

Abstract
EV71 is the primary pathogenic cause of hand-foot-mouth disease (HFMD), but an effective antiviral drug currently is unavailable. Rupintrivir, an inhibitor against human rhinovirus (HRV), has potent antiviral activities against EV71. We determined the high-resolution crystal structures of the EV71 3Cpro/rupintrivir complex, showing that although rupintrivir interacts with EV71 3Cpro similarly to HRV 3Cpro, the C terminus of the inhibitor cannot accommodate the leaving-group pockets of EV71 3Cpro. Our structures reveal that EV71 3Cpro possesses a surface-recessive S2′ pocket that is not present in HRV 3Cpro that contributes to the additional substrate binding affinity. Combined with mutagenic studies, we demonstrated that catalytic Glu71 is irreplaceable for maintaining the overall architecture of the active site and, most importantly, the productive conformation of catalytic His40. We discovered the role of a previously uncharacterized residue, Arg39 of EV71 3Cpro, that can neutralize the negative charge of Glu71, which may subsequently assist deprotonation of His40 during proteolysis.
INTRODUCTION
Hand-foot-mouth disease (HFMD) is a serious rapid-spread disease caused mainly by enterovirus 71 (EV71), coxsackie A16, as well as other enteroviruses (10). In China alone, around 1 million people (mostly children) were diagnosed with HFMD in the past year (http://news.xinhuanet.com/english2010/health/2010-07/01/c_13378827.htm). In severe cases, HFMD can lead to neurological damage with significant fatalities (22). Unfortunately, there are no drugs or vaccines against the disease.
Similarly to other picornavirus, EV71 has a single-stranded RNA genome encoding a large polyprotein precursor that requires proteolytic processing to produce the functional, structural, and replication proteins. The cleavages are dependent mainly on the viral 3C protease (3Cpro). Recent studies demonstrated that EV71 3Cpro can impair the antiviral responses of the infected cell by disruptions of retinoic acid-inducible gene I (RIG-I) and Toll-like receptor 3 (TLR3) signaling pathways (15, 16). Therefore, the protease generally is considered an appealing drug target. Rupintrivir is a peptidomimetic inhibitor designed to target human rhinovirus (HRV) 3Cpro (17). Interestingly, recent studies indicated that the inhibitor is also effective against enteroviruses (6, 13, 24), presumably due to the structural similarity of their 3C proteases. A recent study showed that the interferon (IFN)-mediated antiviral mechanism can be compromised by the proteolytic cleavage of EV71 3Cpro, suggesting that a combination of 3Cpro inhibitor and IFN-α could be an effective treatment for EV71 infection (11). Therefore, it is important to ascertain the mechanism of EV71 3Cpro inhibition at the molecular level, which will benefit further inhibitor optimization.
We carried out structural studies on EV71 3Cpro previously (8). The crystal structure of the unliganded EV71 3Cpro revealed that the protease shares structural similarity with 3C proteases from hepatitis A virus (HAV), foot-and-mouth-disease virus (FMDV), HRV, poliovirus (PV), and coxsackie B virus (CVB) (2, 7, 14, 18, 20). However, one striking difference is that a conserved structural feature, the β-ribbon that is located above the substrate binding cleft and forms parts of S2 to S4 specificity pockets in other picornaviral 3Cpro, adopts an unusual open conformation in EV71 3Cpro. Due to the open β-ribbon conformation, the active site of EV71 3Cpro is very exposed to the solvent, there were poor electron densities to define the conformations of the active site, and in particular there were no electron densities to define the side chain conformation of catalytic Glu71. Nevertheless, the mutagenic study proved that Glu71 is essential for protease activity. Many of the available picornaviral 3Cpro structures demonstrate that the active site of the protease is comprised of a cluster of the catalytically important residues Cys, His, and Asp/Glu that are linked together by an extensive hydrogen bond network, maintaining a geometry similar to that of the Ser-His-Asp catalytic triad found in serine proteases, supporting the hypothesis that picornaviral 3Cpro adopts the catalytic triad mechanism. However, the hypothesis was called into question by the independent structure determinations of HAV 3Cpro (4, 5), in which the catalytic aspartic acid is directed away from the active site, suggesting that the Cys-His dyad is sufficient for proteolytic activity. The dispute reflects that the role of the third member of the catalytic triad has not been fully characterized. The third member of the catalytic triads is always aspartic acid in serine protease, whereas in picornaviral 3Cpro the residue can either be aspartic acid or glutamic acid. The acidic member of the catalytic triad is strictly conserved in picornaviruses. Mutagenic studies showed that any substitutions of this residue, even the conserved mutation from aspartic acid to glutamic acid, resulted in severe damage to catalytic activity (23). However, the structure basis of the conservation remains unclear.
In this work, we show that rupintrivir is a potent inhibitor against EV71 (isolate BJ/CHN/2008), and the drug inhibits the protease activity of EV71 3Cpro in vitro. We determined the high-resolution crystal structures of EV71 3Cpro mutants in complex with rupintrivir, providing precise molecular insights into the substrate recognition and inhibition of protease, which is invaluable for structure-based inhibitor design. Our structural and mutagenic studies of EV71 3Cpro mutants demonstrate the irreplaceable role of catalytic Glu71 and the role of a previously uncharacterized residue, Arg39, that can modulate the charging of Glu71 during proteolysis. Our findings add novel details to the proteolysis mechanism of EV71 3Cpro.
MATERIALS AND METHODS
Constructs and proteins.
The expression and purification of the recombinant wild-type (WT) EV71 3Cpro and the protease mutants were carried out as described previously (8). Briefly, plasmids encoding N-terminal 6× His-tagged EV71 3Cpro mutants were obtained by site-directed mutagenesis (QuikChange). The recombinant proteins were overexpressed in Escherichia coli (Rosetta; Novagen) and purified using Ni-nitrilotriacetic acid (NTA) resin (Qiagen), followed by thrombin (Sigma) digestion to remove the His tag. Additional purifications were carried out to remove thrombin contamination.
