

Abstract
Genome maintenance activities including DNA repair, cell division cycle control, and checkpoint signaling pathways preserve genome integrity and prevent disease. Defects in these pathways cause birth defects, neurodegeneration, premature aging, and cancer. Recent technical advances in functional genomic approaches such as expression profiling, proteomics, and RNA interference (RNAi) technologies have rapidly expanded our knowledge of the proteins that work in these pathways. In this review, we examine the use of these high-throughput methodologies in higher eukaryotic organisms for the interrogation of genome maintenance activities.
Keywords: genome maintenance, RNA interference, DNA repair, interactome, proteomics, expression profiling, cell cycle
Introduction
Approximately 10,000 trillion cell divisions happen in the typical human lifetime. During each cell division cycle, nearly 6.8 billion base pairs of DNA must be replicated and then segregated to each daughter cell. Furthermore, the cell division cycle happens in the context of thousands of DNA lesions that occur in each cell each day. In all but a few specialized cases, the goal is to create and maintain an identical genome for each cell in the body.
Maintaining the genome requires the concerted actions of cellular metabolism, cell cycle, and DNA repair activities that together constitute genome maintenance pathways (Fig. 1). Enzymes like superoxide dismutase protect cellular molecules from the action of reactive oxygen species generated as a byproduct of cellular metabolism, thereby reducing the DNA damage burden in the cell (Ikner and Shiozaki, 2005). DNA lesions that do occur are identified and repaired by multiple DNA repair systems that scan the genome looking for imperfections (Friedberg, 2003). Many proteins work to ensure a single round of DNA replication is completed with minimal errors each cell cycle (Bell and Dutta, 2002). Proper chromosome segregation during mitosis requires spindle assembly, kinetochore attachment, and physical separation (Nasmyth, 2001). All of these genome maintenance activities happen in the context of chromatin necessitating a large cohort of enzymes that control chromatin dynamics (Campos and Reinberg, 2009). Finally, many of these activities are coordinated through signaling pathways that constitute the DNA damage response (DDR). This response regulates transcriptional programs, DNA replication, mitosis, and repair (Harper and Elledge, 2007). Furthermore, it can promote the elimination of damaged cells from the cycling population through either apoptosis or senescence.
Figure 1. Genome maintenance requires the coordination of multiple cellular activities.
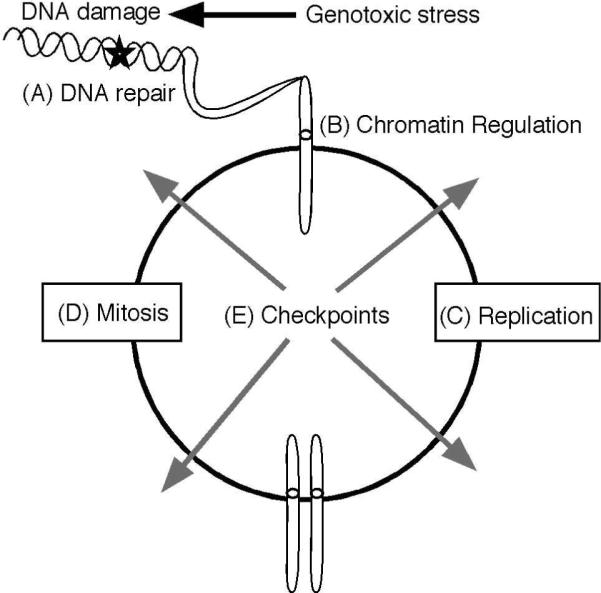
(A) Multiple DNA repair pathways operate to remove DNA lesions caused by endogenous and exogenous genotoxic agents. (B) DNA repair and metabolism occurs in the context of chromatin. Chromatin modifications regulate protein access to the DNA as well as signaling responses to DNA damage. (C) DNA replication must faithfully duplicate the DNA and chromatin structure once and only once per cell division cycle. (D) Proper spindle assembly and chromosome segregation during mitosis ensures each daughter cell receives a complete copy of the genome. (E) Cell cycle checkpoints monitor DNA damage, replication, and mitosis.
Our knowledge of the genome maintenance activities that happen in human cells comes from the study of all kingdoms of life since most of these activities are highly evolutionarily conserved. Genetic and biochemical studies of bacteria provide insights into replication and DNA repair proteins that also operate in eukaryotic systems. Archaebacteria provide a rich resource for structural biologists since it is often easier to purify and crystallize proteins from the thermophilic species. The genetics of simple eukaryotes like Schizosaccharomyces pombe and Saccharomyces cerevisiae that can divide as either haploid or diploid cells provided some of our first mechanistic understanding of the cell division cycle (Hartwell et al., 1970, Nurse, 1975). Higher eukaryotes including the invertebrates Caenorhabditis elegans and Drosophila, as well as the vertebrates Xenopus and zebrafish have unique strengths as research models. For example, Xenopus eggs contain high concentrations of genome maintenance proteins and have been used to understand the mechanisms of DNA damage checkpoints using complex biochemical strategies. Finally, mammalian systems have been historically important and are increasingly providing resources for new discovery.
Some genome maintenance discoveries came directly from human systems through bedside to bench research. This is especially true in the DNA damage response field since many human diseases are caused by mutations in DDR genes. For example, clinicians studying patients with the cancer predisposition disease ataxia telangiectasia described how at the cellular level this disease is characterized by an inability to properly arrest the cell cycle in response to ionizing radiation (Painter and Young, 1980) (Houldsworth and Lavin, 1980). These were some of the first descriptions of cell cycle checkpoints. Positional cloning of the gene responsible for ataxia telangiectasia (ATM), identified one of the most important kinases that signals in response to DNA double-strand breaks (DSB) (Savitsky et al., 1995). Other notable examples include the identification of DSB repair and signaling proteins like breast and ovarian cancer susceptibility protein 1 (BRCA1) and Nijmegen breakage syndrome protein (NBS1) (Futreal et al., 1994, Miki et al., 1994, Carney et al., 1998, Matsuura et al., 1998); the identification of the DNA crosslink repair proteins defective in patients with Fanconi anemia (FANC proteins) (D'Andrea, 2010); and the description of proteins involved in the repair of ultraviolet (UV)-radiation damage via nucleotide excision repair pathways from people with xeroderma pigmentosum (XP proteins) (Cleaver et al., 2009). The primary limitations of this approach for understanding genome maintenance are that it is time and resource-intensive and restricted by the size of the human population.
Yeast systems historically provided the simplest methods to identify genes involved in genome maintenance pathways. Classic mutagenesis screens including the Hartwell and Nurse cell division cycle screens (Hartwell et al., 1970, Nurse, 1975, Nurse et al., 1976), DNA damaging agent sensitivity screens (Birrell et al., 2001, Bennett et al., 2001, Cejka and Jiricny, 2008), and mitotic arrest deficient screens (Li and Murray, 1991, Hoyt et al., 1991) generated lists of yeast strains that were converted to lists of genes as the mutant alleles were identified. As DNA sequencing provided a complete sequence of the yeast genome, these mutagenesis screens gave way to more systematic functional genomic approaches based on libraries of yeast strains with targeted knockouts in every open reading frame (ORF) or tags integrated at every ORF to allow high-throughput protein-protein interaction and localization studies (Giaever et al., Winzeler et al., 1999, Gelperin et al., 2005).
