

Abstract
Glyceryl triacetate (GTA), a compound effective at increasing circulating and tissue levels of acetate was used to treat rats subjected to a continual 28 day intra-ventricular infusion of bacterial lipopolysaccharide (LPS). This model produces a neuroinflammatory injury characterized by global neuroglial activation and a decrease in choline acetyltransferase immunoreactivity in the basal forebrain. During the LPS infusion, rats were given a daily treatment of either water or GTA at a dose of 6g/kg by oral gavage. In parallel experiments free-CoA and acetyl-CoA levels were measured in microwave fixed brains and flash frozen heart, liver, kidney and muscle following a single oral dose of GTA. We found that a single oral dose of GTA significantly increased plasma acetate levels by 15 min and remained elevated for up to 4 hr. At 30 min the acetyl-CoA levels in microwave-fixed brain and flash frozen heart and liver were increased at least 2.2-fold. The concentrations of brain acetyl-CoA was significantly increased between 30 and 45 min following treatment and remained elevated for up to 4 hr. The concentration of free-CoA in brain was significantly decreased compared to controls at 240 min. Immunohistochemical and morphological analysis demonstrated that a daily treatment with GTA significantly reduced the percentage of reactive GFAP-positive astrocytes and activated CD11b-positive microglia by 40–50% in rats subjected to LPS-induced neuroinflammation. Further, in rats subjected to neuroinflammation, GTA significantly increased the number of ChAT-positive cells by 40% in the basal forebrain compared to untreated controls. These data suggest that acetate supplementation increases intermediary short chain acetyl-CoA metabolism and that treatment is potentially anti-inflammatory and neuroprotective with regards to attenuating neuroglial activation and increasing ChAT immunoreactivity in this model.
Introduction
Chronic inflammatory processes (McGeer & McGeer 1995) and disturbances in phospholipid metabolism (Ginsberg et al. 1993, Ginsberg et al. 1998, Farooqui et al. 1997) have a role in the neurodegenerative events associated with inflammation-induced brain injury. In Alzheimer’s disease, for example, affected brain regions not only show characteristic changes in neuroglial reactivity, but also demonstrate significant decreases in esterified fatty acid content in ethanolamine and choline glycerophospholipid and in the content of ether phospholipid when compared to non-affected brain regions (Guan et al. 1999, Han et al. 2001, Ginsberg et al. 1993). A rat model of neuroinflammation produced by the infusion of bacterial lipopolysaccharide into the forth ventricle of the rat brain reproduces many of the inflammatory components found in AD (Hauss-Wegrzyniak et al. 1998), and is a model consistent with the presence of glial-related neurotoxic events (Eddleston & Mucke 1993, Tyagi et al. 2008). LPS infusion induces a progressive and global increase in the number of reactive microglia that results in the sustained formation of inflammatory cytokines, the expression of complement protein, and β-amyloid precursor protein mRNA in the basal forebrain and hippocampus (Hauss-Wegrzyniak et al. 1998). A chronic 30 day infusion of LPS results in the temporal decrease in choline acetyltransferase (ChAT) immunoreactivity, a decrease in hippocampal NMDA R1 receptor density (Willard et al. 1999, Rosi et al. 2004), and impairment of spatial memory when compared to controls (Hauss-Wegrzyniak et al. 2000).
This model of neuroinflammation has been used to identify early and selective alterations in the metabolism of brain arachidonic acid (ARA, 20:4n-6). The turnover and metabolism of brain ARA is increased by 40 % in ethanolamine and choline glycerophospholipid in rats subjected to a low dose (0.5 ng/hr) intra-ventricular infusion of bacterial lipopolysaccharide (LPS) (Rosenberger et al. 2004, Lee et al. 2004, Basselin et al. 2003). The increase in the turnover rates of ARA in this model is specific to esterified brain ARA as the turnover rates of brain docosahexaenoic acid (DHA, 22:6n-3) in all phospholipid classes do not change (Rosenberger et al. 2010). Low-dose infusion of LPS also increases the activity of both ARA-selective secretory and cytosolic phospholipases A2 activity and the levels of prostaglandins E2 and D2 (Rosenberger et al. 2004). Therefore the continual infusion of LPS results in characteristic changes in neuroglia and induces an ARA-selective response consistent with those lipid changes identified in human disorders having a neuroinflammatory component. These findings suggest that identifying brain lipid mediated signaling pathways altered during the consolidation of inflammatory events in brain may be beneficial in identifying potential therapeutic strategies to reduce the pathology associated with neuroinflammation.
In this regard, acetate supplementation is an effective therapy in stimulating myelin deposition (Mathew et al. 2005) and reducing the tremor phenotype in a rat model of Canavan’s disease (Arun et al. 2010b). Further acetate supplementation is effective at maintaining ATP levels in a rat model of traumatic brain injury (Arun et al. 2010a) suggesting that exogenous acetate can enter brain and influence injury progression. Acetate is a widely active precursor that when converted into acetyl-CoA is central to mitochondrial energy supply, fatty acid synthesis, and lipid metabolism (Deutsch et al. 2002). In brain, acetate is converted to acetyl-CoA through the combined actions of nuclear acetyl-CoA synthetase 1 (aceCS1) (Ariyannur et al. 2010) and mitochondrial acetyl-CoA synthetase 2 (aceCS2) (Fujino et al. 2001). Mitochondrial fatty acid metabolism plays a role in the development of secondary injury in traumatized brain, due to evidence that 3-hydroxy-3methylglutaryl-CoA reductase inhibitors (statins) are neuroprotective (Vaughan & Delanty 1999). In models of cerebral ischemia and stroke, statins reduce infarct size (Sacks et al. 1996), up regulate endothelial nitric oxide synthase (Endres et al. 1998), down regulate inducible nitric oxide synthase (Pahan et al. 1997), and attenuate the inflammatory cytokine responses that accompany cerebral ischemia (Weber et al. 1997, Pantoni et al. 1998). All of these responses are neuroprotective and dependent on the metabolism of short chain acyl-CoA. Therefore, in a pathological situation like ischemia, the over stimulation of lipases result in the rapid accumulation of fatty acids and a loss of normal recycling of fatty acid back into the phospholipids through their CoA derivatives (Farooqui et al. 2000).
