

Abstract
Objective
To identify factors associated with frequent severe vaso-occlusive pain crises in a contemporary pediatric cohort of sickle cell anemia (SCA)enrolled in a prospective study of pulmonary hypertension and the hypoxic response in sickle cell disease (SCD).
Study design
Clinical and laboratory characteristics of children with SCA who had ≥3 severe pain crises requiring health care in the preceding year were compared with subjects with <3 such episodes.
Results
Seventy-five children (20%) reported ≥3 severe pain episodes in the preceding year, and 232 (61%) had none. Frequent pain episodes were associated with older age (OR 1.2; 95% CI 1.1–1.3; P<0.0001), α-thalassemia trait (OR 3.5; 1.6–6.7; P=0.002), higher median hemoglobin (OR 1.7; 95% CI: 1.2–2.4; P<0.003) and lower lactate dehydrogenase (LDH) concentration (OR 1.82; 95% CI: 1.07–3.11; P = 0.027). Children with high pain frequency also had an increased iron burden (serum ferritin 480 vs. 198 μg/L; P=0.006) and higher median tricuspid regurgitation jet velocity (2.41 vs. 2.31 m/s; P=0.001). Neither hydroxy urea use nor fetal hemoglobin levels were significantly different according to severe pain history.
Conclusions
In our cohort of children with SCA increasing age was associated with higher frequency of severe pain episodes as were α-thalassemia, iron overload, higher hemoglobin and lower LDH concentration and higher tricuspid regurgitation velocity.
Keywords: Sickle cell anemia, vaso-occlusive crisis, pain
Periodic episodes of severe pain known as vaso-occlusive pain crises are the most common symptom of sickle cell anemia (SCA), adversely impacting the quality of life of the patients and their caregivers. Adults with sickle cell disease show a large inter-patient variability in the frequency and severity of vaso-occlusive pain crisis (1, 2). A high rate of admissions for pain is associated with early mortality in adults with SCA (1). The proximate cause of vaso-occlusive pain crisis is occlusion of the microcirculation of the bone marrow by sickled red blood cells. Factors such as higher hematocrit(1, 3), lower fetal hemoglobin and presence of α-thalassemia trait (4, 5) are associated with increased rate of vaso-occlusive pain crisis, and a high steady-state serum lactate dehydrogenase concentration (LDH) is associated with less frequent vaso-occlusive pain crises in adults with SCA(6).
The risk factors for vaso-occlusive pain crisis in children and adolescents with SCA are not well defined. The Cooperative Study of Sickle Cell Disease (CSSCD) identified dactylitis, severe anemia and leukocytosis in the first two years of life as the risk factors for severe disease including frequent vaso-occlusive pain crises later in life (7), but these factors were not confirmed in a subsequent newborn cohort (8). Panepinto et al reported that older children account for a higher number of admissions and a longer duration of stay for vaso-occlusive pain crises based on hospital discharge data (9). Identification of characteristics associated with vaso-occlusive pain crisis is crucial not only for better understanding of the pathophysiology of pain but also for designing interventions to decrease the frequency of vaso-occlusive pain crisis. Here, we analyzed data from a large, current pediatric SCA cohort receiving contemporary therapy.
Methods
Study participants were children and adolescents(3 to 20 years old)with SCA (homozygous hemoglobin SS) who were enrolled in the prospective multicenter PUSH study. The analysis was restricted to individuals with homozygous hemoglobin S (HbSS) of the cohort due to known differences in the clinical course of HbSS compared with other SCD genotypes such as HbSC and HbSβ+-thalassemia. A total of 381 participants were recruited from Howard University, Children’s National Medical Center, National Institutes of Health and the University of Michigan where they received care for SCD. The institutional review boards of all four participating institutions approved the study protocol. The subjects and/or their legal guardian provided written informed consent to participate in accordance with the Declaration of Helsinki. Trial registered with clinicaltrial.gov (NCT00495638).
