into 25 mM HEPES, pH 7.0, with 100 mM KCl and 0.1 mM ZnSO4. For EcLpxC and PaLpxC proteins, 2 mM dithiothreitol (DTT) was included in the purification buffers. All protein samples for enzymatic assay and crystallography were stored at −80 °C.
Enzymatic Inhibition Assay
UDP-3-O-[(R)-3-hydroxymyristoyl]-N-acetylglucosamine and [α-32P]UDP-3-O-[(R)-3-hydroxymyristoyl]-N-acetylglucosamine were prepared and assayed as previously described (Jackman, et al., 2001). Assays of LpxC activity were performed at 30 °C in 25 mM sodium phosphate, pH 7.4, 1 mg/mL bovine serum albumin, 25 mM KCl and 0.5 mM DTT, in the presence of 5 µM substrate and 0.2 nM EcLpxC, unless noted otherwise. 10% DMSO was included and held constant in assay mixtures. Initial velocities were calculated from the linear portion of reaction progress curves (<10% conversion of substrate to product).
KM and Vmax values were determined by varying the substrate concentration from 0.5 to 50 µM. Data were analyzed using an Eadie-Hofstee plot (Dowd and Riggs, 1965) and by a nonlinear curve-fitting program (KaleidaGraph, Synergy Software); the resultant values were nearly identical within experimental errors. To determine a KI value, LPC-009 concentrations were varied from 12.5 pM to 15 nM. Fractional activity (υi/υ0) versus LPC-009 concentration was plotted and fitted to calculate a KIapp value using the Morrison equation (Copeland, 2005):
| Equation [1] |
where υi is the initial velocity of the reaction in the presence of the inhibitor, υ0 is the initial velocity of the reaction in the absence of the inhibitor, [E]T is the total enzyme concentration, and [I]T is the total inhibitor concentration. A KI value was calculated using
| Equation [2] |
where [S] is the substrate concentration. All measurements were done in triplicates.
MIC Tests
MICs were determined according to the NCCLS protocol (Wikler, et al., 2006) adapted to 96-well plates and LB media. Briefly, 1.0 × 105 bacterial cells in LB medium containing 5% DMSO and various concentrations of the compound were incubated at 37°C for 22 hours. After the incubation, [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide solution (MTT) was added (0.2 mg/mL final concentration) and incubated at 37 °C for another 3 hrs. The MIC was determined as the lowest concentration of an antibiotic that prevented color change (yellow to black).
Crystallization and X-ray Data Collection
Purified EcLpxC (1–300), PaLpxC (1–299, C40S) and AaLpxC (1–275, C181A) were diluted to final concentrations of 10 mg/mL, 12mg/mL and 15 mg/mL, respectively. For PaLpxC, 10 mM zinc sulfate was added. A four-fold molar excess of LPC-009 dissolved in DMSO was added to the diluted protein samples. The protein and inhibitor were incubated at room temperature for 1 hr to obtain a homogenous sample before setting up crystallization screening. EcLpxC/LPC-009 complex crystals were obtained using the hanging drop vapor diffusion method at 17 °C, with the reservoir solution containing 0.1 M HEPES pH 7.5, 1.5 M LiSO4 and 10 mM DTT. AaLpxC/LPC-009 and PaLpxC/LPC-009 complex crystals were crystallized using the sitting drop vapor diffusion method at 20 °C, with reservoir solutions containing 0.1 M Tris pH 8.5, and 2.5 M ammonium phosphate dibasic, and 0.1 M sodium acetate trihydrate pH 5.0, and 2.7 M ammonium nitrate, respectively. All protein-inhibitor complex samples were mixed with equal amounts of reservoir solutions and equilibrated against individual reservoir solutions. The crystals were cryoprotected with perfluoropolyether (PFO-X175/08) before being flash-frozen in liquid nitrogen. Diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory and processed with HKL2000 (Otwinowski and Minor, 1997).
