

Abstract
We used whole-exome sequencing to study three individuals with a distinct condition characterized by short stature, chondrodysplasia with brachydactyly, congenital joint dislocations, cleft palate, and facial dysmorphism. Affected individuals carried homozygous missense mutations in IMPAD1, the gene coding for gPAPP, a Golgi-resident nucleotide phosphatase that hydrolyzes phosphoadenosine phosphate (PAP), the byproduct of sulfotransferase reactions, to AMP. The mutations affected residues in or adjacent to the phosphatase active site and are predicted to impair enzyme activity. A fourth unrelated patient was subsequently found to be homozygous for a premature termination codon in IMPAD1. Impad1 inactivation in mice has previously been shown to produce chondrodysplasia with abnormal joint formation and impaired proteoglycan sulfation. The human chondrodysplasia associated with gPAPP deficiency joins a growing number of skeletoarticular conditions associated with defective synthesis of sulfated proteoglycans, highlighting the importance of proteoglycans in the development of skeletal elements and joints.
Main Text
Among the genetic disorders of the skeleton, there is a cluster of disorders with common features of short stature, dysplasia of skeletal elements, and congenital joint dislocations that includes Larsen syndrome, Desbuquois dysplasia, diastrophic dysplasia, and pseudodiastrophic dysplasia (MIM 150250, 245600, 251450, 222600, 264180, and 603799, respectively). Whereas classic (dominant) Larsen syndrome is caused by mutations in the intracellular filamin B,1,2 recessive Larsen syndrome and diastrophic dysplasia are caused by defective sulfation of proteoglycans.3–6 The function of CANT1 (MIM 613165), the nucleotide phosphatase homozygously mutated in some individuals with Desbuquois syndrome, is not precisely characterized but is likely related to the cellular handling of sugar nucleotides and thus to proteoglycan synthesis.7,8 Here, we report on a condition consisting of congenital joint dislocations, chondrodysplasia with short stature, micrognathia and cleft palate, and a distinctive face that is caused by homozygous mutations in IMPAD1 (NM_017813.3), the gene encoding Golgi-resident PAP phosphatase (gPAPP), which is responsible for the hydrolysis of phosphoadenosine phosphate (PAP) to AMP.9
We first studied three patients from two unrelated consanguineous families. All studies were conducted within a research project approved by the institutional ethics board (see Acknowledgments). Patient 1 was a singleton case born to German parents who were first-degree cousins. She was referred at age 23 for genetic counseling because of unclassified skeletal dysplasia. Her personal history included reduced length at birth with shortening and “deformity” of her limbs (medical records unavailable), micrognathia with congenital posterior cleft of the palate (surgically repaired in infancy), and surgery for recurrent patellar dislocation at age 16. She had been diagnosed with hypoacusis in childhood and had worn hearing aids since that time. Her adult height was 127 cm, and she had shortening of the limbs. Her forehead was high, and her nasal root was broad. There was brachydactyly, most marked in digits I and III bilaterally, and the thumb had a “hitch-hiker” appearance similar to that seen in diastrophic dysplasia (Figure 1D). There was limited supination at the elbow. Both knees showed valgus deformity. The feet were adducted and short, and they lacked arching. The toes were short (Figure 1E). Her development was not formally tested but was estimated to be in the lower range of normal. Radiographic findings are shown in Figure 2. No vertebral changes were observed. Noteworthy is the brachydactyly with inhomogeneity of the size of the metacarpal epiphyses (the epiphysis of metacarpal II being significantly larger), longitudinal splitting of the proximal phalanx of digit I, and fusion of the capitate and hamate bones (Figure 2A).
Figure 1.
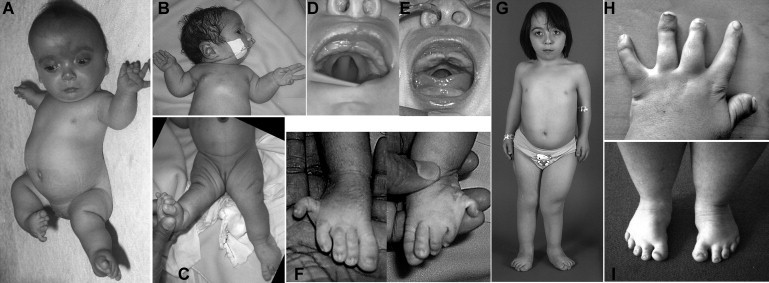
Clinical Features of Individuals with IMPAD1 Mutations
(A) Patient 2 at age 2 months. A high forehead with a midline hemangioma, a broad nasal bridge, a small mouth, short stature, bilateral anterolateral dislocation of the knees, and lateral clinodactyly of the fifth toes can be seen.
