

Abstract
Biomaterial microparticles are commonly utilized as growth factor delivery vehicles to induce chondrogenic differentiation of mesenchymal stem/stromal cells (MSCs). To address whether the presence of microparticles could themselves affect differentiation of MSCs, a 3D co-aggregate system was developed containing an equal volume of human primary bone marrow-derived MSCs and non-degradable RGD-conjugated poly(ethylene glycol) microspheres (PEG-μs). Following TGF-β3 induction, differences in cell phenotype, gene expression and protein localization patterns were found when compared to MSC aggregate cultures devoid of PEG-μs. An outer fibrous layer always found in differentiated MSC aggregate cultures was not formed in the presence of PEG-μs. Type II collagen protein was synthesized by cells in both culture systems, although increased levels of the long (embryonic) procollagen isoforms were found in MSC/PEG-μs aggregates. Ubiquitous deposition of type I and type X collagen proteins was found in MSC/PEG-μs cultures while the expression patterns of these collagens was restricted to specific areas in MSC aggregates. These findings show that MSCs respond differently to TGF-β3 when in a PEG-μs environment due to effects of cell dilution, altered growth factor diffusion and/or cellular interactions with the microspheres. Although not all of the expression patterns pointed toward improved chondrogenic differentiation in the MSC/PEG-μs cultures, the surprisingly large impact of the microparticles themselves should be considered when designing drug delivery/scaffold strategies.
Keywords: Mesenchymal stem cell, polyethylene, microsphere, cartilage tissue engineering, TGF-β, chondrocyte
1. Introduction
Articular cartilage is a highly specialized tissue with important load-bearing functions. Mature tissue consists of a relatively sparse population of chondrocytes embedded within an abundant, well-organized extracellular matrix (ECM) [1]. Type II collagen fibers provide the tissue with tensile strength while proteoglycans (mainly aggrecan) contribute to its compressive load-bearing function. The avascular nature of articular cartilage renders it incapable of self-repair following traumatic, degenerative or inflammatory damage. Subsequently, many tissue engineering strategies are being investigated as a means to generate articular cartilage tissue in vitro or in vivo [2–5]. Common approaches involve the use of multipotent mesenchymal stem/stromal cells (MSCs) in combination with TGF-β superfamily growth factors and three-dimensional (3D) biomaterial scaffold systems [6–9]. Recent advances in the field are now focusing on ways to spatially and temporally control the availability of bioactive growth factor to enhance MSC differentiation. Specifically, the application of biomaterial microspheres as growth factor delivery vehicles for cartilage engineering is currently being explored. Microspheres generated from either poly(lactic-co-glycolic acid) (PLGA) or gelatin have been engineered to release TGF-β1 with somewhat promising effects on improving chondrocyte differentiation in MSC aggregate cultures [10, 11]. Other strategies to attempt to enhance cartilage matrix production have been reported by the application of biodegradable insulin-loaded PLGA microspheres or hyaluronic acid-based hydrogel microparticles releasing BMP-2 [12, 13]. In addition, PLGA microspheres as injectable scaffolds for cartilage tissue engineering are being investigated [14] while other studies are exploring the use of 3D scaffolds generated from protein-loaded microspheres or the combination of bulk hydrogel scaffolds with alginate microspheres to permit slow release of biological factors [15–17]. Based on these current microsphere-based technologies to improve cartilage tissue engineering, questions arise as to the effect of microspheres themselves on the differentiation of MSCs. The goal of this study was to investigate the effect of non-degradable poly(ethylene glycol) microspheres (PEG-μs) on TGF-β3-induced differentiation of human bone marrow derived MSCs by generating co-aggregate 3D cultures of MSCs and PEG-μs. Data obtained was directly compared to MSC aggregate cultures grown in the absence of microspheres; this MSC aggregate system has been well reported in the literature as a common in vitro chondrogenesis assay [18]. The rationale for using PEG-based microspheres is that PEG surfaces are noteworthy for their low degree of protein adsorption and cell adhesion [19] thus providing a relatively non-adhesive background to directly address the architectural effects of microspheres on MSC differentiation. In our system, RGD (arginine-glycine-aspartic acid) peptide was incorporated within the PEG-μs to promote cell adhesion. The reason for doing this stemmed from previous studies showing that cells within PEG scaffolds tended to undergo apoptosis due to lack of adhesion sites [20, 21] and that MSC viability was improved by incorporation of RGD peptide within such scaffolds [20, 22]. As others have shown that microspheres may affect ES cell differentiation [23, 24], we hypothesized that the presence of microspheres would alter the MSC response to TGF-β3 due to effects of cell dilution, altered growth factor diffusion and/or cellular interactions with the microspheres themselves. These studies have important implications in the cartilage tissue engineering field by directly addressing the impact of biomaterials on MSC differentiation and set the foundations for future investigations aimed toward improving cartilage matrix production in vitro and in vivo.
2. Materials and Methods
2.1. Human mesenchymal stem cells
Human bone marrow-derived mesenchymal stem cells (MSCs) from two independent donors were used in this study. One MSC primary cell line was purchased commercially (Lonza, Walkersville Inc.) and the second primary MSC cell line was isolated in-house from a bone marrow aspirate specimen obtained via an IRB-approved core facility at Washington University. Using standardized differentiation assay kits (Lonza), the multipotential capacity of both MSC cell lines (i.e. the ability to differentiate to adipocytes, osteoblasts and chondrocytes) was first confirmed before using the cells for further analyses.
2.2. Synthesis of PEG microspheres
Unless otherwise noted, all reagents were purchased from Sigma-Aldrich. PEG Synthesis:Eight-arm PEG-OH (PEG8-OH; MW=10,000; Shearwater Polymers, Huntsville, AL) was used to synthesize PEG8-vinylsulfone (PEG8-VS) and PEG8-amine as previously described [25]. PEG macromonomers were dissolved separately in 200 mg/mL in Dulbecco’s phosphate buffered saline (1×PBS; 8 mM sodium phosphate, 2 mM potassium phosphate, 140 mM sodium chloride, 10 mM potassium chloride, pH 7.4) and sterile filtered with 0.22 μm syringe filters (Millipore). To generate the microspheres, PEG8-amine solutions were combined with PEG8-VS solutions at a 1:1 ratio [26]. An equal volume of RGD peptide (Ac-GCGYGRGDSPG-NH2; GenScript, Piscataway, NJ; 20 mg/mL; 1.88 mol RGD/mol PEG) in 1×PBS was added to the PEG solutions and incubated at 37°C for 20 min. The PEG solutions were diluted to 20 mg/mL with 1×PBS and 1.5 M sodium sulfate (in 1×PBS) to a final sodium sulfate concentration of 0.6 M at room temperature. The PEG8-VS/PEG8-amine solutions were then incubated above the cloud point at 70°C for 20 min. Suspensions of microspheres (approximately 6.01± 1.98 μm diameter) were subsequently buffer exchanged twice in 1×PBS to remove the sodium sulfate, each time diluting the microsphere solution 3:1 with 1×PBS and titurating, centrifuging at 14,100 × g for 2 min and removing the supernatant.