Recent technological advances including high-density microarrays, mass spectrometry, and especially RNA interference (RNAi), have made functional genomic approaches feasible in diploid organisms including human cells. In many cases, these approaches have been copied from the work done in yeast. For example, cell division cycle (Björklund et al., 2006, Kittler et al., 2007, Kittler et al., 2004, Mukherji et al., 2006), mitotic arrest (Draviam et al., 2007), and DNA damage sensitivity screens (Wang et al., 2009, Smogorzewska et al., 2010, O'Connell et al., 2010, Hurov et al., 2010) have all been completed with RNAi approaches in cell culture (see Table for a list of screens discussed in this review). Some of the experimental advantages of human cell culture such as the ease of imaging are now being exploited to complete novel high content microscopy screens (O'Donnell et al., 2010, Kolas et al., 2007, Doil et al., 2009, Stewart et al., 2009, Lovejoy et al., 2009, Paulsen et al., 2009). Furthermore, other screens like the proteomic identification of hundreds of ATM substrates have been pioneered in human cell culture and only later translated into yeast (Matsuoka et al., 2007, Mu et al., 2007, Stokes et al., 2007, Smolka et al., 2007). Overall, these high-throughput approaches have identified hundreds of new proteins in genome maintenance pathways and rapidly accelerated genome maintenance research.
In this review we will describe functional genomic approaches to identify and characterize genome maintenance pathways. A brief introduction of each approach will be followed by specific examples of their successful use in discovery. We will focus primarily on approaches in diploid, multi-cellular eukaryotic systems, we point readers interested in functional genomics using yeast systems to other excellent reviews (Cagney et al., 2006, Davidson and Brown, 2009).
Gene expression profiling
One of the first functional genomic approaches used in higher eukaryotic organisms was gene expression profiling (Schena et al., 1995). The general idea is to track transcriptional changes in response to a genome maintenance challenge. This is most commonly done using microarrays to measure mRNA abundance. Many investigators analyzed relative mRNA expression levels as a function of cell cycle position, time after exposure to a DNA- or spindle-damaging agent, or genetic background (i.e. p53 overexpression or ATM-deficiency) (Chaudhry et al., 2002, Amundson et al., 2003, Rashi-Elkeles et al., 2006).
There are two rationales for gene expression profiling in the interrogation of genome maintenance pathways. First, genes with activities in a specific cell cycle phase or in response to DNA damage may be more highly expressed in cells under these conditions. Second, the outcome of many genome maintenance pathways such as the DDR depends on a change in transcription, so tracking transcriptional changes can be used as an assay to monitor signaling through these pathways.
A well-studied example is the G1 arrest or apoptosis induced by p53 in response to DNA damage, which is mediated by p53-dependent transcriptional changes in multiple genes (Levine and Oren, 2009, Menendez et al., 2009). DNA damage leads to activation of p53 through post-translational modifications that promote its stabilization. By identifying p53-target genes, investigators have learned how cells regulate the G1-S transition and apoptosis. These studies have been complemented by the identification of p53 response elements throughout the genome using chromatin immunoprecipitation experiments. The reader is directed to several recent reviews to learn more about these methods to study p53 function (Beckerman and Prives, 2010, Menendez et al., 2009).
The limitations of this type of approach are that regulation of genome maintenance pathways often proceeds through transcriptional-independent mechanisms and that many transcriptional changes may be only indirectly related to the process being studied. Proteins like ATM that initiate signaling in response to DNA damage are largely unregulated at the transcriptional level. Also, many transcriptional changes occur as a general reaction to any type of environmental stress so they may not be particularly useful in dissecting a specific pathway (Birrell et al., 2002). Finally, expression profiling provides little information on the function of the identified genes.
High throughput proteomics
Proteomic approaches to studying genome maintenance pathways include techniques that measure protein abundance, protein-protein interactions, post- translational modifications, and/or subcellular localization (Fig. 2). Assays include protein microarrays, yeast two-hybrid, mass spectrometry, and fluorescent imaging. These are inherently more technically difficult, expensive, and lower throughput than expression profiling. However, they offer the significant advantage of easier functional interpretation.
Figure 2. Proteomic approaches are useful to identify genome maintenance proteins.

(A) Protein microarrays can be used to identify post-translational modifications (A1) and protein-protein interactions (A2). For example, Merbl et al., identified substrates of the APC ubiquitin conjugating enzyme using protein microarrays (Merbl and Kirschner, 2009). (A2) Protein interactions can be visualized on protein microarrays using fluorescently tagged proteins as the probe. (B) Yeast 2 hybrid screens detect interactions between bait proteins and prey proteins. Although useful to interrogate pairs of proteins (B1), these screens are easily easily expanded to screen cDNA libraries to generate interactome maps (B2). (C) Affinity purification coupled to mass spectrometry is useful to interrogate protein complexes purified from their native cellular context or as depicted, to identify differential post-translational modifications following genotoxic stress. (D) Analysis of protein localization is a good indicator of function within genome maintenance pathways. (D1) Fluorescence microscopy to examine subcellular localization can identify proteins that change localization in response to genotoxic drugs like hydroxyurea (HU). (D2) Biochemical purification of cellular components coupled to mass spectrometry provides an alternative method of defining proteins that reside in cellular structures relevant to genome maintenance activities.
Protein microarrays
Protein microarrays or protein chips are arrays of proteins or peptides deposited on a glass or silicon surface (MacBeath and Schreiber, 2000). They are useful to measure protein abundance, protein-protein interactions, and post-translational modifications (Fig. 2A). This technology has seen only limited use largely due to issues of cost and robustness of the assays. However, a recent publication on the use of protein microarrays to identify ubiquitination substrates of the anaphase promoting complex (APC) highlights the potential of this emerging technology (Merbl and Kirschner, 2009).
The APC is an E3 ubiquitin ligase that polyubiquitinates proteins to regulate the cell cycle (Peters, 2006). For example, APC-dependent ubiquitination of cyclin B is responsible for the cycling of its abundance and required for exit from mitosis (King et al., 1996, King et al., 1995, Sudakin et al., 1995). The Kirschner group used what they termed extract-based functional assays combined with protein microarrays to define APC substrates (Merbl and Kirschner, 2009). They isolated extracts from HeLa cells that were arrested at the mitotic checkpoint. These extracts catalyzed the ubiquitination of proteins spotted on a solid surface that was detectable using an anti-polyubiquitin antibody. They defined APC substrates by comparing the activities of these extracts on the protein microarray to extracts in which the APC was inactivated. Of the 8,000 proteins arrayed on the chips, they found that 132 were differentially modified.
To validate the methodology for the identification of APC substrates, they selected six mitotic proteins from their list that were previously unknown targets of the APC complex (Nek9, Calm2, p27, RPS6KA4, cyclin G2, and DDA3) and demonstrated through more standard assays that they were indeed APC substrates. In addition, four proteins not previously linked to mitosis (Zap-70, MAP3K11, RPL30, and Dyrk3) were chosen for validation. Strikingly, two of four (RPL30 and Dyrk3) were also shown to be APC substrates. RPL30 is a component of the 60S ribosome. Dyrk3 is a dual specificity protein kinase that has functions in regulating cell survival in response to energy stresses and in modulating erythropoiesis (Guo et al., 2010, Li et al., 2002). It is unclear why these proteins would be regulated by the APC, but their identification in this screening effort points to either an unappreciated function for them in the cell cycle or a broader function for the APC.
This example provides several lessons about the use of protein microarrays. First, the power of working directly with proteins is that results immediately lead to some mechanistic understanding of the pathway. The example also demonstrates that complex cell extracts can be used to recapitulate a genome maintenance activity. This provides several advantages especially when a genome maintenance activity has yet to be fully reconstituted with purified proteins. However, it also illustrates the current limitations of this technology. The most obvious limitation is the current protein chips, which contain only a small fraction of the proteome. It is also difficult to know the percentage of active, folded, and sterically unhindered proteins retained on the microarray.
Yeast two-hybrid and affinity purification combined with mass spectrometry methods for identifying protein-protein interactions
Many genome maintenance pathways depend on assemblies of multi-subunit protein machines. Thus, the identification of protein-protein interactions is a primary method for discovering new genome maintenance proteins and understanding their regulation. While there are many methodologies to accomplish this goal, the standard methods rely on interrogating proteins or complexes individually in a one protein per project fashion. More recently, investigators have worked to convert these approaches into higher throughput formats and to create a full “interactome” description of protein-protein interactions that occur within a cell (Cusick et al., 2005).
