

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder in which the loss of dystrophin causes progressive degeneration of skeletal and cardiac muscle. Potential therapies that carry substantial risk, such as gene and cell-based approaches, must first be tested in animal models, notably the mdx mouse and several dystrophin-deficient breeds of dogs, including golden retriever muscular dystrophy (GRMD). Affected dogs have a more severe phenotype, in keeping with that of DMD, so may better predict disease pathogenesis and treatment efficacy. We and others have developed various phenotypic tests to characterize disease progression in the GRMD model. These biomarkers range from measures of strength and joint contractures to magnetic resonance imaging. Some of these tests are routinely used in clinical veterinary practice, while others require specialized equipment and expertise. By comparing serial measurements from treated and untreated groups, one can document improvement or delayed progression of disease. Potential treatments for DMD may be broadly categorized as molecular, cellular, or pharmacologic. The GRMD model has increasingly been used to assess efficacy of a range of these therapies. While some of these studies have largely provided general proof-of-concept for the treatment under study, others have demonstrated efficacy using the biomarkers discussed. Importantly, just as symptoms in DMD vary among patients, GRMD dogs display remarkable phenotypic variation. While confounding statistical analysis in preclinical trials, this variation offers insight regarding the role that modifier genes play in disease pathogenesis. By correlating functional and mRNA profiling results, gene targets for therapy development can be identified.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder affecting approximately 1 of 3,500-7,500 newborn live human males (Cowan et al 1980) in whom absence of the protein dystrophin causes progressive degeneration of skeletal and cardiac muscle (Hoffman et al 1987). Becker muscular dystrophy (BMD), a less severe clinical form, occurs at approximately one tenth the frequency of DMD (Prior and Bridgeman 2005). BMD mutations have a similar incidence and distribution to those causing DMD but maintain the dystrophin reading frame, allowing for production of a truncated, partially-functional protein. DMD boys are typically confined to wheel chairs before 12 years of age, while those with BMD walk beyond 16 years (Malhotra et al 1988). Approximately two thirds of DMD gene mutations have historically been attributed to large (> 1 exon) deletions (Wulff et al 1989; Prior and Bridgement 2005; Dent et al 2005). These deletions tend to predominate in one of two hotspots, namely the central rod domain around exons 44-53 (~ 80%) and, to a lesser extent (~ 20%), the 5’ terminus (Beggs, 1990; Prior and Bridgement 2005). A recent study in which point mutations identified by sequence analysis were likely over represented found an incidence of only 43% deletions, 11% duplications, and 46% point mutations (Flanigan et al 2009). While DMD classically demonstrates a strong family history, about one third of cases occur due to new mutations (Caskey et al 1980).
Potential therapies that carry substantial risk, such as gene and cell-based approaches, must ideally first be tested in relevant animal models, most notably the mdx mouse (Bulfield et al 1984; Gillis 1999) and several dystrophin-deficient breeds of dogs (Kornegay et al 1988; Cooper et al 1988; Schatzberg et al 1999; Jones et al 2004; Baltzer et al 2007; Smith et al 2011). Because clinical features in dogs are more severe than those in mdx mice, in keeping with the DMD phenotype, preclinical studies in the canine models might be more predictive of disease pathogenesis and treatment outcome. While numerous dog breeds with dystrophin-deficient muscular dystrophy have been characterized clinically, few have been studied at the molecular level. We have conducted extensive studies in a dystrophin-deficient form of muscular dystrophy originally characterized in golden retrievers (GRMD). A mRNA processing error in GRMD dogs results from a single base change in the 3’ consensus splice site of intron 6. Exon 7 is consequently skipped during mRNA processing (Sharp et al 1991). The resulting transcript predicts that the dystrophin reading frame is terminated within its N-terminal domain in exon 8.
DMD gene mutations have also been described in Rottweilers (nonsense mutation in exon 58) (Winand et al 1994 and Winand N, personal communication), German shorthaired pointers (large deletion, essentially amounting to a “dystrophin knock out”) (Schatzberg et al 1999), Welsh corgis (repetitive element-1 [LINE-1] insertion in intron 13) (Smith et al 2011), and Cavalier King Charles Spaniels (missense mutation in the 5’ donor splice site of exon 50 resulting in deletion of exon 50 in mRNA transcripts) (Walmsley et al 2010) (Table 1). Separately, we have identified three additional DMD gene mutations in the Cocker spaniel (deletion of four nucleotides in exon 65, with a reading frame shift predicting a premature stop codon at the site of the deletion), Tibetan terrier (a large deletion of exons 8-29), and Labrador retriever (184 nucleotide [pseudoexon] insertion between exon 19 and exon 20, which results in a premature stop codon at the next codon downstream of the insertion) (Larsen CA et al, unpublished). The Labrador retriever mutation presumably corresponds to one in an earlier report (Smith et al 2007).
Table 1.
DMD Gene Mutations in Dogs
| Breed | Mutation | Reference |
|---|---|---|
| Golden retriever | Splice site point mutation, with skipping of exon 7 in transcript | Sharp et al, 1992 |
| German shorthaired pointer | Deletion encompassing the entire dystrophin gene | Schatzberg et al, 1999 |
| Rottweiler | Nonsense point mutation in exon 58 | Winand et al, 1994 |
| Cavalier King Charles spaniel | Splice site point mutation, with skipping of exon 50 in transcript | Walmsley et al, 2010 |
| Pembroke Welsh corgi | Insertion in intron 13 | Smith et al, 2011 |
| Labrador retriever | Insertion in intron 19 | Smith et al, 2007; current paper |
| Cocker spaniel | Four nucleotide deletion in exon 65 | Current paper |
| Tibetan terrier | Deletion of exons 8-29 | Current paper |
Biomarkers for assessment of the natural history and response to treatment
Clinical course of DMD
The study of muscle diseases has evolved from a classical period in which a diagnosis was based on clinical and pathologic features, to a modern period when muscle biopsies were further characterized through histo- and cytochemical techniques, to the current era of molecular diagnosis (Engel and Osawa 2004; Meola 2005). With the advent of techniques such as multiplex polymerase chain reaction and Southern blotting, DMD can be diagnosed non-invasively without the need for muscle biopsy (Prior and Bridgeman 2005). As a result, baseline and follow-up pathologic data are not typically available to assess disease progression or response to therapy. Other surrogate biomarkers must be utilized to ensure that results of treatment trials are interpreted appropriately. Most DMD natural history studies have included measurements of muscle strength, joint contractures, and timed function tests. Results from these tests are used to track disease progression and offer insight on clinical milestones, such as the loss of ambulation and the need for ventilatory support. Both muscle weakness and joint contractures contribute to postural instability and ultimate loss of ambulation (Vignos 1963). Contracture and muscle strength scores generally correlate and deteriorate synchronously over time (Brooke et al 1983).
Clinical course of GRMD
Several studies have defined the characteristic clinical signs of GRMD (Kornegay et al 1988; Valentine et al 1988; Shimatsu et al 2005; Ambrósio et al 2009) (Figure 1). Signs occur soon after birth because affected pups are often ineffectual sucklers and must be supplemented. As a result, they typically exhibit stunted growth. By 6 weeks of age, the pelvic limbs may be advanced simultaneously and trismus is noted. Dogs subsequently develop a progressively more stilted gait, atrophy of particularly the truncal and temporalis muscles, a plantigrade stance due to hyperextension of the carpal joints and flexion at the tibiotarsal joints, excessive drooling suggesting pharyngeal muscle involvement, and initial lumbar kyphosis that progresses to lordosis. As with DMD, some muscles paradoxically undergo hypertrophy. Aspiration pneumonia may occur due to pharyngeal or esophageal muscle involvement. Cardiac failure due to cardiomyopathy can also occur.
Figure 1. Characteristic plantigrade stance in GRMD dog at 6 months of age.