2.3. Induction of MSCs by TGF-β3 in two different in vitro culture systems
2.3.1. Stem cell aggregate cultures
MSC aggregate cultures each containing 2.5×105 cells were generated as described previously [18]. Aggregates were formed in 15 mL polypropylene tubes following centrifugation at 350 × g for 10 min. Serum-free differentiation medium (500 μL) was added to each cell aggregate. This medium consisted of DMEM, 1% ITS+, 1% sodium pyruvate, 0.1 μM dexamethasone, 40 μg/mL ascorbate and TGF-β3 (10 ng/mL; Lonza, Walkersville, Inc). The lids of the tubes were loosened slightly to allow gas exchange and then incubated at 37°C. Medium was changed every 2 days.
2.3.2. MSC/PEG microsphere co-aggregate cultures
MSCs (2.5×105) were combined with PEG microspheres (1:1 volume ratio of cell: microsphere suspension) in sterile 1.5 mL eppendorf tubes and gently rotated for 3 h at 37°C to ensure uniform mixing of cells with microspheres. MSC/PEG-μs aggregates were formed by centrifugation at 350 × g for 10 min. Pin-holes were punched in the lids of each sample tube to permit gas exchange. Prior to differentiation, MSC/PEG-μs aggregates were cultured for a short time (3 d) at 37°C to maximize cell attachment to the microspheres and permit the cells to acclimate to the microsphere environment. During this time, a medium containing FGF-2 (known to maintain MSCs in a de-differentiated state) [27, 28] was added to the aggregates [DMEM-LG containing 10% FBS (Atlas Biologicals, Fort Collins, CO) 10 ng/mL FGF-2 (Peprotech, Rocky Hill, NJ)]. Medium was then replaced with the same serum-free TGFβ3-containing differentiation medium (500 μL) as described above and replenished every 2 d until time of harvest at 12 d or 28 d. Of note, our experience with MSC culture has shown that the presence of FGF-2 in medium prior to MSC aggregate formation had no effect on how the cells responded to TGF-β3-induced differentiation (unpublished observations). All downstream analyses were carried out in triplicate for each MSC or MSC/PEG-μs aggregate differentiation experiment.
2.4. Analysis of cell viability and phenotype
2.4.1. Viability Assay
Following 12 days and 28 days of TGF-β3 induction, MSC and MSC/PEG-μs aggregate cultures were stained with a combination of calcein AM (2 μM) and ethidium homodimer (4 μM) (Live/Dead cytotoxicity kit; Invitrogen) for 30 min at room temperature. Cultures were rinsed twice in 1×PBS, fixed in 4% paraformaldehyde for 2 h and processed through a gradient of 30%, 50% and 70% ethanol for paraffin embedding. Paraffin sections (10 μm) were obtained for fluorescence analysis. Calcein uptake by live cells was visualized using the fluorescein (FITC) filter set and uptake of ethidium homodimer by dead cells was analyzed using the rhodamine (TRITC) filter set. As a control, MSC/PEG-μs aggregates cultured in the absence of TGF-β3 were analyzed in order to show that ethidium homodimer staining was functional and to prove that calcein or ethidium homodimer did non-specifically stain the microspheres.
2.4.2. Safranin O staining
Paraffin sections of aggregate cultures from day 12 or day 28 of TGF-β3 induction were treated with xylene, rehydrated through decreasing concentrations of ethanol, stained in Weigert’s Hematoxylin for 5 min, washed in running water for 5 min and stained with 0.001% Fast Green for 3 min. Samples were then rinsed in 1% glacial acetic acid, stained in 0.1% Safranin O for 5 min, then dehydrated and cleared by incubation in 95% alcohol, 100% alcohol and xylene. Mounting medium was applied and stained sections were cover-slipped.
2.5. Antibodies
The following antibodies were used for detection of extracellular matrix collagens and α-smooth muscle actin (α-SMA) by immunofluorescence: type I collagen mouse monoclonal antibody (Abcam; ab6308) used at 1/1000 dilution; type II collagen rat polyclonal antisera [29] used at 1/400 dilution; type IIA/IID collagen mouse monoclonal antisera that recognizes epitopes within the exon 2-encoded domain of the IIA and IID procollagen isoforms and used at 1/500 dilution (gift from Dr Linda Sandell); type X collagen mouse polyclonal antibodies used at 1/500 dilution [30]; and α-SMA mouse monoclonal antibodies (Sigma A5228) used at 1/1000 dilution.
2.6. Immunohistochemical analysis of protein localization
MSC or MSC/PEG-μs aggregate cultures were harvested following 12 days or 28 days of TGF-β3 induction, fixed in 4% paraformaldehyde for 2 h, rinsed thoroughly in 1×PBS followed by dehydration in increasing concentrations of ethanol (30%, 50% and 70% ethanol) and then embedded in paraffin. Micro-thin (10 μm) sections were de-paraffinized, rehydrated and blocked with 1% hyaluronidase (Sigma) for 30 min at 37°C. Sections were rinsed with 1×PBS and blocked further with 10% goat serum and then incubated overnight at 4°C with the primary antibody diluted in 2% goat serum. Following 1×PBS washes, sections were incubated with species-specific secondary antibodies conjugated to Alexa-488 dye at a dilution of 1/200 (Invitrogen; A11001, A11006) for 45 min at room temperature. DAPI mounting medium was applied following three rinses in 1×PBS. Human embryonic limb sections (day 54 or day 67 of development) were obtained from the Laboratory of Developmental Biology at University of Washington in Seattle, under an IRB approved protocol. Sections of these tissues were used as positive controls for all collagen antibodies to confirm specificity. A section of human aorta served as a positive control for α-SMA. Negative controls (absence of primary antibody) were also carried out for each experiment. Images were viewed with a Nikon Eclipse E800 fluorescence microscope. The FITC band pass filter set was used to view sections labeled with Alexa 488 dye and the DAPI filter set was used for viewing cell nuclei.