The two most common interactome methodologies are yeast two-hybrid (Y2H) and affinity purification combined with mass spectrometry (Fig. 2B and 2C). Efforts began in yeast systems (Fromont-Racine et al., 1997) and expanded to other organisms. One impediment to using these methods in higher eukaryotes is the need for full-length ORFs cloned into appropriate vectors. Fortunately, orfeome cloning projects using recombination-based vectors are rapidly removing this obstacle. Y2H is a sensitive method to detect binary interactions while affinity purification followed by mass spectrometry is better suited to identify stable protein complexes that occur in the native context.
Vidal and colleagues used interactome mapping to generate a protein-protein interaction map for proteins involved in the DDR in Caenorhabditis elegans (Boulton, 2002). They focused on 75 worm orthologs of known DDR proteins, and initially used Y2H to generate pairwise protein interaction maps among these proteins. From this data they concluded that they had approximately a 50% success rate in identifying predicted interactions. More importantly they used 67 of the genes to perform unbiased proteome-wide Y2H screens. A total of 165 interacting sequences were recovered, many encoding novel proteins not previously linked to the DDR.
This example illustrates both the power of interactome mapping and the current deficiencies in the technologies. The major limitation of the technology is that it is not easily scaled to a high-throughput approach. Thus, we only see a very limited portion of the interactome landscape in any single study. A second problem is the issue of false-positives. Each of the “hits” from a Y2H or mass spectrometry experiment can only be considered a potential protein-protein interaction. Performing these experiments with large numbers of baits helps eliminate the false-positives through the identification of common contaminants. However, validation using an independent method is essential. Finally, false-negatives will be difficult to avoid using any single method so combining results from multiple methods will be important.
Mass spectrometry methods for identifying post-translational modifications
In addition to identifying protein complexes, mass spectrometry can be used to detect post-translational modifications such as phosphorylation sites. Unbiased phosphoproteomic screens have yielded large databases of thousands of phosphorylation sites (see (Yates et al., 2009) and references therein). Typically, these studies employ a phospho-protein enrichment strategy using technologies such as metal affinity chromatography. More recently, they have been combined with quantitative mass spectrometry methodologies to compare cells or tissues from two or more conditions.
This methodology was applied to identify cell cycle-related phosphorylation (Daub et al., 2008). In this study, the authors combined stable isotope labeling in cell culture (SILAC) with affinity purification of kinases using resins attached to five kinase inhibitors. They differentially labeled cells synchronized in S or M phase with isotopes, performed the kinase enrichment strategy, and after trypsinization, enriched phosphopeptides prior to identification by mass spectrometry. This technique yielded 1182 phosphopeptides about 50% of which are upregulated in mitotic cells and 10% upregulated in replicating cells. Many new sites on known mitotic regulatory proteins were described including sites on CDC2, PLK1, and AurB. Further experiments will be needed to understand their functional significance. The large number of changes in phosphopeptides identified indicates that progression through mitosis involves large changes in the phospho-proteome.
A second example comes from the study of the kinases that operate in the DNA damage response. The DDR kinases ATM and ATR have a strong preference for phosphorylating serines or threonines that are followed by glutamines (Kim et al., 1999). This bias can be used to pinpoint putative phosphorylation sites within known substrates. More importantly, it can be exploited to identify novel ATM and ATR substrates. The first example where this strategy was used successfully was our identification of MCM2 and MCM3 as substrates of ATM and ATR (Cortez et al., 2004). In that proof-of-concept study, we raised a phosphopeptide specific antibody to random phosphopeptides that contained a phosphorylated SQ motif. This antibody was then used to immunopurify proteins from damaged cells followed by mass spectrometry to identify the phosphorylated proteins. Another group followed a similar approach to identify MCM3 phosphorylation sites (Shi et al., 2007). Importantly, these proof of concept studies have now been extended to identify hundreds of potential ATM- or ATR-catalyzed phosphorylation sites.
Elledge and colleagues performed the largest study of this kind (Matsuoka et al., 2007). They assembled a large panel of phosphopeptide specific antibodies that recognized phospho-SQ or -TQ epitopes. Reasoning that these antibodies likely cross-react with other phospho-SQ/TQ epitopes, they used them to immunopurify tryptic peptides derived from undamaged and ionizing-radiation treated cells. By performing quantitiative mass spectrometry, they defined over 900 regulated phosphorylation sites in a little over 700 proteins. This study greatly expanded the number of phosphorylated residues identified on known components of the DDR. For example, they identified 27 new sites on the BRCA1, 53BP1, and TOPBP1 proteins. The sites identified on MDC1 have since been defined as ATM-dependent phosphorylation sites and shown to be required for MDC1 to promote 53BP1 foci formation at DNA DSBs (Kolas et al., 2007).
In addition to identifying new sites on known DDR proteins, the method also identified damage-dependent phosphorylation events on proteins not previously implicated in the cellular response to DNA damage. These include proteins linked to RNA processing, gene expression, chromatin remodeling, and developmental processes. Additional studies by other laboratories using similar approaches have further increased the total of number of potential ATM and ATR substrates (Mu et al., 2007, Stokes et al., 2007).
Although further experimentation is necessary to validate that any one of these phosphorylation sites is really catalyzed by ATM/ATR and determine whether these phosphorylations are functionally important, these lists provide a rich resource for investigators using other genomic or reductionist approaches to understanding DDR mechanisms. Finally, this general strategy can be used for other post-translational modifications like methylation, ubiquitylation, sumoylation, or acetylation provided that efficient affinity purifications can be achieved. See Boisvert et al., for an example in which several arginine methylated DNA repair proteins were identified (Boisvert et al., 2003).
The primary drawback of the mass spectrometry approach is that it is biased to identify the most abundant post-translationally modified peptides. Low abundant proteins that are only modified transiently and sub-stoichiometrically are likely to be poorly represented. The second issue is that there may be significant amounts of post-translational modifications that have no or redundant functional consequences. Thus, the catalogue of modifications is only a starting point for reductionist methods of studying function.
Screens examining protein subcellular localization
The subcellular localization of a protein provides a powerful indicator of whether it functions in a genome maintenance pathway. For example, proteins localized to the kinetochore likely regulate chromosome segregation during mitosis. Proteins localized to telomeres may be important in chromosome end replication or protection. Proteins localized to replisomes likely function in DNA replication.
There are two general strategies for using subcellular localization to identify genome maintenance proteins (Fig. 2D). The first is to make a library of tagged cDNAs and examine localization by fluorescence microscopy. The second is to purify cellular structures like telomeres and identify the associated proteins. The first approach has seen only limited application primarily as a secondary screening strategy for other functional genomic approaches. The second approach, however, has seen widespread application with great success.
Dejardin and Kingston developed a technique they called proteomics of isolated chromatin segments (PICh) to selectively purify proteins associated with specific DNA loci (Dejardin and Kingston, 2009). The steps of PICh include formaldehyde-crosslinking cells to crosslink DNA-protein and protein-protein complexes, solubilizing chromatin, denaturing the DNA to allow for DNA probe hybridization, purification of hybrids, elution, and finally analysis of eluted proteins by mass spectrometry. Since DNA hybridization for capture is not sensitive to high concentrations of ionic detergent, PICh allows for stringent capture and purification conditions that reduce non-specific binding of proteins. They applied this technique to identify proteins associated with mammalian telomeres. Their experiments successfully identified 85% of known telomere binding proteins and over 100 new proteins. Several of these are orphan nuclear receptors including COUP-TF2 and TR4. These proteins actually ranked higher on their abundance list than two known shelterin components. Interestingly, these proteins were found in telomere preparations from cells using the ALT method of telomere elongation specifically suggesting they have some function at ALT telomeres.
A few technical challenges must be overcome to use PICh for the classification of chromatin composition relevant to other genome maintenance activities. One of the primary concerns with this methodology is DNA-probe design. In many cases such as damage caused by a genotoxic agent, site-specificity is not available. The other major hurdle is that it may not be easily adapted to identifying proteins at DNA sequences that are found at one or few copies in the genome.