Crystallization and structure determination.
EV71 3Cpro mutants were concentrated to ∼10 mg/ml prior to the addition of rupintrivir (TRC, Inc.) dissolved in methanol. The final molar ratio of protease to inhibitor was 1:5. After a brief incubation on ice, the mixtures were clarified by centrifugation prior to the crystallization trails. The optimal crystallization was achieved by mixing 0.8 μl protein with 1.0 μl reservoir buffer in a hanging drop vapor diffusion system at 295K. The reservoir buffer for mutants H133G and E71A contains 100 mM Tris-HCl (pH 8.3) and 26% polyethylene glycol (PEG)-1000, and the reservoir buffer for mutant E71D contains 0.1 M HEPES (pH 7.3), 10% PEG-4000, and 20% isopropanol. Cryocooling was carried out by soaking the crystals in reservoir solution containing 15% ethylene glycol prior the flash-freezing. Diffraction data were collected at beamline PXIII (Swiss Light Source, Villigen, Switzerland) and were processed with the XDS package (12). The space group identified for the crystals of mutants H133G and E71A was I212121, with 2 molecules per asymmetric unit (ASU). The space group identified for the crystal of mutant E71D was P41212, with 4 molecules per ASU. The initial phases were obtained by Phaser v2.1 (19) using HRV 3Cpro (Protein Data Bank [PDB] no. 1CQQ) as the search model. The electron density map was improved by manual model building using Coot v0.6 (9). The models were refined with PHENIX (1). Data collection and the final model statistics are summarized in Table 1.
Table 1.
Crystallographic data and model statisticsa
| Parameter | Value for: |
||
|---|---|---|---|
| EV71 3Cpro H133G | EV71 3Cpro E71A | EV71 3Cpro E71D | |
| Data collection statistics | |||
| Space group | I212121 | I212121 | P41212 |
| Cell dimensions | |||
| a, b, c (Å) | 69.716, 98.203, 102.619 | 69.785, 97.822, 102.540 | 142.443, 142.443, 69.564 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.000 | 0.800 | 1.54 |
| Resolution (Å) | 1.308 | 1.039 | 1.7 |
| Rsym | 0.071 (0.708) | 0.033 (0.509) | 0.020 (0.055) |
| I/σ(I) | 13.78 (2.55) | 22.22 (2.85) | 22.7 (8.7) |
| Completeness (%) | 98.1 (93.9) | 99.2 (97.6) | 93.3 (79.5) |
| Redundancy | 3.81 | 3.94 | 3.18 |
| Refinement statistics | |||
| Resolution range (Å) | 57.667-1.308 | 25.635-1.039 | 28.06-1.70 |
| No. of reflections | 158,376 | 166,400 | 73,112 |
| Rwork/Rfree | 0.1475/0.1748 | 0.150/0.164 | 0.198/0.254 |
| No. of: | |||
| Proteins | 3,065 | 3,461 | 5,800 |
| Ligands | 2 Rupintrivir | 2 Rupintrivir | 4 Rupintrivir |
| Water molecules | 293 | 393 | 598 |
| Hydrogen molecules | 3,786 | 4,357 | 5,890 |
| Protein B factor avg | 20.93 | 17.08 | 26.68 |
| RMSD | |||
| Bond length (Å) | 0.008 | 0.011 | 0.018 |
| Bond angle (°) | 1.210 | 1.608 | 1.021 |
The highest-resolution shell is shown in parentheses. Rwork = Σh|Fobs − Fcalc|/ΣhFobs, where Fobs and Fcalc are the observed and calculated structure factors. Rfree is Rwork calculated by using a randomly selected 5% sample of reflection data omitted from the refinement. Rsym = ΣhΣj/Ihj − Ih|/ΣhΣjIhj, where Ih is the weighted mean intensity of the symmetry-related reflections. I/σ(I) = mean of intensity/sigma (intensity) of unique reflections after merging symmetry-related reflections. RMSD, root mean square deviations.
Protease activity assay.
The fluorogenic peptide Dabcyl-RTATVQGPSLDFE-Edans (EV71 3Cpro cleaves the peptide bond between the underlined residues), corresponding to the EV71 polyprotein 3B/3C autoprocessing site, was used as the substrate, and recombinant EV71 3Cpro and the protease mutants were use as enzymes in the protease assays. The initial velocities of reactions were plotted as a function of substrate concentrations; the data were fitted to the Michaelis-Menten equation to calculate kinetic parameters Km, maximum rate of metabolism (Vmax), and Km/kcat (Microcal Origin).
Protease inhibition assay.
To evaluate the inhibition of protease activity of EV71 3Cpro by rupintrivir, the 50% inhibitory concentration (IC50) was determined similarly to the method described previously (13). Briefly, the reactions were performed with 4 μM protease and 150 mM fluorogenic peptide (Dabcyl-RTATVQGPSLDFE-Edans, corresponding to the EV71 polyprotein autoprocessing site between 3B and 3C) in a buffer of Tris-HCl (pH 7.0), 200 mM NaCl, 2 mM dithiothreitol (DTT) at 30°C. The initial velocities of the proteolysis were plotted as the function of the rupintrivir concentrations to obtain the IC50 by fitting with the following equation: A(I) = A(0) × {1 − ([I])/([I] + IC50)]}, where A(I) is the enzyme activity with rupintrivir concentration [I] and A(0) is enzyme activity without inhibitor.
Antiviral assay.