The subjects were evaluated at steady state as previously described (10). At enrollment, family-reported history of number and severity of painful episodes in the preceding 12 months was recorded. Episodes of pain that may or may not have required pain medicines but did not prevent normal daily activity were described as mild pain episodes, and painful episodes requiring pain medications and changes in daily activities, such as missing work or school, were described as of moderate severity. Severe vaso-occlusive pain crisis included the episodes that required a visit to emergency department or doctor’s office or extremely severe painful episodes which required hospitalizations. The number of severe vaso-occlusive pain crises requiring health care utilization was used to classify study subjects into frequent and infrequent vaso-occlusive pain crisis groups. History of red cell transfusions in lifetime and hydroxy urea use was also obtained. The diagnosis of SCA was confirmed by DNA sequencing, hemoglobin electrophoresis and/or high-performance liquid chromatography. Doppler echocardiography was used prospectively to estimate systolic pulmonary artery pressure through measurement of the tricuspid regurgitation velocity. Hemoglobin and fetal hemoglobin (HbF) levels, leukocyte, platelet and reticulocyte counts, serum lactate dehydrogenase (LDH), aspartate aminotransferase, and bilirubin were determined at the time of enrollment in the respective clinical laboratories using standard methodologies. The serum ferritin concentration was determined centrally by ELISA. The presence of co-existing α-thalassemia was determined by molecular genetic testing as described by Tan et al(11). Clinical and laboratory characteristics of study subjects who had ≥3 severe and/or extremely severe pain crises requiring health care utilization in the previous year (frequent vaso-occlusive pain crisis group) were compared with subjects who had <3 such pain crises during the previous year (infrequent vaso-occlusive pain crisis group). Information on mild to moderate painful episodes, defined as episodes of pain that did not require hospitalization or ED/unscheduled clinic visit, was also collected. Hospitalizations not related to pain were not included in the analysis.
Statistical analysis
Comparisons were made using the chi square test for categorical variables and the Student t-test for continuous variables (after normal transformation). The association of ≥3 severe pain crises with other variables was determined by multiple logistic regression analysis. We applied a combination of biologic and statistical approaches to select the final model. Variables with P <0.2 in univariate analysis were entered into model unless this was not appropriate because of co-linearity with another variable or implausible causal relationship (Table I). To adjust for co-linearity between LDH and hemoglobin, we applied a pathway analysis to determine variables linking the two. The final logistic model was checked for outliers using standardized residual in the range of +3 to −3. Observations beyond this range were removed from the final model. P values less than 0.05 were considered to be statistically significant. Analyses were performed with STATA 10.1 (Stata Corp College Station, TX, USA). Pathway analysis was done by AMOS 18.0 (IBM corp. Somers, NY).
Table I.
Independent factors associated with frequent severe vaso-occlusive pain crisis (3 or more episodes in last year) in children with SCA
| N=213* | OR (95% CI) | P value |
|---|---|---|
| Age (year) | 1.25 (1.13–1.38) | <0.0001 |
|
| ||
| Alpha-thalassemia: | ||
| Heterozygote (−α/αα) | 4.83 (2.05–11.40) | 0.0003 |
| Homozygote (−α/−α) | 8.79 (1.43–53.86) | 0.019 |
| Trend | 0.004 | |
|
| ||
| TRV (m/sec) | 26.31 (4.06–170.32) | 0.001 |
|
| ||
| Lactate dehydrogenase (natural log)**(U/L) | 0.17 (0.05–0.56) | 0.005 |
Variables entered into model include age, thalassemia, hemoglobin, granulocyte count, MCV, LDH, history of asthma, ferritin and TRV.
Six outliers were removed from model. Area under ROC curve=0.87, P value for goodness of fit=0.6.
Hemoglobin could be substituted for LDH with OR (95% CI) = 1.66 (1.15–2.39, P = 0.006).