A second localization-based screen identified proteins whose localization to chromatin increased after cells were exposed to ionizing radiation (Larsen et al., 2010). Both DSB repair and DDR signaling proteins accumulate at the site of the DSB. This accumulation is often sufficient to observe a significant redistribution of the protein to the chromatin. Biochemical cell fractionation into soluble and chromatin compartments can be used to measure this change in localization. In this example, the authors determined that several subunits of the NuRD (nucleosome remodeling and histone deacetylation) chromatin-remodeling complex (CHD4, CHD3, MTA1 and MTA2) were recruited to chromatin after IR. These proteins have been best studied in the context of transcription where they work to remodel nucleosomes and thereby change gene expression (Knoepfler and Eisenman, 1999). DSB repair, especially homology-directed repair, requires access of the DSB repair proteins to the DNA surrounding the break (Xu and Price, 2011). Chromatin remodeling proteins likely work to regulate this access. Chromatin remodeling proteins are frequently identified in other functional genomic screens for genome maintenance proteins (see below) emphasizing that genome maintenance also requires chromatin maintenance.
RNAi screening
Genetics are difficult in diploid organisms because two copies of a gene must be manipulated to study loss of function phenotypes. Since RNAi-mediated silencing of genes occurs independently of gene copy number, it provides an opportunity to perform loss of function genetic screens in diploid cells. Thus, the genetic screens that were previously limited to organisms like S. cerevisiae and S. pombe can now be done in human, mouse, and insect cell culture or even whole organisms like C. elegans.
There are currently three primary genome-wide RNAi platforms. Small interfering RNAs (siRNAs) are chemically synthesized silencing reagents that can be arrayed in multi-well plates. Formats include individual siRNAs per well or pools of multiple siRNAs targeting a gene in each well. Enzymatically prepared siRNAs (esiRNAs) are produced through enzymatic methodologies instead of chemical synthesis and create arrayed pools of siRNAs targeting each gene. Finally, short-hairpin RNA (shRNA) vectors are DNA vectors encoding RNAs that are processed to yield the silencing RNA. These are often produced in viral vector systems allowing stable integration into the genome of infected cells. Both arrayed and large pool formats of shRNA libraries are available (Fig. 3).
Figure 3. RNAi screens provide a genetic approach to identifying genome maintenance activities.

(A) Pooled format screens facilitate drug hyper-sensitivity screening. (A1) Barcoded shRNA vector libraries are introduced into a population of cells, (A2) split into treated and untreated groups, (A3) grown for several generations to allow selection against a subset of the shRNA expressing cells; and (A4) analyzed by sequencing or microarray technology to identify differences in shRNA abundance. (B) Arrayed format RNAi libraries can be used both for (B1) hyper-sensitivity or (B2) high-content screens.
Drug hypersensitivity screens
Designing an RNAi screen begins with an assay suitable for high-throughput screening. The simplest screen designs relevant to genome maintenance pathways are drug hypersensitivity screens. The rationale is that by treating cells with a drug that places a high burden on a specific genome maintenance pathway, researchers can identify genes whose loss of function causes hypersensitivity to the drug. These types of screens have been used in many genetically tractable organisms like yeast for decades, are simple to execute, and are compatible with all RNAi formats.
Since drug hypersensitivity screens rely on growth inhibition or cell death as the primary assay, they are compatible with pooled libraries of shRNAs. In this format, a population of cells is infected at a low multiplicity of infection such that each cell harbors only a single shRNA. Since the shRNA should produce a stable knockdown, the cell population can be split in half, treated with a drug, and then examined for the relative abundance of each shRNA in the two populations. If a shRNA silences a gene required for cells to survive or proliferate in the presence of the drug, then that shRNA will be lost from the treated cell population but retained in the control population. The abundance of each shRNA can be determined by sequencing the shRNAs recovered from the two populations. Alternatively, microarrays can be used to detect barcodes incorporated into the shRNA library.
Using a pooled hypersensitivity screen, Elledge and colleagues sought to identify novel components of the DDR using cellular sensitivity to ionizing radiation as the assay (Wang et al., 2009, Hurov et al., 2010). The screen conducted in the human U2OS osteosarcoma cell line used a library of approximately 75,000 shRNA vectors pooled into six sub-libraries. 850 shRNAs targeting 813 genes were identified that caused significant hypersensitivity to ionizing radiation. These genes were grouped into functional categories using the PANTHER (Thomas et al., 2003). Unclassified genes accounted for 30% of the gene set. Multiple genes known to function in DDR networks were identified, and two new components of the DSB repair and DNA damage checkpoint signaling pathways were examined at the mechanistic level, NBA1 and the Triple T complex. NBA1 localizes to sites of DNA damage, associates with BRCA1 and is required for the recruitment of BRCA1 to damage sites (Wang et al., 2009). The Triple T complex consists of three proteins TTI1, TTI2, and TEL2 that regulates the stability of the phosphoinositide-3-kinase-related protein kinases ATM and ATR (Hurov et al., 2010).
Drug hypersensitivity screens can also be performed using libraries of arrayed shRNA, siRNAs or esiRNAs. Arrayed hypersensitivity screens measure the effects of single gene knockdown on cells in each well of a multi-well plate. Individual or pools of siRNAs targeting a single gene are typically introduced into cells by reverse transfection using lipid-based methods. Reagents that change color or fluorescence relative to the number of proliferating cells in a well can be used as a measure of cell viability. Microplate spectrophotometers enable multi-well processing of this assay endpoint.
Whitehurst and colleagues used an arrayed RNAi hyper-sensitivity screen to identify gene functions that drive the cellular response to paclitaxel in lung cancer cells (Whitehurst et al., 2007). Paclitaxel is a chemotherapeutic drug that targets microtubules to inhibit mitosis and is commonly used to treat patients with non-small lung cancers. Four unique siRNAs targeting each of 21,127 genes were transfected into a non-small lung cancer cell line in a one gene per well format with each well containing a pool of four siRNAs. A concentration below the half maximal inhibitory concentration for paclitaxel was selected to enrich for genes that when silenced rendered cells hypersensitive to low doses of drug. The screen was completed in triplicate and normalizations to internal plate controls were made to eliminate plate-to-plate technical variations.
To identify significant hits, a statistical algorithm was applied to calculated viability ratios yielding an estimated false discovery rate below 5%. As validation for their analysis, siRNAs targeting core proteasome components were identified as hits and proteasome inhibitors have already been demonstrated to enhance the sensitivity of paclitaxel in cancer cells (Lynch and Kim, 2005). They identified 87 genes that fit their criteria and reconfirmed that silencing caused sensitivity to paclitaxel with four additional siRNAs in pools and individually. Importantly, they confirmed these affects across multiple cell lines and defined genes that when silenced specifically sensitized cancer cells to low doses of paclitaxel compared to normal cell lines. Thus, the hits from this screen are potential drug targets for lung cancer since they selectively sensitize cancer cells and not normal cells (Nezi and Musacchio, 2009, Jordan and Wilson, 2004).
These screens offer several lessons about RNAi-based functional genomics approaches. It is striking that in the shRNA pooled screen for IR sensitivity the vast majority of genes found in the primary screen were only identified by a single shRNA despite multiple shRNAs per gene being present in the library (Wang et al., 2009, Hurov et al., 2010). This suggests that the shRNA targeting was inefficient, or a large number of the hits were due to off-target effects. The pooled screens require that a single shRNA vector inserted into the cellular genome produces sufficient shRNA to silence the gene effectively. Even with optimal vector designs, this requirement may not often be fulfilled. This issue will increase the false-negatives in the screen. Off-target effects leading to false-positives are also a concern necessitating that further follow-up experimentation be performed to rule them out. Finally, the large pooled format cannot ensure that every shRNA vector is equally represented or expressed in the library.