Because acetate supplementation is effective at increasing circulating and tissue levels of acetate, and is effective at reducing injury deficits in rat models of traumatic brain injury (Arun et al. 2010a) and Canavan’s disease (Arun et al. 2010b), we proposed that acetate supplementation acts through a mechanism involving short chain acetyl-CoA metabolism and the inhibition of the brain inflammatory reaction. To test this hypothesis, we quantified brain and tissue levels of acetyl-CoA and free CoA and measured the effect of acetate supplementation on neuroglia reactivity and cholinergic cell loss in a rat model of neuroinflammation. These studies demonstrate that GTA-derived acetate is converted to acetyl-CoA in brain, liver, and heart suggesting that acetate supplementation does stimulate intermediary short chain acyl-CoA metabolism. Further, treatment with oral glyceryl triacetate significantly reduced neuroglial reactivity and increased to control levels ChAT immunoreactivity in a rat model of neuroinflammation. These data suggest that a therapeutic mechanism of action of acetate supplementation works by stimulating short chain acyl-CoA metabolism that results in the reduction of neuroglial activation.
METHODS
Reagents
p-bromophenacylbromide, 18-crown-6, coenzyme A, acetyl-CoA, proprionyl-CoA, butyryl-CoA, glyceryl triacetate, buffers, and fixative solutions were purchased from Sigma (St. Louis, MO). HPLC-grade acetonitrile, methanol, and 2-propranol, and other reagents were obtained from EMD Chemicals (Gibbstown, NJ) and oligonucleotide purification cartridges were purchased from Applied Biosystems (Foster, CA). Normal horse serum, normal goat serum, biotinylated horse anti-6 mouse immunoglobulin G and Vectastain kits were obtained from Vector Laboratories (Burlingame, CA). An antibody recognizing specifically CD11b, the rat homolog of the human C3bi complement receptor and a marker for microglia and monocytes, was obtained from AbD Serotec (Raleigh, NC). Anti- glial fibrillary acidic protein (GFAP) antibody, 3,3′-diaminobenzidine tetrahydrochloride (DAB), anti- choline acetyl transferase (ChAT) antibody, and a rat adsorbed biotinylated goat anti-rabbit immunoglobulin G were purchased from Millipore (Temecula, CA). Permount™ was obtained from Fisher Scientific (Pittsburgh, PA) and absolute ethanol was purchased from Pharmco (Brookfield, CT).
Animal Studies
This study was conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 80-23) under an animal protocol approved by the University of North Dakota ACUC (Protocol #0706-7) using male Sprague-Dawley rats (Charles River, Wilmington, MA). Rats used to determine plasma acetate pharmacokinetics were anesthetized with 3% halothane (Halocarbon, River Edge, NJ) and polyethylene catheters (PE 50, Becton Dickinson, Sparks, MD) filled with sodium heparin (100 IU) were implanted into the right femoral artery and vein. The skin was closed with surgical clips and 1% lidocaine was applied to the wound. The hindquarters of the rats were loosely secured to wooden block with strapping tape then the rats were allowed to recover from anesthesia for 3 hr. Glycerol triacetate (6 g/kg) was administered via gastric gavage to recovered rats. Following treatment arterial blood samples taken at predetermined time points and used to measure plasma acetate levels. To measure tissue levels of acetate, acetyl-CoA, and free CoA rats were deeply anesthetized with an intravenous injection of pentobarbital (50 mg/kg) then immediately subjected to head-focused microwave irradiation to stop brain metabolism. The heart, liver, muscle, and kidney samples were removed following microwave irradiation and flash frozen in liquid nitrogen. The post-mortem interval for these samples did not exceed 3 min and were stored at negative 80° C until use. Rats used to determine the effect of acetate supplementation on LPS-induced neuroinflammation were subjected to surgeries in which cannula (Model 3280PM, Plastics One, Roanoke, VA) connected to subcutaneous osmotic mini-pumps (Model 2004, Durect Corp. Cupertino, CA) were surgically implanted into the 4th ventricle of the rat brain. The surgical placement of the cannula and osmotic pump was performed on male Sprague Dawley rats weighing between 160–180 g (Charles River Laboratories, Portage, MI) as described (Hauss-Wegrzyniak et al. 1998). Isoflurane (3%) by inhalation (Butler Animal Health Supply, Dublin, OH) was used to induce and maintain anesthesia during the surgery. A solution of LPS (5.0 ng/hr dissolved in aCSF) or aCSF (Harvard Apparatus, Holliston, MA) was infused continually at a rate of 0.25 μL/hr via the osmotic mini-pump for a period of 28 days. The concentration of endotoxin used in these studies is based on unpublished results showing that 5.0 ng/hr results in significant neuroglia activation and cholinergic cell loss above control treated rats and is consistent with previous studies demonstrating a selective increase in ARA metabolism using in this model (Rosenberger et al. 2004, Lee et al. 2004, Basselin et al. 2003). During the infusion period, rats were treated daily with either GTA or water at a dose of 6 g/kg by gastric gavage. All rats were allowed to acclimate in our facility for at least seven days prior to inclusion in the study and were maintained on a constant 12 hr light cycle. Rats were fed a standard 2018 Teklad Global 18% protein rodent diet (Harlan, Madison, WI) ab libidum.
Fixation and Sectioning
Rats were anesthetized with sodium pentobarbital (50 mg/kg) and euthanized with a cardiac perfusion of 0.9% saline containing 1000 units/L sodium heparin (Baxter, Deerfield, IL). Fixation was performed using a periodate-lysine-paraformaldehyde (PLP) solution containing 4% formaldehyde, 0.01 M sodium periodate, and 0.1 M lysine dissolved in 0.05 M phosphate buffer (pH 7.4). The rat brains were post-fixed for 2–3 days in the PLP fixation solution at 4° C before a 12 hr equilibration at 4° C in a 0.1 M phosphate buffer (pH 7.2) solution containing 20% sucrose. Sucrose-impregnated brains were frozen in isopentane (Alfa Aesar, Ward Hill, MA) cooled to −50° C on dry ice. The frozen brains were mounted on cryostat pedestals with M-1 embedding matrix (Lipshaw, Pittsburgh, PA) and allowed to equilibrate at −20° C before sectioning. A cryostat (IEC, Needham Hts., MA) was used to cut 20 μm serial coronal brain sections, which were mounted on gelatin-coated glass slides. The mounted sections were stored in slide cases at room temperature until use.
