

Stacking interactions between aromatic amino acids and nucleic acid bases are common in complexes formed with single-stranded nucleic acids, including RNA-protein complexes.1 Investigations involving non-polar base analogs have revealed the ability of dispersive forces and solvation effects to stabilize stacking interactions in helical structures and nucleic acid-protein complexes.2 A useful characteristic of aromatic groups is the ability to vary their electronic properties. Although investigations with small molecules suggest that varying electronic properties of aromatic groups affects the strength of stacking interactions,3 such correlations in biological systems have been unclear,2a, 4 and studies with RNA-protein complexes have not been conducted.
The RNA Recognition Motif (RRM) is the most common RNA binding domain in higher vertebrates and is found in all organisms.5a The most conserved interactions between RRMs and RNA are stacking interactions. Thus, we have used an RRM-RNA complex to vary the electronic properties of aromatic groups involved in stacking interactions. We have found that variation of the pattern of fluorination of a base analog in stem loop 2 (SL2) of U1 snRNA changes the stability of the complex formed with the N-terminal RRM of the U1A protein by 4.3 kcal/mol. High-level calculations suggest a correlation between the strength of the stacking interaction and the stability of the U1A-SL2 RNA complex.
U1A is a component of the U1 snRNP, a subunit of the spliceosome, and regulates polyadenylation of U1A and other pre-mRNAs.5b–5e The focus of these investigations is the conserved stacking interaction formed between Tyr13 of the U1A protein and C5 of SL2 RNA (Figure 1). Hydrogen bonds are formed between the NH2 of C5 and the side chain of Gln85 and the amide backbone of Tyr86 and between O2 and N3 of C5 and the amide backbone of Lys88.5f The substitution of Tyr13 with Phe or Thr results in a 3.6 or 4.6 kcal/mol destabilization of the complex, respectively,5g while the substitution of C5 with U results in a 2.3 kcal/mol destabilization of the complex.5h
Figure 1.
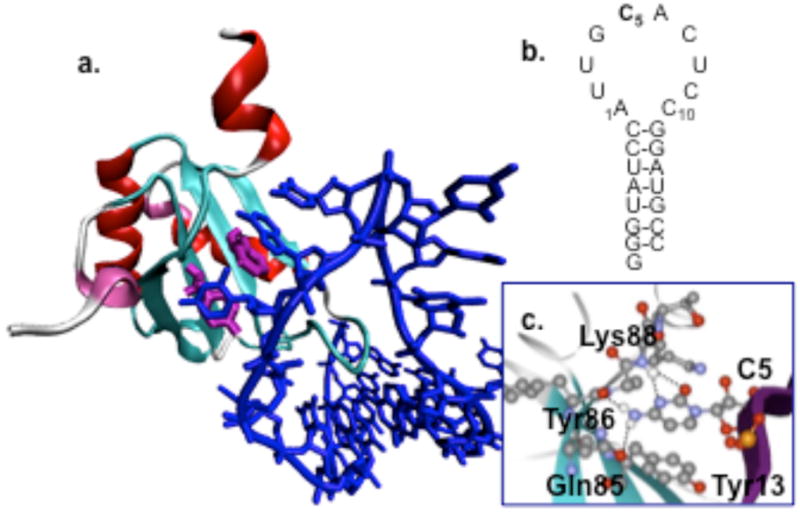
(a) The U1A-SL2 RNA complex from the X-ray cocrystal structure.5f (b) SL2 RNA. (c) An expanded view of the structure.
To investigate whether varying the electronic character of the aromatic rings involved in stacking interactions could be used to modulate the stability of the U1A-RNA complex, we replaced C5 with a series of phenyl base analogs containing different fluorine substitution patterns (Figure 2). Fluorine reduces electron density in the pi system, but is small and forms considerably weaker hydrogen bonds than the natural bases.4a, 6 These alterations may perturb the structure of the free RNA or the complex and thus, affect binding affinity independently of the stacking interaction with Tyr13. However, structural studies have shown little perturbation of helical structure upon incorporation of phenyl-derived base analogs.7 We reasoned that structural perturbations would be likely to be relatively constant across the series of phenyl base analogs, allowing an evaluation of the sensitivity of complex stability to varying electronic properties.