(B and C) Patient 4 at age 2 days, with severe retrognathia, limited extension of the elbows, and knee dislocation.
(D and E) The median posterior palatal cleft in patients 2 and 4.
(F) The proximal position and lateral deviation of the fifth toes in patient 4 (compare with A).
(G) Patient 2 at age 9 yrs. Her height was 97 cm and thus far below the 3rd percentile for her age. The hands and feet are short, there are multiple surgical scars, and the right leg is shorter than the left because of hip dislocation.
(H and I) Hands and feet of patient 1 at age 23 years. Note the marked brachydactyly associated with shortened metacarpals, as well as the proximal insertion and radial deviation of digit I (so-called “hitch-hiker thumb”). The feet are short, with forefoot adduction.
Figure 2.
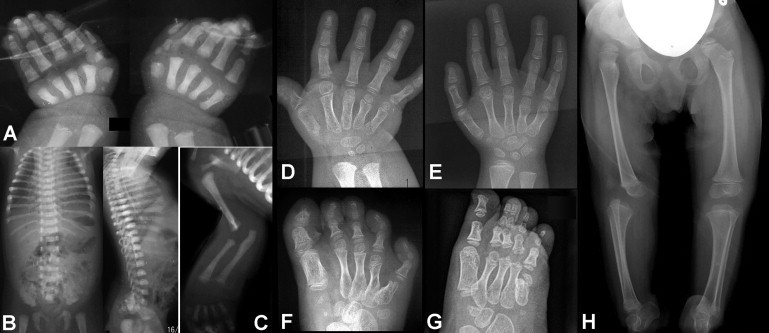
Radiographic Features of Individuals with IMPAD1 Mutations
(A) Hand X-rays of patient 4 at birth show shortening of all metacarpals, the presence of an accessory ossification center at the base of metacarpal 1 (left hand), and splitting of the proximal phalanx of digit I into two parts.
(B and C) Findings in patient 4, recorded at birth: there is mild platyspondyly but no coronal clefting, and there is elbow joint dysplasia.
(D and E) Hand X-rays of patient 1 (D; age, 2.5 yrs) and patient 2 (E; age, 4 yrs), showing fusion between the hamate and capitate bones, short metacarpals, irregular sizes of the distal metacarpal epiphyses (note the large size of the epiphysis of metacarpal II), and longitudinal splitting of the proximal and distal phalanx of digit 1 (D). The findings are reminiscent of those in Desbuquois dysplasia.8,22,31
(F and G) Foot radiographs of patients 1 and 2, respectively (same ages as the patients in A and B). There are supernumerary tarsal bones; all metatarsal bones are shortened, with metatarsal V being particularly short; note again the difference in size of the distal metatarsal epiphyses in (D). Skeletal age is not accelerated. Clinodactyly of toe V in patient 2 has been corrected by casting.
(H) Pelvis and legs of patient 2 at age 3 yrs, showing proximal dislocation of the right femur and persistence of the anterior dislocation of the right knee. Dislocation of the left knee was also present but was not appreciable on this film.
Patients 2 and 3 were two girls, both offspring of a consanguineous couple of Turkish origin. There was a third, unaffected sister. Patient 2 was born with short stature, micrognathia and posterior cleft palate (the Pierre Robin sequence), bilateral knee dislocation, and marked lateral deviation of her fifth toes. Her forehead was high, her eyes were prominent, her nose and mouth were small, and her mandible was small (Figures 1A and 1C). Coronal craniosynostosis was diagnosed at 1 year of age. Both craniosynostosis and knee dislocations were surgically repaired with various procedures at different ages. There was mild hypoacusis, but speech development was normal. Her intellectual development was moderately delayed. Radiographic findings are shown in Figure 2. The most significant findings were short metacarpals, irregular size of the metacarpal epiphyses, hamate-capitate fusion, and supernumerary carpal ossification centers (Figure 2B); however, carpal age at birth and in the first two years of life was not accelerated. There was dysplasia of the hip acetabulum and the proximal femoral epiphyses, as well as dislocation of the knees (Figure 2E). A moderate reduction of the intervertebral spaces was observed, but unlike in Desbusquois dysplasia or recessive Larsen syndrome, vertebral clefts were not observed. Her younger sister, patient 3, was also born with short stature, micrognathia with a median posterior palatal cleft, and bilateral subluxation of her knees. Both of her fifth toes were laterally deviated. Although her facial appearance resembles that of her affected sister, with a high forehead and a flat face, she has no signs of craniosynostosis. At follow-up examination at the age 2 years, her psychomotor development was within the normal range but her stature was markedly below the third percentile. Radiographic findings were similar to those of her older sister, including brachydactyly, irregular metacarpal epiphyses, and reduction of intervertebral spaces but no vertebral clefting. Subsequently, we were consulted about a newborn for whom a Desbuquois dysplasia was suspected; the newborn (patient 4) had been born at 36 weeks, with a length of 42 cm and a head circumference of 33 cm, to healthy parents from the Kerala region of India. Growth retardation had been noted in the sixth month of pregnancy via ultrasonography. At birth, he was noted to have prominent eyes, a flat face, severe micrognathia with posterior cleft palate, dislocation of both knees, limited extension at both elbows, and a peculiar abduction (lateral deviation) of the fifth toes bilaterally (Figure 1). Radiographic examination showed short metacarpals and phalanges with an accessory ossification center at the base of the first metacarpal in the left hand, whereas the right hand showed splitting of the first metacarpal in two parts (Figure 2). There was moderate shortening of the long bones and subluxation of the radial heads. The vertebral contour was irregular, but clefting was not observed. Unfortunately, the boy succumbed to aspiration pneumonia as a complication of his palatal cleft on day 5. The similarity of this boy's clinical and radiographic phenotype with that of the previous three patients prompted a direct mutation analysis of IMPAD1 (see below). The clinical features of the four patients are summarized in Table S1 (available online).