Antigen Retrieval
The tissue sections were rehydrated in 0.1 M Tris-buffered saline, pH 7.6 (TBS). An antigen retrieval procedure was performed to enhance antibody binding efficiency (Costa et al. 1986, Shi et al. 1991). Briefly, the re-hydrated mounted tissue sections were submerged in plastic jars containing 10 mM Tris buffer, pH 9.0 with 1 mM EDTA and 0.05% Tween® 20 (BDH, West Chester, PA). The samples were heated to 100° C in a pressure cooker for 3 min. After heating, the slides were allowed to cool to room temperature then re-equilibrated by submersion in TBS at room temperature.
Immunohistochemistry
To visualize activated microglial cells, a monoclonal antibody recognizing CD11b (final dilution 1:1000) was used to identify the CR3 complement receptor expressed on microglia (Gehrmann et al. 1992). Astrocytes were visualized using a monoclonal antibody recognizing GFAP (final dilution 1:400) and cholinergic neurons were visualized with a monoclonal antibody recognizing ChAT (final dilution 1:1000). Following antigen retrieval, endogenous peroxidase was blocked with 0.3% H202 in TBS for 30 min followed by three 10 min washes in 0.1 M TBS, pH 7.6 + 0.025% Triton™ X-100 (EDM, Gibbstown, NJ) (TNT buffer). The sections were then incubated for 1 hr in blocking solution containing 1% bovine serum albumin (BSA), 0.1% Triton™ X-100 and 2% horse serum in TBS (CD11b and GFAP antibody) or 10% normal goat serum in TBS (ChAT antibody). The serial sections were incubated with primary antibody for 72 hr (CD11b), 48 hr (ChAT), or 24 hr (GFAP) at 4° C in hydration chambers. After incubation the sections were washed three times with TNT buffer. The sections exposed to the ChAT antibody were incubated for 2 hr in a 1:500 solution of rat-adsorbed, goat anti-rabbit biotinylated secondary antibody. Sections exposed to the CD11b or GFAP antibodies were incubated for 2 hr in a 1:200 solution of rat-adsorbed, horse anti-mouse biotinylated secondary antibody. All secondary antibodies were diluted in appropriate blocking solutions listed above. At 2 hr all sections were washed three times in TNT buffer then incubated with in an avidin-biotin complex (ABC) mixture for 1 hr. The ABC mixture was washed off with TBS then immunoreactivity was detected with a 0.05% solution of 3, 3′-diaminobenzidine tetrahydrochloride (DAB) dissolved in TBS containing 0.015% H202. The samples were allowed to incubate in the DAB solution for 5–8 min until a brown precipitate was apparent on the tissue sections. The reaction was stopped by washing the sections with TBS. Parallel measurements were performed in the absence of primary and secondary antibodies to rule out non-specific antibody binding. Sections were dehydrated with increasing concentrations of ethanol/water solutions (70, 95, and 100% ethanol by vol.), then equilibrated in a Xylene/Ethanol (1:1, by Vol.) followed by 100% Xylene. The dehydrated samples were prepared for analysis by affixing glass covers held in place with Permount™. Sections were analyzed using an Olympus Model BX50 light microscope equipped with Spot™ Model 2.3.1 imaging system and version 3.4.5 software (Leeds, Minneapolis, MN).
Cell counting
Three independent quantitative image cell count analysis studies was performed on 10 parallel sections from six different animals per treatment group within a defined distance of 200 μm that encompassed the basal forebrain. By using a 20x objective, a grid field of 500 × 500 μm was developed that encompassed six specific regions, left and right medial septum (MS), left and right striatum, and left and right horizontal diagonal bands of Broca (hDBB) within the basal forebrain. Anatomical landmarks were used to define, and standardize, the location of counting frames as described (Traissard et al. 2007). In the striatum, all ChAT-positive neurons were counted in the left and right hemispheres. For each section at least three independent observations were made and cell counts were averaged as a reading for each animal. The numbers of positive neurons was determined separately for each region and were summed. Quantitative morphological image analysis of CD11b-positive microglia and GFAP-positive astrocytes cell counts were performed on sections as defined above. A morphological criterion of activated microglia was determined by classifying individual microglial cells based on morphology as described (Davis et al. 1994, Ayoub & Salm 2003). The assessment of astrocyte activation was determined by morphological differences in cellular hypertrophy and cytoplasmic enlargement between naïve and activated astrocytes (Ridet et al. 1997, Baldwin & Scheff 1996, Amaducci et al. 1981). For each section at least three independent observations were made and cell counts based on morphological assessment were averaged as a reading frame for each animal. The number of CD11b- and GFAP-positive microglia and astrocytes was determined separately for each reading frame and the totals were summed.
Plasma Extraction and measurement of circulating levels of acetate
Arterial blood samples collected at predetermined times following GTA treatment were centrifuged at 10,000 × g for 4 min at room temperature to isolate plasma. An aliquot of plasma was extracted in methanol/water (90:10, by Vol.) by mixing for 5 min. The extracted samples were centrifuged for 10 min at 1,000 × g (27° C) to pellet protein and 100 μL of the supernatant, equivalent to 10 μL plasma, was used to measure plasma acetate as described (Persson et al. 1991). Aliquots of the tissue supernatant were used to measure tissue concentrations of acetate as following conversion to their phenacyl ester derivatives as described below. Phenacyl estersof acetate were prepared using p-bromophenacyl-8 reagent (0.1 mmol/ml p-bromophenacylbromide and 0.005 mmol/ml crown ether in acetonitrile) and analyzed as described (Durst et al. 1975). Briefly, 100 μl of acetonitrile and 100 μl of p-bromophenacyl-8 reagent were added to the plasma extracts and the samples were vortex-mixed then incubated at 80° C for 1 hr with constant stirring. Following the incubation, the vials were cooled to 4° C then the solvent was removed under a steady-stream of nitrogen at 45° C and reconstituted in 100 μL acetonitrile. Plasma levels of acetate were analyzed via HPLC.