The ability of compounds to protect cells against infection was measured by a dye reduction method. Briefly, RD cells were plated onto 96-well plates at a density of 104 cells per well and cultured overnight. Cells were infected with EV71 at a multiplicity of infection (MOI) of 0.01 or mock infected with medium only. Three days later, 20 μl of MTS lysis buffer (CellTiter 96 Aqueous; Promega) was added to the test plates, and the amount of formazan produced was quantified spectrophotometrically at a test reference of 490 nm. Data were expressed as the percentage of formazan produced in compound-treated cells compared to formazan produced in wells of uninfected, compound-free cells. The 50% effective concentration (EC50) was calculated as the concentration of compound that increased the percentage of formazan production in infected, compound-treated cells to 50% of that produced by uninfected, compound-free cells.
Western blot analysis.
The ability of rupintrivir to inhibit viral protein accumulation was determined by using RD cells grown in Dulbecco's modified essential medium (DMEM) supplemented with 10% fetal calf serum. The confluent cells in 24-well plates were infected with EV71 at an MOI of 0.1 in the absence or presence of rupintrivir at 0, 1, and 10 nM. Twenty-four h postinfection, total cell lysate was harvested and subjected to 12% SDS-PAGE and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane and probed with a mouse monoclonal antibody against EV71 VP1 and anti-β-actin antibody(1:2,000). An IRDye 680-conjugated goat anti-mouse antibody (1:10,000) was used as the secondary antibody and was detected by the Odessy system.
Real-time reverse transcription-PCR.
Primers 5′-AGTATGATTGAGACACGCTG-3′ and ′-GCAACAAAAGTGAACTCTGC-3′ were used to amplify the region of 233 nucleotides (nt) from nt 2622 to 2854 in EV71 isolate BJ/CHN/2008 coding sequence (CDS). An RNAeasy Minikit (Qiagen, Germany) was used for RNA extraction according to the manufacturer's instructions. A reverse transcription (RT) reaction was carried out by using Superscript III reverse transcriptase (Invitrogen) in a 20-μl reaction mixture by using 1.2 μg total RNA. Real-time PCR was conducted using an ABI Prism 7000 rReal-time PCR system (Applied Biosystems) according to the manufacturer's instructions. Reactions were performed in a 50-μl volume that contained 2 μl cDNA, 1 μl of each primer, and 25 μl power SYBR green PCR master mix (Applied Biosystems). Relative quantities of viral RNA were calculated by using GAPDH as the inner control.
Accession number.
Coordinates and structure factors have been deposited in the PDB under accession numbers 3QZQ, 3R0F, and 3QZR.
RESULTS AND DISCUSSION
Structure determination of EV71 3Cpro mutants in complex with rupintrivir.
To elucidate the structural basis of the inhibition of EV71 3Cpro by rupintrivir, we determined the structures of EV71 3Cpro/rupintrivir complexes. Due to the practical difficulty in obtaining the measurable crystals from the WT EV71 3Cpro/rupintrivir complex, we chose EV71 3Cpro mutant H133G/rupintrivir complex for structural studies. Mutant H133G has close-to-WT enzyme activity and substrate specificity (8), suggesting that the mutant maintains conformations for substrate binding and hydrolysis similar to those of the WT protease. The crystal of the EV71 3Cpro mutant H133G/rupintrivir complex belongs to space group I212121, contained two H133G/rupintrivir complexes per asymmetric unit, and diffracted X rays to a resolution limit of 1.3 Å. To reveal the role of the third member of the catalytic triad, Glu71, we further crystallized two inactive EV71 3Cpro mutants, E71A and E71D, in complex with rupintrivir. The crystal of the EV71 3Cpro mutant E71A/rupintrivir complex belongs to space group I212121, contained two E71A/rupintrivir complexes per asymmetric unit, and diffracted X rays to a resolution limit of 1.0 Å. The crystal of the EV71 3Cpro mutant E71D/rupintrivir complex belongs to space group P41212, contained four E71D/rupintrivir complexes per asymmetric unit, and diffracted X rays to a resolution limit of 1.7 Å. These three crystal structures of EV71 3Cpro mutant/rupintrivir complexes were solved by molecular replacement using the HRV 3Cpro/rupintrivir structure (PDB no. 1CQQ) as the search model. The initial phases were improved by manual model building, especially at the protease active sites and the bound rupintrivir molecules, resulting in final atomic models with good refinement as well as stereochemical quality (Table 1).
Conformational changes associated with inhibitor binding.
The structures of the rupintrivir-bound form of EV71 3Cpro mutants adopt the typical chymotrypsin-like fold that is similar to that of the unliganded protease structure described in detail previously (8) (Fig. 1A and B). However, the unliganded protease maintains a much more relaxed fold (average B factor, 83.5 Å2) than the tight folds of EV71 3Cpro mutant/rupintrivir complexes (average B factor, 17.1 to 20.7 Å2). In particular, the catalytically important loop of amino acids (aa) 141 to 147 is disordered in the unliganded protease (8) (Fig. 1A). Upon the binding of rupintrivir, the loop of aa 141 to 147 was restored to a well-defined conformation in which the backbone NH groups of Gly145 and Cys147 formed the optimally configured oxyanion hole. In the unliganded EV71 3Cpro structure, the β-ribbon adopts an unusual open conformation which likely is contributed by the crystallization artifact (8). In this study, we obtained the crystals of EV71 3Cpro mutant/rupintrivir complexes in which the crystal-packing interactions are different from that found in the crystal of unliganded EV71 3Cpro. As anticipated, the β-ribbon (aa 123 to 133) restores the canonical close conformation. The ribbon folds over the substrate binding cleft, similar to that observed for other picornaviral 3C proteases (Fig. 1B). Thus, the open and closed β-ribbon conformations were observed in different crystal forms of EV71 3Cpro, supporting our hypothesis that the β-ribbon is highly mobile and can undergo flipping movement around the hinge residues Gly123 and His133 during substrate recognition.
Fig. 1.