Previous sequencing studies had ruled out DTDST (SLC26A2 [MIM 606718]), CHST3 (MIM 603799), and CANT1 mutations as the cause of the phenotype in patients 1–3. We then decided to use a whole-exome sequencing approach to identify the cause of this disorder under the assumptions of an autosomal-recessive inheritance and all patients having a mutation in the same gene. We sequenced the exome (∼21,000 genes) of all three patients by using the SureSelect Human All Exon 50Mb Kit (Agilent, Santa Clara, CA, USA) and multiplexed analysis on a SOLiD 4 System sequencing slide (Life Technologies, Carlsbad, CA, USA). We obtained an average of 4.3 Gb of mappable sequence data per patient. Color space reads were mapped to the hg19 reference genome with the SOLiD BioScope version 1.3, which utilizes an iterative mapping approach. On average, 82% of bases came from the targeted genome, resulting in a mean coverage of 62-fold (Table S2). Eighty-one percent of the targeted exons were covered more than ten times (Figure S1). Single-nucleotide variants were subsequently called by the Di-Bayes algorithm with high call stringency.10 Small insertions and deletions were detected with the use of the SOLiD Small Indel Tool. Called variants were then combined and annotated with a custom analysis pipeline.
After initial filtering (excluding variants that had five or fewer reads or accounted for 20% or less of all reads; see Figure S1 and Table S2), on average, 10,693 genetic variants were identified, including 5185 nonsynonymous and canonical splice-site variants (Table S2). We then applied a prioritization scheme to identify the pathogenic mutation, similar to what was done in recent studies.11–13 For recessive diseases, it is possible that pathogenic mutations are listed as benign polymorphisms in dbSNP or in our in-house control data set because of unaffected individuals carrying the variant in a heterozygous state. However, given the rare incidence of this type of dysplasia, we considered this unlikely. Hence, we excluded known dbSNP132 variants as well as variants from our in-house variant database. At the time of this study, this in-house database contained variants from 177 in-house “exomes,” which contributed a total of 332,849 variants. The filtering step that used these data further reduced the number of candidate mutations by more than 98%, to a total of 136 variants per individual (range 100 – 160; Table S2). Next, we prioritized these variants on the basis of the expectation of an autosomal-recessive mode of inheritance and the assumption, based on parental consanguinity, of a common ancestral allele. On average, 14 homozygous variants were detected per individual. Interestingly, only a single gene, IMPAD1 (NM_017813.3), contained variants in all three patients. For patient 1, over 95% of reads showed a c.529G>A transition, predicting a p.Asp177Asn substitution, whereas patients 2 and 3 showed a c.547A>C transversion, predicting p.Thr183Pro, in over 95% of reads (Figure 3A). For a control cohort, we used the results from 679 control individuals from the 1000 Genomes Project, as well as the 177 exomes present in the in-house database. Thereby, the presence of either one of the two mutations (as well as of the mutation subsequently identified in patient 4; see below) was excluded in 1712 control chromosomes, indicating that they are not common or rare polymorphisms. Conventional bidirectional Sanger sequencing confirmed homozygosity for these changes in patients 1–3, as well as heterozygosity in the parents of the affected siblings (Figure 3B and Figure S2). For patient 4, who was referred later, direct sequencing of the exons and splice junctions of IMPAD1 identified homozygosity for a c.559C>T transition in exon 3, predicting a premature termination codon (Arg187Stop) (see Table S4 and Figure S3). Parental DNA samples of patients 1 and 4 were unavailable for testing.