2.7. RNA extraction and real time PCR
RNA was extracted from MSC or MSC/PEG-μs aggregate cultures at day 28 of differentiation using Trizol reagent (Invitrogen) following the manufacturer’s instructions. For each experiment, three aggregate cultures were pooled together to ensure sufficient RNA concentrations for downstream analysis. RNA concentration and quality was determined by a NanoDrop spectrophotometer (ThermoScientific). RNA (1μg) was reverse transcribed with the Superscript II reverse transcriptase kit (Invitrogen; 18064-014) following the manufacturer’s instructions. Resulting cDNA (20 μL) was diluted to 100 μL with dH2O to give a final concentration of approximately 10 ng/μL. cDNA (3 ng) was mixed with 2× SYBR Green master mix for real time PCR analysis (Applied Biosystems; 4309155). Primers used to amplify each gene of interest are shown in Supplemental Table 1. Primer sequences were either designed with Primer Express® software (Applied Biosystems) or selected from peer-reviewed publications (Supplemental Table 1). The specificity of primers was confirmed by analysis of dissociation curves before carrying out each experiment. For normalization purposes, 18S rRNA was used as the housekeeping gene. Real time PCR reactions were carried out on a 7500 Fast Real Time PCR system (Applied Biosystems). Expression levels of each gene were calculated from data obtained using the ΔΔCT method where the MSC aggregate sample was set as the calibrator. Fold changes in gene expression levels in MSCs cultured with PEG-μs were calculated and the two sample Student’s t-test was applied to measure significance (n=6; p < 0.05).
2.8. Alternative transcript quantitative PCR to measure COL2A1 mRNA isoforms
A method to quantify absolute levels of each alternatively-spliced mRNA isoform of the human α1(II) COL2A1 gene (IIA, IIB and IID) has been previously described [31]. A slightly modified and improved method was used in the present study. Custom Taqman® assays (Applied Biosystem™ by Life Technologies Corp., Carlsbad, CA) were designed using Primer Express® software (Applied Biosystems) to specifically anneal on either side of each mRNA isoform splice site. FAM™/MGB probe oligonucleotides that specifically anneal to exon-exon junctions corresponding to each COL2A1 mRNA isoform, were also designed (Supplemental Table 2). A chemically synthesized DNA insert containing all of the relevant primer and probe binding sites (referred to as a multi-amplicon standard, MAS) was cloned into pUC57 (GenScript USA Inc., Piscataway, NJ). The human α1(II) MAS plasmid also contained an 18S rRNA amplicon for normalization of each sample to 18S rRNA. The MAS was used to generate standard curves for absolute quantification of each collagen splice form, as well as 18S rRNA. A ten-fold dilution series of the MAS plasmid, spanning from 3 × 108 to 3 × 104 copies per 2.5 μL sample was prepared and the same dilution series was used in each of the four qPCR assays (IIA, IID, IIB and 18S rRNA). Aliquots of cDNA synthesized from 1 μg of total MSC or MSC/PEG-μs aggregate culture RNA was diluted to provide sufficient volume for analysis of each of the three splice forms in triplicate. Samples for 18S rRNA quantification were diluted 1/2500 to stay within the linear range of the MAS standard dilution series. Real time quantitative PCR was performed using an ABI 7500 Fast Real-Time PCR System (Applied Biosystems). Absolute IIA, IIB, and IID isoform values were multiplied by a normalization factor based on the absolute 18S rRNA value. The factor was calculated by dividing the 18S rRNA mean copy number by the absolute 18S copy number for each sample. Each of the A, B, and D absolute copy number values were multiplied by this factor to normalize to the mean 18S rRNA copy number.
3. Results
3.1. MSC/PEG-μs aggregate formation and cell viability
MSC/PEG-μs co-aggregate cultures were formed as illustrated in Figure 1. The co-aggregates were successfully generated if RGD peptides were present on the PEG microspheres (Supplemental Fig. 1). In the absence of RGD peptides, MSC and PEG-μs separated into distinct layers due to buoyancy differences (not shown). Figure 2 shows that after 28 days of TGF-β3 induction, differentiated MSCs were still uniformly distributed within the MES/PEG-μs aggregate cultures and that the majority of cells were viable as shown by positive calcein staining (Fig. 2B). The majority of cells within the MSC aggregate cultures were also viable (Fig. 2A). MSCs cultured with PEG-μs in the absence of TGF-β3 for 28 days were non-viable as shown by the presence of incorporated ethidium homodimer (Fig. 2C). This result also confirms that calcein or ethidium homodimer did not not-specifically stain the PEG microparticles.
Figure 1. Generation of MSC/PEG-μs aggregates.
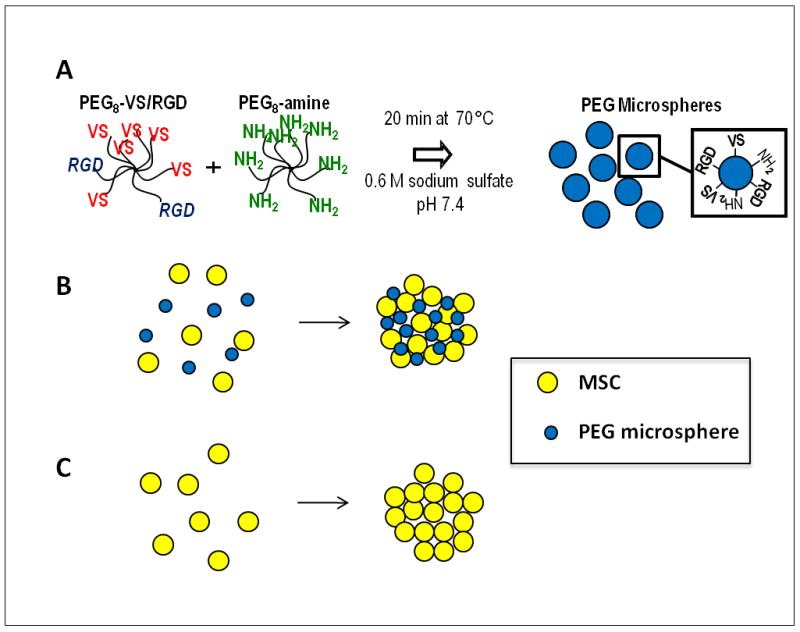
(A) Procedure for generation of PEG microspheres (PEG-μs) of approximately 6.01±1.98 μm in diameter containing RGD peptides (Ac-GCGYGRGDSPG-NH2) by phase inversion polymerization. (B) Microspheres and MSCs were mixed 1:1 (v/v) and rotated for 3 h to allow attachment of the cells to the microspheres. This was followed by centrifugation to form the aggregates. (C) As a control, MSC aggregates without PEG-μs were also generated.
Figure 2. Cell viability within aggregate cultures by co-staining with calcein and ethidium homodimer.

The Live/Dead staining reagent contains a combination of both calcein and ethidium homodimer vital dyes. Fluorescence images demonstrated numerous calcein-stained cells (green, viable) within MSC aggregate cultures (A) or MSC/PEG-μs aggregate cultures (B) following 28 days of TGF-β3 induction. In contrast, only ethidium homodimer-positive cells (red, non viable) are shown in fluorescent images of MSC/PEG-μs aggregates cultured for 28 days in the absence of TGF-β3 (C). Scale bars = 200 μm.