The arrayed screens likely suffer less from the issue of insufficient targeting. Algorithms for designing siRNAs are sufficiently robust to yield high percentages of highly effective siRNAs. Furthermore, they are transfected into the cells at very high copy number. However, off-target effects are also a concern in this format. In the paclitaxel arrayed screen on-target effects were confirmed by validating hits from the pooled siRNA primary screen with four individual siRNAs (Whitehurst et al., 2007). Alternatively, if the library is arrayed such that several individual siRNAs are used independently, then the results can be quickly scanned for genes that have at least two siRNAs yielding the same hypersensitive phenotype. This raises the largest obstacle to the arrayed screens – cost. Typically, whole genome screens are done in formats where small pools of siRNAs targeting a single gene are combined into a single well. This saves money in performing the screen but necessitates de-convolution of the pools to eliminate off-target effects. The arrayed screens also require specialized liquid handling systems that may not be available to a typical laboratory.
Synthetic genetic interaction screens
Synthetic genetic interactions arise when the phenotype from the loss of function of two genes are compared to the phenotypes of the individual gene effects. Often these interactions take the form of synthetic lethality. Synthetic genetic interaction screens in yeast are currently being done using synthetic genetic array (SGA) technologies and are very useful for dissecting complex biological pathways (Pan et al., 2006, Baryshnikova et al., 2010). SGA provides a format for the high-throughput construction of double mutants at the genome-wide level. Similar methodologies are not available in higher eukaryotes. Instead, investigators are turning to three approaches to perform synthetic genetic interaction screens: drug sensitivity screens, screens combining RNAi molecules targeting two different genes, and screens using matched pairs of cell lines in which a specific gene is mutated or wild-type.
The first involves using selective drugs in combination with RNAi to target proteins in the cell and is particularly useful in identifying drug targets or genetic backgrounds where a drug might be useful in treating cancer. For example, several synthetic lethal screens have been performed with recently developed inhibitors of the poly-ADP ribose polymerase (PARP) enzymes (Lord et al., 2008, Wiltshire et al., 2010, Turner et al., 2008). These inhibitors selectively kill cells deficient in homologous recombination (HR) based repair mechanisms. Thus, they are being developed for treating BRCA-deficient tumors since BRCA1 and BRCA2 are required for HR repair. Investigators reasoned that loss of function mutations in other genes are likely to yield defects in HR and performed synthetic lethal screens with PARP inhibitors to identify these genes. These screens not only define novel HR genes but also may point to additional clinical settings where PARP inhibitors can be useful.
The alternative two approaches to perform synthetic genetic interaction screens do not use selective drugs. The first is to combine two RNAi molecules targeting different genes. The second is to use matched pairs of cell lines in which a specific gene is mutated or wild-type. The first approach may be more quickly applied to any gene but suffers from issues of whether the simultaneous knockdown of two genes is as efficient as the single knockdown. It also suffers from the potential for synthetic off-target effects, which may be very difficult to control. Thus, the superior results that can be obtained from the second approach using matched pairs of cell lines with a defined loss of function mutation at the gene level may be worth the added time and cost of generating the cell lines.
High content RNAi screens
Hypersensitivity and synthetic lethal screens have relatively low information content because cell proliferation or viability is not a phenotype specific to genome maintenance pathways. Thus, large numbers of genes are identified in these types of screens. Even in yeast, these screens often identify large percentages of the genome as being required to survive or proliferate in response to a drug. Thus, higher-content assays are increasingly being employed. Essentially, anything that can be imaged can be tested using high content microscopy (HCM). High-resolution images can be generated rapidly in microtiter plates containing up to 1536 wells. Image processing software can yield information as varied as DNA content, cell size, and protein localization. Thus high content screening can be used to interrogate specific genome maintenance pathways at high resolution, and the screens typically yield smaller numbers of “hits”. These types of screens are best suited to arrayed RNAi library formats.
One example of a high content screen relevant to genome maintenance is a screen to define cell division cycle genes in human cells (Kittler et al., 2004). In this study, video microscopy was used to measure changes in the timing of cell division in HeLa cells in real time. In addition, fixed cells were stained for tubulin and DNA to visualize physical aberrations in mitotic structures. Screening a set of esiRNAs representing 5,305 genes led to the classification of genes with functional roles in spindle formation, mitotic arrest, and cytokinesis, including 7 genes with no previous assigned function and 23 with annotated functions other than cell division. The functional readouts for this study were specific and facilitated direct classification of gene groups.
Additional examples of high content screens include several recent screens that studied the DNA damage response by microscopy. Two screens were reported that used the detection of phosphorylated ATM/ATR substrates as the primary assay (Lovejoy et al., 2009, Paulsen et al., 2009). The largest of these screens used an siRNA library targeting the entire genome and visualized phosphorylation of the histone variant H2AX. H2AX is dispersed throughout the genome in approximately 10% of nucleosomes (Bonner et al., 2008). Phosphorylation of H2AX at serine 139 (γH2AX) by ATM and ATR is a well-defined early event at or near the site of DDR activation (Rogakou et al., 1998, Burma et al., 2001, Ward and Chen, 2001). γH2AX spreads kilobases of DNA beyond the break site, which allows visualization of bright γH2AX nuclear foci by microscopy (Rogakou et al., 1999). γH2AX has classically been used as a marker for DNA DSBs but is also phosphorylated at stalled replication forks and perhaps at other lesions where ATR is active.
The Cimprich group monitored γH2AX phosphorylation following gene silencing in the absence of any added genotoxic agent with the rationale that genes involved in genome maintenance pathways like DNA repair or replication prevent DNA damage from accumulating in cells (Paulsen et al., 2009). Silencing these genes would increase the DNA damage burden, thereby increasing the activation of ATM or ATR and phosphorylation of H2AX. The authors grouped their results into confidence levels defined by the strength of the phenotype, reproducibility, and numbers of individual siRNAs that yielded the same results. They defined 581 genes as high confidence hits. In addition to genes that function in the known genome maintenance pathways, they identified genes predicted to function in many other cellular metabolism activities including a large group of RNA processing proteins. After further investigation, they determined that at least a subset of these genes are needed to prevent RNA-DNA hybrids from forming during transcription which presumably lead to RNA polymerase stalling followed by collisions with the replication machinery in S-phase. Importantly, RNA processing proteins were also enriched in ATM/ATR substrate identification screens (Matsuoka et al., 2007) suggesting that RNA processing is a critical component of genome maintenance..
DNA damage also causes the recruitment of many DDR proteins to intranuclear foci that are easily visualized by immunofluoresence, including p53 binding protein (53BP1) (Schultz et al., 2000, Rappold et al., 2001). 53BP1 localizes to sites of DNA DSBs and functions in DSB repair via end joining (Iwabuchi et al., 2003, Ward et al., 2004, Nakamura et al., 2006, Manis et al., 2004, Difilippantonio et al., 2008, Xie et al., 2007). The recruitment of 53BP1 to DNA damage foci depends on γH2AX and MDC1 (FitzGerald et al., 2009). The underlying mechanisms of focal recruitment and retention of these DNA repair factors at DNA breaks has been a major question in the field. RNAi screens based on imaging 53BP1 localization were designed to address this question and led to the discovery of RNF8 and RNF168, which are ubiquitn conjugating enzymes that regulate the chromatin microenvironment important for DNA DSB repair (Kolas et al., 2007, Doil et al., 2009, Stewart et al., 2009).
Durocher and colleagues identified RNF8 in a genome-wide siRNA screen for the abundance of 53BP1 foci in U2OS cells 24hrs after IR (Kolas et al., 2007). They reasoned that gene functions required upstream of 53BP1 foci formation at DSBs would result in significant changes in abundance of 53BP1 foci when silenced. Pools of four siRNAs per gene were arrayed in 384 well formats and transfected into cells in quadruplicate. Using the high throughput acquisition and analysis capabilities of the Opera imaging system, they identified 500 siRNAs resulting in significant reduction in 53BP1 foci compared to controls. RNF8 was the top scoring hit following known upstream regulators of 53BP1 like γH2AX and MDC1. In a follow up study, 59 candidate genes from the original screen were targeted with additional esiRNAs to further validate 53BP1 loss. RNF168 was the top hit in this secondary validation and was subsequently defined to act downstream of RNF8, to assemble K63-linked ubiquitin chains at sites of DNA damage (Stewart et al., 2009).