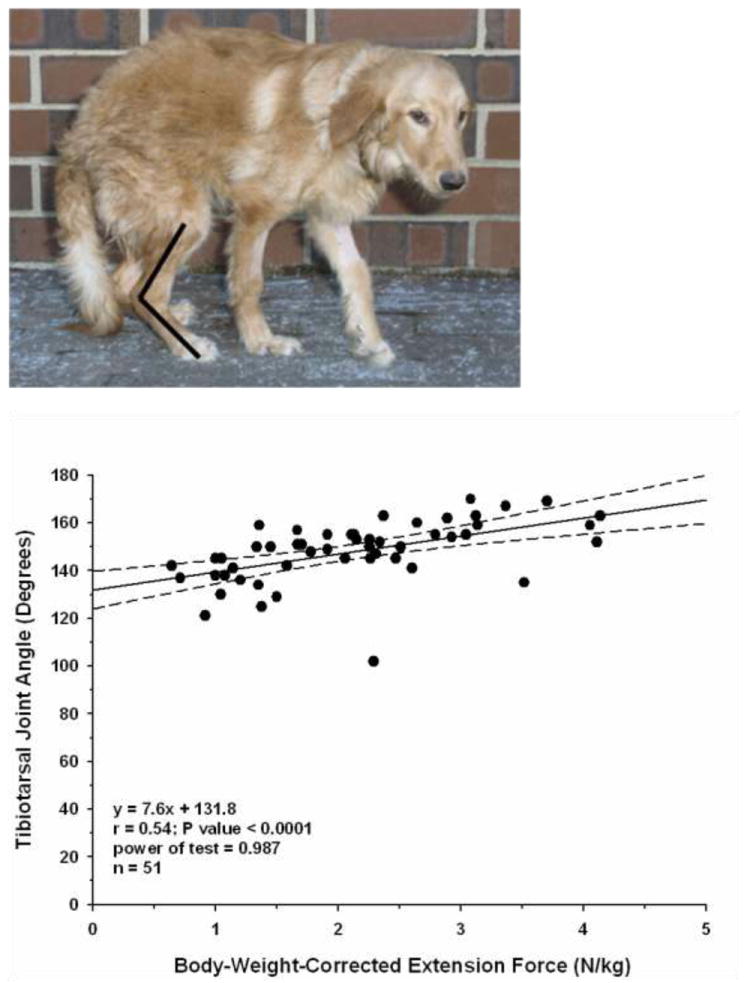
Pelvic limbs are shifted forward. The angle formed by the flexor surface of the tibiotarsal joint (black lines) is approximately 110° versus the 140° angle of normal standing dogs. There is hyperextension of the carpus.
Importantly, there can be marked variation in disease severity in GRMD, with some dogs dying soon after birth due to extreme respiratory compromise and others demonstrating a remarkably mild phenotype (Ambrósio et al 2008). In their early study of the clinical signs of GRMD, Valentine et al suggested that homozygous females and smaller affected dogs might have milder signs. Smaller beagle crosses with the GRMD mutation also may have a less severe phenotype (Shimatsu et al 2003; Yugeta et al 2006). Supporting a potential influence of either gender or body weight, female mdx mice have a milder phenotype (Salimena et al 2004) and one DMD boy with growth hormone deficiency had less severe signs (Zatz et al 1981). Effects due to gender or body weight could influence interpretation of GRMD preclinical studies in which both males and females of variable size are used.
With regard to body size, DMD treatment trials directed at reducing growth hormone have failed to support a relationship with disease severity (Griggs et al 1990). Moreover, in a separate GRMD study, lower growth hormone levels actually tended to correlate with a more severe phenotype (de Lima et al 2007). In reviewing our own phenotypic data, we found that larger GRMD dogs, if anything, have milder signs at 6 months of age. Tibiotarsal joint angle and body weight correlated directly, indicating that larger dogs had less severe postural instability. Larger GRMD dogs also had significantly smaller cranial sartorius muscles and trended towards lower flexion force values, both of which predict a less severe phenotype. This negative correlation between body size and phenotype likely reflects stunting rather than cause and effect. We believe the milder clinical signs initially reported in beagle crosses probably reflect the ameliorating effects of outbreeding, as Labrador retrievers with the GRMD mutation also had a less severe phenotype (Miyazato et al 2011). Similarly, in assessing the effects of gender in our own colony, results from phenotypic tests did not differ between homozygous female and heterozygous male GRMD dogs (Kornegay et al 2011). Thus, we have been unable to document an effect of either gender or body size in GRMD.
Biomarkers in DMD and GRMD
To better utilize the GRMD model in therapeutic trials, we and others have developed various phenotypic tests to objectively characterize disease progression. Results from functional tests tend to correlate with one another and with other clinicopathologic features (Figure 2). By comparing serial measurements from treated and untreated groups, one can document improvement or delayed progression of disease. In keeping with the phenotypic variation seen clinically, functional outcome values have also varied considerably, even among dogs within the same litter, suggesting that modifier genes significantly influence the phenotype (Kornegay et al 1994a, 1994b; Kornegay et al 1999). Importantly, phenotypic variation confounds data analysis, requiring larger group sizes to demonstrate significance. The effects of phenotypic variation on statistical analysis can be offset by establishing baseline outcome values prior to treatment so that each dog serves as its own control. With localized treatments, the effect of phenotypic variation is less of a concern because the untreated opposite limb can serve as the control.
Figure 2. Correlations among phenotypic tests in GRMD dogs.
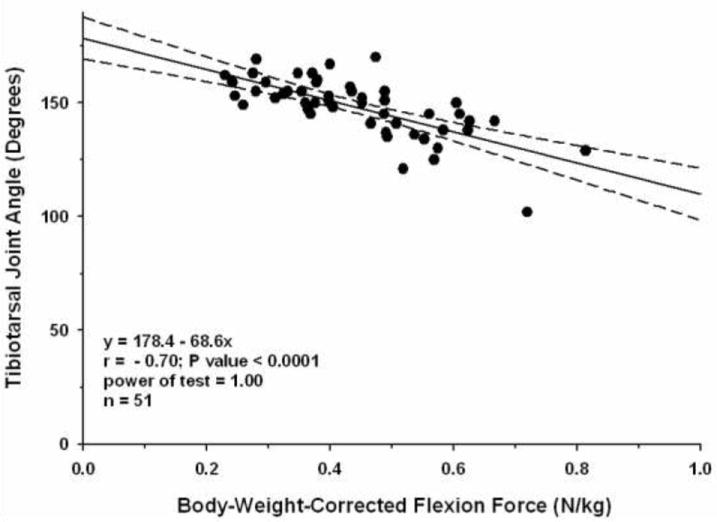
Scattergrams with regression lines drawn to show correlations between tibiotarsal joint angle (vertical axis in both) and body-weight-corrected tibiotarsal joint isometric tetanic extension (A) and flexion (B) force in 51 GRMD dogs at 6 months of age. Joint angle correlates strongly (p < 0.0001) with both parameters. The correlation is direct (r is positive) for extension force and inverse (r is negative) for flexion force.
Composite and group severity scores
Composite scores that take into account somewhat subjective numerical values assigned to particular clinical signs (Sampaolesi et al 2006; (Thibaud et al 2007); Rouger et al 2011) or overall disease phenotype (Ambrósio et al 2009) have been described and utilized in preclinical GRMD trials. With the composite score method, a variety of postural and gait changes were assessed and scored 0, 1, and 2 (normal, mild, and severe). The scores were added to indicate the degree of dysfunction (Sampaolesi et al 2006; Thibaud et al 2007). A later study used this same method but also assigned scores for functional categories (ptyalism, breathing, etc) not directly related to gait and posture (Rouger et al 2011). In another paper, dogs were subdivided into three functional groups (mild, moderate, and severe) based principally on postural changes (Ambrosio et al 2009). While these endpoints could have value in broadly categorizing general therapeutic trends, additional more objective tests should be utilized to document therapeutic gain.
Gait
Stephan Blot’s research group at Maisons-Alfort has utilized accelerometry to objectively characterize the slow, short-stepped, and swaying gait of adult GRMD dogs (Barthélémy et al 2009). Several quantitative indices, including total power, force and regularity of accelerations, stride length and speed (normalized for the dog’s height at the withers), stride frequency, and cranio-caudal power were significantly decreased, while medio-lateral power was increased, in GRMD versus normal dogs. Results tended to parallel those achieved with composite clinical scores discussed above. A subsequent longitudinal study showed that gait changes could be identified in GRMD dogs as early as 2 months of age and then evolved with age (Barthélémy et al 2011).
We have assessed two-dimensional gait kinematics of the stifle and tibiotarsal joints in adult GRMD dogs (Marsh et al 2010). They walked significantly slower and had more extended stifle and less flexed tibiotarsal joints than normal dogs; range of motion was comparable in the stifle but reduced in the tarsus. These changes should be interpreted in light of the joint angle data collected at 6 months of age (discussed below). While the finding of greater extension of the stifle is consistent with these data, the tibiotarsal joints of GRMD dogs typically are more (rather than less) flexed. This discrepancy may relate to the differing ages and phenotype of the two groups, as the kinematic data came from older, mildly-affected dogs.
The 6-minute-walk test has become a standard outcome parameter in DMD patients (MacDonald et al 2010) and has also been used to assess dogs with heart disease (Boddy et al 2004). In their study of accelerometry, Barthélémy et al indicated that GRMD dogs had difficulty completing the test. Our group has no experience with the 6-minute walk test.