Figure 2.
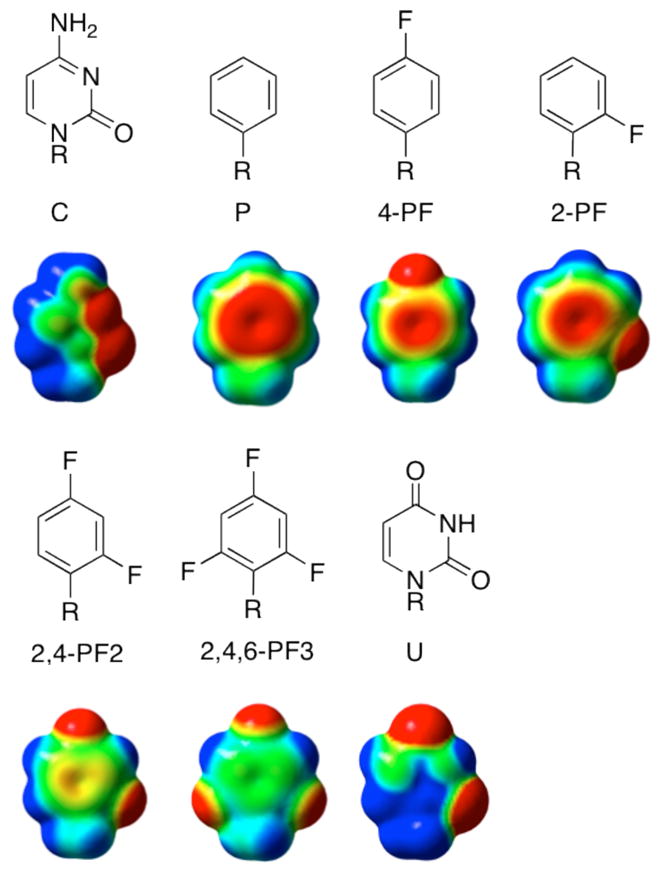
Phenyl and fluorinated phenyl base analogs. Below each structure is shown calculated electrostatic surface potentials for each base or base analog. The electrostatic surface potentials were calculated for structures in which R=Me, using MP2 6-31G*(0.25)//B3LYP6-31G(d).
Phosphoramidites of the phenyl base analogs were synthesized and incorporated into SL2 RNA.6c, 7b, 8 The synthesis of the phosphoramidite of the 2,4,6-trifluorophenyl base analog has not previously been reported. The stability of SL2 RNA, measured by temperature melt analyses, was not altered by these base analogs. SL2 RNAs substituted with phenyl and 4-flurophenyl base analogs bound with similar weak affinities to U1A protein (Table 1). Substitution of C5 with 2,4-difluorophenyl or 2-fluorophenyl groups resulted in a large gain of binding energy compared to phenyl substitution, 4.3 and 3.7 kcal/mol, respectively. 2- and 4-fluorophenyl base analogs have similar polarizability, size, and hydrophobicity, yet the stabilities of complexes containing these two analogs differ by 2.8 kcal/mol. It is unlikely that a hydrogen bond formed between 2-F and the amide backbone of Lys88 or another residue is responsible for the large difference in stability of the complexes because hydrogen bonds with F are weak.4a, 6 It is also unlikely that unfavorable interactions involving 4-F are responsible for the decreased stability of this complex because the most stable complex in the series contains the 2,4-difluorophenyl base analog.
Table 1.