3.2. Altered cell phenotype and proteoglycan expression patterns in MSC/PEG-μs aggregate cultures
Dramatic differences in cell phenotype were noted between both culture systems following 28 days of TGF-β3 induction (Fig. 3). A distinctive outer fibrous layer containing elongated fibroblast-like cells was present in the MSC aggregate cultures as expected (Fig. 3A and 3C); this layer was not present in the MSC/PEG-μs aggregate cultures (Fig. 3B and 3D). However, an outer layer of elongated-shaped cells (approximately 1–2 cells thick) was shown to surround the MSC/PEG-μs cultures (Fig. 3D). Positive Safranin-O staining patterns (indicating proteoglycan deposition) were restricted to the inner two-thirds of the MSC aggregate cultures (Fig. 3A), while positive staining was found throughout the entire MSC/PEG-μs cultures (Fig. 3B). There was an apparent gradient in cell shape within the MSC aggregate cultures that varied from fibroblastic-like cells in the outer layers to more rounded chondrocyte-like cells embedded within lacunae in the proteoglycan-rich areas (Fig. 3C). In the MSC/PEG-μs cultures, cell shape appeared more homogeneous throughout the entire co-aggregate cultures when compared to the MSC aggregate cultures (Fig. 3D). These findings related to cell shape and proteoglycan staining patterns were similar after 12 days of TGF-β3 induction (Supplemental Fig. 2). We also showed in a control experiment (MSC/PEG-μs aggregates cultured for 12 days or 28 days in the absence of TGF-β3) a lack of Safranin-O staining, thus confirming no non-specific binding of Safranin-O to the PEG microspheres (Supplemental Fig. 3).
Figure 3. Proteoglycan synthesis.

Safranin-O stained MSC (A) and MSC/PEG-μs (B) aggregates following 28 days of TGF-β3 induction. Panels C and D are higher magnification images of the boxed regions in A and B, respectively. Scale bars = 100 μm. Safranin-O staining patterns following 12 days of TGF-β3 induction are shown in Supplemental Figure 2.
3.3. Protein expression patterns of type II collagen and type X collagen
Immunolocalizations were carried out using a polyclonal antibody that recognizes an epitope in the triple helical collagen domain common to all type II collagen isoforms (“total” type II collagen). A monoclonal antibody recognizing epitopes within the alternatively-spliced exon 2-encoded cysteine-rich domain of the type II procollagen amino propeptide was also used. These isoforms represent the “embryonic/chondroprogenitor” forms of type II procollagen (type IIA and type IID) [31]. Following 28 days of TGF-β3 induction, “total” type II collagen staining was present throughout the MSC aggregrate sections, except for the very outer fibrous layer (Fig. 4A), while ubiquitous expression was found throughout the entire MSC/PEG-μs aggregate cultures (Fig 4B). Protein expression of the type IIA/IID isoforms was lower in the MSC aggregate culture (Fig. 4C) and restricted to the non-fibrous regions of the aggregates that showed positive staining for “total” type II collagen (Fig. 4A). In the presence of microspheres, more homogeneous expression of type IIA/IID isoforms was found in the MSC/PEG-μs aggregates (Fig 4D). At day 12 of TGF-β3 induction, the first signs of type II collagen deposition were noted at the very outer regions of the MSC/PEG-μs cultures (Supplemental Fig. 4). Type X collagen (a common marker of hypertrophic chondrocytes) was restricted to the central region of the MSC aggregates following 28 days of TGF-β3 induction as previously reported in other studies [32, 33](Fig. 4E). At the same time point of differentiation, expression was more ubiquitously distributed throughout the MSC/PEG-μs cultures, although less staining was noted at the very outer regions and central regions of the aggregates (Fig. 4F). At day 12 of TGF-β3 induction, type X collagen expression was not yet detectable in MSC/PEG-μs aggregates while some expression was noted in the central region of the MSC aggregates at this earlier time point (results not shown).
Figure 4. Localization of type II collagen and type X collagen.

Antibodies against the fibrillar portion of type II collagen (total COL II) (A and B) or the exon 2-encoded protein domain in the amino propeptide of type II collagen (COL IIA/IID) (C and D) were used. Type X collagen (COL X) localization is shown in panels E and F. Fluorescence immunohistochemistry was carried out on micro-thin (10 μm) paraffin sections of MSC aggregate cultures (A, C, E) or MSC/PEG-μs aggregate cultures (B, D, F) that had been induced for 28 days with TGF-β3. Antibody staining (green), with DAPI nuclear stain (blue). Scale bars = 100 μm.
3.4. Localization of α-smooth muscle actin and type I collagen proteins
Differences between the two MSC differentiation culture systems was further highlighted by the protein expression patterns of α-smooth muscle actin (α-SMA) and type I collagen. Strong positive staining for α-SMA was found inside cells localized at the outer fibrous region in addition to regions near the center of the MSC aggregate culture (Fig. 5A). In the presence of microspheres, expression of α-SMA was found inside some differentiated MSCs, but not restricted to any specific region (Fig. 5B). As expected, the presence of type I collagen protein was confined to the outer fibrous layer of the MSC aggregate cultures (Fig. 5C) while a more ubiquitous staining pattern was seen throughout the whole MSC/PEG-μs aggregate cultures (Fig 5D). As with type II collagen, the first indication of type I collagen deposition was noted at the very outer regions of the MSC/PEG-μs aggregates following 12 days of TGF-β3 treatment (Supplemental Fig. 4).
Figure 5. Localization of α-SMA and type I collagen.

Fluorescence immunohistochemistry was carried out on micro-thin (10μm) paraffin sections of MSC aggregate cultures (A, C) or MSC/PEG-μs aggregate cultures (B, D) that had been induced for 28 days with TGF-β3. α-SMA distribution is shown in panels A and B; type I collagen (COL I) localization is shown in panels C and D. Antibody staining (green), with DAPI nuclear stain (blue). Scale bars = 100 μm.
3.5. Gene expression levels in MSC and MSC/PEG-μs aggregate cultures
Expression of a range of cartilage matrix genes was analyzed by real time PCR and data were compared between MSC and MSC/PEG-μs aggregate cultures after 28 days of TGF-β3 treatment. Levels of COL2A1 mRNA (using primers that recognize all spliced isoforms of the gene) were higher in cells from the MSC/PEG-μs differentiated cultures while mRNA levels of COL1A1 and COL10A1 were lower when compared to cells in MSC aggregate cultures (Fig. 6A). Using a specific TaqMan-based assay [31] to distinguish and quantitate levels of the different COL2A1 spliced mRNA isoforms (IIA, IIB and IID mRNA), significantly higher levels of the IIA and IID exon 2-containing mRNA isoforms and lower levels of IIB mRNAs were found in cells that had been cultured with PEG-μs. As expected, primary human articular chondrocytes (used as a positive control for the assay) expressed almost exclusively the mature IIB isoform of COL2A1 (Fig. 6B).