Results
Data were available in 381 children and adolescents with HbSS. Eighty-four (32%) individuals had co-existing α-thalassemia. Forty-three percent of the participants were on hydroxy urea and 18% were on chronic red blood cell transfusion therapy. Twenty-eight percent reported receiving more that 10 red blood cell transfusions in their lifetime. Vaso-occlusive pain crisis was the most common morbidity (39%) followed by acute chest syndrome (32%).
Clinical and laboratory characteristics according to frequency of severe vaso-occlusive pain crises requiring health care utilization
Table II shows that 75 children (20%) had ≥3 severe pain crises in the preceding 12 months, and 232 (61%) subjects had no admissions or ED visits for vaso-occlusive pain crises during this time. Frequency of severe vaso-occlusive pain crisis correlated with frequency of mild to moderate painful episodes (Spearman correlation coefficient 0.58; P<0.0001). The subjects in the frequent vaso-occlusive pain crisis group were older, had higher hemoglobin concentrations and neutrophil counts and lower serum LDH and aspartate aminotransferase (AST) concentrations (Table II). Median tricuspid regurgitation velocity (TRV) was higher in the frequent vaso-occlusive pain crisis group (2.4 vs. 2.3 m/s; p=0.0003) and the proportion of children with TRV>2.59 m/s showed a parallel trend (19.4% vs. 11.5%; p=0.09). History of asthma was more prevalent in the high pain group (37% vs. 23%; P=0.008) as was history of acute chest syndrome (61% vs. 25%; P<0.0001). Deficiency of glucose-6-phosphate dehydrogenase, and histories of priapism and stroke were not different between the groups.
Table II.
Clinical and laboratory characteristics of children with SCA according to frequency of severe vaso-occlusive pain crisis*
| Characteristics | Frequency of severe vaso-occlusive pain crisis in preceding 12 months | P Value | |||
|---|---|---|---|---|---|
| < 3 episodes | ≥ 3 episodes | ||||
| n | Results | n | Results | ||
| Age, years | 306 | 11 (6–16) | 75 | 15 (11–19) | <0.0001 |
| Female sex, no. (%) | 306 | 146 (48%) | 75 | 38 (51%) | 0.6 |
| Hydroxyurea therapy, No. (%) | 302 | 122 (40%) | 75 | 39 (52%) | 0.07 |
| Chronic transfusion therapy, No. (%) | 300 | 50 (17%) | 74 | 17 (23%) | 0.2 |
| History of Asthma, No. (%) | 302 | 68 (23%) | 75 | 28 (37%) | 0.008 |
| History of acute chest syndrome, No (%) | 301 | 76 (25%) | 75 | 46 (61%) | <0.0001 |
| History of priapism, No. (%) | 145 | 26 (18%) | 37 | 8 (22%) | 0.6 |
| History of stroke, No. (%) | 302 | 37 (12%) | 75 | 11 (15%) | 0.6 |
| Alpha thalassemia trait, No. (%) | 205 | 56 (27%) | 55 | 27 (49%) | 0.002 |
| Hemoglobin, g/dl | 296 | 8.5 (7.6–9.5) | 72 | 9.0 (8.3–9.6) | 0.008 |
| Mean corpuscular volume, fL | 295 | 86 (80–92) | 72 | 89 (83–95) | 0.0333 |
| White blood cells, ×109/L | 295 | 11.1 (8.3–13.6) | 72 | 11.1 (8.7–13.5) | 0.3 |
| Absolute neutrophil count, 1000/μL | 295 | 4.8 (3.5–7.1) | 71 | 5.7 (4.0–8.6) | 0.038 |
| Platelets, ×109/L | 295 | 399 (331–493) | 72 | 402 (295–495) | 0.8 |
| Reticulocytes, ×109/L | 288 | 9.3 (6.2–15.7) | 72 | 9.0 (6.1–11.8) | 0.6 |
| Lactate dehydrogenase, U/L | 262 | 467 (354–597) | 67 | 358 (268–518) | 0.0004 |
| Aspartate aminotransferase, U/L | 294 | 47 (35–59) | 72 | 36 (31–53) | 0.005 |
| Total bilirubin, mg/dL | 294 | 2.6 (1.7–4.1) | 72 | 2.5 (1.8–3.4) | 0.9 |
| Ferritin | 250 | 198 (88–808) | 66 | 480 (125–1282) | 0.006 |
| Hemoglobin F (%) | 154 | 12 (6–19) | 38 | 10 (5–13) | 0.13 |
| Hemoglobin Oxygen saturation (%) | 288 | 98 (96–99) | 72 | 98 (97–99) | 0.041 |
| TRV (m/sec) | 265 | 2.3 (2.1–2.5) | 71 | 2.4 (2.3–2.5) | 0.0003 |
| TRV>2.59 (m/sec) | 265 | 30 (11.3%) | 71 | 14 (19.7%) | 0.06 |
Results are in median and interquartile range unless otherwise indicated.