Short Chain acyl-CoA Extraction
Tissue concentration of short chain acyl-CoA were measured using microwave-fixed rat brain and flash frozen heart, liver, kidney and muscle samples as described (Deutsch et al. 2002). Briefly, tissue samples (~ 600 mg) were added to a 7 mL Tenbroeck homogenizer containing internal standard (proprionyl-CoA). The samples were immediately homogenized in 3 mL of ice cold 12% perchloric acid, transferred to a 15 mL plastic conical tube and mixed for 2 minutes. The homogenate was then centrifuged at 2,600 × g for 10 minutes at 4° C. The supernatant was transferred into a 50 mL conical tube and 16 mL of 75 mM KH2PO4 was added. The solution was mixed gently by hand and then placed in a 10 mL syringe and filtered through an activated OPC column at a rate of 1.0 ml/min or 10–15 drops/min. The bound short chain acyl-CoA esters and CoA were then washed with 1 mL of 25 mM KH2PO4. The acyl-CoA esters and CoA were eluted off the OPC with 0.3 mL of 2-propanol/1mM acetic acid (75:25, by Vol.). The samples were dried with a nitrogen evaporator at 45° C then re-suspended in 100 uL of 4% 2-propanol 16% methanol in 25 mM and analyzed using HPLC.
Statistical Analysis
Statistical analysis was performed on all data comparing controls to either GTA-treated rats or rats treated with GTA and LPS with a One-way ANOVA with Tukey’s post-test or an Unpaired T-test with Welch’s corrected post-test when appropriate using GraphPad InStat® statistical software (Version 3.06 for Windows, GraphPad Software, San Diego CA). All data are presented as means ± SEM or means ± SD in which statistical significance was set at p ≤ 0.05.
RESULTS
Plasma acetate and its conversion to acetyl-CoA in tissue
Arterial blood samples taken from rats given a single oral gavage of glyceryl triacetate (6 g/kg) were used to demonstrate that GTA was converted to acetate and distributed to the plasma. These measurements showed that the plasma acetate levels in treated rats were increased by 15 min following the oral administration of GTA (6 g/kg) and that the plasma concentration of acetate reached a maximum of 190 μmol/mL at 30 min (Figure 1). The concentration of plasma acetate in control rats treated with water (6 g/kg) ranged from 2 to 6 μmol/mL at all post-treatment times measured (data not shown). The circulating levels of acetate in GTA-treated rats remained significantly elevated compared to control-treated rats up to 4 hr post-treatment. These data suggest that GTA is hydrolyzed to glycerol and acetate in the stomach and small intestine consistent with known routes of fat digestion in the human (Hamosh et al. 1981). To demonstrate that circulating acetate is distributed throughout the body and converted to acetyl-CoA we measured free-CoA and acetyl-CoA in microwave fixed brain and flash frozen heart, kidney, liver, and muscle samples 30 min following a single oral dose of water or GTA (6 g/kg) (Table 1). These measurements showed that acetyl-CoA levels in brain, heart, and liver were significantly increased by 2.2-, 2.6-, and 2.2-fold over control rats having tissue concentrations of 5.7 ± 1.9, 36.3 ± 17.4, and 60.1 ± 12.9 μg/g tissue, respectively. The concentration of acetyl-CoA in kidney and muscle samples did not significantly differ from control treated rats. To determine the temporal effect that GTA-treatment has on brain short chain acetyl-CoA metabolism, we measured the free-CoA and acetyl-CoA levels in microwaved brain of rats treated with either water or GTA (6 g/kg) (Figure 2). These measurements showed that GTA treatment significantly increased brain levels of acetyl-CoA above control levels by 30 min and remained significantly elevated for up to 4 hr post-treatment. The levels of free-CoA in GTA treated rats remained unchanged up to 75 min, however, a significant decrease in the concentration of free-CoA was found at 4 hr. These data demonstrate the acetate derived from the oral administration of GTA results in a significant increase in the tissue levels of acetyl-CoA and that in brain acetyl-CoA levels remain significantly elevated for up to 4 hr.
Figure 1.
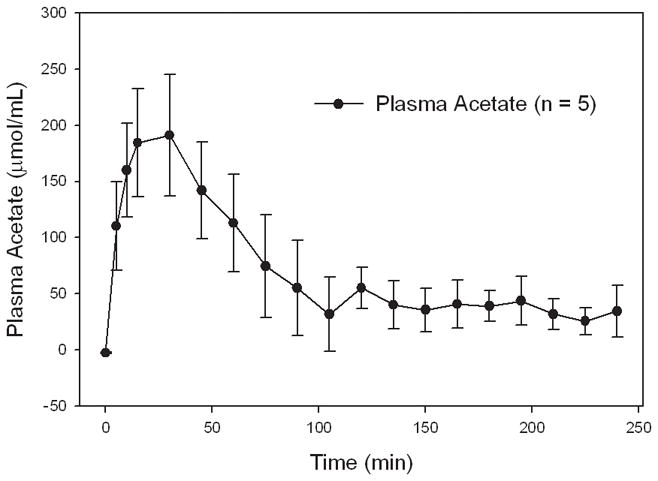
Plasma acetate level in rats treated with a single oral dose on glyceryl triacetate. Values represent the means ± SD (n = 5).
Table 1.
Effect of a single oral dose of glyceryl triacetate on tissue levels of free CoA and acetyl-CoA compared to control-treated rats.
| Free-CoA |
Acetyl-CoA |
||||
|---|---|---|---|---|---|
| μg/g tissue (30 min post-treatment) | |||||
| Tissue | Control | GTA | Control | GTA | Fold Increase |
| Brain (microwave fixed) | 16.5 ± 1.5 | 16.4 ± 4.9 | 2.6 ± 1.0 | *5.7 ± 1.9 | 2.2 |
| Heart | 30.8 ± 3.4 | 28.4 ± 2.2 | 13.7 ± 4.9 | *36.3 ± 17.4 | 2.6 |
| Kidney | 44.7 ± 7.7 | 46.7 ± 5.8 | 9.9 ± 2.6 | 9.8 ± 3.4 | - |
| Liver | 78.9 ± 3.9 | 76.4 ± 13.7 | 27.2 ± 6.1 | *60.1 ± 12.9 | 2.2 |
| Muscle | 2.0 ± 0.5 | 1.9 ± 0.6 | 3.4 ± 2.1 | 3.5 ± 1.2 | - |
Values represent the means ± SD (n=8) of free CoA and acetyl-CoA from microwaved-fixed brain and flash frozen heart, kidney, liver, and muscle samples at 30 min following a single oral dose of water or GTA (6 g/kg). Fold increase was calculated comparing acetyl-CoA levels in GTA-treated rats to acetyl-CoA levels in water-treated rats. The asterisks (*) represents values that were significantly different from control values as determined using an Unpaired T-test with Welch’s corrected post-test (*, P ≤ 0.05).