Figure 6. Collagen gene expression in MSC and MSC/PEG-μs aggregate cultures following 28 days of TGF-β3 induction.

(A) Levels of genes encoding type II collagen (COL2A1), type I collagen (COL1A1) and type X collagen (COL10A1) in MSC/PEG-μs cultures were expressed as Log 10 values of fold change when compared to expression in MSC aggregate cultures. (B) Using a TaqMan-based assay, levels of each COL2A1 isoform (IIA, IIB, IID) was expressed as a percentage of the total COL2A1 isoforms for both culture systems. Expression levels in human articular chondrocytes (HAC) are also shown. All differences in expression levels of each COL2A1 isoform between MSC/PEG-μs cultures and the MSC aggregate cultures were calculated as significant based on the two-sample Student’s t-test (n=6; * indicates p < 0.05 versus MSC aggregates).
Analysis of common, non-collagenous cartilage gene markers was also carried out. Levels of mRNA expression of aggrecan (AGAN), cartilage oligomeric matrix protein (COMP) and transcription factor SOX9 were higher in differentiated MSCs cultured in the presence of PEG-μs, while levels of versican (VCAN) and lubricin (PRG-4) were lower when compared to expression in MSC differentiated aggregate cultures. There was no significant change in decorin (DCN) mRNA expression (Fig. 7A). Levels of genes commonly expressed by pre-hypertrophic chondrocytes (MMP-13), hypertrophic chondrocytes (Runx2) and osteoblast-like cells (osteocalcin and osterix) were all found to be significantly lower in differentiated MSC/PEG-μs cultures as was expression of MMP-7, N-cadherin and α-smooth muscle actin (Fig. 7B).
Figure 7. Gene expression in MSC and MSC/PEG-μs aggregate cultures following 28 days of TGF-β3 induction.

(A) Genes encoding the cartilage matrix components aggrecan (AGAN), versican (VCAN), lubricin (PRG-4), cartilage oligomeric matrix protein (COMP) and decorin (DCN) and transcription factor SOX9 were analyzed. (B) Also investigated were genes encoding alpha-smooth muscle actin (α-SMA), N-Cadherin (NCAD), matrix metalloproteinase-7 (MMP-7), MMP-13, osteocalcin (OCN), Runx2 and osterix (OSX) (B). Expression levels of all genes in MSC/PEG-μs cultures are represented as Log 10 fold changes when compared to expression in MSC aggregate cultures. All differences in expression levels between the two cultures systems were calculated as significant (except for DCN) based on the two-sample Student’s t-test (n=6; * indicates p < 0.05 versus MSC aggregates).
4. Discussion
A number of groups are investigating aggregates consisting of cells and drug-delivering microparticles. The overall hypothesis is that growth factor diffusion into cell spheroids is a limiting factor in the differentiation of stem cells. Current strategies have evolved from techniques using microparticles made from degradable poly(esters) [34–38]. Microparticles based on chondroitin sulfate, agarose and gelatin have also been developed to release growth factors to affect cell differentiation. These microspheres were incorporated within mouse embryoid bodies resulting in good cell viability. However, variable effects on embryonic stem (ES) cell differentiation were found depending on the biomaterial composition of the microspheres [23, 24]. Similar approaches are now currently being investigated in the cartilage tissue engineering field, utilizing microspheres as growth factor delivery vehicles to induce differentiation of multipotent mesenchymal stem/stromal cells (MSCs) or to induce matrix synthesis of primary chondrocytes [10–17]. As was shown for ES cell differentiation [23, 24], we hypothesized that the presence of microspheres themselves may affect how MSCs differentiate in response to growth factor. To test this hypothesis, a 3D co-aggregate culture system was developed consisting of human primary bone marrow-derived MSCs and non-degradable poly(ethylene glycol) microspheres (PEG-μs). Formation of PEG-μs is a novel application of phase inversion first described by Elbert and colleagues [39]. This is a rapid, highly-scalable process where microsphere density and size can be independently controlled. Unlike other methods that produce small (1–3 micron) hydrogel microparticles, the synthetic process does not involve the use of organic solvents or surfactants, potentially enhancing biocompatibility. PEG-μs have been utilized to form bioactive modular scaffolds containing HepG2 hepatoma cells utilizing hydrogel microspheres as building blocks [40]. Other strategies combining cells and hydrogel microparticles have been recently reviewed [41].
The current study observed the cumulative effects of dilution of cells with microspheres and adhesion of adjacent cells through a linear RGD peptide. This linear RGD peptide attached to PEG was previously shown to promote cell adhesion, primarily through the αVβ3 integrin [25]. In future studies, we may be able to tune the buoyancy of the microspheres to match those of the cells to deconvolute the effects of cell adhesion versus cell dilution [26, 42]. However, it may be that some form of cell attachment is necessary to permit cell viability since it has been reported that MSCs within PEG bulk hydrogels undergo apoptosis due to lack of adhesion sites [20, 21].
Dramatic differences were found compared to MSC aggregates grown in the absence of PEG-μs. A characteristic feature of TGF-β-induced MSC aggregate cultures is the formation of a distinct outer fibrous layer consisting of elongated cells that synthesize and deposit a type I collagen-rich extracellular matrix [32, 33]. We also showed that a subset of these cells express α-smooth muscle actin (α-SMA), further highlighting the heterogeneity of this culture system. In addition to TGF-β induction, other studies have shown that cyclic stretch and cell contraction can induce α-SMA synthesis in MSCs [43–46]. Therefore, the combination of growth factor induction and tension effects on cells may be other explanations for positive α-SMA staining within the fibrous region of the MSC aggregate cultures. When MSCs are grown in the presence of PEG-μs, this outer fibrous layer is not formed, perhaps due to effects of cell dilution, inhibition of cell-cell contacts and paracrine signaling, or altered cellular responses by direct binding of the cells to the microspheres through integrin receptors. In the presence of microspheres, a proteoglycan matrix was generated by the differentiating MSCs throughout the aggregate rather than in the interior alone. In addition, using an antibody that recognizes the triple helical domain of type II collagen, ubiquitous expression was found, further suggesting deposition of a cartilage-like matrix. However, more detailed analysis of alternatively-spliced isoforms of type II collagen at the mRNA and protein level revealed slightly increased amounts of the longer isoforms (type IIA and IID) [31] when MSCs were cultured with PEG-μs. These protein isoforms contain an exon 2-encoded cysteine-rich domain in the amino propeptide of type II procollagen and are predominantly expressed by chondroprogenitor cells. As chondrocytes differentiate, exon 2 is removed, forming the type IIB collagen isoform [31, 47]. Our data thus suggests that cells bound to PEG-μs are not as far advanced in the chondrogenic differentiation pathway compared to those in the microsphere-free aggregate cultures. It may be that a longer time in culture is needed to alter the COL2A1 splicing patterns or that removal of the microspheres is required to produce a more mature type II collagen matrix. Further evidence for the presence of less differentiated chondrocytes is shown by the ubiquitous expression of type I collagen in the PEG-μs co-aggregate cultures. Detailed morphological examination of MSC aggregate cultures has shown that type I collagen is expressed throughout these aggregate cultures at earlier time points (7 days) of TGF-β induction and expression of type I collagen then decreases over time as type II collagen expression increases [33, 48]. Type I collagen is also known to be expressed in vivo during cartilage development at sites where the exon 2-containing type II collagen isoforms are being expressed by chondroprogenitor cells [49].