Figure 3.
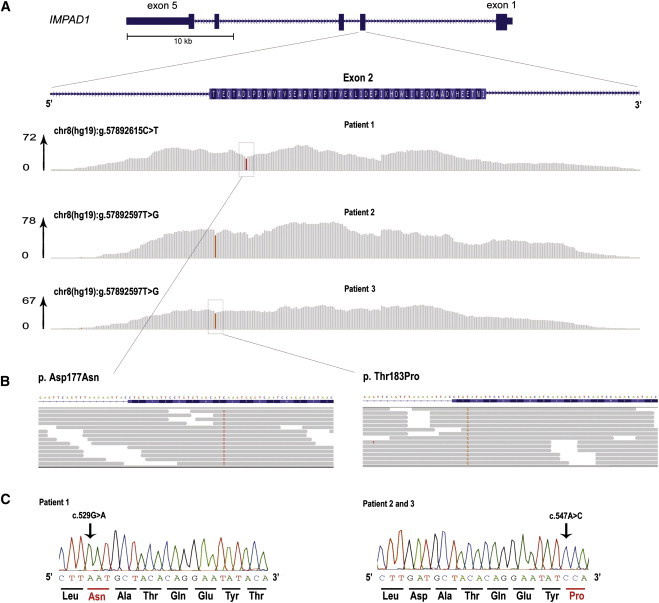
Identification of Homozygous Mutations in IMPAD1 by Exome Sequencing in Patients 1, 2, and 3
(A) Schematic representation of IMPAD1 with detailed overview of exon 2, containing the identified mutations. Note that the coding sequence of IMPAD1 is on the reverse strand of the chromosome 8 sequence, so that the chromosomal sequence corresponds to the noncoding strand. The per-base coverage is provided with an axis to the right indicating the maximum coverage obtained per individual. Coverage represented in gray indicates that the sequence data in the patient showed the wild-type base, whereas colored bases indicate the detection of variants. The relative height of the color is indicative of the presence of variant in the homozygous state. Top: patient 2. Middle: patient 3. Bottom: patient 1.
(B) Detailed zoom-in of reads showing the homozygous missense mutations. Grey arrows indicate the mapped sequence reads, with colored bases indicating nucleotides deviating from the wild-type genomic sequence. In the nucleotides of interest, >95% of reads show the mutated sequence.
(C) Sanger sequencing validation of the homozygous mutations in the patients 1–3. Note that the orientation is presented from the 5′ to the 3′ end, representing the coding sequence.
Whereas the Grantham scores for the amino acid substitutions indicate only a small biochemical change (p.Asp177Asn: Grantham score 23; p.Thr183Pro: Grantham score 38), evolutionary measures at the base-pair level suggest a strong conservation (c.529G>A: PhyloP score 4.75; c.547A>C: PhyloP score 3.91). Moreover, both residues are located within the phosphatase domain of IMPAD1 and are completely conserved throughout evolution (Figures 4A and 4B). In the UniProt database, IMPAD1 is still listed as a probable myoinositol monophosphatase on the basis of the original description,14 and IMPAD1 residues 176–179 are predicted to be substrate binding on the basis of similarity with other phosphatases.
Figure 4.
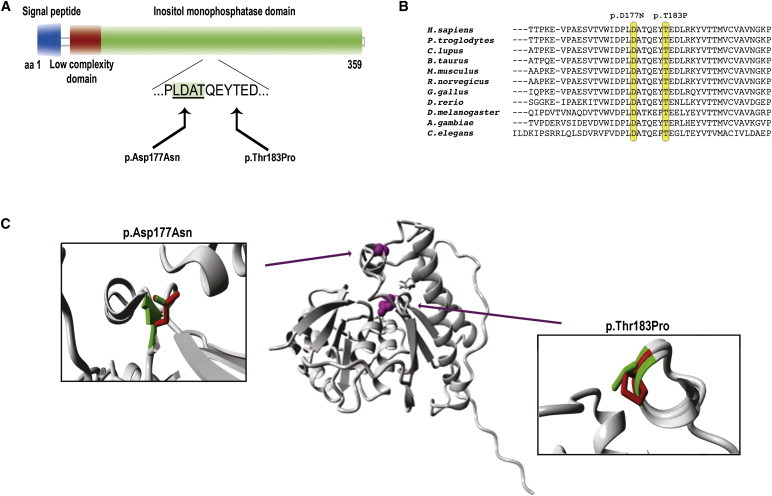
Schematic Representation and Protein Modeling of IMPAD1 p.Asp177Asn and p.Thr183Pro Mutations
(A) Schematic representation of the predicted effect at the protein level. Domains and functions are provided in different colors, with the binding domain harboring the mutations indicated by an underlined protein sequence in a green shaded box.