Joint angles
We previously reported that 6-month-old GRMD dogs have abnormally acute (contracted) tibiotarsal joint angles while positioned in dorsal recumbency for force measurements (Kornegay et al 1994a, 1994b). Other investigators have subsequently described methods to measure joint angles at maximal flexion and extension in normal dogs (Jaegger et al 2002; Nicholson et al 2007). The method of Jaegger et al is now utilized to measure pelvic limb joint angles for ongoing natural history and preclinical trials in our laboratory. Based on our preliminary natural history findings collected at 6 months of age, GRMD dogs tend to have more restricted maximal flexion of the hip joint (~ 60° normal vs. ~ 80° GRMD), increased maximal stifle extension (~145° normal vs. ~ 155° GRMD), and more acute maximal tibiotarsal flexion (~ 45° normal vs. ~ 35° GRMD). To objectively characterize the cranioventral shift of the pelvis typically seen in GRMD dogs, we also measure the angle formed by two lines extending cranially from the tuber ischium, one drawn parallel to the lumbar spine and the other extending to the midpoint of the tuber coxae. Consistent with a previous report in which the pelvis was shown to shift into essentially a vertical position in some severely-affected dogs (Brumitt et al 2006), this angle has been larger in GRMD (~50°) versus normal (~ 35°) dogs at 6 months of age. While these pelvic limb joint angle data are preliminary and correlations have not yet been completed, we believe proximal joint and postural changes may contribute to the characteristic plantigrade tarsal stance in GRMD, just as relative sparing of proximal flexor muscles plays a role in distal limb flexor contractures in DMD (Vignos and Archibald 1960; Siegel et al 1968). Indeed, cranial sartorius circumference corrected for body weight correlates negatively with tibiotarsal joint angle, as measured by our original method (Kornegay et al 2003). This suggests that the hypertrophied muscle might play a role analogous to iliotibial band tightness in DMD (Rideau 1984).
Force/Torque measurements
We have demonstrated that GRMD dogs have decreased strength of individual (Kornegay et al 1994a) and grouped (Kornegay et al 1999) muscles. In our first study, tension was measured longitudinally in the peroneus longus, using a tendon suture to secure the muscle to a transducer. Tension was clearly lower than normal at 3 months of age but approached normal values by 6 months (Kornegay et al 1994a). Subsequent studies have focused principally on measurement of torque generated by the tibiotarsal joint (Kornegay et al 1999). For initial studies, force values were not corrected for the lever arm (metatarsus) length, and were reported in Newtons. We now multiply the force value by the length of the metatarsus (Newton-meters) (torque). The peroneal and tibial nerves are stimulated percutaneously so that the paw pulls (peroneal nerve, flexion) or pushes against (tibial nerve, extension) a lever interfaced with a force transducer. In our initial study, force values were measured at 3, 4.5, 6, and 12 months of age. Absolute and body-weight-corrected GRMD twitch and tetanic force values were lower than normal at all ages (P<0.01 for most). However, tibiotarsal flexion and extension were differentially affected. Flexion values were especially low at 3 months, whereas extension was affected more at later ages. Several other GRMD findings differed from normal. The twitch/tetany ratio was generally lower; post-tetanic potentiation for flexion values was less marked; and extension relaxation and contraction times were longer. The consistency of GRMD values was studied to determine which measurements would be most useful in evaluating treatment outcome. Standard deviation was proportionally greater for GRMD versus normal recordings. More consistent values were seen for tetany versus twitch and for flexion versus extension. Left and right limb tetanic flexion values did not differ in GRMD; extension values were more variable. These results suggested that measurement of tibiotarsal tetanic flexion force should be most useful to document therapeutic benefit in GRMD dogs. Groups of 15 and five would be necessary to demonstrate differences of 0.2 and 0.4 in the means of treated and untreated GRMD dogs at 6 months of age, with associated powers of 0.824 and 0.856, respectively (Sigma Stat, Jandel Scientific, 2591 Kerner Blvd., San Rafael, CA, USA). Values for extension force were more variable, suggesting that larger group sizes would be required to demonstrate significance. However, extension force was used as an outcome parameter to show that electromyographic (EMG) versus anatomic guided injection of botulinum toxin achieved an ~ 20-25% greater effect (p < 0.05) in three normal beagle dogs (Childers et al 1998). We have also demonstrated therapeutic benefit using both increased extension (~ 60%) and paradoxical decreased flexion (~ 40%) (both p < 0.05) with prednisone using a group size of six GRMD dogs (Liu et al 2004).
Eccentric contraction decrement
Dystrophin serves to buttress the muscle cell membrane. In the absence of dystrophin, the membrane is prone to tearing during minimal exercise. Dystrophic muscles are particularly prone to injury subsequent to eccentric (lengthening) contractions (Edwards et al 1983). As an example, mdx mice have normal or slightly-higher absolute force measurements but demonstrate greater-than-normal force decrement after eccentric muscle contractions (Moens et al 1994). In an initial study of GRMD dogs, we induced eccentric contractions in flexor muscles of the cranial tibial compartment of the pelvic limb by stimulating the sciatic trunk in the mid-thigh area (Childers et al 2002). This caused contraction of both the tibiotarsal joint flexors and extensors. Because the extensors are more powerful, eccentric (lengthening) contractions were induced in the flexors. More recently, we have developed a technique whereby the common peroneal nerve is stimulated while simultaneously extending the tibiotarsal joint with a servomotor (Aurora Scientific) coupled to a lever arm (Tegler et al 2011; Childers et al 2011) (Figure 3). Movement of the lever arm is controlled by use of a computer and customized LabView software (Aurora Scientific). Eccentric contractions are induced using square wave pulses of 100 μsec duration in a tetanic run for 700 ms at a frequency of 50 Hz. The contraction is held isometric at Lo for the first 500 ms. For the final 200 ms, the muscles of the cranial tibial compartment are stretched by the servomotor at 0.5 Lo/s, such that the muscles are displaced 0.1 Lo. Thus, the muscles of the cranial tibial compartment are repeatedly stretched to induce mechanical damage. Three sets of 10 stretches are performed. Each individual stretch is separated by 5 seconds and a four minute rest is provided between the three sets. Contraction-induced injury is quantified by the force (torque) deficit (Fd) using the following equation: Fd = (Po before stretch - Po after stretch / Po before stretch) × 100. We have shown that the decrement in normal dogs after 30 stretches does not exceed ~ 25%, while that of GRMD dogs can be as high as ~ 85% (Tegler et al 2011). The sequence of decrement over the groups of contractions needs to be more critically evaluated to determine the optimal point where normal and GRMD dogs can be distinguished.
Figure 3. Eccentric contraction decrement.
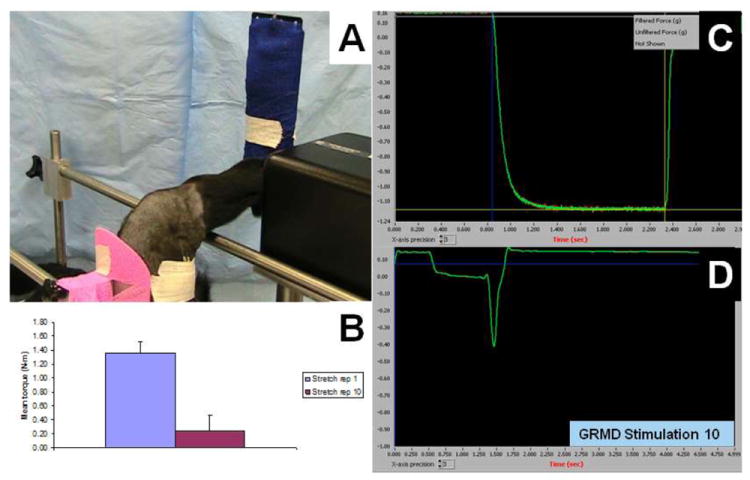
A. Left pelvic limb of a 6-month-old GRMD dog immobilized in a stereotactic frame, with needles (covered by tape) positioned to stimulate the peroneal nerve. B. Histogram showing mean torque (N-m) generated by tetanic tibiotarsal joint flexion during a series of 10 tetanic contractions. At the conclusion of each tetanic contraction, these maximally-stimulated cranial tibial compartment muscles were forcibly stretched by the servomotor as the tibiotarsal joint was extended a further 30 degrees (see D). The mean value of the initial contractions (blue bar) was ~ 1.4 N-m, while the tenth contraction (red bar) was ~ 0.2 N-m, representing an 85% decrement. C. The characteristic tetanic mechanical potential produced by tibiotarsal joint flexion is illustrated. D. The tenth single tetanic mechanical potential of the series is followed by a sharp further deflection due to the eccentric contraction induced by the servomotor, with an immediate return to baseline.