Overall structure of EV71 3Cpro/rupintrivir complex. (A) Ribbon model of unliganded EV71 3Cpro with annotated secondary structures. The β-ribbon adopts the open conformation. (B) Ribbon model of rupintrivir (carbon is in orange) binding to EV71 3Cpro with annotated secondary structures. The β-ribbon adopts the closed conformation. (C) Rupintrivir bound to EV71 3Cpro. The active site of the protease is rendered as a semitransparent solvent-accessible surface color coded at residues (involving substrate binding) according to amino acid conservation among the picornaviral 3Cpro of known structure. Residues indicated in red are invariant. Increasing amino acid variation at residues is indicated by progressively fading red to white, with white indicating the least conserved residues. The carbons of the bound rupintrivir are colored in white; the ordered waters are red spheres. (D) Structure-based multiple-sequence alignment of 3C proteases from different picornaviruses. The secondary structure is shown at the top. Invariant residues are white with a red background; conserved residues are shown in red font. The residues involved in the interactions with rupintrivir are marked below the sequences. Residues contacting with P1′, P1, P2, P3, and P4 of rupintrivir are blue, red, purple, brown, and green bars, respectively. (E) Structural formula of rupintrivir color coded as described for panel D. Residues involving the binding with rupintrivir are listed and color coded.
Previous studies suggested that the substrate specificity of 3Cpro is achieved by the highly coupled mechanism for substrate recognition and catalysis (17, 18). Evidently, the structural studies of EV71 3Cpro support this idea. EV71 3Cpro is a highly specific protease that preferentially recognizes the peptide substrate with a Q-G junction in which P1-Q is absolutely required for digestion. The P2 to P4 residues of the substrate also contribute significantly to the substrate specificity (8). The unliganded EV71 3Cpro adopts a highly flexible fold (diffracted only to 3.0 Å) in which the catalytically important loops are disordered and the substrate binding pockets (P1 to P4) are not completely formed, representing a nonproductive conformation. In stark contrast, the relaxed fold of the unliganded protease can be dramatically stabilized by the binding of the inhibitor (diffracted to 1.0 to 1.7 Å). The inhibitor-induced conformational changes involve the disorder-to-order transition of the catalytically important loops, which eventually lead to the productive conformation and stable protease-inhibitor complex.
Interactions between rupintrivir and EV71 3Cpro.
Recent studies showed that rupintrivir is effective against EV6 and EV9, with an EC50 of 43 and 15 nM, respectively (6). In our antiviral assays, we found that rupintrivir has potent antiviral activities against EV71 (BJ/CHN/2008), with an EC50 of ∼1 nM, which is in the same range as the EC50 measured for HRV (Fig. 2). In our protease inhibition assays, rupintrivir inhibited the protease activity of EV71 3Cpro with an IC50 of 2.3 ± 0.5 μM. In the structure of EV71 3Cpro mutant H133G/rupintrivir complex, rupintrivir is linked to EV71 3Cpro by a covalent bond (1.98 Å) between the Sγ atom of Cys147 and the electrophilic β-carbon of the inhibitor. The overall binding mode of rupintrivir to EV71 3Cpro is similar to that observed in the HRV 3Cpro/rupintrivir structure (17). One striking difference is that the C-terminal ethyl ester of rupintrivir does not attach to the leaving-group side pockets in EV71 3Cpro (Fig. 3A and D). Compared to rupintrivir bound to HRV 3Cpro, the ethyl ester group of rupintrivir bound to EV71 3Cpro rotated around the bond between α-carbon and β-carbon by ∼78o and adopted an unusual cis conformation, and thus the C terminus of the inhibitor is directed ∼5 Å away, where it can preferentially reach the solvent. Consequently, the carbonyl oxygen of the ethyl ester is positioned ∼2.5 Å away from the oxyanion hole. Interestingly, the C terminus of inhibitor TG-0204998 bound to CVB 3Cpro (PDB no. 2zu3) also doesn't accommodate the leaving-group side pocket (14) (Fig. 3A and D, image 3). These data suggest that the leaving-group pockets of EV71 3Cpro and CVB 3Cpro are structurally different from those of HRV 3Cpro. Structure-based sequence alignment showed that although almost the entire area of the substrate binding cleft of EV71 3Cpro is constituted by invariant or conserved residues (Fig. 1C and D), the residues forming the leaving-group side of the active site are much less conserved. In the HRV 3Cpro/rupintrivir structure, the ethyl ester of rupintrivir accommodates the leaving-group side pockets formed by a loop of aa 22 to 25 and a tight loop of aa 105 to 111. In EV71 3Cpro, the corresponding loop of aa 105 to 111 also maintains a tight fold (average B, 17.9 Å2; that for the protein is 20.7 Å2); however, the main-chain and side-chain conformations are very different from their counterparts in HRV 3Cpro (Fig. 3B). For example, in HRV 3Cpro the tip of the loop of aa 105 to 111 is oriented toward the oxyanion hole. Residue Asn107, located on the tip of the loop, provides an electrostatic interaction to the carbonyl oxygen of ethyl ester of rupintrivir, thus stabilizing the C-terminal portion of the inhibitor inside S1′ pocket. In contrast, the tip of the loop of aa 105 to 111 is pointing away from the oxyanion hole in EV71 3Cpro. The corresponding residue Glu107 is located ∼10 Å away, with its side chain pointing up to the solvent. Surprisingly, the loop of aa 105 to 111 defines a recessive molecular surface located next to the S1′ pocket in EV71 3Cpro (Fig. 3D, image 1). The shallow pit is constituted by residue Thr106 (as the floor) and the hydrophobic parts of residues His24, Phe25, His108, M109, Gly145, and Cys147 (as the walls). This feature is not available in HRV 3Cpro (Fig. 3D, image 2) but is present in CVB 3Cpro (Fig. 3D, image 3), suggesting that EV71 3Cpro and CVB 3Cpro are structurally more related. The superimposition of FMDV 3Cpro/peptide (PDB no. 2wv4) complex and EV71 3Cpro reveals that the shallow pit of EV71 3Cpro could accommodate the hydrophobic P2′ leucine side chain, indicating that the pit serves as the S2′ pocket (Fig. 3D, image 4). In summary, our data suggest that in 3C proteases like EV71 3Cpro and CVB 3Cpro, the substrate specificity determinants extend well beyond the core region (S1′ to S4 pockets) that rupintrivir was originally designed to occupy. The putative surface-recessive S2′ pocket may contribute to improving the inhibitor binding affinity. As evidenced by the inhibitors studies of CVB 3Cpro, the addition of an extra cyclopropyl group to P1′ of the inhibitor could enhance inhibitor efficacy by 4-fold (14).