Figure 2.

Time-dependent changes in the concentrations of brain acetyl-CoA (A) and free CoA (B) following a single oral dose of glyceryl triacetate in microwave fixed rat brain. Values represent the means ± SD in units of μg/g brain (n=9 per group). Statistical analysis was performed comparing the brain concentration of brain acetyl-CoA and free CoA in GTA-treated rats to rats treated with a single oral dose of water (6 g/kg) (*, P ≤ 0.05).
Attenuation of neuroglia activation in GTA-treated rats
To test our main hypothesis that acetate supplementation imparts its neuroprotective effect by attenuating inflammatory events in the brain we quantified the number of activated microglia and reactive astrocytes in rats subjected to LPS-induced neuroinflammation (Figure 3a and 3b). Following a 28 day intra-ventricular infusion of aCSF we found that the percentage of activated microglia did not differ significantly from rats treated daily with either water or GTA (6 g/kg) (Figure 3c). In rats subjected to a continual infusion of 5.0 ng/hr LPS we found a statistically significant 10-fold increase in the percentage of activated microglia as determined by quantitative morphological analysis. Further, rats subjected to both LPS-induced neuroinflammation and a daily treatment with GTA by oral gavage showed that the percentage of activated CD11b-positive microglia was significantly reduced by 3-fold compared to rats subjected to neuroinflammation and given water. The percentage of activated microglia remained significantly elevated compared to aCSF control rats treated with GTA. These data suggest that a daily oral gavage of GTA significantly attenuates, but does not completely block, the activation of microglia in rats subjected to LPS-induced neuroinflammation.
Figure 3.


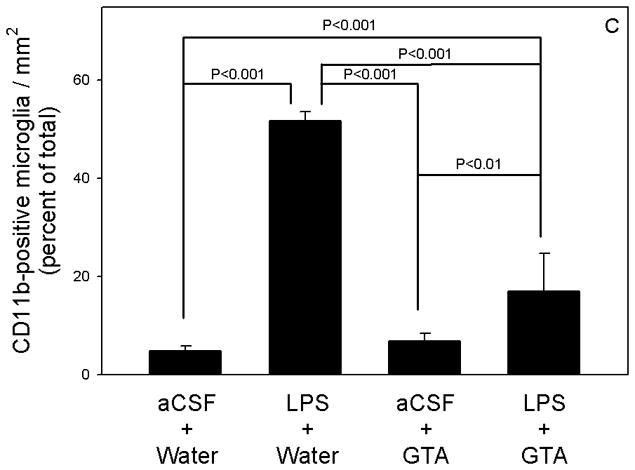
Quantification of activated microglia in control, LPS-treated, GTA-treated, and LPS+GTA-treated rats. Representative images of CD11b-positive microglia in control (panel A, scale bar is equal to 100 μm) and LPS-treated (panel B) rat brain. The bar graph (panel C) represents the averaged percent of total activated CD11b-positive microglia as determined from three independent measurements. A one-way analysis or variance was performed comparing the percentage change in activated microglia in the four different groups. All values represent the means ± SEM (n = 6 samples per group).
Because LPS-induced neuroinflammation also results in a dose-dependent increase in the percentage of reactive astrocytes (unpublished results) we quantified the percentage of reactive astrocytes in rats subjected to aCSF or LPS and treated with a daily dose of either water or GTA (6 g/kg) (Figure 4a and 4b). The measurements showed that the daily oral gavage with either water or GTA did not significantly increase the percentage of reactive astrocytes in rats subjected to a 28 day continual intra-ventricular infusion of aCSF (Figure 4c). Rats given a daily oral gavage of water and subjected to LPS-induced neuroinflammation demonstrated a significant 4-fold increase in the percentage of reactive astrocytes above aCSF control rats treated with water. On the other hand, rats given a daily oral dose of GTA and subjected to LPS-induced neuroinflammation demonstrated that the percentage of reactive astrocytes was reduced to levels that were not significantly different from aCSF control rats treated with GTA, but remained significantly elevated compared to aCSF control rats treated with water. Because the initiating event of LPS-induced neuroinflammation is thought to occur through LPS binding to the toll-like receptor 4 (TLR4) complex on microglial cells (Latz et al. 2003, Buchanan et al. 2010), our data suggest that the anti-inflammatory activity of acetyl-CoA derived from acetate supplementation results in the attenuation of LPS-induced microglial activation and the subsequent reactive astrogliosis.
Figure 4.



Quantification of reactive astroglia in control, LPS-treated, GTA-treated, and LPS+GTA-treated rats. Representative images of GFAP-positive astroglia in control (panel A, scale bar is equal to 100 μm) and LPS-treated (panel B) rat brain. The bar graph (panel C) represents the averaged percent of total reactive GFAP-positive astroglia as determined from three independent measurements. A one-way analysis or variance was performed comparing the percentage change in reactive astroglia in the four different groups. All values represent the means ± SEM (n = 6 samples per group).
GTA restores basal forebrain ChAT immunoreactivity in rats subjected to neuroinflammation
One of the main hallmarks associated with this model is the temporal decrease in cholinergic immunoreactivity in the basal forebrain indicating a decrease in cholinergic activity (Hauss-Wegrzyniak et al. 1998). To determine the effect that acetate supplementation had on basal forebrain ChAT immunoreactivity we compared the number of cholinergic cells in aCSF control rats and LPS-treated rats given either a daily oral dose of water or GTA (6 g/kg) (Figure 5a and 5b). These measurements show that aCSF control rats given a daily oral dose of either water or GTA did not result in a significant decline in the number of ChAT-positive cells in the basal forebrain (Figure 5c). Rats subjected to LPS-induced neuroinflammation that were given a daily oral dose of water demonstrated a significant 2-fold decline in the number of ChAT-positive cells within the basal forebrain. The number of ChAT-positive neurons in those rats given a daily oral dose of GTA and subjected to LPS-induced neuroinflammation was significantly greater then rats subjected to LPS-induced neuroinflammation, and did not differ significantly from aCSF control rats given either water or GTA. These data show that acetate supplementation can increase ChAT immunoreactivity in the basal forebrain of rats subjected to neuroinflammation suggesting that acetate supplementation can recover acetylcholine esterase activity by stimulating acetylcholine formation or by reducing cholinergic cell loss.