(B) Evolutionary conservation of the protein sequence throughout evolution. Mutated amino acids are indicated by yellow boxes.
(C) Overview of the protein in ribbon presentation. Middle: the protein is colored gray, and the side chains of the mutated residue are colored magenta and shown as small balls. Left and right: Close-up of the p.Thr183Pro and p.Asp177Asn mutations. Side chains of both the wild-type and the mutant residue are shown and are colored green and red, respectively.
Because the three-dimensional structure of gPAPP (Uniprot entry Q9NX62) is unknown, protein modeling was performed for the mutations p.Asp177Asn and p.Thr183Pro via two approaches. The HOPE program15 was able to build a model by using PDB file 2WEF, which shares a 26% sequence identity with IMPAD1. In parallel, three-dimensional models for the wild-type IMPAD1, as well as those for polypeptides containing either one of the two mutations, were obtained from the ESyPred3D site,16 also with 2WEF used as a template, and analyzed with the use of the SPDBV software version 4.0.1. Molecular modeling via both programs shows that Asp177 and Thr183 are located in, and close to, the substrate binding pocket (see Figure S4). For p.Asp177Asn, the loss of the negatively charged amino acid is expected to affect the binding of an essential magnesium ion, thereby most likely directly disturbing the phosphatase activity (Figure 4C, Figure S4). Thr183 is located at the C terminus of an alpha helix that contains important substrate binding site residues at its N-terminal side, including residue Asp177. Replacing Thr183 by proline will disturb this helix because proline has no free hydrogen in its backbone (Figure 4C, Figure S4). The helix-breaker effect of proline resulting from its rigidity is well known.17 The p.Arg187Stop mutation identified in patient 4 predicts the truncation of the protein with the loss of 173 amino acid residues (out of a total of 359) at the carboxy-terminal end; if the corresponding mRNA escapes nonsense-mediated decay, the truncation is likely to have a major effect on the folding of the protein.
Impad1 inactivation has been associated with skeletal dysplasia and abnormal joint formation in mice by three independent groups. Mitchell et al. generated mice bearing gene-trap alleles and observed a short limb and trunk phenotype produced by the trapping of the gene for a membrane protein with similarity to inositol monophosphatases.14 Sohaskey et al. studied this mouse strain in more depth and observed a severe chondrodysplasia including ectopic interphalangeal joints, leading to the denomination of the phenotype as “JAWS” for “joints abnormal with splitting.”18 They observed delayed and disorganized maturation of growth-plate chondrocytes, impaired sulfation of chondroitin, and abnormal metabolism of the chondroitin sulfate proteoglycan aggrecan, but a functional characterization of the specific enzymatic function of Impad1 was not performed.18 Frederick et al. explored the enzymatic properties of Impad1 in vitro and showed that when targeted to the Golgi, Impad1 had a strong phosphoadenosine phosphate 3-nucleotidase (or PAP phosphatase, PAPP) activity.9 Subsequently, they made use of a different Impad1 gene-trap cell line to generate transgenic mice and noted that the chondrodysplasia phenotype was associated with markedly reduced sulfation of chondroitin and heparan proteoglycans.9 To explain the sulfation defect associated with gPAPP inactivation, they suggested that the physiologic role of gPAPP might be to remove the PAP that remains after the transfer of the high-energy sulfate group from PAPS to the sulfation substrate (such as the nascent chondroitin chains) in the Golgi (Figure 5).9 Hydrolysis of PAP to AMP and phosphate would prevent product inhibition on the sulfotransferase reactions and possibly allow for reverse transport of AMP from the Golgi to the cytoplasm (Figure 5).9 Both groups of researchers placed their findings in the context of skeletal dysplasias related to proteoglycan undersulfation and suggested that the Impad1-inactivated mouse was a possible model for a human disorder yet to be identified.9,18
Figure 5.