The expression of type X collagen within both MSC/PEG-μs and MSC aggregate cultures suggests the presence of terminally-differentiated hypertrophic chondrocytes. Levels of COL10A1 mRNA were found to be lower in PEG-μs cultures following 28 days of TGF-β3 induction; however, there was more ubiquitous expression of protein identified in the MSC/PEG-μs cultures when compared to MSC aggregate cultures. These immunohistochemical analyses are not quantitative in terms of measuring protein amounts and it may be that mRNA expression levels do not reflect protein expression at the end-stage time point of the differentiation assay. Future studies to investigate mRNA and protein levels at various time points up to and including day 28 will be informative. In any case, the presence of type X collagen protein suggests that there may be a mixture of chondrocytes at various stages of differentiation within both aggregate culture systems. It has been shown in MSC aggregate cultures that type X collagen synthesis, in many cases, actually precedes that of type II collagen [32, 33, 48, 50]. This does not follow the natural course of events in vivo during cartilage formation where type II collagen-expressing chondrocytes are always formed before the appearance of type X collagen-expressing hypertrophic cells. Also, we have shown that some MSC populations express low levels of COL10A1 mRNA even before TGF-β induction [51]. It may be that MSCs extracted from a bone marrow niche are primed to express type X collagen. The concern with the presence of type X collagen is the potential of inducing bone formation in vivo due to apoptosis of hypertrophic cells followed by vascular invasion and endochondral ossification; this would not be desirable for cartilage tissue engineering. The lower mRNA expression levels of pre-hypertrophic, hypertrophic and osteoblast markers (MMP-13, Runx2, osteocalcin, osterix) within the MSC/PEG-μs aggregate cultures is therefore a promising finding in our studies. Also, type X collagen has in fact been identified as a component of articular cartilage that does not contain hypertrophic chondrocytes [52]. Thus, the effect of having type X collagen present in these aggregate cultures, or indeed in any MSC/biomaterial scaffold system, remains to be fully elucidated. Implantation of differentiated MSC/PEG-μs aggregates into subcutaneous regions of SCID mice could be carried out to determine if vascular invasion/bone formation will occur following at least 6 weeks in this ex vivo environment as has been shown in other studies [50].
Although microspheres are commonly utilized as growth factor delivery vehicles, data from the present study highlight the fact that the biological consequences of biomaterial/MSC interactions must be considered. The effect by these microparticles may be multi-faceted. For example, the presence of microspheres might affect diffusion of the exogenous growth factor as well as affect paracrine or autocrine factors produced by cells within the aggregate cultures. The microspheres may also interfere with cell-cell adhesion, paracrine signaling or alter the ability of the cells to exert tension on the secreted ECM. In our studies, the finding of lower expression levels of N-cadherin by differentiating MSCs when cultured in the presence of PEG-μs certainly suggests disruption of cell-cell contacts. The effect of RGD-mediated attachment should also be taken into consideration in addition to the fact that the PEG-μs in the present study were non-degradable. It may be that degrading the microspheres over time will improve matrix deposition and perhaps alter the collagen make-up of the matrix. Attractive future approaches will be to engineer matrix metalloproteinase (MMP) peptide substrates within the microspheres. This type of strategy has been carried out in other studies utilizing bulk PEG hydrogels. One group found enhanced chondrocyte differentiation when an MMP-13 cleavage site was incorporated into the RGD peptide [22] while another group demonstrated improved type II collagen matrix deposition by engineering MMP-7-degradable scaffolds [53]. We have shown expression of both MMP-7 and MMP-13 mRNA in MSC/PEG-μs cultures, albeit at lower levels than in MSC aggregates. However, a more detailed time course experiment is required to determine MMP expression levels in TGF-β-induced MSC/PEG-μs aggregate cultures to better determine a suitable strategy to develop bioresponsive microspheres more finely tuned to the differentiation of MSCs. The potential now exists to develop a more intricate system where MSCs can be grown in 3D cultures containing microspheres with distinct properties (i.e. microspheres with cell attachment sites, degradable peptides and/or growth factor delivery function) with the ultimate goal of improving cartilage tissue engineering strategies.
5. Conclusions
Microspheres are often used as growth factor delivery vehicles to induce MSC differentiation. However, the present study demonstrates that biomaterials themselves may have a strong impact on differentiation. Compared to MSC aggregates alone, MSC/PEG-μs aggregates were not surrounded by a fibrous layer and exhibited differential expression of a number of markers of chondrocyte differentiation. Although the mechanism for these changes were not elucidated in this study, the potential effects of cell dilution, loss of cell-cell contacts and paracrine signaling, and signaling through integrin receptors should be considered in developing drug delivery systems for cell differentiation. The results also suggest methods to engineer the differentiation response of MSCs through design of biomaterials.