Multiple logistic regression analysis of severe vaso-occlusive pain crisis
Lower natural log LDH (OR 0.17, 95% CI: 0.05–0.56, P=0.005), older age, α-globin gene deletion (P for trend =0.004), and higher TRV each were independently associated with more frequent severe pain episodes (Table I). In this model higher hemoglobin concentration could substitute for low LDH and was associated with more frequent severe pain episodes (OR 1.66; 95% CI: 1.15–2.39; P=0.006). In a subgroup analysis among patients without α-thalassemia trait, higher natural log serum LDH was associated with less frequent pain crisis (OR 0.05, 95% CI: 0.01–0.37, P = 0.003), and age and TRV were independently associated with more frequent severe pain. Higher hemoglobin concentration was associated with more frequent pain crisis in this model if substituted for LDH (OR= 1.82; 95% CI: 1.07–3.11; P = 0.027) (Table III). In the α-thalassemia trait subgroup, only older age was significantly associated with higher risk of frequent severe pain (Table III).
Table III.
Independent factors associated with frequent severe vaso-occlusive pain crisis (3 or more episodes in last year) in children with SCA with and without alpha thalassemia
| Characteristics | OR (95% CI) | P value |
|---|---|---|
| No alpha-thalassemia; N=146 (excluding 6 outliers) | ||
| Age (year) | 1.46 (1.17–1.82) | 0.001 |
| Lactate dehydrogenase (natural log)*(U/L) | 0.05 (0.01–0.37) | 0.003 |
| TRV (m/sec) | 209.97 (5.95–7411.90) | 0.003 |
| Alpha-thalassemia trait; N=83 | ||
| Age (year) | 1.15 (1.03–1.27) | 0.011 |
Hemoglobin could replace LDH with OR (95% CI) = 1.82 (1.07–3.11, P = 0.027).
Pathway analysis
We used a pathway analysis (Figure) to confirm the results of the logistic regression analyses. In this pathway, older age, α-thalassemia trait and higher TRV were associated with more frequent pain episodes, and higher LDH was associated with less frequent pain episodes. Additionally LDH was associated with higher TRV and α-thalassemia was associated with lower LDH. All P values in this pathway were <0.04. This pathway was supported by the data (root mean square error approximation (RMSEA) = 0.01, 90% CI=0.0–0.08). In this pathway the relationship between hemoglobin concentration and severe pain episodes was not significant (P = 0.6), so LDH replaced hemoglobin in the final pathway. In addition although high TRV was associated with more frequent pain crises the direction of causation between pain and TRV could not be differentiated by this analysis.
Figure 1.