Figure 5.



Quantification cholinergic cell loss in control, LPS-treated, GTA-treated, and LPS+GTA-treated rats. Representative images of ChAT-positive cells found in the basal forebrain in control (panel A, scale bar is equal to 100 μm) and LPS-treated (panel B) rat brain. The bar graph (panel C) represents the averaged total number of activated ChAT-positive cell in the basal forebrain as determined from three independent measurements. A one-way analysis or variance was performed comparing the percentage change in activated microglia in the four different groups. All values represent the means ± SEM (n = 6 samples per group).
DISCUSSION
At the onset of these studies we proposed that acetate supplementation acts through a mechanism involving short chain acetyl-CoA metabolism and inhibition of the brain inflammatory reaction. To test this hypothesis, we quantified the conversion of acetate to acetyl-CoA in brain and measured the effectiveness of acetate to reduce established markers of neuroinflammation. These studies demonstrate that GTA-derived acetate is converted to acetyl-CoA in brain, liver, and heart. Further, daily treatment with oral GTA significantly reduced neuroglial reactivity and cholinergic cell loss in this model of neuroinflammation.
The importance of energy supplementation as a therapeutic strategy for neurodegenerative disorders has long been appreciated (Mayne et al. 2001, Guzman & Blazquez 2004, Cullingford 2004). Energy homeostasis is achieved by the integration of lipid energy metabolism with protein and carbohydrate metabolism (Fukao et al. 2004). However, the brain cannot utilize substantial amounts of fatty acid for energy. In contrast, ketone bodies enter the brain and can serve as a major source of energy during fasting (Owen et al. 1967). Similarly, acetate enters the brain and is converted to acetyl-CoA via acetyl-CoA synthetases (Ariyannur et al. 2010, Fujino et al. 2001). In this scenario, mitochondrial acetate requires a single step conversion to acetyl-CoA. This increase in acetyl-CoA can increase the subsequent TCA cycle intermediate flux to generate ATP and phosphocreatine, be exported out of the mitochondria as citrate for fatty acid synthesis, or used to supply glycogen synthesis.
Because mitochondria play a major role in cellular bioenergetics, intermediary metabolism, calcium buffering, signaling cascades, and apoptosis (Nicholls 2002), the structure and function of mitochondria are tightly coupled and their performance relies on mitochondrial integrity (Morais & De Strooper 2010). Mitochondrial toxins selectively cause oxidative phosphorylation deficits and impairment of ATP production. The “slow or weak progressive excitotoxicity” theory for neurodegeneration advocates a primary defect in mitochondrial energy metabolism (Albin & Greenamyre 1992, Beal 1992). Inability to maintain cellular ATP has been linked to stimulation of excitotoxicity, oxidative mitochondrial damage, and aging (Novelli et al. 1988). Normal ATP levels are required to drive sodium/potassium ATPase and thus maintain the resting membrane potential of neurons. However, cellular energy disruption, resulting from ATP depletion, leads to a partial depolarization of neurons, activation of NMDA receptors, and subsequent excitotoxic neural cell-death (Novelli et al. 1988). Lower cellular energy stores can also disturb mitochondrial calcium buffering capacity, increase reactive oxygen species generation, and induce apoptosis (Hartley et al. 1993). Failure to maintain normal mitochondrial metabolism can initiate numerous detrimental processes leading to free radical generation, mutation of mitochondrial DNA, and activation of nitric oxide synthase that can further augment neuroinflammation (Beal 1995). Thus, it is important to maintain energy stores and mitochondrial metabolism in order to limit the progression of neurodegenerative events.
Mitochondrial dysfunction and neuroinflammation are thought to synergistically activate a cycle of deleterious events leading to neuronal death (Di Filippo et al. 2010). Microglia, the primary mediators of neuroinflammation, release potentially harmful factors, including reactive oxygen species and pro-inflammatory cytokines, that can damage mitochondria (Block et al. 2007). It is known that neural mechanisms are involved in inflammatory responses in brain (Tyagi et al. 2008) and that the outcome of certain neurodegenerative disorders are influenced by the balance between pro- and anti-inflammatory mediators (O’Shea et al. 2002). Because the metabolism of acetate in the brain is primarily centered in the astrocyte (Hertz et al. 2007, Muir et al. 1986) and that astrocytes influence microglial activation (Kim et al. 2010), suggests that the protective effects of acetate supplementation in this model may occur by a disruption of inflammatory neuroglia communication. The results showing the ability of acetate to completely attenuate LPS-induced astroglia activation to control levels, while only reducing microglia activation supports this premise. Mitochondrial disruption in microglia cell culture stimulated with LPS disrupts the balance between pro- and anti-inflammatory cytokine production suggesting that mitochondrial integrity is necessary to support injury resolution (Ferger et al. 2010). Furthermore, primary mitochondrial dysfunction resulting from exposure to neurotoxins induce microglial activation (Di Filippo et al. 2010) and several lines of evidence have demonstrated independently that microglial activation and mitochondrial dysfunction directly trigger neurodegeneration (Block et al. 2007, Beal 2005). Pharmacological inhibition of key mitochondrial components is also widely used to induce neuroinflammation and neurodegeneration in experimental models (Di Filippo et al. 2006). Taken together, these data provide a reasonable rationale to suggest that impaired mitochondrial metabolism and its influence on neuroglial communication may be an essential link between neuroinflammation and neurodegeneration.