Cardiac Evaluation
Specific mutations in the dystrophin gene cause distinct cardiac syndromes in DMD (Cox et al 1997; Muntoni 2003; Jefferies et al 2005; Connuck et al 2008), BMD (Nigro G et al 1995; Melacini et al 1996; Saito et al 1996; Holloway et al 2008), and X-linked dilated cardiomyopathy (XLDCM) (Ferlini et al 1999). The severity of cardiac dysfunction in these conditions varies from subclinical disease to fatal congestive heart failure. There tends to be an inverse relationship between skeletal and cardiac muscle involvement (Saito et al 1996; Connuck et al 2008). DMD and BMD carriers may also have cardiomyopthy (Hoogerwaard et al 1999; Holloway et al 2008).
Electrocardiographic (Moise et al 1991; Yugeta et al 2006), echocardiographic (Moise et al 1991; Chetboul et al, 2004; Yugeta et al 2006), radionuclide angiographic (Devaux et al 1993), cardiac MRI (Kellman et al, 2009), and cardiac pathologic (Valentine et al 1989a) studies have been completed in dogs with GRMD. Additional longitudinal studies have been completed in dogs expressing both the GRMD and Labrador retriever insertion mutations (Fine et al 2011). Given that the dystrophin gene mutation can influence cardiac phenotype (Jefferies et al 2005), it’s not clear whether changes in these dogs mirror those of GRMD. GRMD dogs have ventricular arrhythmias and increased Q/R ratios due to characteristic deep Q waves on ECG evaluation (Moise et al 1991; Yugeta et al 2006); evidence of ventricular dilation and decreased fractional shortening, together with increased echogenicity that corresponds to mineralization at necropsy, with two dimensional and M-mode echocardiography (Moise et al 1991; Yugeta et al 2006); decreased radial and longitudinal left ventricular motions with tissue Doppler imaging (Chetboul et al, 2004); and myocardial fibrosis and mineralization on pathologic evaluation as early as 6.5 months of age (Valentine et al, 1989a). Electrocardiographic changes are seen at 6 months and progress with age (Moise et al 1991; Yugeta et al 2006). Ventricular measurements and shortening fraction determined with two dimensional and M-mode echocardiography are usually within the normal range over the first two years of life (Moise et al 1991; Yugeta et al 2006). The original echocardiographic studies done by Moise et al identified areas of increased echogenicity within the posterobasal left ventricular wall in 6-month-old GRMD dogs, with progression over time to the apex of the heart (Moise et al 1991). Areas of increased echogenicity were not seen in a later study of 6- to 21-month-old GRMD dogs crossed to beagles, (Yugeta et al 2006) in keeping with a trend for outbreeding to lessen severity of the GRMD phenotype (Kornegay et al, 2011). On pathologic evaluation, fibrosis is most pronounced in the left ventricular papillary muscle and apical left ventricular free wall (Valentine et al 1989a). Fatty deposition in seen in older dogs. Mineralization appears to be more pronounced than in DMD. Analogous ECG and echocardiographic changes progressed over the course of 3, 6, and 12 month evaluations in the GRMD-Labrador retriever muscular dystrophy crossbred dogs, suggesting that such markers can be used in preclinical trials (Fine et al 2011).
Working with investigators at NHLBI, we have completed preliminary cardiac MRI studies on two GRMD dogs and a Welsh corgi with the intron 13 insertion mutation (Kellman et al 2009). These studies focused on defining the degree of fibrosis and fat deposition within the myocardium using multi-echo Dixon methods both pre- and post-late gadolinium enhancement (LGE) and a segmented phase sensitive inversion recovery (PSIR) turbo FLASH sequence. Retrogated cardiac cine true-fast imaging with a steady-state precession (FISP) sequence was used to assess left ventricular function. Intramural fat was clearly seen with pre-contrast, fat-separated images. Corresponding hyper-enhanced lesions were noted with LGE. Intramural fibrosis, distinguished from fat on the LGE water phase image, was also evident. Left ventricular ejection fractions were reduced in the two GRMD dogs (22% and 26%) and normal in the Welsh corgi (53%), approximating values seen with echocardiography (Table 2).
Table 2.
Cardiac Findings in GRMD Dogs
| Dog | Age | Physical | ECG Findings | Echo Findings | Diagnosis | Treatment |
|---|---|---|---|---|---|---|
| 11 | 9 Y | III/VI L apical murmur | Sinus arrhythmia; QRS 65 ms | LVD, LAE, hyperechoic areas, mitral regurgitation; FS 21.9%; EF 44.5% | DCM | Enalapril, atenolol, fish oil, taurine, carnitine, |
| 2 | 4 Y | III/VI L Apical Murmur | Sinus arrhythmia, deep Q waves, QRS 70 ms, left-sided VPCs, fusion beats | LVE, LAE, heterogeneous myocardium, FS = 22.4%., EF = 44.4% | DCM | EnalApril, atenolol, fish oil, taurine, carnitine |
| 3 | 2 Y | No murmur | LVEDD 4.13 cm, LA 2.17 cm, hyperechoic area IVS, normal function (FS 38%, EF 68%) | Normal | None | |
| 4 | 3 Y | No Murmur | Sinus arrhythmia, Q waves present | LVD, LVEDD 4.23 cm, FS 15%, EF 33%, mottled heterogeneous myocardium | DCM | Enalapril, Lasix (taper), atenolol bridge with pimobendan |
| 5 | 5 Y | II/VI L Apical Murmur | Deep Q waves | Normal LVEDD and LA, FS 15.7%, EF 33.8%, ESVI 53 ml/m2, mitral regurgitation | DCM | Enalapril, |
| 6 | 22 M | No Murmur | Deep Q waves, R wave 2.9 mV | Normal LVEDD and LA. FS 14%, EF 30% | DCM | Enalapril, atenolol |
| 7 | 13 M | No Murmur | Normal | Normal, EF 41%, EF 73.5% | Normal | |
| 8 | 7 M | No murmur | Q wave 0.4 mV | LVE DD 2.8, LVESD 2.03 cm, LA 1.84 cm, FS 27.6%, EF 55.4% | Low norm systolic function | None |
| 92 | 7 Y | III/VI L Apical Murmur | Sinus rhythm, deep Q waves, QRS 80 ms | LVD, LVEDD 4.85 cm, mitral regurg, LAE 2.42 cm, FS 10%, EF 21% | DCM; CHF, pneumonia | Enalapril, Lasix, pimobendan, clavamox |
| 101,3 | 9 Y | No murmur | Sinus arrhythmia, deep Q waves | Normal LVEDD, FS 37%, EF 68%, Normal EDVI and ESVI | Mild DMD | None |
| 11 | 2 Y | No murmur | Sinus arrhythmia; deep Q waves | LVD, LVEDD 4.3 cm, normal LA 2.06 cm; hyperechoic areas; Normal systolic function - FS 31%, EF 60% | Abnormal structure; normal function | None |
| 124 | 9 Y | No murmur | Sinus arrhythmia; rare supraventricular complexes (SVCs); deep Q waves | LVEDD 2.0 cm, LA 0.57 cm, FS 39%, EF 72%, mild LVH 0.9 cm. | Near normal structure/function; rare SVCs | None |
Abbreviations: DCM – Dilated cardiomyopathy; LVD – left ventricular dilation; LAE – left atrial enlargement; LVE – left ventricular enlargement; LVEDD – left ventricular end-diastolic diameter; FS – left ventricular fractional shortening; EF – left ventricular ejection fraction; EDVI – end-diastolic volume index; ESVI – end-systolic volume index; SVCs – supraventricular contractions
Littermates
The dog developed acute dyspnea; thoracic radiographs showed an alveolar pattern most consistent with aspiration pneumonia; the dog died and on necropsy had cardiomegaly consistent with DCM and pneumonia
Thoracic radiographs – Mild LAE, pulmonary venous distension, no pulmonary edema.
Welsh corgi with intronic insertion (Smith et al 2011)
We have conducted cardiac evaluations, including ECG and echocardiography, on 11 GRMD dogs ranging from 7 months to 9 years of age at North Carolina State University (Kornegay JN et al, unpublished) (Table 2). Most were breeding males without clinical heart disease; nine had abnormalities on ECG and/or echocardiography (Figure 4). A diagnosis of dilated cardiomyopathy (DCM) was made in seven of them. Several dogs had systolic murmurs best heard in the left apical area. Sinus arrhythmia, occasional premature ventricular contractions, prolonged (> 60 ms) QRS complexes, and deep Q waves were noted on ECG. Structural and functional changes were seen with echocardiography. In particular, the myocardium was mottled, consistent with fatty deposition and fibrosis, and there was cardiomegaly, as evidenced by chamber dilation/enlargement. Functional indices, such as fractional shortening and ejection fraction, were often decreased. Two of the dogs had mitral regurgitation. Several of the dogs with DCM have been treated with ACE inhibitors (enalapril), β blockers (atenolol), and/or phosphodiesterase inhibitors (pimobendan).