Lukas and colleagues also identified RNF168 in a screen based on similar rationale and functional readout but using distinct screening methodologies (Doil et al., 2009). They applied a protocol that was developed for screening siRNAs spotted on chambered slides (Erfle et al., 2007). This technique represents another method for siRNA arrayed screens, siRNA microarrays. In this particular screen, U2OS were seeded in chambers containing 384 individual siRNA spots that were embedded in optimized transfection mixtures. After three days, the central area of each spot, containing approximately 150 cells, was analyzed for number of 53BP1 foci. A total of 21541 human genes were targeted with multiple siRNAs. When the screen was published, 15000 genes had been analyzed and 42 had at least one siRNA that resulted in reduced 53BP1 foci. Only three of 42 hits had two siRNAs score positive, MDC1, RNF8, and the previously uncharacterized RING finger protein, RNF168. These results may reflect a current disadvantage of this methodology, a high false negative rate. This problem may be due to inefficient siRNA knockdown or the small numbers of cells that were analyzed per data point. Increasing replicates or cells analyzed may suffice for reducing false-negatives.
RNAi screens in multi-cellular organisms
The benefits of RNAi genetic screens are not limited to cell culture. RNAi screens in the C. elegans worm provide a method for analyzing loss of function phenotypes in an intact animal. Delivery of RNAi into worms is performed either by injection of dsRNA directly into head or tail, soaking the worms in a solution of RNAi molecules, or by feeding animals bacteria that express dsRNA targeting a specific gene (Kamath and Ahringer, 2003). The effects of RNAi in C.elegans can be analyzed by whole mount staining or other phenotypic assays.
A genome-wide RNAi screen in C. elegans identified genes that protect animal cells against IR (van Haaften et al., 2006). The screen was completed with worm larvae cultured in the presence of bacteria expressing RNAi molecules. 19000 C. elegans genes were screened in an arrayed, multi-well format. Worm cultures were exposed to IR and subsequently imaged to measure for IR-induced cell cycle arrest and apoptosis in germ cells. After undergoing stringent filtering using dose response analyses, 45 genes emerged as IR protective genes. 43 of the 45 have clear human homologs emphasizing the extent to which the DDR pathway is conserved across evolution. Genes were cataloged based on their previously known functions, requirement for resistance to other DNA damaging agents, and homology across human, fly, and yeast.
Importantly, the human homologs of seven genes (ATM, ITGA6, NIPBL, CAND1/TIP120, NOB1, WWP2, and TOPBP1) were targeted by siRNA and measured for IR sensitivity, and all seven were confirmed as genes required for IR resistance in human cells. Several of these genes like ATM and TOPBP1 are not surprising since they were already known to function in DNA damage response pathways. However, less clear was why NOB1 (a protein implicated in ribosome assembly) or WWP2 (a ubiquitin ligase) were identified. Interestingly, WWP2 has since been shown to function as an E3 ligase for PTEN (Maddika et al., 2011). This may provide a link to the DNA damage response since PTEN function may modulate DNA repair activities (Chalmers et al., 2010).
Another C. elegans screen used a gfp-LacZ reporter of frameshifts and small insertions/deletions to screen for DNA instability in somatic cells (Pothof et al., 2003). This method enabled visual screening of cells harboring mutations that reverted the out of frame LacZ gene to the correct reading frame. The authors found 61 out of 16,606 genes screened resulted in a mutator phenotype. The orthologs of the DNA mismatch repair genes were among this list as would be expected. In addition to DNA repair genes, they found genes such as the histone deacetylases HDAC1 and HDAC2 which regulate chromatin organization and genes like cdc5 and cdc14 that regulate cell cycle checkpoints. 29 of the genes were not described previously but several have interesting protein domains such as exonuclease, histone acetyltransferase, and SET domains suggesting they could be involved in DNA metabolism.
General considerations about RNAi screens
RNAi screens provide an increasingly important source of discovery for genome maintenance researchers. A question like how do cells repair a double-strand break can be approached at multiple levels with RNAi screens. The simplest but least specific may be the ionizing radiation sensitivity screens. The most specific, but also most technologically difficult will be screens based on direct measurements of repair. In between are the high content imaging screens scoring for markers like γH2AX or 53BP1 foci. The choice of screen assay in part determines the optimal screening platform. In practice, the most useful information may come from combining the results of multiple screens. Certainly the confidence that any previously unstudied gene functions in a DSB repair pathway is significantly increased by the independent identification of that gene in more than one screen.
Eliminating false-positives is critical for the results of the screen to be useful. The most common source of false-positives is off-target effects of the RNAi. Since silencing does not require perfect base pair complementarity between the siRNA and the mRNA, each RNAi molecule has the potential to silence multiple genes. The most common method to eliminate off-target effects is to require that more than one RNAi molecule targeting the same gene yield the same phenotypic consequence. If antibodies to the targeted protein are available then correlating protein knockdown efficiency with strength of phenotype adds further confidence. Finally, complementation of the knockdown phenotype with a resistant cDNA is the best method of demonstrating an on-target effect. However, this approach is not feasible in a high-throughput format.
RNAi screens likely suffer from high rates of false-negatives. Even when two investigators use similar methodologies, the overlap in gene hits is often low. False-negatives come from inefficient knockdown, cell type specific effects, and deficiencies in the genome-wide libraries. The cell-type specific effects are likely to be large since most screens use tumor-derived cells that have large numbers of genetic abnormalities already. These differences can be exploited especially in synthetic lethal type screens where the important question is what makes two or more cell types behave differently.
All of these RNAi screens depend on a statistical determination of what is a “hit”. Defining the hits requires a measurement of difference from the control, variability, and elimination of off-target effects. Many different statistical methods have been developed but determining biological significance requires more in-depth analyses. Functional annotation programs such as PANTHER can help but these issues point out the need for secondary screens to validate hits and provide further guidance on future directions. Ideally, these secondary screens should also be amenable to high-throughput assays. Ultimately, more traditional reductionist approaches are needed to fully validate and understand the results of any RNAi screen.
Conclusions
Expression profiling, proteomics, and RNAi screening methodologies have proven successful in the investigation of genome maintenance activities. When considering the power of these methodologies for discovery, it is important to realize that in many cases they were made with first generation technologies. In the next ten years we can expect the development of arrayed full-genome epitope-tagged cDNA libraries for faster genome-wide proteomics; protein microarray chips will expand to include a much higher percentage of the proteome; and RNAi libraries will continue to improve.
The amount of data being generated is quickly outpacing the ability of a single investigator to assimilate. Thus, bioinformatic analyses must be developed alongside of improving high-throughput screening technologies. Better databases to store, report, search, and integrate data sets from RNAi screens are also needed.
The rapidly expanding data sets from expression profiling, proteomics, and RNAi screening methods are generating lists of new genome maintenance proteins. These lists contain genes that function in pathways like chromatin regulation that have obvious links to the DNA metabolic processes that maintain the genome. But they also contain evidence that many other intracellular pathways impact genome integrity. Our challenge is to complete the catalogue and explain at a mechanistic level how these activities are integrated within the cell to protect genome stability and prevent disease.
Table.