Fig. 2.
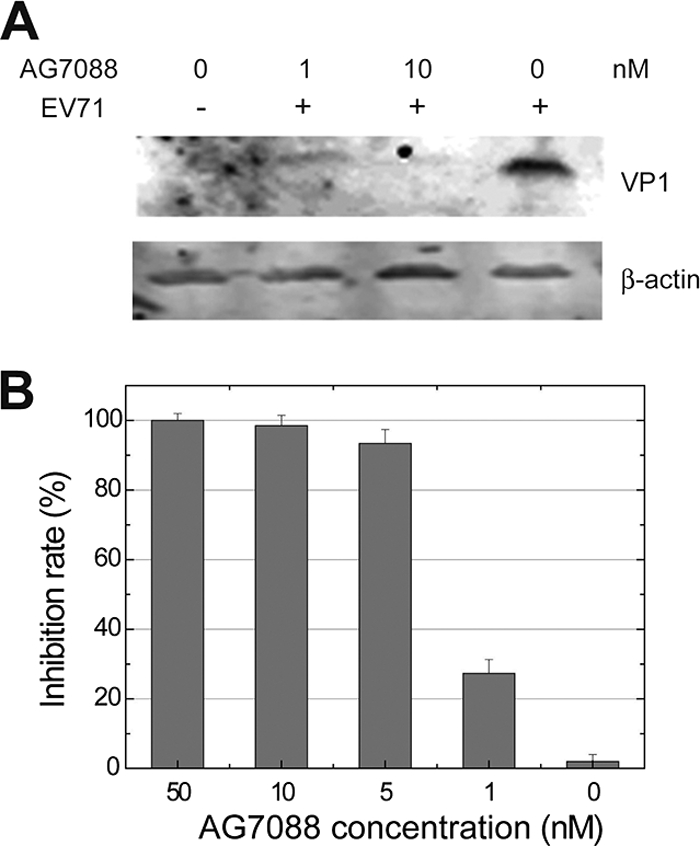
Inhibition of EV71 by rupintrivir (AG7088). The efficacy of rupintrivir against EV71 was evaluated in antiviral assays. The data showed that rupintrivir was effective against EV71 (isolate BJ/CHN/2008) with a mean EC50 of ∼1 nM, which is comparable to the antiviral activities of rupintrivir against HRV or other picornaviruses. No obvious cytotoxicity was found by adding rupintrivir to RD cells. The antiviral activity of rupintrivir was further demonstrated using Western blotting and RT-PCR analyses. RD cells infected with EV71 showed less quantity of VP1 protein than that detected in the presence rupintrivir (1 or 10 nM). RT-PCR experiments detecting the quantities of viral RNAs in RD cells show that rupintrivir suppresses EV71 replication in a dose-dependent manner, which is consistent with the results from antiviral assays. (A) Cell lysates (40 μg protein per lane) were prepared from either mock-infected (lane 1) or EV71-infected RD cells at 24 h postinfection and resolved with 12% SDS-PAGE. Western blot analysis for VP1 or β-actin was conducted. Lane 1, mock-infected cells not treated with rupintrivir (AG7088); lanes 2 to 4, cells were treated with 1, 10, or 0 nM AG7088, respectively. The bands corresponding to EV71 VP1 are indicated. The expression of β-actin was used to control equal protein loading. (B) Cells seeded in 24-well plates were treated with different concentrations of AG7088 and infected with EV71 at an MOI of 0.1. Twenty-four h postinfection viral RNAs were extracted from cells, and real-time RT-PCR was performed. The inhibition rate (percent inhibition of viral RNA) is plotted as a function of inhibitor concentrations. The plotted data are averages from triplicates.
Fig. 3.

Interactions between EV71 3Cpro and rupintrivir. (A) Superimposition of the stick models of EV71 3Cpro (carbon in orange), HRV 3Cpro (carbon in gray; PDB no. 1cqq), and CVB 3Cpro(carbon in teal; PDB no. 2zu3) bound by different inhibitors. The superimposed oxyanion holes are indicated with an arrow. (B) Stick model of EV71 3Cpro/rupintrivir (carbon in orange) superimposed with the stick model of HRV 3Cpro/rupintrivir (carbon in gray; PDB no. 1cqq). Residues on the differently orientated tight loops (aa 105 to 111) are labeled. The dashed lines indicate the electrostatic interaction between Asn107 and the C-terminal carbonyl oxygen of rupintrivir in HRV 3Cpro. (C) Stick model of EV71 3Cpro/rupintrivir (carbon in orange) and the superimposed stick model of HRV 3Cpro/rupintrivir (carbon in gray). The 4-fluoroPhe side chain of P2 of rupintrivir is buried more shallowly in the S2 pocket in EV71 3Cpro than in HRV 3Cpro. The basic residues Arg39 and Lys130 form the closed distal end of the S2 pocket in EV71 3Cpro and interact with the side chain fluoride of the P2 residue of rupintrivir. (D) Electrostatic surface of proteases active sites bound by inhibitors with annotated substrate specificity pockets. The position of the shallow pit defined by a loop of aa 105 to 111 is indicated with a black arrow. Image 1 shows that the distal end of the S2 specificity pocket is closed in EV71 3Cpro, whereas the distal end of the S2 pocket is open in other picornaviral 3C proteases; image 2, rupintrivir bound to EV71 3Cpro; image 3, rupintrivir bound to HRV 3Cpro (PDB no. 1cqq); image 4, inhibitor TG-0204998 bound to CVB 3Cpro (PDB no. 2zu3). The peptide substrate of FMDV 3Cpro (PDB no. 2wv4) is modeled in the substrate binding cleft in EV71 3Cpro by superimposition, showing that the shallow pit next to the S1′ pocket can serve as the S2′ pocket accommodating the hydrophobic P2′ side chain.