Figure 4. GRMD cardiac studies.
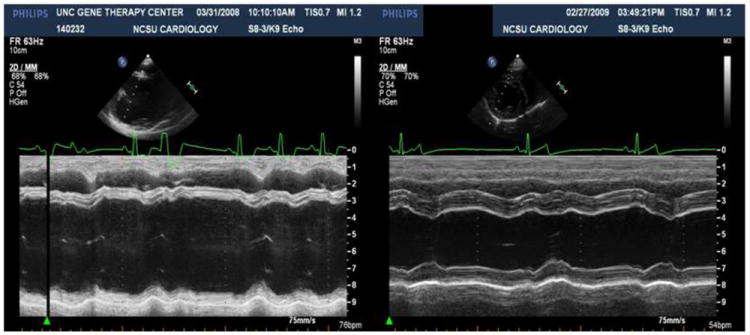
M-mode echocardiogram from a 4-year-old, male GRMD dog (left; Dog 2 in Table 2) showing a marked reduction in ventricular contractility at systole compared to a normal dog (right). Measurements from M-mode study for GRMD dog for Diastole/Systole: IVS 0.55 cm/0.88 cm; LV 5.75 cm/4.46 cm; LVPW 0.68 cm/0.86 cm; LV Vol 163.1 ml/90.6 ml. LV FS = 22.4%. LV EF = 44.4%.
We saw acute death, presumably due to an arrhythmia, in a GRMD carrier that had dramatic myocardial fibrosis at necropsy. This prompted us to complete a prospective study of cardiac function, including Holter monitoring and necropsy of a group of GRMD carriers. Some dogs had dramatic ventricular arrhythmias and all had myocardial fibrosis at necropsy (Kane et al submitted).
Cardiac pathologic studies have been performed on a number of GRMD dogs from our colony that died or were euthanized. Gross and histologic findings have been consistent with those described previously (Valentine et al, 1989a). Cardiomegaly, due particularly to left ventricular dilation, is seen grossly. Mineralization and focal to generalized fibrosis are present on histologic evaluation.
Skeletal muscle magnetic resonance imaging (MRI)
MRI has been used increasingly to provide meaningful data on the natural history and response to therapy of a number of diseases, including DMD (Lamminen 1990; Liu et al 1993; Marden et al 2005; Lovitt et al 2006). Principal MRI changes in DMD include an increase in T2 and decrease in T1 relaxation times due to accumulation of fat in affected muscles and an associated increase in whole body fat and decrease in muscle mass. Objective grading systems allow data to be compared over the course of the disease (Lamminen 1990; Liu et al 1993). Results from these grading systems have correlated with those of clinical functional tests. One serial study suggested that MRI is more sensitive than functional tests in predicting disease progression (Liu et al 1993). MRI has also been used to monitor DMD disease progression in treatment trials (Karpati et al 1993; Miller et al 1997).
The potential role of MRI as a biomarker in GRMD has been reported in both natural history and preclinical papers. In the first study, the thoracic limbs of 2-month-old GRMD and normal dogs were scanned at 4 T (Thibaud et al 2007). GRMD dogs had an abnormally-high T2-weighted/T1-weighted signal ratio, greater T2-weighted image heterogeneity, and more pronounced signal enhancement post-contrast. An additional study of 3-month-old GRMD dogs showed increased signal intensity on T2-weighted images in which the fat signal was suppressed, increased T2 values, and greater enhancement with gadolinium, all consistent with inflammation associated with early necrosis (Kobayashi et al 2009). T2 signal was decreased in GRMD dogs treated systemically with morpholinos compared to age-matched untreated dogs, supporting the notion that MRI can be used to track improvement in longitudinal studies (Yokota et al 2009). An additional study showed that increased signal on T2-weighted images could be used to track sites of AAV-micro-dystrophin construct injection (Wang et al 2010). As discussed further below, we have also seen increased signal intensity in muscles associated with an apparent innate immune response in dogs treated with AAV-9 and a codon-optimized human mini-dystrophin (Kornegay et al 2010). Moreover, MRI has been used to track the distribution of fluid in dogs treated with AAV-mini-dystrophin constructs delivered by regional limb delivery.
We have completed MRI studies in ~ 50 GRMD dogs (Wang et al 2011; Kornegay JN et al, unpublished). Some of these have been longitudinal studies and others have been at single time points. Consistent with published studies, signal-intense lesions presumably corresponding to fluid accumulation in necrotic lesions have been seen on fat suppressed, T2-weighted images in younger dogs, while increased fat deposition has been seen at later ages (Figure 5). The severity of these changes has varied among muscles. We have been particularly interested in assessing volumetric and T2 signal values longitudinally in GRMD dogs between 3 and 12 months of age, as this time frame has most commonly been used for preclinical studies by our group and others. Water and fat values are calculated separately on T2-weighted images, so as to gain insight on their relative contribution to the T2 signal (data not reported here).
Figure 5. GRMD MRI Studies.
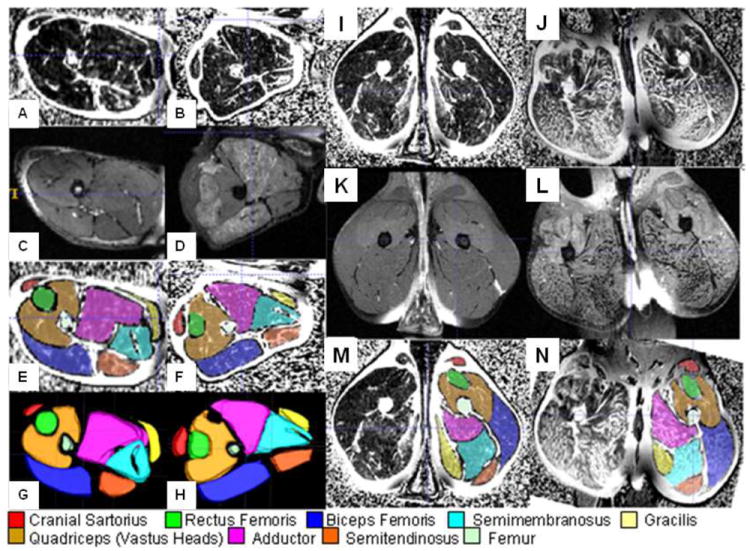
The four panels from left to right are MRI images from a 2-month-old GRMD carrier (A,C,E,G) and affected littermate (B,D,F,H) and 5-year-old GRMD carrier (I,K,M) and affected dog (J,L,N). A, B, I, and J are TSE-fat percentage and C, D, K, and L are TSE-fat saturation. Transverse sections of muscle have been segmented in E, F, M, and N for region-of-interest measurements and are shown in three dimensions in G and H (2-month-old dogs only). Note, particularly, the signal-intense lesions in several muscles in D and J, representing fluid accumulation, acutely, and fatty change, chronically, respectively. Signal-intense lesions seen in J reverse with fat saturation in L. Segmentation was done using ITK-SNAP (http://www.itksnap.org/pmwiki/pmwiki.php) (Yushkevich et al 2006).
Preliminary results have been evaluated in five GRMD and 10 age-matched normal dogs in which six proximal pelvic limb muscles (semitendinosus, rectus femoris, cranial sartorius, adductor magnus, gracilis, and biceps femoris) were evaluated as part of an ongoing natural history study. T2 values were higher than normal in each of the six GRMD muscles at all ages and these values declined in both groups from 3 to the 9-12 month age group (Table 3; Figure 6). Body-weight-corrected muscle volumes varied, with the GRMD cranial sartorius being markedly larger than normal at 6 and 9-12 months (Table 4), as we have previously reported (Kornegay et al 2003). The GRMD semitendinosus muscle was also larger, while the rectus femoris and biceps femoris were smaller. Body-weight-corrected volumetric values for the gracilis and adductor muscles were comparable in GRMD versus normal dogs.
Table 3.