Binding affinity of wild type and modified RNAs to U1A and calculated properties of the free base analogs.
| SL2 RNA | Kd (M)a | ΔG (kcal/mol) | ΔΔG (kcal/mol)a | Log Pc(water-octanol) | Polarizability (Å3)d | Surface Area (Å2)e | Dipole moment (D) f | Quadrupole moment (B) |
|---|---|---|---|---|---|---|---|---|
| Wild type | 3 ± 2 × 10−10 | −13.1 ± 0.3 | ||||||
| P | 4 ± 3 × 10−6 | −7.4 ± 0.3 | 5.7 | 2.20 | 9.5 | 222.83 | 0.34 | −10.03 |
| 4-PF | 9 ± 1 × 10−7 | −8.3 ± 0.1 | 4.8 | 2.34 | 9.5 | 223.69 | 2.76 | −6.27 |
| 2-PF | 8 ± 6 × 10−9 | −11.1 ± 0.4 | 2.0 | 2.34 | 9.5 | 223.68 | 2.16 | −7.75 |
| 2,4-PF2 | 3 ± 1 × 10−9 | −11.7 ± 0.2 | 1.4 | 2.48 | 9.5 | 225.52 | 2.63 | −3.10 |
| 2,4,6-PF3 | 1.5 ± 0.4 × 10−7 | −9.3 ± 0.2 | 3.8 | 2.62 | 9.5 | 228.27 | 0.52 | 0.82 |
Kd values were measured by gel mobility shift assays and are the average of at least three independent experiments.
ΔΔG is the difference in affinity between the indicated complex and the wild type complex.
RHF/6-31G**//RHF6-31G** was used with methylated base analogs.
Base analogs contained a hydrogen in place of ribose.
Calculations used methylated bases in water using the PCM with Gaussian 03 using MP2 6-31G* (0.25)//B3LYP 6-31G(d).
We calculated properties of the fluoroaryl groups that often affect stacking interactions, including log P,2b, 9 polarizability, surface area, quadrupole moment, and dipole moment, to reveal whether the origins of the effects of fluorophenyl substitutions on complex stability may be related to effects on stacking interactions (Table 1). The changes in binding affinities do not correlate well with any single parameter, even when taking into account the direction of the dipole moment.
To evaluate stacking interactions between the base analogs and tyrosine, while effectively including many of the parameters in Table 1 simultaneously, we performed high level ab initio calculations using the polarizable continuum model (PCM) with Gaussian 03 using MP2 6-31G*(0.25). These types of calculations have been found to be accurate in reproducing trends in stacking affinities of nucleic acid bases.10 The calculations were performed on the isolated tyrosine side chain and fluorophenyl group interacting within a geometry similar to that found in the X-ray structure of the complex. The calculated energies (ΔΔEs) reproduce the significant differences in stability between complexes containing phenyl or 4-fluorophenyl base analogs and those containing 2-fluorophenyl or 2,4-difluorophenyl base analogs (Table 2). Not surprisingly, the magnitudes of the experimentally determined ΔΔG’s and the calculated ΔΔE’s differ, which could result from changes in the structure of the complex or RNA upon incorporation of the base analogs and/or entropic effects that are not included in the calculations. Nevertheless, the calculations suggest that differences in stacking interactions may contribute to the sensitivity of complex stability to fluorination position.
Table 2.
Comparison of measured binding affinities and calculated stacking energies with Tyr13.
| RNA | ΔG (kcal/mol) | ΔΔG (kcal/mol)a | Stacking energy (kcal/mol)b,c | ΔΔE (kcal/mol)a |
|---|---|---|---|---|
| P | −7.4 ± 0.3 | 0 | −1.84 | 0 |
| 4-PF | −8.3 ± 0.1 | −0.9 | −2.57 | −0.7 |
| 2-PF | −11.1 ± 0.4 | −3.7 | −3.43 | −1.6 |
| 2,4-PF2 | −11.7 ± 0.2 | −4.3 | −4.18 | −2.3 |
| 2,4,6-PF3 | −9.3 ± 0.2 | −1.9 | −4.09 | −2.2 |
ΔΔG and ΔΔE are the differences in energy of the complexes containing P and the indicated base analog.
Calculations were performed in water using the PCM with Gaussian 03 using MP2 6-31G*(0.25)//B3LYP6-31G(d).