Compared to that of the HRV 3Cpro/rupintrivir structure, the 4-fluorophenylalanine (4-fluoroPhe) side chain of the P2 residue of rupintrivir has far fewer Van der Waals contacts with the S2 specificity pocket in EV71 3Cpro. The aromatic side chain rotates ∼21° around the Cα-Cβ bond, thus the side-chain fluoride moves up ∼2 Å to the solvent, where it contacts with the residues at the distal end of the S2 pocket (Fig. 3C). Residues Arg39 and Lys130 are located at the back of the S2 pocket, forming an enclosed distal end of the S2 pocket, whereas in other picornaviral 3Cpro the distal end of S2 pockets usually is open to the solvent (Fig. 3D, images 1 to 3). The studies of HRV 3Cpro inhibitors showed that the structural variabilities at the distal ends of the S2 pockets are pronounced; however, this doesn't affect the efficacy of the inhibitors (17). Whether or not the basic residues located at the distal end of the S2 pocket affect the inhibitor efficacy against EV71 3Cpro requires further studies.
The structure of the active site.
In the structure of the unliganded EV71 3Cpro, there were insufficient electron densities to define the conformation of the Glu71 side chain. When EV71 3Cpro is bound by rupintrivir, the side chain of Glu71 adopts a well-defined conformation in which the carboxylate group is folded in the antiorientation to accept a hydrogen bond from the Nδ1 atom of His40. The long glutamic acid side chain is further stabilized by the hydrogen bonds denoted by the backbone NH of His40 and by the Nδ2 atom of Asn69. Strikingly, a conserved residue, Arg39, provides several unusual hydrogen bonds to the carboxylate of Glu71 (Fig. 4A). In particular, hydrogen bonds were found between the syn-oriented lone pair of electrons of the carboxylate of Glu71 and the Nη2 atom of Arg39 and between Oε1 of Glu71 and the Nε atom of Arg39. Moreover, the carbonyl group of Glu71 accepts a hydrogen bond from the NH group of Arg39, and the NH group of Glu71 donates a hydrogen bond to the Oδ1 atom of Asn69. Thus, all possible hydrogen bonding interactions for the side chain and main chain of Glu71 are fully satisfied by the surrounding residues. The data indicate that Glu71 side chain acts as a negative charge center that joins the catalytically important residues as well as other conserved residues to form a hydrogen bonding network of the active site. These directional hydrogen bonds define the geometric specificity for glutamic acid side chains, providing the structural basis for the strict conservation of this residue. We previously found that the E71A mutation of EV71 3Cpro abrogates the protease activity (8). We determined the crystal structure of this mutant, showing that when the carboxylate of Glu71 is removed, the hydrogen bond network of the active site collapses completely (Fig. 4B). There were poor electron densities for the side chain of Arg39. Residue Asn69 and the upstream loop of aa 61 to 69 adopt alternative conformations. Importantly, catalytic His40 displays two conformations. The first conformation of His40 is similar to the productive conformation seen in most picornaviral 3Cpro; however, this conformation accounted for only 39% occupancy. The second conformation the His40 side chain rotated around the Cα-Cβ bond ∼84o away, where the imidazole side chain is positioned outside the hydrogen bonding distance to Cys147, indicating a nonproductive conformation that accounted for 61% occupancy. The data suggest that the negatively charged carboxylate side chain of Glu71 is crucial for maintaining the overall architecture of the active site and stabilizing the productive conformation of His40. Interestingly, similarly disordered active-site conformations were previously observed in structural studies of HAV 3Cpro and FMDV 3Cpro (2, 21).
Fig. 4.

Structure of the active sites. (A, B, and C) Final 2Fo-Fc electron densities (1.5-s contour) at the active site and the superimposed final model of EV71 3Cpro mutants H133G, E71A, and E71D. The hydrogen bond network of the active site is indicated with dashed lines, and each hydrogen bond is numbered. Residues involved in the hydrogen bond network of the active site are annotated. Mutated residues are indicated in red. (D) Stick model of the EV71 3Cpro mutant E71D active site (carbon in orange) and the superimposed FMDV 3Cpro active site (carbon in gray). Catalytically important residues are indicated. Distances between catalytic His and Asp are indicated with dashed lines.