Mean T2 Map Intensity of Selected Muscles
| GRMD Dogs | Normal Littermates | |||||
|---|---|---|---|---|---|---|
| 3 month | 6 month | 9 month | 3 month | 6 month | 9 month | |
| Cranial Sartorius | 60.18 | 49.43 | 40.38 | 47.21 | 41.72 | 37.87 |
| Rectus Femoris | 63.01 | 51.29 | 45.12 | 41.43 | 36.75 | 31.08 |
| Semitendinosus | 61.47 | 52.36 | 46.80 | 42.83 | 39.22 | 33.31 |
| Biceps Femoris | 56.34 | 51.89 | 46.80 | 45.62 | 39.85 | 33.31 |
| Gracilis | 62.89 | 52.53 | 45.98 | 44.92 | 39.66 | 34.29 |
| Adductor Magnus | 58.33 | 50.40 | 46.69 | 47.07 | 40.23 | 34.22 |
Figure 6. T2 maps from GRMD and normal dogs.
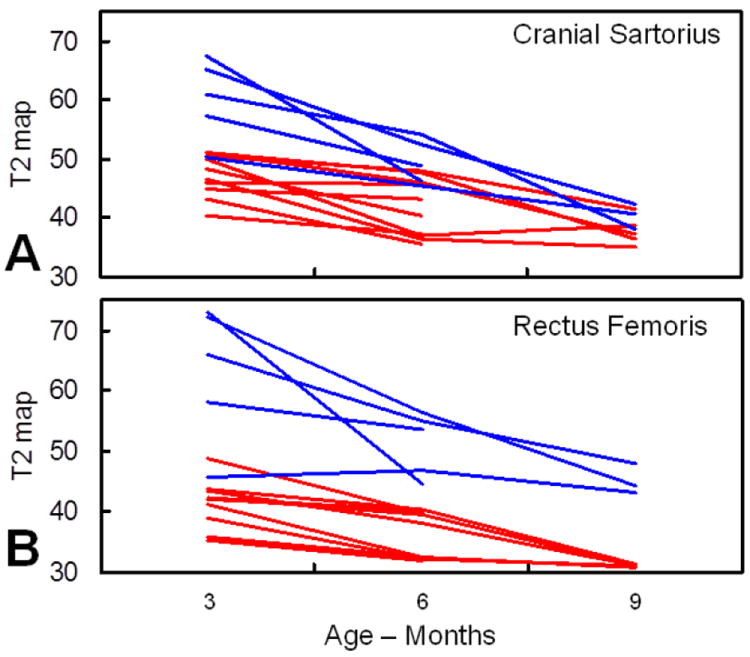
Comparison of the mean intensity of the cranial sartorius (A) and rectus femoris (B) muscles in T2 map of 5 GRMD (blue lines) and 10 normal littermates (red lines), at 3, 6, and 9-12 months of age. GRMD values tend to be higher at each age in both muscles but those for the cranial sartorius overlap with normal at 6 months and even more so at 9-12 months.
Table 4.
Mean Body-Weight Corrected Volumes of Selected Muscles
| GRMD Dogs (mm3/kg) | Normal Littermates (mm3/kg) | |||||
|---|---|---|---|---|---|---|
| 3 month | 6 month | 9 month | 3 month | 6 month | 9 month | |
| Cranial Sartorius | 505.10 | 1044.21 | 1633.38 | 412.27 | 537.84 | 716.77 |
| Rectus Femoris | 978.11 | 1356.97 | 1497.90 | 980.72 | 1559.34 | 1837.08 |
| Semitendinosus | 1931.42 | 2582.10 | 3237.04 | 1568.09 | 2356.96 | 2966.04 |
| Biceps Femoris | 3945.90 | 5623.97 | 6399.88 | 4112.48 | 6397.20 | 7925.80 |
| Gracilis | 1600.85 | 1742.43 | 2198.14 | 1230.59 | 1741.51 | 2142.67 |
| Adductor Magnus | 5374.01 | 8044.26 | 8000.80 | 5314.52 | 7478.24 | 8825.25 |
Electromyographic and single fiber studies
Electromyography (EMG) can be used to objectively characterize muscle in both natural history and treatment studies. As with other myopathies, DMD is characterized by small, polyphasic, short-duration motor unit potentials (MUPs), with associated spontaneous activity ranging from fibrillation potentials to complex repetitive discharges (CRDs) (Buchthal et al 1963; Desmedt and Borenstein 1976; Emeryk-Szajewska and Kopeć 2008). EMG was performed in the two original GRMD dogs studied by our group at 3, 5, 7, and 34 months of age (Kornegay et al 1988). There were persistent, spontaneous, high-frequency discharges of constant amplitude, with frequency components of up to 2000 Hz on power spectral analysis. In a subsequent study to which we contributed, seven GRMD dogs produced in the Cornell colony were evaluated between the ages of 6 weeks and 5.5 years of age (Valentine et al 1989b). Spontaneous activity, consisting primarily of CRDs, positive sharp waves, and polyphasic potentials, was found in all dogs, particularly after 10 weeks. Motor unit potentials were of short duration and polyphasic. Abnormalities were rare at 6 weeks but easily detected at 8 to 10 weeks. Taken together, these changes were consistent with those seen in DMD. However, the CRDs were much more prominent in affected dogs.
Details of MU architecture and function are not adequately quantified by conventional EMG. In contrast, quantitative EMG (Nandedkar et al 1995) allows one to map objective motor unit parameters over the natural course of disease or in response to treatment. Despite these diagnostic advantages, quantitative measures of the motor unit have not been assessed in animal models of muscular dystrophy and only on a limited basis in DMD patients. In an ongoing natural history study, we have performed concentric needle EMG and multi-motor unit potential (Multi-MUP) analysis on the cranial sartorius, cranial tibialis, gastrocnemius lateral head, and vastus lateralis muscles of a total of 10 GRMD and 15 normal dogs between 2 months and 4 years of age (Bhavaraju-Sanka R, Kornegay JN, and Howard JF, unpublished). Data collected included presence and severity of spontaneous activity and MUP amplitude, duration, turns, phases and polyphasicity. GRMD dogs exhibited widespread complex repetitive discharges at all ages that often made it hard to perform multi-MUP analysis. When data from all ages were compared, multi-MUP analysis showed reduced MUP duration and amplitude with smaller MUP areas in GRMD versus controls. In comparing data from three each GRMD and normal dogs at 2-3 and 9-12 months age, differences in MUP duration and area were evident at both ages, while amplitude normalized somewhat in the older dogs.
Single fiber EMG (SFEMG) action potentials can be recorded from individual muscle fibers (Hilton et al 1985; Sanders and Stalberg 1996). Two particular parameters, jitter and fiber density, offer insight into mechanisms contributing to neuromuscular disease. Jitter refers to the variable speed with which nerve impulses are transmitted across motor endplates and is typically expressed as the mean of consecutive differences (MCD) of interpotential intervals. Up to 40% of DMD patients have increased jitter, due presumably to abnormal motor endplates associated with diseased or regenerating muscle fibers (Sanders and Stalberg 1996; Hilton et al 1985). Fiber density is defined based on the number of individual myofiber action potentials recorded at one time with the SFEMG electrode. DMD patients have increased fiber density, most likely as a function of myofiber proliferation/regeneration in response to the dystrophic process (Hilton et al 1985; Sanders and Stalberg 1996). Building on an earlier study of SFEMG in normal dogs (Hopkins et al 1993), we have assessed stimulation (s)SFEMG in the peroneus longus and cranial tibialis muscles of most of the dogs included in the quantitative EMG studies (Figure 7). Dogs were anesthetized and ten to twenty fibers were recorded from each muscle and neuromuscular jitter (MCD) was measured. The affected dogs exhibited increased jitter in both muscles, with MCD values of ~ 30 versus ~ 15 msec in normal dogs. There was also more impulse blocking in the affected dogs. In addition, an increase in the fiber density estimate was inferred by the greater number of action potentials at each stimulation site [data not presented; see Figure 7].
Figure 7. Representative sSFEMG potentials from the peroneus longus muscle from normal (A) and GRMD (B) dogs.