The stacking energy was obtained by subtracting the Basis Set Superposition Error (BSSE) energy calculated in vacuum from the stacking energy in water. For these calculations the methylene of tyrosine and the ribose were replaced with methyl groups.
Previous investigations of fluorinated phenyl base analogs in DNA and RNA helices have suggested that stacking interactions can be modulated by the position of fluorination. Incorporation of a series of fluorinated phenyl base analogs into the 5′-end of a DNA helix resulted in a maximum stabilization of the duplex of 1.3 kcal/mol.2a Similar to the results discussed in this paper, correlations between quadrupole moment, dipole moment, surface area, or the number of fluorines substituted were not observed. The authors suggested an important role for dispersive effects along with dipolar interactions for DNA base stacking. Investigations of RNA duplexes containing 2-fluorophenyl, 4-fluorophenyl, or 2,4-difluorophenyl base analogs paired with non-polar or natural bases suggested the stacking energy with natural bases increased with increasing fluorination with a difference of 0.9 kcal/mol between the stacking of phenyl and 2,4-difluorophenyl and that 2-fluorophenyl may form slightly stronger stacking interactions than 4-fluorophenyl (0.2–1 kcal/mol).6c,8b The results of these investigations are consistent with those reported here on the U1A-RNA complex and taken together, they reveal a complex relationship between fluorination and the stability of biological complexes.
In conclusion, we have shown that the stability of an RRM-RNA complex is surprisingly sensitive to the position of fluorine substitution of a phenyl base analog in the RNA target site. Changing the position of one fluorine group alters the stability of the complex by 2.8 kcal/mol. The correlation between experimental results and high level calculations suggests that changes in the stacking interaction may contribute to the observed alteration of complex stability. These results support an approach of modifying the electronic character of aromatic groups to modulate the stability of complexes involving stacking interactions.
Supplementary Material
Acknowledgments
We are grateful to Prof. Petersson and Prof. Novick for discussions on the calculations and to Prof. K. Nagai for the expression vector for U1A. Funding was provided by NSF (MCB-1019958), NIH (GM-056857) and the Petroleum Research Fund.
Footnotes
SUPPORTING INFORMATION. Description of syntheses, binding reactions, and calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Allers J, Shamoo Y. J Mol Biol. 2001;311:75. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]; (b) Baker CM, Grant GH. Biopolymers. 2007;85:456. doi: 10.1002/bip.20682. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lai JS, Qu J, Kool ET. Angew Chem. 2003;42:5973. doi: 10.1002/anie.200352531. [DOI] [PubMed] [Google Scholar]; (b) Guckian KM, Schweitzer BA, Ren RX, Sheils CJ, Tahmassebi DC, Kool ET. J Am Chem Soc. 2000;122:2213. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Cozzi F, Ponzini F, Annunziata R, Cinquini M, Siegel JS. Angew Chem. 1995;34:1019. [Google Scholar]; (b) Collins SK, El-Azizi Y, Schmitzer AR. J Org Chem. 2007;72:6397. doi: 10.1021/jo070568r. [DOI] [PubMed] [Google Scholar]; (c) Zakarian JE, El-Azizi Y, Collins SK. Org Lett. 2008;10:2927. doi: 10.1021/ol800821f. [DOI] [PubMed] [Google Scholar]; (d) Cockroft SL, Hunter CA, Lawson KR, Perkins J, Urch CJ. J Am Chem Soc. 2005;127:8594. doi: 10.1021/ja050880n. [DOI] [PubMed] [Google Scholar]; (e) Shu L, Müri M, Krupkea R, Mayor M. Org Biomol Chem. 2009;7:1081–1092. doi: 10.1039/b817274a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kopitz H, Zivkovic A, Engels JW, Gohlke H. Chem Bio Chem. 2008;9:2619. doi: 10.1002/cbic.200800461. [DOI] [PubMed] [Google Scholar]; (b) Waters ML. Biopolymers. 