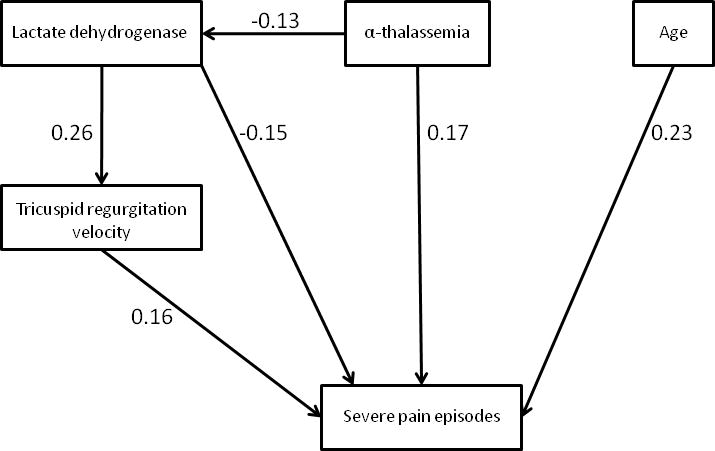
Pathway analysis of pain episode frequency in children with sickle cell anemia. Model was adequately fitted to data with root mean square error approximation (RMSEA) =0.01 (90% CI=0.0–0.08). Model chi-square is 4.2 (P=0.4, Degree of freedom = 4). Standardized beta are shown in the figure. All p values are < 0.04. A similar pathway in which severe pain was a predictor of higher tricuspid regurgitation velocity was not significantly different from this pathway as a result the direction of causation between pain and TRV could not be differentiated by this analysis.
Discussion
Our study of associated factors for pain in children and adolescent with SCA contributes new observations while replicating several of the factors previously reported in adults with SCD. Contrary to the popular perception among health care providers that children with SCA are frequently seen in the ED or hospitalized for vaso-occlusive pain crisis, about two thirds of our study population did not report any episodes of vaso-occlusive pain crisis that required health care utilization in the 12 months preceding the study enrollment. A subset (20%) of children reported more than three episodes during the preceding 12 months which is higher compared with CSSCD results where 1.5–6.9 % of 0–19 years olds with SCA experienced ≥ 3 pain episodes per patient-year (1). This discrepancy could be due to predominance of younger study subjects in CSSCD, differences in the methodology of data collection as well as selection or recall bias in our study. Increasing age was associated with more frequent vaso-occlusive pain crises consistent with the CSSCD analysis showing an increase in pain rates as patients grew older from 0 to 30 years (1). There was a trend toward more hydroxy urea use in children with more frequent pain, suggesting that history of hydroxy urea therapy defines a group of children with more severe disease. Unlike a previously reported association of lower pain episodes in patients on a chronic transfusion program(12)absence of lower frequency of VOCs in the transfused group in our cohort might be due to inadequacy of transfusion as suggested by hemoglobin S level of < 50% in only 33% of those on chronic transfusion and available hemoglobin electrophoresis data.
The observed association of higher vaso-occlusive pain crisis frequency with histories of asthma and acute chest syndrome in this cohort may be related to common pathways of pathogenesis of these three conditions. Severe painful episodes are a known risk factor for developing acute chest syndrome (13), and asthma-associated inflammatory pathways along with airway obstruction, ventilation-perfusion mismatch, and hypoxia have been proposed to contribute to local tissue hypoxia and to promote sickling of red blood cells, factors which may contribute to the initiation of an episode of vaso-occlusive pain crisis (14–17). Results of other studies evaluating the association between asthma and pain in children with SCA have been mixed. An association was reported between asthma and more frequent painful episodes in African-American children with SCA but a similar association could not be reproduced in French children with SCA (18, 19). Other than acute chest syndrome and asthma, frequency of pain did not correlate with the prevalence of other severe SCA-associated co-morbidities in the present study, such as stroke, and priapism. Thus, frequent vaso-occlusive pain crisis by itself may not be an adequate marker of other co-morbidities in children with SCA. The finding of higher median TRV being significantly associated with frequent pain deserves further investigation. Even though the median TRVs in both the groups were normal and the absolute difference between the groups was small the pathway analysis suggests that high LDH is directly associated with both less pain and high TRV, and TRV in turn has an independent association with more frequent pain.