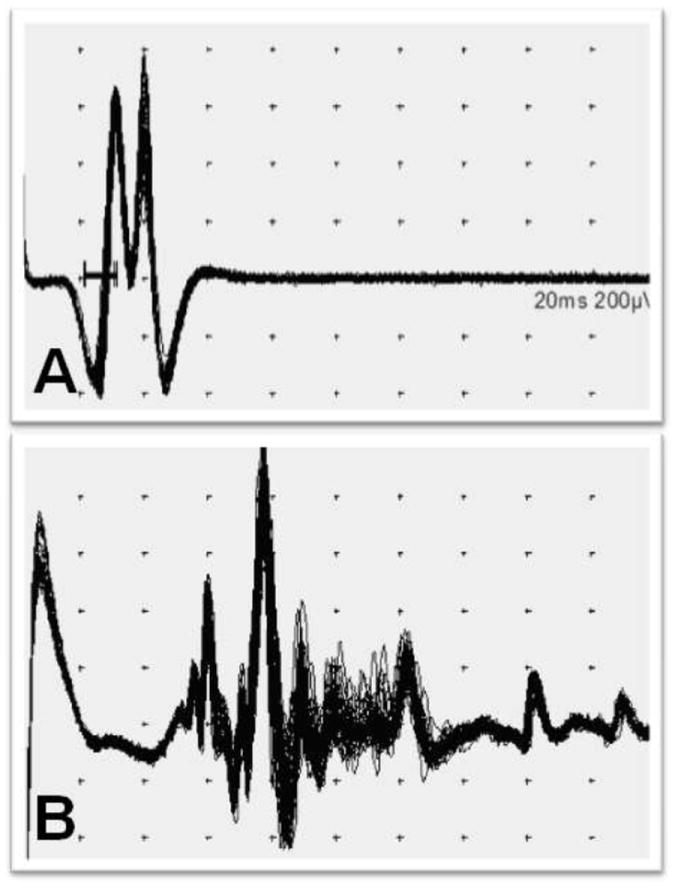
The normal dog has minimal neuromuscular jitter and only a few action potentials at the acquisition site, while the GRMD dog has increased neuromuscular jitter and multiple action potentials.
Pathologic studies
Gross and histopathologic lesions vary remarkably among dystrophin-deficient muscles (Kornegay et al 1988; Valentine et al 1990; Childers et al 2001; Nguyen et al 2002; Kornegay et al 2003). At the light microscopic level, there is a pattern of small group myofiber necrosis and regeneration, which is typical of dystrophin-deficient muscular dystrophy across species. Fiber size variation occurs due to the combined effects of individual myofiber hypertrophy and a marked increase in small regenerating fibers. Necrotic myofibers typically are in various states of degeneration, ranging from swollen, granular (so-called hyaline or hypercontracted) myofibers that stain intensely eosinophilic with H&E to fragmented fibers that are undergoing necrosis and mineralization (calcification), with an associated infiltrate of macrophages (myophagocytosis) and other mononuclear inflammatory cells. As necrotic material is removed by the monocyte-marcrophage system, only the basal lamina of affected myofibers may remain, resulting in empty sarcolemmal tubes. On longitudinal section, necrosis occurs segmentally along the length of the myofiber, in keeping with the domain of individual nuclei. Regenerating fibers tend to occur in clusters, often closely associated with areas of necrosis, and are small with prominent nuclei/nucleoli and basophilic cytoplasm. Sarcolemmal nuclei may be located centrally within the myofiber (so-called central or internal nuclei) rather than peripherally. On histochemical stains, type II fibers are preferentially affected, with an associated predominance of type 1 fibers and fiber type grouping (Kornegay et al 1988). Intermediate fiber types, apparently reflecting immaturity associated with regeneration, occur commonly in neonatal GRMD muscles (Nguyen et al 2002). In a prednisone preclinical trial in GRMD, we attributed reduced numbers of fetal-myosin positive fibers in treated dogs to a reduction in necrosis and accompanying decrease in demand for regeneration (Liu et al 2004) Electron microscopic changes evident in affected fibers include disruption of myofibrillar organization, dilatation of sarcoplasmic reticulum, increasedglycogen, and both hyperplasia and hypertrophy of mitochondria (Kornegay et al 1988; Valentine et al 1990).
The pattern of muscle involvement must be carefully considered in assessing functional and pathologic results from preclinical trials. As with DMD and the mdx mouse, the extraoccular muscles are largely spared in GRMD (Valentine et al 1990; Nguyen et al 2002). In sharp contrast, muscles that are used heavily in utero and early in life, such as the tongue, diaphragm, and limb flexors (including the sartorius, semitendinosus, and cranial tibialis), demonstrate acute necrosis (Valentine et al 1990; Nguyen et al 2002). Other muscles are spared early in life and develop lesions later. This delayed pattern of necrosis is more typical of extensor muscles, which presumably do not become severely affected until dogs begin walking and muscles such as the quadriceps femoris experience eccentric contractions. Interestingly, the rectus femoris responds differently than the other three heads of the quadriceps, as it shows severe early lesions, probably because of its origin from the pelvis and dual role as a hip flexor and stifle extensor. The long digital extensor of the pelvic limb also has a dual function as a digital extensor and tibiotarsal flexor. Unlike the cranial tibialis which is a strict tibiotarsal flexor and undergoes moderate (Nguyen et al 2002) to severe (Valentine et al 1990) necrosis in neonates, the long digital extensor was unaffected in one study of GRMD neonatal lesions (Nguyen et al 2002). Thus, again, the flexor function seemed to drive the pattern of early necrosis.
Muscles that undergo early necrosis may then regenerate and even hypertrophy. This pattern is perhaps best represented by the cranial sartorius. We have shown it seemingly recovers remarkably from the initial necrosis and then can reach two to three times normal size when corrected for body weight (Kornegay et al 2003). Based on morphometric histological studies, the cranial sartorius demonstrates true hypertrophy in dogs aged 4-10 months. Muscle is then replaced by fat and connective tissue, giving a pattern more in keeping with the pseudohypertrophy that has classically been described in DMD. Recent imaging studies in DMD have suggested that some muscles, including the sartorius (Marden et al 2005), are relatively spared or even hypertrophied. Moreover, the gastrocnemius, which has typically been thought to exhibit pseudohypertrophy, may initially undergo true hypertrophy in some patients (Cros et al 1989). We believe true hypertrophy of the cranial sartorius and other muscles may have clinical significance, in that imbalances between differentially-affected agonist and antagonist muscles can contribute to contractures (see earlier discussion under joint angles).
Use of the GRMD model in treatment development for DMD
Preclinical trials
Potential treatments for DMD may be broadly categorized as molecular, cellular, or pharmacologic (Chakkalakal et al 2005; Goyenvalle et al 2011). The two most common molecular approaches are gene therapy, whereby the dystrophin gene is introduced into muscle either locally or through the vasculature, generally through use of plasmids or viral vectors; and gene correction, which involves introduction of oligonucleotides (chimeric or antisense) to induce either inherent repair mechanisms or exon skipping to reestablish the correct nucleotide sequence (reading frame). With cell-based therapies, normal cells such as myoblasts or stem cells are transplanted into diseased muscle. Pharmacologic approaches do not deliver the defective gene and/or protein to the diseased muscle(s). Instead, specific pathogenetic mechanisms that contribute to the dystrophic phenotype are targeted. Examples include compounds that reduce inflammation (NF-κB inhibition), increase muscle mass (insulin-like growth factor, myostatin inhibition), read through stop codons in the defective dystrophin gene (aminoglycoside antibiotics), or increase production of utrophin (the autosomal homologue of dystrophin). Essentially all preclinical studies conducted thus far in dystrophin-deficient dogs have been completed in the GRMD model (Table 5).
Table 5.
Preclinical Treatment Trials in GRMD Dogs
| Plasmid and Vector-Based Gene Therapy | |
|---|---|
| Plasmid/full length dystrophin and mini-dystrophin | Howell et al 1998a |
| Adenovirus/mini-dystrophin | Howell et al 1998b |
| Adenovirus/utrophin | Cerletti et al 2003 |
| AAV/mini-dystrophin | Wang et al 2007a, b; Yuasa et al 2007; Kornegay et al 2010 |
| Gene Repair | |
| Chimeric oligonucleotides | Bartlett et al 2000 |
| Antisense oligonucleotides | Yokota et al 2009 |
| Cell Therapy | |
| Hematopoietic stem cells | Dell’Agnola et al 2004 |
| Mesoangioblasts | Sampaolesi et al 2006 |
| Dental pulp cells | Kerkis et al 2008 |
| Muscle stem cells | Rouger et al 2011 |
| Human adipose-derived mesenchymal stromal cells | Vieira et al 2011 |
| Umbilical cord mesenchymal stromal cells | Zucconi et al 2011 |
| Pharmacologic | |
| Prednisone | Liu et al 2004 |
| Membrane-sealing poloxamer | Townsend et al 2010 |
| AAV/myostatin peptide | Bish et al 2011 |
| Calpain inhibition | Childers et al in press |
Plasmid- and vector-based gene therapy
Howell et al showed that GRMD myofibers can be transduced for at least 14 days using plasmids containing full length dystrophin and mini-dystrophin cDNA (Howell et al 1998a). Adenovirus-mediated mini-dystrophin (Howell et al 1998b) and utrophin (Cerletti et al 2003) transfer have also been achieved in immunosuppressed GRMD dogs. In other GRMD studies, dystrophin minigene transfer with an adeno-associated virus (AAV) vector was limited by a marked immunologic response to viral capsid antigens (Wang et al 2007a) that could be blocked with brief immunosuppression (Wang et al 2007b). Others have suggested that the immune response occurs against components of the construct, including dystrophin itself (Yuasa et al 2007).