Examples of RNAi screens for genome maintenance discussed in text
| Format | Genotoxic challenge | Cell Type | RNAi type | Library size (RNAi; genes targeted) | Hits | Genome Maintenance Activity | Reference |
|---|---|---|---|---|---|---|---|
| Pooled-hype rsensitivity | IR | U2OS | barcoded shRNA | 74,905; 32,293 | 813 | DNA damage response to DSB | Hurov et al., 2010 |
| Arrayed-hypersensitivity | paclitaxel | NCI-H1155 | siRNAs | 84,508; 21,127 | 87 | mitosis; spindle assembly checkpoint | Whitehurst et al., 2007 |
| Arrayed-synthetic lethal | PARP | CAL51 | siRNAs | 3,116; 779 | 6 | homologous recombination; DSB repair | Turner et al., 2008 |
| Arrayed-synthetic lethal | PARP | CAL51 | siRNAs | 460; 230 | 5 | homologous recombination; DSB repair | Lord et al., 2008 |
| Arrayed-synthetic lethal | PARP | U2OS | siRNAs | ~250; 73 | 29 | homologous recombination; DSB repair | Wiltshire et al., 2008 |
| Cell cycle image screen | none | HeLa | esiRNAs | 5,305 genes | 37 | cell cycle; cytokinesis; spindle assembly | Kttler et al., 2004 |
| H2AX image screen | none | HeLa | siRNAs | ~84,000; ~21,000 | 581 | spontaneous DSB prevention | Paulsen et al., 2009 |
| H2AX image screen | none | HeLa, U2OS | shRNAs, siRNAs | 6386; 2287 | 93 | spontaneous DSB prevention | Lovejoy et al., 2009 |
| 53BP1 image screen | IR | U2OS | siRNAs | genome-wide | 500 | DSB-induced chromatin microenvironment | Kolas et al., 2007 |
| 53BP1 image screen | none | U2OS | siRNA microarrays | ~50000; 21,541 | 42 | DSB-induced chromatin microenvironment | Doil et al., 2009 |
| Organism | IR | C. elegans | RNAi food | 19,000 genes | 45 | IR-induced cell cycle arrest; apoptosis | van Haaften et al., 2006 |
| Organism | none | C. elegans | RNAi food | 16,757; 16,606 | 61 | spontaneous mutation prevention | Pothof et al., 2003 |
Acknowledgements
We thank Edward Nam and Bianca Sirbu for critical reading of the manuscript.
Footnotes
Declaration of Interest Functional genomics research in the Cortez laboratory is supported by NIH grant CA136933, the Vanderbilt-Ingram Cancer Center grant CA06485, and the Susan G. Komen for the Cure grant KG080227.
References
- Amundson SA, Bittner M, Fornace AJ., Jr. Functional genomics as a window on radiation stress signaling. Oncogene. 2003;22:5828–33. doi: 10.1038/sj.onc.1206681. [DOI] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ, Myers CL, Andrews B, Boone C. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Methods Enzymol. 2010;470:145–79. doi: 10.1016/S0076-6879(10)70007-0. [DOI] [PubMed] [Google Scholar]
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–74. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Bennett CB, Lewis LK, Karthikeyan G, Lobachev KS, Jin YH, Sterling JF, Snipe JR, Resnick MA. Genes required for ionizing radiation resistance in yeast. Nat Genet. 2001;29:426–34. doi: 10.1038/ng778. [DOI] [PubMed] [Google Scholar]
- Birrell GW, Brown JA, Wu HI, Giaever G, Chu AM, Davis RW, Brown JM. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. Proc Natl Acad Sci U S A. 2002;99:8778–83. doi: 10.1073/pnas.132275199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell GW, Giaever G, Chu AM, Davis RW, Brown JM. A genome-wide screen in Saccharomyces cerevisiae for genes affecting UV radiation sensitivity. Proc Natl Acad Sci U S A. 2001;98:12608–13. doi: 10.1073/pnas.231366398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund M, Taipale M, Varjosalo M, Saharinen J, Lahdenperä J, Taipale J. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature. 2006;439:1009–1013. doi: 10.1038/nature04469. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, Cote J, Boulanger MC, Richard S. A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics. 2003;2:1319–30. doi: 10.1074/mcp.M300088-MCP200. [DOI] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ. Combined Functional Genomic Maps of the C. elegans DNA Damage Response. Science. 2002;295:127–131. doi: 10.1126/science.1065986. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Cagney G, Alvaro D, Reid RJD, Thorpe PH, Rothstein R, Krogan NJ. Functional genomics of the yeast DNA-damage response. Genome Biol. 2006;7:233. doi: 10.1186/gb-2006-7-9-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–86. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Cejka P, Jiricny J. Interplay of DNA repair pathways controls methylation damage toxicity in Saccharomyces cerevisiae. Genetics. 2008;179:1835–44. doi: 10.1534/genetics.108.089979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol. 2010;20:274–81. doi: 10.1016/j.semradonc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Chaudhry MA, Chodosh LA, Mckenna WG, Muschel RJ. Gene expression profiling of HeLa cells in G1 or G2 phases. Oncogene. 2002;21:1934–42. doi: 10.1038/sj.onc.1205264. [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10:756–68. doi: 10.1038/nrg2663. [DOI] [PubMed] [Google Scholar]
- Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci U S A. 2004;101:10078–83. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick ME, Klitgord N, Vidal M, Hill DE. Interactome: gateway into systems biology. Hum Mol Genet. 2005;14(Spec No. 2):R171–81. doi: 10.1093/hmg/ddi335. [DOI] [PubMed] [Google Scholar]
- D'andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med. 2010;362:1909–19. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Korner R, Greff Z, Keri G, Stemmann O, Mann M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell. 2008;31:438–48. doi: 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Davidson MB, Brown GW. Dissecting the DNA damage response using functional genomics approaches in S. cerevisiae. DNA Repair (Amst) 2009;8:1110–7. doi: 10.1016/j.dnarep.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Dejardin J, Kingston RE. Purification of proteins associated with specific genomic Loci. Cell. 2009;136:175–86. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, Sleckman BP, Nussenzweig A. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–33. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, Lukas J, Lukas C. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- Draviam VM, Stegmeier F, Nalepa G, Sowa ME, Chen J, Liang A, Hannon GJ, Sorger PK, Harper JW, Elledge SJ. A functional genomic screen identifies a role for TAO1 kinase in spindle-checkpoint signalling. Nat Cell Biol. 2007;9:556–64. doi: 10.1038/ncb1569. [DOI] [PubMed] [Google Scholar]
- Erfle H, Neumann B, Liebel U, Rogers P, Held M, Walter T, Ellenberg J, Pepperkok R. Reverse transfection on cell arrays for high content screening microscopy. Nat Protoc. 2007;2:392–9. doi: 10.1038/nprot.2006.483. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JE, Grenon M, Lowndes NF. 53BP1: function and mechanisms of focal recruitment. Biochem Soc Trans. 2009;37:897–904. doi: 10.1042/BST0370897. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. DNA damage and repair. Nature. 2003;421:436–40. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- Fromont-Racine M, Rain JC, Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat Genet. 1997;16:277–82. doi: 10.1038/ng0797-277. [DOI] [PubMed] [Google Scholar]
- Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–2. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, Gerstein M, Dumont ME, Phizicky EM, Snyder M, Grayhack EJ. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–26. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, Labonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–32. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Culotti J, Reid B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc Natl Acad Sci U S A. 1970;66:352–9. doi: 10.1073/pnas.66.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houldsworth J, Lavin MF. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980;8:3709–20. doi: 10.1093/nar/8.16.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–17. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Hurov KE, Cotta-Ramusino C, Elledge SJ. A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes & Development. 2010;24:1939–50. doi: 10.1101/gad.1934210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikner A, Shiozaki K. Yeast signaling pathways in the oxidative stress response. Mutat Res. 2005;569:13–27. doi: 10.1016/j.mrfmmm.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, Date T, Doherty AJ. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem. 2003;278:36487–95. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–21. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–43. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–9. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–88. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Kittler R, Pelletier L, Heninger A-K, Slabicki M, Theis M, Miroslaw L, Poser I, Lawo S, Grabner H, Kozak K, Wagner J, Surendranath V, Richter C, Bowen W, Jackson AL, Habermann B, Hyman AA, Buchholz F. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol. 2007;9:1401–1412. doi: 10.1038/ncb1659. [DOI] [PubMed] [Google Scholar]