In conclusion, GTA-derived acetate significantly increases the brain levels of acetyl-CoA and is effective at reducing neuroglial activation and restoring ChAT immunoreactivity to control levels in a rat model of LPS-induced neuroinflammation. Our results support those studies demonstrating that acetate supplementation is effective at reducing tremor associated with Canavan’s disease and at reducing injury in a rat model of traumatic brain injury. The results outlined in this study suggest that another plausible mechanism of action to be considered in parallel to alteration in brain energy supply and lipid synthesis should also include acetates influence on inflammatory phenotype.
Acknowledgments
This publication was made possible by a Grant from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) (# P20RR17699-05).
Abbreviations
- GTA
glyceryl triacetate
- aCSF
artificial cerebral spinal fluid
- LPS
lipopolysaccharide
- ChAT
choline acetyltransferase
- GFAP
glial fibrillary acidic protein
- CoA
co-enzyme A
References
- Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42:733–738. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- Amaducci L, Forno KI, Eng LF. Glial fibrillary acidic protein in cryogenic lesions of the rat brain. Neurosci Lett. 1981;21:27–32. doi: 10.1016/0304-3940(81)90052-5. [DOI] [PubMed] [Google Scholar]
- Ariyannur PS, Moffett JR, Madhavarao CN, Arun P, Vishnu N, Jacobowitz DM, Hallows WC, Denu JM, Namboodiri AM. Nuclear-cytoplasmic localization of acetyl coenzyme a synthetase-1 in the rat brain. J Comp Neurol. 2010;518:2952–2977. doi: 10.1002/cne.22373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Ariyannur PS, Moffett JR, Xing G, Hamilton K, Grunberg NE, Ives JA, Namboodiri AM. Metabolic acetate therapy for the treatment of traumatic brain injury. J Neurotrauma. 2010a;27:293–298. doi: 10.1089/neu.2009.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Madhavarao CN, Moffett JR, et al. Metabolic acetate therapy improves phenotype in the tremor rat model of Canavan disease. J Inherit Metab Dis. 2010b;33:195–210. doi: 10.1007/s10545-010-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub AE, Salm AK. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J Neurosci. 2003;23:7759–7766. doi: 10.1523/JNEUROSCI.23-21-07759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Scheff SW. Intermediate filament change in astrocytes following mild cortical contusion. Glia. 1996;16:266–275. doi: 10.1002/(SICI)1098-1136(199603)16:3<266::AID-GLIA9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Basselin M, Chang L, Seemann R, Bell JM, Rapoport SI. Chronic lithium administration potentiates brain arachidonic acid signaling at rest and during cholinergic activation in awake rats. J Neurochem. 2003;85:1553–1562. doi: 10.1046/j.1471-4159.2003.01811.x. [DOI] [PubMed] [Google Scholar]
- Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PP, Jacobsson B, Collins VP, Biberfeld P. Unmasking antigen determinants in amyloid. J Histochem Cytochem. 1986;34:1683–1685. doi: 10.1177/34.12.2431032. [DOI] [PubMed] [Google Scholar]
- Cullingford TE. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot Essent Fatty Acids. 2004;70:253–264. doi: 10.1016/j.plefa.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Davis EJ, Foster TD, Thomas WE. Cellular forms and functions of brain microglia. Brain Res Bull. 1994;34:73–78. doi: 10.1016/0361-9230(94)90189-9. [DOI] [PubMed] [Google Scholar]
- Deutsch J, Rapoport SI, Rosenberger TA. Coenzyme A and short-chain acyl-CoA species in control and ischemic rat brain. Neurochem Res. 2002;27:1577–1582. doi: 10.1023/a:1021614422668. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis. 2010;20(Suppl 2):S369–379. doi: 10.3233/JAD-2010-100543. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Picconi B, Costa C, Bagetta V, Tantucci M, Parnetti L, Calabresi P. Pathways of neurodegeneration and experimental models of basal ganglia disorders: downstream effects of mitochondrial inhibition. Eur J Pharmacol. 2006;545:65–72. doi: 10.1016/j.ejphar.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Durst HD, Milano M, Kikta EJ, Jr, Connelly SA, Grushka E. Phenacyl esters of fatty acids via crown ether catalysts for enhanced ultraviolet detection in liquid chromatography. Anal Chem. 1975;47:1797–1801. doi: 10.1021/ac60361a025. [DOI] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:8880–8885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA, Farooqui T. Deacylation and reacylation of neural membrane glycerophospholipids. J Mol Neurosci. 2000;14:123–135. doi: 10.1385/jmn:14:3:123. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Rapoport SI, Horrocks LA. Membrane phospholipid alterations in Alzheimer’s disease: deficiency of ethanolamine plasmalogens. Neurochem Res. 1997;22:523–527. doi: 10.1023/a:1027380331807. [DOI] [PubMed] [Google Scholar]
- Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, Ludolph AC, Witting A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J Neuroinflammation. 2010;7:45. doi: 10.1186/1742-2094-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem. 2001;276:11420–11426. doi: 10.1074/jbc.M008782200. [DOI] [PubMed] [Google Scholar]
- Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. 2004;70:243–251. doi: 10.1016/j.plefa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Bonnekoh P, Miyazawa T, Hossmann KA, Kreutzberg GW. Immunocytochemical study of an early microglial activation in ischemia. J Cereb Blood Flow Metab. 1992;12:257–269. doi: 10.1038/jcbfm.1992.36. [DOI] [PubMed] [Google Scholar]
- Ginsberg L, Atack JR, Rapoport SI, Gershfeld NL. Evidence for a membrane lipid defect in Alzheimer disease. Mol Chem Neuropathol. 1993;19:37–46. doi: 10.1007/BF03160167. [DOI] [PubMed] [Google Scholar]
- Ginsberg L, Xuereb JH, Gershfeld NL. Membrane instability, plasmalogen content, and Alzheimer’s disease. J Neurochem. 1998;70:2533–2538. doi: 10.1046/j.1471-4159.1998.70062533.x. [DOI] [PubMed] [Google Scholar]
- Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G, Sindelar PJ. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:740–747. doi: 10.1097/00005072-199907000-00008. [DOI] [PubMed] [Google Scholar]