In the mutagenic analysis of FMDV 3Cpro, a protease with the Cys-His-Asp catalytic triad, the replacement of the catalytic Asp84 by either Ala or Glu accounted for severe damage to the protease activity, suggesting that both the availability of carboxylate and the side chain geometry of Asp84 are important (23). We determined the structure of EV71 3Cpro mutant E71D, which is void of protease activity (data not shown). The structure shows that when Glu71 is replaced by Asp, the side chain is simply too short to reach the catalytic histidine and thus cannot provide enough electrostatic stabilization to the residue (Fig. 4C). Residue His40 of mutant E71D again adopts two conformations, similar to those of mutant E71A. The productive His40 conformation accounted for 45% occupancy, and the nonproductive conformation accounted for 55% occupancy in mutant E71D. Superimposing the FMDV 3Cpro structure onto the EV71 3Cpro mutant E71D structure reveals that in FMDV 3Cpro, the side chain of Asp84 has sufficient length to maintain a syn-oriented hydrogen bond with His46 (Fig. 4D). This is accomplished by positioning the Cα of Asp84 ∼1 Å closer to the Cα of His46. The distance between the Cα atoms of Asp84-His46 is 6.21 Å, whereas in EV71 3Cpro structures the corresponding distances are 7.09 (H133G), 7.12 (E71D), and 7.24 Å (E71A), showing that the Cα distances between the catalytic His-Glu in EV71 3Cpro are, on average, longer than the distances between the catalytic His-Asp in FMDV 3Cpro. We further measured the corresponding distances in other proteases. As listed in Table 2, the Cα distance between the catalytic His-Glu is 7.09 to 7.34 Å, which is in general longer than the distances between the catalytic His-Asp (6.18 to 6.42 Å).
Table 2.
Cα distances between catalytic histidine and glutamic acid/aspartic acid
| Protease | PDB no. | Catalytic triad | Cα distance between the catalytic His and Glu/Asp (Å) |
|---|---|---|---|
| EV71 3Cpro | 3R0F | Cys-His-Glu | 7.09 |
| HRV 3Cpro | 1CQQ | Cys-His-Glu | 7.14 |
| CVB 3Cpro | 2VB0 | Cys-His-Glu | 7.34 |
| PV 3Cpro | 1L1N | Cys-His-Glu | 7.19 |
| FMDV 3Cpro | 2J92 | Cys-His-Asp | 6.21 |
| HAV 3Cpro | 2H9H | Cys-His-Asp | 6.18 |
| Α-lytic | 1SSX | Ser-His-Asp | 6.25 |
| Trypsin | 2AGG | Ser-His-Asp | 6.42 |
In summary, our data support the hypothesis that picornaviral 3Cpro requires a full catalytic triad for proteolysis. We further characterized the significant role of the third member of the catalytic triad. Both the carboxylate functionality and glutamic acid side chain geometry of Glu71 are critical for maintaining the overall architecture of the active site and stabilizing the productive conformation of the catalytic histidine, therefore the residue is irreplaceable for protease activity. The folding of picornaviral 3Cpro and serine proteases are similar overall; however, they can be further divided into two structurally different subgroups that support either Asp or Glu as the acidic member of the catalytic triads. The choice of employing Asp or Glu is correlated to the Cα position of Asp/Glu relative to the positions of the catalytic histidine.
The role of residues Asn69 and Arg39.
In EV71 3Cpro structures, residue Asn69 forms a hydrogen bond with catalytic Glu71. Our mutagenesis studies showed that mutations N69S and N69D, in which the hydrogen bond between Asn69 and Glu71 is disrupted, could significantly reduce the Km/kcat values (Fig. 5), suggesting that this hydrogen bond is important to the proteolysis activity of EV71 3Cpro. A similar feature was observed in many picornaviral proteases with the Cys-His-Glu catalytic triad, such as PV 3Cpro, HRV 3Cpro, and CVB 3Cpro (14, 18, 20), but was not found in picornaviral proteases with Cys-His-Asp triads (4, 5, 7, 21), suggesting that the hydrogen bond donated by residue Asn69 serves to further stabilize the conformation of the relatively longer glutamic acid side chain, which tends to be more flexible than the aspartic acid counterpart in Cys-His-Asp triads. However, the residue Asn69 is not strictly conserved in picornaviral 3C proteases even in PEV 3Cpro, BEV 3Cpro, and HRV 3Cpro from a different HRV serotype (17) (Fig. 1D), which also possessed the Cys-His-Glu catalytic triad. Therefore, the additional stabilization of the catalytic glutamic side chain may not be required for the proteolysis activity of picornaviral 3C proteases with the Cys-His-Glu catalytic triad.
Fig. 5.

Mutagenic studies of residues Arg39 and Asn69. Specificity constants (kcat/Km) of WT EV71 3Cpro and mutants R39K, R39E, R39T, N69S, and N69D are shown. Error bars represent standard errors from the nonlinear regression analysis. The data show that besides the canonical catalytic triad, the residues Asn69 and Arg39 also are important to proteolysis.