- Kittler R, Putz G, Pelletier L, Poser I, Heninger A-K, Drechsel D, Fischer S, Konstantinova I, Habermann B, Grabner H, Yaspo M-L, Himmelbauer H, Korn B, Neugebauer K, Pisabarro MT, Buchholz F. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature. 2004;432:1036–40. doi: 10.1038/nature03159. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Eisenman RN. Sin meets NuRD and other tails of repression. Cell. 1999;99:447–50. doi: 10.1016/s0092-8674(00)81531-7. [DOI] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, Danielsen JMR, Menard P, Sand JC, Stucki M, Lukas C, Bartek J, Andersen JS, Lukas J. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. The Journal of Cell Biology. 2010;190:731–40. doi: 10.1083/jcb.200912135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Zhao S, Karur V, Wojchowski DM. DYRK3 activation, engagement of protein kinase A/cAMP response element-binding protein, and modulation of progenitor cell survival. J Biol Chem. 2002;277:47052–60. doi: 10.1074/jbc.M205374200. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–31. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Mcdonald S, Swift S, Turner NC, Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 2008;7:2010–9. doi: 10.1016/j.dnarep.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao R, Ye F, Sirbu BM, Titus LC, Shyr Y, Cortez D. Functional genomic screens identify CINP as a genome maintenance protein. Proc Natl Acad Sci USA. 2009;106:19304–9. doi: 10.1073/pnas.0909345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch T, Jr., Kim E. Optimizing chemotherapy and targeted agent combinations in NSCLC. Lung Cancer. 2005;50(Suppl 2):S25–32. [PubMed] [Google Scholar]
- Macbeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–3. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- Maddika S, Kavela S, Rani N, Palicharla VR, Pokorny JL, Sarkaria JN, Chen J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat Cell Biol. 2011 doi: 10.1038/ncb2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–7. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, Mcdonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, Smeets D, Solder B, Belohradsky BH, Der Kaloustian VM, Oshimura M, Isomura M, Nakamura Y, Komatsu K. Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet. 1998;19:179–81. doi: 10.1038/549. [DOI] [PubMed] [Google Scholar]
- Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- Merbl Y, Kirschner MW. Large-scale detection of ubiquitination substrates using cell extracts and protein microarrays. Proc Natl Acad Sci U S A. 2009;106:2543–8. doi: 10.1073/pnas.0812892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Mu J-J, Wang Y, Luo H, Leng M, Zhang J, Yang T, Besusso D, Jung SY, Qin J. A Proteomic Analysis of Ataxia Telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) Substrates Identifies the Ubiquitin-Proteasome System as a Regulator for DNA Damage Checkpoints. Journal of Biological Chemistry. 2007;282:17330–17334. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- Mukherji M, Bell R, Supekova L, Wang Y, Orth AP, Batalov S, Miraglia L, Huesken D, Lange J, Martin C, Sahasrabudhe S, Reinhardt M, Natt F, Hall J, Mickanin C, Labow M, Chanda SK, Cho CY, Schultz PG. Genome-wide functional analysis of human cell-cycle regulators. Proc Natl Acad Sci USA. 2006;103:14819–24. doi: 10.1073/pnas.0604320103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006;5:741–9. doi: 10.1016/j.dnarep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu Rev Genet. 2001;35:673–745. doi: 10.1146/annurev.genet.35.102401.091334. [DOI] [PubMed] [Google Scholar]
- Nezi L, Musacchio A. Sister chromatid tension and the spindle assembly checkpoint. Curr Opin Cell Biol. 2009;21:785–95. doi: 10.1016/j.ceb.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Nurse P. Genetic control of cell size at cell division in yeast. Nature. 1975;256:547–51. doi: 10.1038/256547a0. [DOI] [PubMed] [Google Scholar]
- Nurse P, Thuriaux P, Nasmyth K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol Gen Genet. 1976;146:167–78. doi: 10.1007/BF00268085. [DOI] [PubMed] [Google Scholar]
- O'connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ, Harper JW. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Mol Cell. 2010;40:645–57. doi: 10.1016/j.molcel.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'donnell L, Panier S, Wildenhain J, Tkach JM, Al-Hakim A, Landry M-C, Escribano-Diaz C, Szilard RK, Young JTF, Munro M, Canny MD, Kolas NK, Zhang W, Harding SM, Ylanko J, Mendez M, Mullin M, Sun T, Habermann B, Datti A, Bristow RG, Gingras A-C, Tyers MD, Brown GW, Durocher D. The MMS22L-TONSL Complex Mediates Recovery from Replication Stress and Homologous Recombination. Molecular Cell. 2010 doi: 10.1016/j.molcel.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc Natl Acad Sci U S A. 1980;77:7315–7. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–81. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee M-C, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE. A Genome-wide siRNA Screen Reveals Diverse Cellular Processes and Pathways that Mediate Genome Stability. Molecular Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–56. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Pothof J, Van Haaften G, Thijssen K, Kamath RS, Fraser AG, Ahringer J, Plasterk RH, Tijsterman M. Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev. 2003;17:443–8. doi: 10.1101/gad.1060703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–20. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashi-Elkeles S, Elkon R, Weizman N, Linhart C, Amariglio N, Sternberg G, Rechavi G, Barzilai A, Shamir R, Shiloh Y. Parallel induction of ATM-dependent pro- and antiapoptotic signals in response to ionizing radiation in murine lymphoid tissue. Oncogene. 2006;25:1584–92. doi: 10.1038/sj.onc.1209189. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–90. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Dodson GE, Mukhopadhyay PS, Shanware NP, Trinh AT, Tibbetts RS. Identification of carboxyl-terminal MCM3 phosphorylation sites using polyreactive phosphospecific antibodies. J Biol Chem. 2007;282:9236–43. doi: 10.1074/jbc.M609256200. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiácovo MP. A Genetic Screen Identifies FAN1, a Fanconi Anemia-Associated Nuclease Necessary for DNA Interstrand Crosslink Repair. Mol Cell. 2010;39:36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen S-H, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007;104:10364–9. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, Oldreive C, Wildenhain J, Tagliaferro A, Pelletier L, Taubenheim N, Durandy A, Byrd PJ, Stankovic T, Taylor AMR, Durocher D. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, Yang V, Beausoleil SA, Gygi SP, Livingstone M, Zhang H, Polakiewicz RD, Comb MJ. Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci USA. 2007;104:19855–60. doi: 10.1073/pnas.0707579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–97. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–41. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–1377. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Haaften G, Romeijn R, Pothof J, Koole W, Mullenders LHF, Pastink A, Plasterk RHA, Tijsterman M. Identification of conserved pathways of DNA-damage response and radiation protection by genome-wide RNAi. Current Biology. 2006;16:1344–50. doi: 10.1016/j.cub.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes & Development. 2009;23:729–739. doi: 10.1101/gad.1770309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. 53BP1 is required for class switch recombination. J Cell Biol. 2004;165:459–64. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, Xie X-J, White MA. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- Wiltshire TD, Lovejoy CA, Wang T, Xia F, O'connor MJ, Cortez D. Sensitivity to Poly(ADP-ribose) Polymerase (PARP) Inhibition Identifies Ubiquitin-specific Peptidase 11 (USP11) as a Regulator of DNA Double-strand Break Repair. Journal of Biological Chemistry. 2010;285:14565–14571. doi: 10.1074/jbc.M110.104745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-Macdonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Xie A, Hartlerode A, Stucki M, Odate S, Puget N, Kwok A, Nagaraju G, Yan C, Alt FW, Chen J, Jackson SP, Scully R. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol Cell. 2007;28:1045–57. doi: 10.1016/j.molcel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle. 2011;10:261–7. doi: 10.4161/cc.10.2.14543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]