Higher hemoglobin and lower LDH concentration was associated with high risk for pain, possibly due to increased blood viscosity that might promote vaso-occlusion and pain (20). The fetal hemoglobin concentration was higher in the low pain group but not to a statistically significant extent. Fetal hemoglobin has been shown to have a pain protective effect by reducing polymerization of hemoglobin S and thus reducing sickling of red cells and vaso-occlusion (1, 21, 22). This anticipated association in our study cohort could have been confounded by nearly half of children in both groups being on hydroxy urea, which increases fetal hemoglobin levels(1, 21, 22). Co-inheritance of alpha thalassemia with SS, reported to blunt the increase in fetal hemoglobin induced by hydroxyurea (23), was significantly more prevalent in the more frequent pain group, complicating the expected relationships between hydroxyurea, fetal hemoglobin and pain. The higher serum ferritin concentration in the frequent vaso-occlusive pain crisis group is probably a reflection of increased iron burden as a result of the greater number of red blood cell transfusions in this group because it is not unusual for individuals with SCA to receive red blood cell transfusion for exacerbation of anemia, a frequently observed complication associated with vaso-occlusive pain crises(24). However, serum ferritin elevation due to chronic inflammation might also play a role. Higher LDH and AST, markers of hemolysis in SCD(25, 26), were associated with less frequent vaso-occlusive pain crises. The relatively lower rate of pain in individuals with higher LDH and AST is consistent with the clinical observation that individuals with a higher hemolytic rate experience mild vaso-occlusive disease (6, 27, 28). Although it is not possible epidemiologically to distinguish the highly correlated effects of lower hemoglobin, high LDH has been proposed as a marker of reduced bioavailability of nitric oxide (NO) caused by scavenging of NO by cell free hemoglobin. Interestingly, in the non-SCD population and animal models, lower NO production has been associated with reduced perception and experience of pain(29). Further investigation is warranted to test whether relative NO deficiency in the high LDH group could contribute to decreased pain sensitivity and lower frequency of severe pain in this group.
One limitation of this study is its inability to correlate risk factors of severe vaso-occlusive pain crisis with measures of daily pain. Recently, Smith et al have reported that over half of the adults with SCD report pain over 50% of the days and for many of these episodes of pain medical care utilization does not occur (2). If a pattern similar to adults exists in the children and adolescents with SCA, then our data analysis of frequency of severe vaso-occlusive pain crisis underestimates the actual frequency of pain in the study population. Nevertheless the utility of monitoring ED and hospital utilization as a measure of pain has been validated by its successful use as an entry criteria and primary outcome variable in the Multi-center Study of Hydroxyurea (30). Another limitation of the study is the self-reported medical history, with a risk of inaccurate recall of ED visits and hospitalization. Finally, the analysis demonstrates associated markers and does not prove predictive risk factors or causality.
Acknowledgments
Supported in part by NHLBI(grants 2 R25 HL003679 and 1 R01 HL079912), Howard University GCRC, NCRR(grant 2MOI RR10284-10), NIH, Bethesda, MD, and the intramural research program of the National Institutes of Health.
We acknowledge the contributions of research coordinators Marlene Peters-Lawrence, Angela Rock, Margaret Fadojutimi-Akinsiku, Erin Yeagley, Sheronda Brown, Mary Yeaney, Paula Ross and protocol managers Mary K. Hall and Varbah Grigsby. We thank the patients and their families for their participation in this study.
Footnotes
Trial registered with clinicaltrial.gov (NCT00495638).