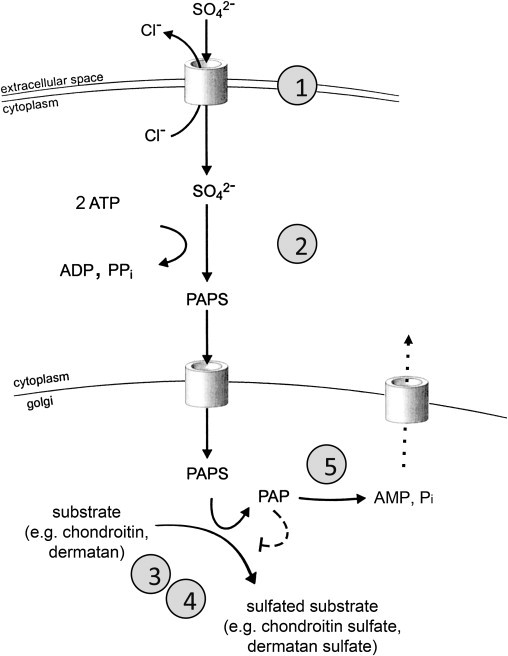
Schematic Representation of Sulfate Activation and Sulfation Pathways
Extracellular sulfate is taken up from the extracellular space through a sulfate-chloride exchanger (SLC26A2, DTDST) and subsequently activated to its high-energy form, phosophoadenosine phosphosulphate (PAPS). A PAPS translocase that shuttles PAPS from the cytoplasm to the endoplasmic reticulum has been identified in zebrafish. PAPS is the sulfate donor for most sulfotransferase reactions both in the cytoplasm and in the endoplasmic reticulum and Golgi complex, such as those required for sulphation of nascent proteoglycans. Inhibition of sulfotransferase reactions by PAP (dashed line) has been demonstrated in vitro. Efflux of AMP from the endoplasmic reticulum through a transporter or exchanger (dotted line) has been postulated but not demonstrated. Known genetic defects leading to human osteoarticular phenotypes are marked by circled numbers: 1, SLC26A2/DTDST (sulfate transporter); 2, PAPSS (PAPS synthase); 3, CHST3 (chondroitin 6-sulfotransferase); 4, CHST14 (dermatan 4-sulfotransferase); 5, IMPAD1 (Golgi-resident phosphoadenosine phosphate phosphatase, gPAPP).
We now report on the human phenotype associated with IMPAD1 mutations and confirm the predictions made on the basis of the mouse knockouts and the biochemical studies. Although the phenotype in our patients is distinct, the differential diagnosis has included disorders associated with disturbed proteoglycan synthesis and sulfation such as diastrophic dysplasia, recessive Larsen syndrome, and Desbuquois dysplasia. Longitudinal splitting of phalanges, as seen in the JAWS mouse model and in our patient 1, can be seen in severe DTDST disorders, as well as in disorders of chondroitin sulfate synthesis (CHSY1 [MIM 608183]),19,20 whereas the presence of additional phalangeal ossification centers is the hallmark of CANT1-related Desbuquois dysplasia.21 The a priori differential diagnostic considerations in our patients; the findings of different, highly disruptive substitutions in IMPAD1 in three unrelated families; and the similarity between the clinical and radiographic findings in our patients and the findings in the existing mouse models leave no doubt as to the causative role of the IMPAD1 mutations and allow us to delineate a nosologic entity that we suggest be designated as “chondrodysplasia with joint dislocations, gPAPP type” (in style with the most recent nosology)22 or, more simply, gPAPP deficiency.
IMPAD1 analysis should be included along with CHST3 and CANT1 in the diagnostic algorithm for infants presenting with congenital dislocations, chondrodysplasia, and short stature. It seems that the differential diagnosis of this entity must be made in consideration with diastrophic dysplasia, Desbuquois dysplasia, and recessive Larsen syndrome. The absence of significant vertebral anomalies may help in distinguishing gPAPP deficiency from Desbuquois syndrome and from CHST3 deficiency, and the presence of knee dislocations and a facial phenotype may distinguish it from diastrophic dysplasia. However, the identification of additional individuals with gPAPP deficiency, particularly by screening of those who have tested negative for CHST3 and CANT1 mutations, may further delineate the phenotypic spectrum resulting from IMPAD1 mutations.
Desbuquois dysplasia has been considered to be heterogeneous with respect to the presence or absence of “typical” hand abnormalities.21,23 However, CANT1 mutations are found in both the group with and the group without typical hand abnormalities,7,8 and some individuals in both phenotypic groups are negative for CANT1 mutations. Although the precise role of CANT1 in the pathogenesis of Desbuquois dysplasia remains to be elucidated, the notion that both CANT1 and IMPAD1 are nucleotide phosphatases further supports a role of CANT1 in proteoglycan synthesis.