- Guzman M, Blazquez C. Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot Essent Fatty Acids. 2004;70:287–292. doi: 10.1016/j.plefa.2003.05.001. [DOI] [PubMed] [Google Scholar]
- Hamosh M, Scanlon JW, Ganot D, Likel M, Scanlon KB, Hamosh P. Fat digestion in the newborn. Characterization of lipase in gastric aspirates of premature and term infants. J Clin Invest. 1981;67:838–846. doi: 10.1172/JCI110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Holtzman DM, McKeel DW., Jr Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem. 2001;77:1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res. 1998;780:294–303. doi: 10.1016/s0006-8993(97)01215-8. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Vannucchi MG, Wenk GL. Behavioral and ultrastructural changes induced by chronic neuroinflammation in young rats. Brain Res. 2000;859:157–166. doi: 10.1016/s0006-8993(00)01999-5. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Kim JH, Min KJ, Seol W, Jou I, Joe EH. Astrocytes in injury states rapidly produce anti-inflammatory factors and attenuate microglial inflammatory responses. J Neurochem. 2010;115:1161–1171. doi: 10.1111/j.1471-4159.2010.07004.x. [DOI] [PubMed] [Google Scholar]
- Latz E, Visintin A, Lien E, Fitzgerald KA, Espevik T, Golenbock DT. The LPS receptor generates inflammatory signals from the cell surface. J Endotoxin Res. 2003;9:375–380. doi: 10.1179/096805103225003303. [DOI] [PubMed] [Google Scholar]
- Lee H, Villacreses NE, Rapoport SI, Rosenberger TA. In vivo imaging detects a transient increase in brain arachidonic acid metabolism: A potential marker of neuroinflammation. J Neurochem. 2004;91:936–945. doi: 10.1111/j.1471-4159.2004.02786.x. [DOI] [PubMed] [Google Scholar]
- Mathew R, Arun P, Madhavarao CN, Moffett JR, Namboodiri MA. Progress toward acetate supplementation therapy for Canavan disease: glyceryl triacetate administration increases acetate, but not N-acetylaspartate, levels in brain. J Pharmacol Exp Ther. 2005;315:297–303. doi: 10.1124/jpet.105.087536. [DOI] [PubMed] [Google Scholar]
- Mayne M, Fotheringham J, Yan HJ, Power C, Del Bigio MR, Peeling J, Geiger JD. Adenosine A2A receptor activation reduces proinflammatory events and decreases cell death following intracerebral hemorrhage. Ann Neurol. 2001;49:727–735. doi: 10.1002/ana.1010. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- Morais VA, De Strooper B. Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J Alzheimers Dis. 2010;20(Suppl 2):S255–263. doi: 10.3233/JAD-2010-100345. [DOI] [PubMed] [Google Scholar]
- Muir D, Berl S, Clarke DD. Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res. 1986;380:336–340. doi: 10.1016/0006-8993(86)90231-3. [DOI] [PubMed] [Google Scholar]
- Nicholls D. Mitochondrial bioenergetics, aging, and aging-related disease. Sci Aging Knowledge Environ. 2002;2002:pe12. doi: 10.1126/sageke.2002.31.pe12. [DOI] [PubMed] [Google Scholar]
- Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2:37–45. doi: 10.1038/nri702. [DOI] [PubMed] [Google Scholar]
- Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF., Jr Brain metabolism during fasting. J Clin Invest. 1967;46:1589–1595. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoni L, Sarti C, Inzitari D. Cytokines and cell adhesion molecules in cerebral ischemia: experimental bases and therapeutic perspectives. Arterioscel Thromb Vasc Biol. 1998;18:503–513. doi: 10.1161/01.atv.18.4.503. [DOI] [PubMed] [Google Scholar]
- Persson M, Bleiberg B, Kiss D, Miles J. Measurement of plasma acetate kinetics using high-performance liquid chromatography. Anal Biochem. 1991;198:149–153. doi: 10.1016/0003-2697(91)90520-4. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rosenberger TA, Villacreses NE, Hovda JT, Bosetti F, Weerasinghe G, Wine RN, Harry GJ, Rapoport SI. Rat brain arachidonic acid metabolism is increased by a six-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J Neurochem. 2004;88:1168–1178. doi: 10.1046/j.1471-4159.2003.02246.x. [DOI] [PubMed] [Google Scholar]
- Rosenberger TA, Villacreses NE, Weis MT, Rapoport SI. Rat brain docosahexaenoic acid metabolism is not altered by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. Neurochem Int. 2010;56:501–507. doi: 10.1016/j.neuint.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S, Ramirez-Amaya V, Hauss-Wegrzyniak B, Wenk GL. Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors. J Neuroinflammation. 2004;1:12. doi: 10.1186/1742-2094-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- Shi SR, Key ME, Kalra KL. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 1991;39:741–748. doi: 10.1177/39.6.1709656. [DOI] [PubMed] [Google Scholar]
- Traissard N, Herbeaux K, Cosquer B, Jeltsch H, Ferry B, Galani R, Pernon A, Majchrzak M, Cassel JC. Combined damage to entorhinal cortex and cholinergic basal forebrain neurons, two early neurodegenerative features accompanying Alzheimer’s disease: effects on locomotor activity and memory functions in rats. Neuropsychopharmacology. 2007;32:851–871. doi: 10.1038/sj.npp.1301116. [DOI] [PubMed] [Google Scholar]
- Tyagi E, Agrawal R, Nath C, Shukla R. Influence of LPS-induced neuroinflammation on acetylcholinesterase activity in rat brain. J Neuroimmunol. 2008;205:51–56. doi: 10.1016/j.jneuroim.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke. 1999;30:1969–1973. doi: 10.1161/01.str.30.9.1969. [DOI] [PubMed] [Google Scholar]
- Weber C, Erl W, Weber KS, Weber PC. HMG-CoA reductase inhibitors decrease CD11b expression and CD11b- dependent adhesion of monocytes to endothelium and reduce increased adhesiveness of monocytes isolated from patients with hypercholesterolemia. J Am Coll Cardiol. 1997;30:1212–1217. doi: 10.1016/s0735-1097(97)00324-0. [DOI] [PubMed] [Google Scholar]
- Willard LB, Hauss-Wegrzyniak B, Wenk GL. Pathological and biochemical consequences of acute and chronic neuroinflammation within the basal forebrain cholinergic system of rats. Neuroscience. 1999;88:193–200. doi: 10.1016/s0306-4522(98)00216-4. [DOI] [PubMed] [Google Scholar]