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Deepika S. Darbari, Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD
Onyinye Onyekwere, Center for Sickle Cell Disease, Howard University, Washington, DC
Mehdi Nouraie, Center for Sickle Cell Disease, Howard University, Washington, DC
Caterina P. Minniti, Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD
Lori Luchtman-Jones, Division of Pediatric Hematology and Oncology, Children’s National Medical Center, Washington, DC
Sohail Rana, Department of Pediatrics, Howard University Hospital, Washington, DC
Craig Sable, Division of Pediatric Cardiology, Children’s National Medical Center, Washington, DC
Gregory Ensing, Division of Pediatric Cardiology University of Michigan, Ann Arbor, MI
Niti Dham, Division of Pediatric Cardiology, Children’s National Medical Center, Washington, DC
Andrew Campbell, Division of Pediatric Hematology, University of Michigan, Ann Arbor, MI
Manuel Arteta, Division of Pediatric Pulmonology, University of Michigan, Ann Arbor, MI
Mark Gladwin, Allergy and Critical Care Medicine University of Pittsburgh Medical Center, Pittsburgh, PA
Oswaldo Castro, Center for Sickle Cell Disease, Howard University, Washington, DC
James G. Taylor, VI, Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD
Gregory J. Kato, Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD
Victor Gordeuk, Center for Sickle Cell Disease, Howard University, Washington, DC
References
- 1.Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11–6. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- 2.Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148:94–101. doi: 10.7326/0003-4819-148-2-200801150-00004. [DOI] [PubMed] [Google Scholar]
- 3.Baum KF, Dunn DT, Maude GH, Serjeant GR. The painful crisis of homozygous sickle cell disease. A study of the risk factors. Arch Intern Med. 1987;147:1231–4. [PubMed] [Google Scholar]
- 4.Billett HH, Nagel RL, Fabry ME. Paradoxical increase of painful crises in sickle cell patients with alpha-thalassemia. Blood. 1995;86:4382. [PubMed] [Google Scholar]
- 5.Gill FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. 1995;86:776–83. [PubMed] [Google Scholar]
- 6.Taylor JGt, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT, Steinberg MH. Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with less frequent vasoocclusive pain. PLoS One. 2008;3:e2095. doi: 10.1371/journal.pone.0002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller ST, Sleeper LA, Pegelow CH, Enos LE, Wang WC, Weiner SJ, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342:83–9. doi: 10.1056/NEJM200001133420203. [DOI] [PubMed] [Google Scholar]
- 8.Quinn CT, Lee NJ, Shull EP, Ahmad N, Rogers ZR, Buchanan GR. Prediction of adverse outcomes in children with sickle cell anemia: a study of the Dallas Newborn Cohort. Blood. 2008;111:544–8. doi: 10.1182/blood-2007-07-100719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panepinto JA, Brousseau DC, Hillery CA, Scott JP. Variation in hospitalizations and hospital length of stay in children with vaso-occlusive crises in sickle cell disease. Pediatr Blood Cancer. 2005;44:182–6. doi: 10.1002/pbc.20180. [DOI] [PubMed] [Google Scholar]
- 10.Minniti CP, Sable C, Campbell A, Rana S, Ensing G, Dham N, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica. 2009;94:340–7. doi: 10.3324/haematol.13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98:250–1. doi: 10.1182/blood.v98.1.250. [DOI] [PubMed] [Google Scholar]
- 12.Miller ST, Wright E, Abboud M, Berman B, Files B, Scher CD, et al. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. J Pediatr. 2001;139:785–9. doi: 10.1067/mpd.2001.119593. [DOI] [PubMed] [Google Scholar]
- 13.Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855–65. doi: 10.1056/NEJM200006223422502. [DOI] [PubMed] [Google Scholar]