IMPAD1 deficiency can be added to the growing list of defects of proteoglycan synthesis associated with a skeletal and articular phenotype, a list that includes B4GALT7 (MIM 604327),24,25 EXT1 (MIM 608177), EXT2 (MIM 608210),26 and CHSY1,19,20 and those affecting proteoglycan sulfation, such as DTDST,3,4 PAPSS (MIM 603005),27 CHST3,5,6,28 and CHST14 (MIM 608429).29,30 The large number of enzymes involved in proteoglycan synthesis and sulfation suggests that more such genetic disorders may exist. More broadly, this study provides additional proof that exome sequencing can be instrumental in finding the remaining genes associated with clinical and molecularly heterogeneous disorders, especially when combined with careful phenotypic characterization and homogeneous patient selection.
Acknowledgments
The excellent technical work of Mme Carole Chiesa-Buzzi is gratefully acknowledged. This work has been supported by the Swiss National Science Foundation (grant no. FN 310030-132940/BONAFE Luisa, for which approval of the Institutional Ethics Committee has been obtained); by the Fonds de Recherche du Departement Medico-Chirurgical de Pédiatrie, CHUV, Lausanne; by the German Bundesministerium für Bildung und Forschung (SKELNET project), by the Deutsche Forschungsgemeinschaft (La 1381/1-3), and the Netherlands Organization for Health Research and Development (ZonMW grants 917-66-363 and 911-08-025 to J.A.V., and 916-86-016 to L.E.L.M.V.). A.S.-F. is supported by the Leenaards Foundation, Lausanne, Switzerland.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
EsyPred3D protein structure prediction site, http://www.fundp.ac.be/sciences/biologie/urbm/bioinfo/esypred/
HOPE protein structure prediction site, http://www.cmbi.ru.nl/hope/home
IGV browser: http://www.broadinstitute.org/igv
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SPDBV software, http://spdbv.vital-it.ch/
UniProt database, http://www.uniprot.org/uniprot/Q9NX62
References
- 1.Krakow D., Robertson S.P., King L.M., Morgan T., Sebald E.T., Bertolotto C., Wachsmann-Hogiu S., Acuna D., Shapiro S.S., Takafuta T. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat. Genet. 2004;36:405–410. doi: 10.1038/ng1319. [DOI] [PubMed] [Google Scholar]
- 2.Bicknell L.S., Farrington-Rock C., Shafeghati Y., Rump P., Alanay Y., Alembik Y., Al-Madani N., Firth H., Karimi-Nejad M.H., Kim C.A. A molecular and clinical study of Larsen syndrome caused by mutations in FLNB. J. Med. Genet. 2007;44:89–98. doi: 10.1136/jmg.2006.043687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi A., Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum. Mutat. 2001;17:159–171. doi: 10.1002/humu.1. [DOI] [PubMed] [Google Scholar]
- 4.Superti-Furga A. Defects in sulfate metabolism and skeletal dysplasias. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., Vogelstein B., Childs B., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; New York: 2001. pp. 5189–5201. [Google Scholar]
- 5.Hermanns P., Unger S., Rossi A., Perez-Aytes A., Cortina H., Bonafé L., Boccone L., Setzu V., Dutoit M., Sangiorgi L. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 2008;82:1368–1374. doi: 10.1016/j.ajhg.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unger S., Lausch E., Rossi A., Mégarbané A., Sillence D., Alcausin M., Aytes A., Mendoza-Londono R., Nampoothiri S., Afroze B. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. A. 2010;152A:2543–2549. doi: 10.1002/ajmg.a.33641. [DOI] [PubMed] [Google Scholar]
- 7.Huber C., Oulès B., Bertoli M., Chami M., Fradin M., Alanay Y., Al-Gazali L.I., Ausems M.G., Bitoun P., Cavalcanti D.P. Identification of CANT1 mutations in Desbuquois dysplasia. Am. J. Hum. Genet. 2009;85:706–710. doi: 10.1016/j.ajhg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuichi T., Dai J., Cho T.J., Sakazume S., Ikema M., Matsui Y., Baynam G., Nagai T., Miyake N., Matsumoto N. CANT1 mutation is also responsible for Desbuquois dysplasia, type 2 and Kim variant. J. Med. Genet. 2011;48:32–37. doi: 10.1136/jmg.2010.080226. [DOI] [PubMed] [Google Scholar]