Model Building and Refinement
Molecular replacement with the program PHASER (McCoy, et al., 2007) was used to obtain the initial phases of three LpxC-inhibitor complex structures. The structures of the AaLpxC/TU-514 complex (PDB entry: 2GO4) and the PaLpxC/BB-78485 complex (PDB entry: 2VES) and a homology model of EcLpxC derived from the PaLpxC/BB-78485 complex structure were used as search models for AaLpxC/LPC-009, PaLpxC/LPC-009 and EcLpxC/LPC-009 complexes, respectively. To avoid phase bias, molecular replacement was performed iteratively, starting from partially refined structures not containing regions of large structural variations (i.e., Insert I and Insert II). The EcLpxC/LPC-009 crystal contains an un-interpretable electron density at the packing interface, which is possibly an impurity from the chemical synthesis of LPC-009. The excellent quality of the electron density of LPC-009 allows an unambiguous interpretation of its binding mode in the active site. Two rotameric conformations of the LPC-009 threonyl moiety were observed in the PaLpxC complex. Water molecules were added using PHENIX (Zwart, et al., 2008) and verified with COOT (Emsley and Cowtan, 2004). The final model was obtained after iterative cycles of manual model building with COOT and refinement using PHENIX. MOLPROBITY (Davis, et al., 2007) was used to evaluate the quality of the refined structure. The statistics for the three complexes are shown in Table 1.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by National Institutes of Health Grants AI055588 (to P.Z.), and GM-51310 (to C.RH.R). X.L. is supported by the China Scholarship Council. Use of Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source (APS), Argonne National Laboratory is supported by Duke University. We would like to thank Jon Werner-Allen for stimulating discussions, and Nathan Nicely, Louis Metzger, James Phillips, and Mike Hast for assistance with crystal screening and data collection.
Footnotes
Accession Numbers
Structure factors and coordinates for EcLpxC/LPC-009, AaLpxC/LPC-009 and PaLpxC/LPC-009 complexes have been deposited in the RCSB Protein Data Bank with the accession codes 3P3G, 3P3C, and 3P3E, respectively.
REFERENCES
- Anderson NH, Bowan J, Erwin A, Harvood E, Kline T, Mdluli K, Pfister KB, Shawar R, Wagman A, Yabannavar A. Antibacterial Agents. Emeryville, CA: Chiron; 2004. WO 2004/062601 A2. [Google Scholar]
- Baldwin KP, Simons RS, Scheiman DA, Lattimer R, Tessier CA, Youngs WJ. Synthesis, characterization, and crystal structures of two strained cyclic diacetylenes and their precursors. J Chemical Crystallography. 1998;28:353–360. [Google Scholar]
- Barb AW, Jiang L, Raetz CR, Zhou P. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: Time-dependent inhibition and specificity in ligand binding. Proc Natl Acad Sci U S A. 2007;104:18433–18438. doi: 10.1073/pnas.0709412104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb AW, McClerren AL, Snehelatha K, Reynolds CM, Zhou P, Raetz CR. Inhibition of Lipid A Biosynthesis as the Primary Mechanism of CHIR-090 Antibiotic Activity in Escherichia coli. Biochemistry. 2007;46:3793–3802. doi: 10.1021/bi6025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb AW, Zhou P. Mechanism and inhibition of LpxC: an essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr Pharm Biotechnol. 2008;9:9–15. doi: 10.2174/138920108783497668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L, Dawson A, Hunter WN. The nucleotide-binding site of Aquifex aeolicus LpxC. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2006;62:1082–1086. doi: 10.1107/S1744309106041893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins BE, McClerren AL, Jiang L, Li X, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: insights into the mechanism and inhibitor design. Biochemistry. 2005;44:1114–1126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery. Hoboken, NJ: John Wiley & Sons, Inc.; 2005. [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobler MR, Lenoir F, Parker DT, Peng Y, Piizi G, Wattanasin S. Organic Compounds for Applications in Bacterial Infections Treatment. Novartis AG: 2010. WO 2010/031750 A1. [Google Scholar]
- Dowd JE, Riggs DS. A Comparison of Estimates of Michaelis-Menten Kinetic Constants from Various Linear Transformations. J Biol Chem. 1965;240:863–869. [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gennadios HA, Christianson DW. Binding of Uridine 5'-Diphosphate in the "Basic Patch" of the Zinc Deacetylase LpxC and Implications for Substrate Binding. Biochemistry. 2006;45:15216–15223. doi: 10.1021/bi0619021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennadios HA, Whittington DA, Li X, Fierke CA, Christianson DW. Mechanistic Inferences from the Binding of Ligands to LpxC, a Metal-Dependent Deacetylase. Biochemistry. 2006;45:7940–7948. doi: 10.1021/bi060823m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J Biol Chem. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, Tahir SH, Hindsgaul O, Raetz CRH. Antibacterial agents that target lipid A biosynthesis in Gram-negative bacteria. Inhibition of diverse UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylases by substrate analogs containing zinc binding motifs. J Biol Chem. 2000;275:11002–11009. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- Jackman JE, Raetz CRH, Fierke CA. Site-directed mutagenesis of the bacterial metalloamidase UDP-(3-O- acyl)-N-acetylglucosamine deacetylase (LpxC). Identification of the zinc binding site. Biochemistry. 2001;40:514–523. doi: 10.1021/bi001872g. [DOI] [PubMed] [Google Scholar]