Supplementary Material
Acknowledgments
We thank Crystal Idleburg for histology services, Elizabeth DeLassus and John Freeman for microscopy assistance, Dr Gary Gibson for the type X collagen antibody and Dr Linda Sandell for type IIA/D monoclonal antisera. Human tissue was obtained from The Laboratory of Developmental Biology at the University of Washington, Seattle; this lab is supported by NIH Award Number 5R24HD000836 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. Work in the present study was funded in part by the Arthritis Foundation (Investigator Award, AM), NIH grant R21RR025397 (AM and TMH) and NIH grant R01HL085364 (DLE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ulrich-Vinther M, Maloney MD, Schwarz EM, Rosier R, O’Keefe RJ. Articular cartilage biology. J Am Acad Orthop Surg. 2003;11:421–30. doi: 10.5435/00124635-200311000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Hollander AP, Dickinson SC, Kafienah W. Stem cells and cartilage development: complexities of a simple tissue. Stem Cells. 2010;28:1992–6. doi: 10.1002/stem.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daher RJ, Chahine NO, Greenberg AS, Sgaglione NA, Grande DA. New methods to diagnose and treat cartilage degeneration. Nat Rev Rheumatol. 2009;5:599–607. doi: 10.1038/nrrheum.2009.204. [DOI] [PubMed] [Google Scholar]
- 4.Nelson L, Fairclough J, Archer CW. Use of stem cells in the biological repair of articular cartilage. Expert Opin Biol Th. 2010;10:43–55. doi: 10.1517/14712590903321470. [DOI] [PubMed] [Google Scholar]
- 5.Keeney M, Lai JH, Yang F. Recent progress in cartilage tissue engineering. Curr Opin Biotechnol. 2011 doi: 10.1016/j.copbio.2011.04.003. Available from URL: http://www.ncbi.nlm.nih.gov/pubmed/21531126. [DOI] [PubMed]
- 6.Toh WS, Spector M, Lee EH, Cao T. Biomaterial-Mediated Delivery of Microenvironmental Cues for Repair and Regeneration of Articular Cartilage. Mol Pharm. 2011 doi: 10.1021/mp100437a. Available from URL: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21500855. [DOI] [PubMed]
- 7.Spiller KL, Maher SA, Lowman AM. Hydrogels for the Repair of Articular Cartilage Defects. Tissue Eng Part B Rev. 2011 doi: 10.1089/ten.TEB.2011.0077. Available from URL: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21510824. [DOI] [PMC free article] [PubMed]
- 8.Vinatier C, Mrugala D, Jorgensen C, Guicheux J, Noel D. Cartilage engineering: a crucial combination of cells, biomaterials and biofactors. Trends Biotechnol. 2009;27:307–14. doi: 10.1016/j.tibtech.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Place ES, Evans ND, Stevens MM. Complexity in biomaterials for tissue engineering. Nat Mater. 2009;8:457–70. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 10.Han Y, Wei Y, Wang S, Song Y. Cartilage regeneration using adipose-derived stem cells and the controlled-released hybrid microspheres. Joint Bone Spine. 2010;77:27–31. doi: 10.1016/j.jbspin.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Solorio LD, Fu AS, Hernandez-Irizarry R, Alsberg E. Chondrogenic differentiation of human mesenchymal stem cell aggregates via controlled release of TGF-beta1 from incorporated polymer microspheres. J Biomed Mater Res A. 2010;92:1139–44. doi: 10.1002/jbm.a.32440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreas K, Zehbe R, Kazubek M, Grzeschik K, Sternberg N, Baumler H, et al. Biodegradable insulin-loaded PLGA microspheres fabricated by three different emulsification techniques: investigation for cartilage tissue engineering. Acta Biomater. 2011;7:1485–95. doi: 10.1016/j.actbio.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Jha AK, Yang W, Kirn-Safran CB, Farach-Carson MC, Jia X. Perlecan domain I-conjugated, hyaluronic acid-based hydrogel particles for enhanced chondrogenic differentiation via BMP-2 release. Biomaterials. 2009;30:6964–75. doi: 10.1016/j.biomaterials.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang SW, La WG, Kim BS. Open macroporous poly(lactic-co-glycolic Acid) microspheres as an injectable scaffold for cartilage tissue engineering. J Biomater Sci Polym Ed. 2009;20:399–409. doi: 10.1163/156856209X412236. [DOI] [PubMed] [Google Scholar]
- 15.Jaklenec A, Wan E, Murray ME, Mathiowitz E. Novel scaffolds fabricated from protein-loaded microspheres for tissue engineering. Biomaterials. 2008;29:185–92. doi: 10.1016/j.biomaterials.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 16.Jaklenec A, Hinckfuss A, Bilgen B, Ciombor DM, Aaron R, Mathiowitz E. Sequential release of bioactive IGF-I and TGF-beta 1 from PLGA microsphere-based scaffolds. Biomaterials. 2008;29:1518–25. doi: 10.1016/j.biomaterials.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Scholten PM, Ng KW, Joh K, Serino LP, Warren RF, Torzilli PA, et al. A semi-degradable composite scaffold for articular cartilage defects. J Biomed Mater Res A. 2011 doi: 10.1002/jbm.a.33005. Available from URL: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=21308980. [DOI] [PMC free article] [PubMed]
- 18.Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp Cell Res. 1998;238:265–72. doi: 10.1006/excr.1997.3858. [DOI] [PubMed] [Google Scholar]
- 19.Scott EA, Nichols MD, Cordova LH, George BJ, Jun YS, Elbert DL. Protein adsorption and cell adhesion on nanoscale bioactive coatings formed from poly(ethylene glycol) and albumin microgels. Biomaterials. 2008;29:4481–93. doi: 10.1016/j.biomaterials.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuttelman CR, Tripodi MC, Anseth KS. Synthetic hydrogel niches that promote hMSC viability. Matrix Biol. 2005;24:208–18. doi: 10.1016/j.matbio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–7. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 22.Salinas CN, Anseth KS. The influence of the RGD peptide motif and its contextual presentation in PEG gels on human mesenchymal stem cell viability. J Tissue Eng Regen Med. 2008;2:296–304. doi: 10.1002/term.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim JJ, Hammoudi TM, Bratt-Leal AM, Hamilton SK, Kepple KL, Bloodworth NC, et al. Development of nano- and microscale chondroitin sulfate particles for controlled growth factor delivery. Acta Biomater. 2011;7:986–95. doi: 10.1016/j.actbio.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bratt-Leal AM, Carpenedo RL, Ungrin MD, Zandstra PW, McDevitt TC. Incorporation of biomaterials in multicellular aggregates modulates pluripotent stem cell differentiation. Biomaterials. 2011;32:48–56. doi: 10.1016/j.biomaterials.2010.08.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wacker BK, Alford SK, Scott EA, Das Thakur M, Longmore GD, Elbert DL. Endothelial cell migration on RGD-peptide-containing PEG hydrogels in the presence of sphingosine 1-phosphate. Biophys J. 2008;94:273–85. doi: 10.1529/biophysj.107.109074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roam JL, Xu H, Nguyen PK, Elbert DL. The formation of protein concentration gradients mediated by density differences of poly(ethylene glycol) microspheres. Biomaterials. 2010;31:8642–50. doi: 10.1016/j.biomaterials.2010.07.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaragosi LE, Ailhaud G, Dani C. Autocrine fibroblast growth factor 2 signaling is critical for self-renewal of human multipotent adipose-derived stem cells. Stem Cells. 2006;24:2412–9. doi: 10.1634/stemcells.2006-0006. [DOI] [PubMed] [Google Scholar]