- 9.Frederick J.P., Tafari A.T., Wu S.M., Megosh L.C., Chiou S.T., Irving R.P., York J.D. A role for a lithium-inhibited Golgi nucleotidase in skeletal development and sulfation. Proc. Natl. Acad. Sci. USA. 2008;105:11605–11612. doi: 10.1073/pnas.0801182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marth G.T., Korf I., Yandell M.D., Yeh R.T., Gu Z., Zakeri H., Stitziel N.O., Hillier L., Kwok P.Y., Gish W.R. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- 11.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 13.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell K.J., Pinson K.I., Kelly O.G., Brennan J., Zupicich J., Scherz P., Leighton P.A., Goodrich L.V., Lu X., Avery B.J. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat. Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- 15.Venselaar H., Te Beek T.A., Kuipers R.K., Hekkelman M.L., Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert C., Léonard N., De Bolle X., Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabartty A., Kortemme T., Baldwin R.L. Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 1994;3:843–852. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sohaskey M.L., Yu J., Diaz M.A., Plaas A.H., Harland R.M. JAWS coordinates chondrogenesis and synovial joint positioning. Development. 2008;135:2215–2220. doi: 10.1242/dev.019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y., Laue K., Temtamy S., Aglan M., Kotan L.D., Yigit G., Canan H., Pawlik B., Nürnberg G., Wakeling E.L. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of BMP signaling. Am. J. Hum. Genet. 2010;87:757–767. doi: 10.1016/j.ajhg.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian J., Ling L., Shboul M., Lee H., O'Connor B., Merriman B., Nelson S.F., Cool S., Ababneh O.H., Al-Hadidy A. Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling. Am. J. Hum. Genet. 2010;87:768–778. doi: 10.1016/j.ajhg.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faivre L., Cormier-Daire V., Eliott A.M., Field F., Munnich A., Maroteaux P., Le Merrer M., Lachman R. Desbuquois dysplasia, a reevaluation with abnormal and “normal” hands: radiographic manifestations. Am. J. Med. Genet. A. 2004;124A:48–53. doi: 10.1002/ajmg.a.20440. [DOI] [PubMed] [Google Scholar]
- 22.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., Lemerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011 doi: 10.1002/ajmg.a.33909. Published online March 15, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faivre L., Le Merrer M., Zerres K., Ben Hariz M., Scheffer D., Young I.D., Maroteaux P., Munnich A., Cormier-Daire V. Clinical and genetic heterogeneity in Desbuquois dysplasia. Am. J. Med. Genet. A. 2004;128A:29–32. doi: 10.1002/ajmg.a.30042. [DOI] [PubMed] [Google Scholar]
- 24.Faiyaz-Ul-Haque M., Zaidi S.H., Al-Ali M., Al-Mureikhi M.S., Kennedy S., Al-Thani G., Tsui L.C., Teebi A.S. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am. J. Med. Genet. A. 2004;128A:39–45. doi: 10.1002/ajmg.a.30005. [DOI] [PubMed] [Google Scholar]
- 25.Götte M., Kresse H. Defective glycosaminoglycan substitution of decorin in a patient with progeroid syndrome is a direct consequence of two point mutations in the galactosyltransferase I (beta4GalT-7) gene. Biochem. Genet. 2005;43:65–77. doi: 10.1007/s10528-005-1068-2. [DOI] [PubMed] [Google Scholar]
- 26.Jennes I., Pedrini E., Zuntini M., Mordenti M., Balkassmi S., Asteggiano C.G., Casey B., Bakker B., Sangiorgi L., Wuyts W. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb) Hum. Mutat. 2009;30:1620–1627. doi: 10.1002/humu.21123. [DOI] [PubMed] [Google Scholar]
- 27.Faiyaz ul Haque M., King L.M., Krakow D., Cantor R.M., Rusiniak M.E., Swank R.T., Superti-Furga A., Haque S., Abbas H., Ahmad W. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat. Genet. 1998;20:157–162. doi: 10.1038/2458. [DOI] [PubMed] [Google Scholar]
- 28.Thiele H., Sakano M., Kitagawa H., Sugahara K., Rajab A., Höhne W., Ritter H., Leschik G., Nürnberg P., Mundlos S. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA. 2004;101:10155–10160. doi: 10.1073/pnas.0400334101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dündar M., Müller T., Zhang Q., Pan J., Steinmann B., Vodopiutz J., Gruber R., Sonoda T., Krabichler B., Utermann G. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009;85:873–882. doi: 10.1016/j.ajhg.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyake N., Kosho T., Mizumoto S., Furuichi T., Hatamochi A., Nagashima Y., Arai E., Takahashi K., Kawamura R., Wakui K. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010;31:966–974. doi: 10.1002/humu.21300. [DOI] [PubMed] [Google Scholar]
- 31.Kim O.H., Nishimura G., Song H.R., Matsui Y., Sakazume S., Yamada M., Narumi Y., Alanay Y., Unger S., Cho T.J. A variant of Desbuquois dysplasia characterized by advanced carpal bone age, short metacarpals, and elongated phalanges: report of seven cases. Am. J. Med. Genet. A. 2010;152A:875–885. doi: 10.1002/ajmg.a.33347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.