We observed novel hydrogen bonds between Arg39 and Glu71, suggesting that residue Arg39 is involved in the proteolysis catalyzed by EV71 3Cpro. To test this idea, we performed a site-directed mutagenesis study. We found that while EV71 3Cpro mutant R39K retained a fraction of the WT protease activity, mutants R39E and R39T lost nearly all protease activities (Fig. 5), suggesting that the positive charging of the Arg39 side chain is important to the proteolytic activity. In the unliganded EV71 3Cpro structure, similar hydrogen bonds were not observed, suggesting that the hydrogen bonds between Arg39 and Glu71 are not maintained throughout the proteolytic steps. Thus, residue Arg39 can adopt at least two different side chain conformations: when the Arg39 side chain is away from Glu71, Glu71 is more negatively charged, but when the Arg39 side chain is close to Glu71 (Fig. 4A), the positively charged guanidinium group of Arg39 can neutralize the charging of the Glu71 side chain. Taken together, our data suggest a role of Arg39 in the proteolysis reaction catalyzed by EV71 3Cpro (Fig. 6). According to the serine protease mechanism, in the acylation or deacylation half of the proteolytic reaction there are two connected proteolytic steps. First, the nucleophilic attacks by Cys147 or hydrolytic water results in the formation of a tetrahedral intermediate, and His40 needs to act as a base to accept a proton from Cys147 or water, forming an His40-H+ intermediate. Second, His40-H+ will act as an acid to protonate the leaving group or Cys147 so that the tetrahedral intermediates collapse with the expulsion of the products. We believe that Arg39 can function as a neutralizer to assist the transition from the first to the second step. When His40 accepts a proton, His40-H+ is stabilized by the negative charging of Glu71. This transition state can be destabilized by the presence of Arg39, which acts as a neutralizer to minimize the charge of Glu71. As a result, the electrostatic interaction between Glu71 and His40-H+ is weakened, which then favors the deprotonation of His40-H+. The moving His mechanism originally was proposed for serine protease in an attempt to explain why His-H+ must be repositioned near the leaving group and the catalytic His must undergo some movements, such as a flipping His mechanism, to protonate the leaving group (3). A similar mechanism may exist in EV71 3Cpro due to the structural similarity. In this work, we found that the conformation of His40 is strictly controlled by the electrostatic interaction from Glu71 in EV71 3Cpro (Fig. 4A). Therefore, only in the presence of the neutralizer Arg39 can the interaction between Glu71 and His40 can be temporarily relaxed; in the mean time, the side chain of the catalytic His will have a better chance to reposition. Residue Arg39 is conserved in some picornaviral 3C proteases, such as CVA16 3Cpro, CVB 3Cpro, SEV 3Cpro, FMDC 3Cpro, etc. A conservative substitution is found in PEV 3Cpro, in which Arg39 is replaced by Lys39 (Fig. 1D). However, in PV 3Cpro, HRV 3Cpro, and HAV 3Cpro, the corresponding residue is the uncharged threonine or serine, which cannot function as the neutralizer for the catalytic glutamic acid, suggesting that the neutralizer mechanism does not exist for all picornaviral 3C proteases.
Fig. 6.

Proteolysis mechanism of EV71 3Cpro involving the neutralizer Arg39. The catalytically important residues are black. The substrate is green. The electron relays are indicated by red arrows. Thicker dashed lines indicate stronger interactions, and thinner dashed lines indicate weaker interactions.
Crystal structures for picornaviral 3C proteases indicate that members of this family of proteases share similar overall active-site architectures, suggesting a common mechanism for proteolysis. Our findings suggest that in addition to a common mechanism, different 3C proteases adopt slightly different mechanisms in proteolytic reactions involving residues that assist in catalysis.
ACKNOWLEDGMENTS
We thank SLS (Swiss Light Source) for beam time allowance and the staff of beamline X06DA (PXIII) at SLS for help with data collection.
This work was supported by the National Basic Research Program of China (973 Program), no. 2011CB504902; National Natural Science Foundation of China, program no. 81071349; and an intramural grant from the Institute of Pathogen Biology, Chinese Academy of Medical Sciences, and Beijing Union Medical College (no. 2008IPB201) to S.C.
Footnotes
Published ahead of print on 3 August 2011.
REFERENCES
- 1. Adams P. D., et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allaire M., Chernaia M. M., Malcolm B. A., James M. N. 1994. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature 369:72–76 [DOI] [PubMed] [Google Scholar]
- 3. Ash E. L., et al. 2000. Unusual 1H NMR chemical shifts support (His) C(epsilon) 1.O==C H-bond: proposal for reaction-driven ring flip mechanism in serine protease catalysis. Proc. Natl. Acad. Sci. U. S. A. 97:10371–10376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergmann E. M., et al. 1999. Crystal structure of an inhibitor complex of the 3C proteinase from hepatitis A virus (HAV) and implications for the polyprotein processing in HAV. Virology 265:153–163 [DOI] [PubMed] [Google Scholar]
- 5. Bergmann E. M., Mosimann S. C., Chernaia M. M., Malcolm B. A., James M. N. 1997. The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J. Virol. 71:2436–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Binford S. L., et al. 2005. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 49:619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Birtley J. R., et al. 2005. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 280:11520–11527 [DOI] [PubMed] [Google Scholar]
- 8. Cui S., et al. 2011. Crystal structure of human enterovirus 71 3C protease. J. Mol. Biol. 408:449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 10. Hu Y. F., et al. Complete genome analysis of coxsackievirus A2, A4, A5, and A10 strains isolated from hand-foot-and-mouth disease patients in china revealing frequent recombination of human enterovirus A. J. Clin. Microbiol. 49:2426–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hung H. C., et al. 2011. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 203:1784–1790 [DOI] [PubMed] [Google Scholar]
- 12. Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuo C. J., et al. 2008. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem. 16:7388–7398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee C. C., et al. 2009. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 284:7646–7655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lei X., et al. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84:8051–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lei X., et al. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by toll-like receptor 3. J. Virol. [Epub ahead of print.] doi:10.1128/JVI.00447–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthews D. A., et al. 1999. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U. S. A. 96:11000–11007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matthews D. A., et al. 1994. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77:761–771 [DOI] [PubMed] [Google Scholar]
- 19. McCoy A. J., et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mosimann S. C., Cherney M. M., Sia S., Plotch S., James M. N. 1997. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 273:1032–1047 [DOI] [PubMed] [Google Scholar]
- 21. Sweeney T. R., Roque-Rosell N., Birtley J. R., Leatherbarrow R. J., Curry S. 2007. Structural and mutagenic analysis of foot-and-mouth disease virus 3C protease reveals the role of the beta-ribbon in proteolysis. J. Virol. 81:115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. World, Health Organization 2008. Report on the hand, foot and mouth disease outbreak in Fuyang City, Anhui province and the prevention and control in China (May 2008). World Health Organization Representative Office in China, Beijing, China [Google Scholar]
- 23. Zhang X. N., et al. 2010. Rupintrivir is a promising candidate for treating severe cases of Enterovirus-71 infection. World J. Gastroenterol. 16:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zunszain P. A., et al. 2010. Insights into cleavage specificity from the crystal structure of foot-and-mouth disease virus 3C protease complexed with a peptide substrate. J. Mol. Biol. 395:375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]