- 28.Coutu DL, Francois M, Galipeau J. Inhibition of cellular senescence by developmentally regulated FGF-receptors in mesenchymal stem cells. Blood. 2011 doi: 10.1182/blood-2010-12-321539. Available from URL: http://www.ncbi.nlm.nih.gov/pubmed/21527526. [DOI] [PubMed]
- 29.Cremer MA, Kang AH. Collagen-induced arthritis in rodents: a review of immunity to type II collagen with emphasis on the importance of molecular conformation and structure. Int Rev Immunol. 1988;4:65–81. doi: 10.3109/08830188809044771. [DOI] [PubMed] [Google Scholar]
- 30.Gibson G, Lin DL, Francki K, Caterson B, Foster B. Type X collagen is colocalized with a proteoglycan epitope to form distinct morphological structures in bovine growth cartilage. Bone. 1996;19:307–15. doi: 10.1016/s8756-3282(96)00222-0. [DOI] [PubMed] [Google Scholar]
- 31.McAlinden A, Johnstone B, Kollar J, Kazmi N, Hering TM. Expression of two novel alternatively spliced COL2A1 isoforms during chondrocyte differentiation. Matrix Biol. 2008;27:254–66. doi: 10.1016/j.matbio.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barry F, Boynton RE, Liu B, Murphy JM. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: differentiation-dependent gene expression of matrix components. Exp Cell Res. 2001;268:189–200. doi: 10.1006/excr.2001.5278. [DOI] [PubMed] [Google Scholar]
- 33.Yoo JU, Barthel TS, Nishimura K, Solchaga L, Caplan AI, Goldberg VM, et al. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J Bone Joint Surg Am. 1998;80:1745–57. doi: 10.2106/00004623-199812000-00004. [DOI] [PubMed] [Google Scholar]
- 34.Mahoney MJ, Saltzman WM. Transplantation of brain cells assembled around a programmable synthetic microenvironment. Nat Biotechnol. 2001;19:934–9. doi: 10.1038/nbt1001-934. [DOI] [PubMed] [Google Scholar]
- 35.Ferreira L, Squier T, Park H, Choe H, Kohane DS, Langer R. Human embryoid bodies containing nano- and microparticulate delivery vehicles. Adv Mater. 2008;20:2285. [Google Scholar]
- 36.Bouffi C, Thomas O, Bony C, Giteau A, Venier-Julienne MC, Jorgensen C, et al. The role of pharmacologically active microcarriers releasing TGF-beta3 in cartilage formation in vivo by mesenchymal stem cells. Biomaterials. 2010;31:6485–93. doi: 10.1016/j.biomaterials.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Carpenedo RL, Bratt-Leal AM, Marklein RA, Seaman SA, Bowen NJ, McDonald JF, et al. Homogeneous and organized differentiation within embryoid bodies induced by microsphere-mediated delivery of small molecules. Biomaterials. 2009;30:2507–15. doi: 10.1016/j.biomaterials.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carpenedo RL, Seaman SA, McDevitt TC. Microsphere size effects on embryoid body incorporation and embryonic stem cell differentiation. J Biomed Mater Res A. 2010;94:466–75. doi: 10.1002/jbm.a.32710. [DOI] [PubMed] [Google Scholar]
- 39.Elbert DL. Liquid-liquid two-phase systems for the production of porous hydrogels and hydrogel microspheres for biomedical applications: A tutorial review. Acta Biomater. 2011;7:31–56. doi: 10.1016/j.actbio.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott EA, Nichols MD, Kuntz-Willits R, Elbert DL. Modular scaffolds assembled around living cells using poly(ethylene glycol) microspheres with macroporation via a non-cytotoxic porogen. Acta Biomater. 2010;6:29–38. doi: 10.1016/j.actbio.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elbert DL. Bottom-up tissue engineering. Curr Opin Biotechnol. 2011 doi: 10.1016/j.copbio.2011.04.001. Available from URL: http://www.ncbi.nlm.nih.gov/pubmed/21524904. [DOI] [PMC free article] [PubMed]
- 42.Nichols MD, Scott EA, Elbert DL. Factors affecting size and swelling of poly(ethylene glycol) microspheres formed in aqueous sodium sulfate solutions without surfactants. Biomaterials. 2009;30:5283–91. doi: 10.1016/j.biomaterials.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narita Y, Yamawaki A, Kagami H, Ueda M, Ueda Y. Effects of transforming growth factor-beta 1 and ascorbic acid on differentiation of human bone-marrow-derived mesenchymal stem cells into smooth muscle cell lineage. Cell Tissue Res. 2008;333:449–59. doi: 10.1007/s00441-008-0654-0. [DOI] [PubMed] [Google Scholar]
- 44.Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, et al. Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells. 2010;28:734–42. doi: 10.1002/stem.319. [DOI] [PubMed] [Google Scholar]
- 45.Kinner B, Zaleskas JM, Spector M. Regulation of smooth muscle actin expression and contraction in adult human mesenchymal stem cells. Exp Cell Res. 2002;278:72–83. doi: 10.1006/excr.2002.5561. [DOI] [PubMed] [Google Scholar]
- 46.Ghazanfari S, Tafazzoli-Shadpour M, Shokrgozar MA. Effects of cyclic stretch on proliferation of mesenchymal stem cells and their differentiation to smooth muscle cells. Biochem Biophys Res Commun. 2009;388:601–5. doi: 10.1016/j.bbrc.2009.08.072. [DOI] [PubMed] [Google Scholar]
- 47.Ryan MC, Sandell LJ. Differential expression of a cysteine-rich domain in the amino-terminal propeptide of type II (cartilage) procollagen by alternative splicing of mRNA. J Biol Chem. 1990;265:10334–9. [PubMed] [Google Scholar]
- 48.Ichinose S, Tagami M, Muneta T, Sekiya I. Morphological examination during in vitro cartilage formation by human mesenchymal stem cells. Cell Tissue Res. 2005;322:217–26. doi: 10.1007/s00441-005-1140-6. [DOI] [PubMed] [Google Scholar]
- 49.Nalin AM, Greenlee TK, Jr, Sandell LJ. Collagen gene expression during development of avian synovial joints: transient expression of types II and XI collagen genes in the joint capsule. Dev Dyn. 1995;203:352–62. doi: 10.1002/aja.1002030307. [DOI] [PubMed] [Google Scholar]
- 50.Pelttari K, Winter A, Steck E, Goetzke K, Hennig T, Ochs BG, et al. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006;54:3254–66. doi: 10.1002/art.22136. [DOI] [PubMed] [Google Scholar]
- 51.Rich JT, Rosova I, Nolta JA, Myckatyn TM, Sandell LJ, McAlinden A. Upregulation of Runx2 and Osterix during in vitro chondrogenesis of human adipose-derived stromal cells. Biochem Biophys Res Commun. 2008;372:230–5. doi: 10.1016/j.bbrc.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rucklidge GJ, Milne G, Robins SP. Collagen type X: a component of the surface of normal human, pig, and rat articular cartilage. Biochem Biophys Res Commun. 1996;224:297–302. doi: 10.1006/bbrc.1996.1024. [DOI] [PubMed] [Google Scholar]
- 53.Bahney CS, Hsu CW, Yoo JU, West JL, Johnstone B. A bioresponsive hydrogel tuned to chondrogenesis of human mesenchymal stem cells. FASEB J. 2011;25:1486–96. doi: 10.1096/fj.10-165514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.