2004;76:435. doi: 10.1002/bip.20144. [DOI] [PubMed] [Google Scholar]; (c) Zahn A, Brotschi C, Leumann CJ. Chem Eur J. 2005;11:2125. doi: 10.1002/chem.200401128. [DOI] [PubMed] [Google Scholar]; (d) Mathis G, Hunziker J. Angew Chem Int Ed. 2002;41:3203. doi: 10.1002/1521-3773(20020902)41:17<3203::AID-ANIE3203>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; (e) Gorske BC, Blackwell HE. J Am Chem Soc. 2006;128:14378. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- 5.(a) Cléry A, Blatter M, Allain FHT. Curr Opin Struct Biol. 2008;18:290. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]; (b) Green MR. Annu Rev Cell Biol. 1991;7:559. doi: 10.1146/annurev.cb.07.110191.003015. [DOI] [PubMed] [Google Scholar]; (c) Boelens WC, Jansen EJR, van Venrooij WJ, Stripecke R, Mattaj IW, Gunderson SI. Cell. 1993;72:881. doi: 10.1016/0092-8674(93)90577-d. [DOI] [PubMed] [Google Scholar]; (d) Liang S, Lutz CS. RNA. 2006;12:111. doi: 10.1261/rna.2213506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Phillips C, Pachikara N, Gunderson SI. Mol Cell Biol. 2004;24:6162. doi: 10.1128/MCB.24.14.6162-6171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Nature. 1994;372:432. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]; (g) Kranz JK, Lu J, Hall KB. Protein Sci. 1996;5:1567. doi: 10.1002/pro.5560050812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Showalter SA, Hall KB. J Mol Biol. 2004;335:465. doi: 10.1016/j.jmb.2003.10.055. [DOI] [PubMed] [Google Scholar]
- 6.(a) Zhou P, Zou J, Tian F, Shang Z. J Chem Inf Model. 2009;49:2344. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]; (b) Kool ET, Sintim HO. Chem Comm. 2006:3665. doi: 10.1039/b605414e. [DOI] [PubMed] [Google Scholar]; (c) Parsch J, Engels JW. J Am Chem Soc. 2002;124:5664. doi: 10.1021/ja012116g. [DOI] [PubMed] [Google Scholar]; (d) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 7.(a) Guckian KM, Krugh TR, Kool ET. Nature Struct Biol. 1998;5:954. doi: 10.1038/2930. [DOI] [PubMed] [Google Scholar]; (b) Minasov G, Matulic-Adamic J, Wilds CJ, Haeberli P, Usamn N, Beigelman L, Egli M. RNA. 2000;6:1516. doi: 10.1017/s1355838200001114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guckian KM, Krugh TR, Kool ET. J Am Chem Soc. 2000;122:6841. doi: 10.1021/ja994164v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zacharias M, Engels JW. Nucleic Acids Res. 2004;32:6304. doi: 10.1093/nar/gkh971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Jensen SH, Limberg G, Pedersen C. Carbohydrate Res. 1997;302:109. [Google Scholar]; (b) Parsch J, Engels JW. Helv Chim Acta. 2000;83:1791. [Google Scholar]
- 9.Ghose AK, Crippen GM. J Comp Chem. 1986;7:565. [Google Scholar]
- 10.(a) Sponer J, Hobza P. Chem Phys Lett. 1997;267:263. [Google Scholar]; (b) Sponer J, Leszczynski J, Hobza P. Biopolymers. 2002;61:3. doi: 10.1002/1097-0282(2001)61:1<3::AID-BIP10048>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (c) Churchill CDM, Wetmore SD. J Phys Chem B. 2009;113:16046. doi: 10.1021/jp907887y. [DOI] [PubMed] [Google Scholar]; (d) Rutlege LR, Durst HF, Wetmore SD. J Chem Theory Comput. 2009;5:1400. doi: 10.1021/ct800567q. [DOI] [PubMed] [Google Scholar]; (e) Gung BW, Amicangelo JC. J Org Chem. 2006;71:9261. doi: 10.1021/jo061235h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.