- 14.Setty BN, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxaemia in sickle cell disease: biomarker modulation and relevance to pathophysiology. Lancet. 2003;362:1450–5. doi: 10.1016/S0140-6736(03)14689-2. [DOI] [PubMed] [Google Scholar]
- 15.Jennings JE, Ramkumar T, Mao J, Boyd J, Castro M, Field JJ, et al. Elevated urinary leukotriene E4 levels are associated with hospitalization for pain in children with sickle cell disease. Am J Hematol. 2008;83:640–3. doi: 10.1002/ajh.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Field JJ, DeBaun MR. Asthma and sickle cell disease: two distinct diseases or part of the same process? Hematology Am Soc Hematol Educ Program. 2009:45–53. doi: 10.1182/asheducation-2009.1.45. [DOI] [PubMed] [Google Scholar]
- 17.Field JJ, Strunk RC, Knight-Perry JE, Blinder MA, Townsend RR, DeBaun MR. Urinary cysteinyl leukotriene E4 significantly increases during pain in children and adults with sickle cell disease. Am J Hematol. 2009;84:231–3. doi: 10.1002/ajh.21370. [DOI] [PubMed] [Google Scholar]
- 18.Bernaudin F, Strunk RC, Kamdem A, Arnaud C, An P, Torres M, et al. Asthma is associated with acute chest syndrome, but not with an increased rate of hospitalization for pain among children in France with sickle cell anemia: a retrospective cohort study. Haematologica. 2008;93:1917–8. doi: 10.3324/haematol.13090. [DOI] [PubMed] [Google Scholar]
- 19.Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with acute chest syndrome and pain in children with sickle cell anemia. Blood. 2006;108:2923–7. doi: 10.1182/blood-2006-01-011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Embury SH. The interaction of coexistent alpha-thalassemia and sickle cell anemia: a model for the clinical and cellular results of diminished polymerization? Ann N Y Acad Sci. 1985;445:37–44. doi: 10.1111/j.1749-6632.1985.tb17173.x. [DOI] [PubMed] [Google Scholar]
- 21.Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992;79:2555–65. [PubMed] [Google Scholar]
- 22.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–22. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 23.Vasavda N, Badiger S, Rees D, Height S, Howard J, Thein SL. The presence of alpha-thalassaemia trait blunts the response to hydroxycarbamide in patients with sickle cell disease. Br J Haematol. 2008;143:589–92. doi: 10.1111/j.1365-2141.2008.07375.x. [DOI] [PubMed] [Google Scholar]
- 24.Ballas SK, Smith ED. Red blood cell changes during the evolution of the sickle cell painful crisis. Blood. 1992;79:2154–63. [PubMed] [Google Scholar]
- 25.Neely CL, Wajima T, Kraus AP, Diggs LW, Barreras L. Lactic acid dehydrogenase activity and plasma hemoglobin elevations in sickle cell disease. Am J Clin Pathol. 1969;52:167–9. doi: 10.1093/ajcp/52.2.167. [DOI] [PubMed] [Google Scholar]
- 26.Kato GJ, McGowan V, Machado RF, Little JA, Taylor Jt, Morris CR, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006;107:2279–85. doi: 10.1182/blood-2005-06-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ballas SK. Sickle cell anemia with few painful crises is characterized by decreased red cell deformability and increased number of dense cells. Am J Hematol. 1991;36:122–30. doi: 10.1002/ajh.2830360211. [DOI] [PubMed] [Google Scholar]
- 28.Alexander N, Higgs D, Dover G, Serjeant GR. Are there clinical phenotypes of homozygous sickle cell disease? Br J Haematol. 2004;126:606–11. doi: 10.1111/j.1365-2141.2004.05025.x. [DOI] [PubMed] [Google Scholar]
- 29.Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269–77. doi: 10.1038/nm1490. [DOI] [PubMed] [Google Scholar]
- 30.Charache S, Terrin ML, Moore RD, Dover GJ, McMahon RP, Barton FB, et al. Design of the multicenter study of hydroxyurea in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea. Control Clin Trials. 1995;16:432–46. doi: 10.1016/s0197-2456(95)00098-4. [DOI] [PubMed] [Google Scholar]