- Kline T, Andersen NH, Harwood EA, Bowman J, Malanda A, Endsley S, Erwin AL, Doyle M, Fong S, Harris AL, et al. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J Med Chem. 2002;45:3112–3129. doi: 10.1021/jm010579r. [DOI] [PubMed] [Google Scholar]
- Mansoor UF, Reddy P, Adulla P, Siddiqui MA. Hydantoin Derivatives Useful As Antibacterial Agents. Schering Corporation; 2008. WO 2008/027466 A1. [Google Scholar]
- Mansoor UF, Reddy PA, Siddiqui MA. Urea Derivatives As Antibacterial Agents. Scherring Coporation; 2010. WO 2010/017060 A1. [Google Scholar]
- McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CR. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry. 2005;44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochalkin I, Knafels JD, Lightle S. Crystal structure of LpxC from Pseudomonas aeruginosa complexed with the potent BB-78485 inhibitor. Protein Sci. 2008;17:450–457. doi: 10.1110/ps.073324108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser H, Lu Q, Patten PA, Wang D, Kasar R, Kaldor S, Patterson BD. Antibacterial Agents. South San Francisco, CA: Achaogen, Inc.; 2008. WO 2008/154642 A2. [Google Scholar]
- Nicolaou KC, Zipkin RE, Petasis NA. The endiandric acid cascade. Electrocyclizations in organic synthesis. 3. "Biomimetic" approach to endiandric acids A–G. Synthesis of precursors. J Am Chem Soc. 1982;104:5558–5560. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:301–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pirrung MC, Tumey LN, McClerren AL, Raetz CRH. High-throughput catch-and-release synthesis of oxazoline hydroxamates. Structure-activity relationships in novel inhibitors of Escherichia coli LpxC: in vitro enzyme inhibition and antibacterial properties. J Am Chem Soc. 2003;125:1575–1586. doi: 10.1021/ja0209114. [DOI] [PubMed] [Google Scholar]
- Raetz CRH. Enzymes of lipid A biosynthesis: targets for the design of new antibiotics. Prog Clin Biol Res. 1998;397:1–14. [PubMed] [Google Scholar]
- Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in Gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju BG, O'Dowd H, Gao H, Patel DV, Trias J. N-hydroxyamide Derivatives Possessing Antibacterial Activity. NY (US): Pfizer Inc.; 2010. US 7,691,843 B2. [Google Scholar]
- Robins LI, Williams AH, Raetz CR. Structural basis for the sugar nucleotide and acyl-chain selectivity of Leptospira interrogans LpxA. Biochemistry. 2009;48:6191–6201. doi: 10.1021/bi900629e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Gennadios HA, Whittington DA, Christianson DW. Amphipathic benzoic acid derivatives: Synthesis and binding in the hydrophobic tunnel of the zinc deacetylase LpxC. Bioorg Med Chem. 2007;15:2617–2623. doi: 10.1016/j.bmc.2007.01.044. [DOI] [PubMed] [Google Scholar]
- Siddiqui MA, Mansoor UF, Reddy PA, Madison VS. Compounds for the Treatment of Inflamatory Disorders and Microbial Diseases. NJ, USA: Schering Corporation : 2007. WO 2007/064749 A1. [Google Scholar]
- Takashima H, Suga Y, Urabe H, Tsuruta R, Kotsubo H, Oohori R, Kawaguchi Y. Novel Hydroxamic Acid Derivative Having Naphthyridine-N-oxide. Taisho Pharmaceutical Co., LTD; 2010. WO 2010/024356 A1. [Google Scholar]
- Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc Natl Acad Sci U S A. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikler MA, Low DE, Cockerill FR, Sheehan DJ, Craig WA, Tenover FC, Dudley MN. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved Standard-seventh edition (M7-A7) (Clinical and Laboratory Standards Institute (Formally NCCLS)) 2006 [Google Scholar]
- Williams AH, Raetz CR. Structural basis for the acyl chain selectivity and mechanism of UDP-N-acetylglucosamine acyltransferase. Proc Natl Acad Sci U S A. 2007;104:13543–13550. doi: 10.1073/pnas.0705833104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga M, Ushiki Y, Tsuruta R, Urabe H, Tanikawa T, Tanabe K, Baba Y, Yokotani M, Kawaguchi Y, Kotsubo H, et al. Novel Hydroxamic Acid Derivative. Taisho Pharmaceutical Co., LTD.; 2008. WO 2008/105515 A1. [Google Scholar]
- Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, et al. Automated structure solution with the PHENIX suite. Methods Mol Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.