We have completed a series of studies of AAV-mini-dystrophin constructs in GRMD dogs to simultaneously extend mdx studies (Wang et al 2009) and clarify the source of the immune response in GRMD dogs (J Li, JR Bogan, DJ Bogan, JN Kornegay, and X Xiao, unpublished). Our initial studies focused on demonstrating the immunologic reaction to constructs. A total of 6 GRMD and 8 normal dogs were injected intramuscularly with AAV1 or 2 constructs containing the Lac-Z reporter gene or human mini-dystrophin gene. Preliminary data from direct muscle injection showed a greater immunologic reaction than that encountered in the mdx mouse. The degree of immunologic reaction was reduced when the canine versus human mini-dystrophin gene was used. These results indicated that direct intramuscular injection of AAV1 and 2 vectors caused a marked immune response not observed in mice, suggesting species differences to AAV-mediated local intramuscular gene transfer.
We have subsequently delivered similar AAV-based mini-dystrophin gene constructs via an afferent transvenular retrograde extravasation (ATVRX; regional limb) method for somatic gene transfer to muscles with favorable results. A total of 33 (20 normal and 13 GRMD) dogs have been injected using various constructs and perfusion volumes/rates. AAV-8 or 9 and canine mini-dystrophin have been used in the most recent studies (Li et al 2009). Widespread mini-dystrophin gene expression has been seen in muscles of the perfused limb without immunosuppression, establishing the utility of AAV-mediated regional limb therapy in GRMD. The distribution of the perfusion fluid has been mapped with MRI (Figure 8). Some of these dogs have also been evaluated with the functional biomarkers discussed above. Isometric tibiotarsal extension force was intermediate between GRMD and normal natural history values in three GRMD dogs treated with AAV-8 or 9-minidystrophin constructs. Values had also normalized somewhat in the contralateral limb, suggesting a systemic effect (see further below). However, the degree of eccentric contraction decrement did not improve in the treated GRMD dogs. The remarkable differences in cellular immunity between local intramuscular injection and vasculature-mediated regional limb gene transfer by AAV vectors suggest that the gene delivery route is an important factor to be considered for clinical studies. With these promising results, a parallel phase 1 safety study of regional limb delivery of saline was initiated in adult muscular dystrophy patients (Fan et al 2011). This study has demonstrated overall safety, with minimal side effects.
Figure 8. MRI 50 minutes after limb perfusion of an 11-week-old GRMD dog.
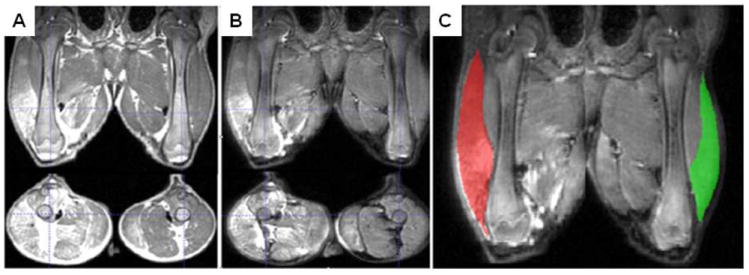
T2-weighted TSE MR images without (A) and with (B, C) fat saturation. Composite images have been created to orient the left and right limbs in the same plane. As a result, some proximal structures such as the colon are duplicated. Craniocaudal (top) and transverse (bottom) sections proximal to the stifle are seen in A and B. In each image, the perfused limb is on the left. Increased signal intensity is consistently seen in the perfused limb. Intensity could be either fat or fluid in A but is due to fluid alone in the fat saturation images in B and C. Intensity is greatest in the distal third of the femur and is particularly prominent in the biceps femoris muscle which is highlighted in C. In the fat saturation images in C, the perfused (red) and non-perfused (green) biceps femoris muscles were segmented for quantitative measurements. The volume and signal intensity of the biceps femoris were proportionally higher in the perfused versus non-perfused limb. Segmentation was done using ITK-SNAP (http://www.itksnap.org/pmwiki/pmwiki.php) (Yushkevich et al 2006).
Ultimately, gene therapy strategies must extend to systemic delivery to achieve widespread muscle transduction. Initial canine studies have utilized neonatal dogs in an effort to minimize the immune response and achieve widespread delivery. Working with the Duan laboratory, we demonstrated widespread muscle transduction of a human placental alkaline phosphatase reporter gene carried by an AAV-9 vector administered intravenously without immunosuppression (Yue et al 2008). Subsequently, in collaboration with Xiao Xiao, GRMD dogs given a single intravenous injection of an AAV9 vector carrying a human codon-optimized human mini-dystrophin gene showed generalized muscle transduction in ~15% to nearly 100% of myofibers (Kornegay et al 2010). However, these pups showed delayed growth and pelvic limb muscle atrophy and contractures, seemingly due to an innate immune response, exacerbated by the use of a high dose of codon-optimized human mini-dystrophin. Marked increased signal intensity, compatible with inflammation, was seen with T2-fat suppressed MRI in the vastus heads of the quadriceps femoris and adductor muscles of these dogs. Analogous selective involvement of the vastus heads, with sparing of the rectus femoris, is seen in humans with sporadic inclusion myositis (Phillips et al 2001). Similarly, dogs with Neospora caninum infection develop hyperextension of the stifle (knee) (genu recurvatum) secondary to quadriceps myositis and contractures (Knowler and Wheeler 1995). Surprisingly, with some of the older GRMD dogs receiving mini-dystrophin via regional limb delivery, there was dramatic body-wide mini-dystrophin gene expression, apparently due to leakage beyond the tourniquet (Li et al 2009). This prompted us to initiate studies of systemic AAV-mini-dystrophin gene delivery in GRMD dogs at a few months of age. Preliminary results have been promising, as widespread and persistent gene expression was seen in one dog treated with an AAV-9 mini-dystrophin construct.
Gene repair
Chimeric oligonucleotides were used to induce normal host cell mismatch repair mechanisms to correct the splice site mutation in a GRMD dog (Bartlett et al 2000). Antisense oligonucleotides (morpholino) given intravenously in three GRMD dogs (Japanese beagle cross) over a period of up to 5.5 months resulted in functional and pathologic improvement, plus reduced T2 signal intensity on MRI (Yokota et al 2009). A limited clinical trial of a mopholino-based exon skipping strategy in DMD patients showed modest dystrophin expression in muscle fibers (Cirak et al 2011).
Cell-based therapies
While much attention has shifted to viral based gene therapy, there continues to be considerable interest in cellular approaches, in part because of the need for cell replacement in more chronic cases. A variety of adult, fetal, and embryonic stem cells can potentially contribute to regeneration of diseased muscle. Myoblasts and their parent satellite cells were the obvious choice for initial studies. Partridge et al showed that donor myoblasts could fuse with host cells in a murine muscle injury model (Partridge et al 1978) and restore dystrophin expression in the mdx mouse (Partridge et al 1989). Human DMD clinical trials were ultimately conducted in the 1990’s (Tremblay et al 1993; Miller et al 1997). Follow-up studies of treated patients didn’t show functional gains, although some donor cells could be detected in the muscles (Gussoni et al 1997). Further clinical trials were halted. The limited success of these trials could be attributed to a high degree of initial cell death due presumably to the combined effects of poor blood supply to the injected cell bolus and immune rejection, plus limited migration of the surviving injected cells (Tremblay et al 1993; Fan et al 1996; Gussoni et al 1997; Smythe et al 2001; Partridge 2002; Skuk et al 2007).
We characterized canine myoblasts and conducted transplantation studies in the GRMD model during the 1990s (Prattis et al 1993; Nakagaki et al 1994a, 1994b). However, we were unable to achieve significant implantation of myoblasts in GRMD dogs. In the context of these studies, we developed an autologous myoblast transplantation model in which cells were labeled with fluorescent microspheres and injected in preirradiated 3-month-old, normal dogs as part of a cocktail that included the muscle toxin notexin and methylene blue. In subsequent studies at 7 and 24 days post-transplantation, large groups of injected cells formed fascicles and expressed ATPase (Figure 9), providing support for the general principle that myoblasts could implant and differentiate in dogs (Kornegay et al 1992).
Figure 9. Autologous myoblast transplantation model in normal dogs.
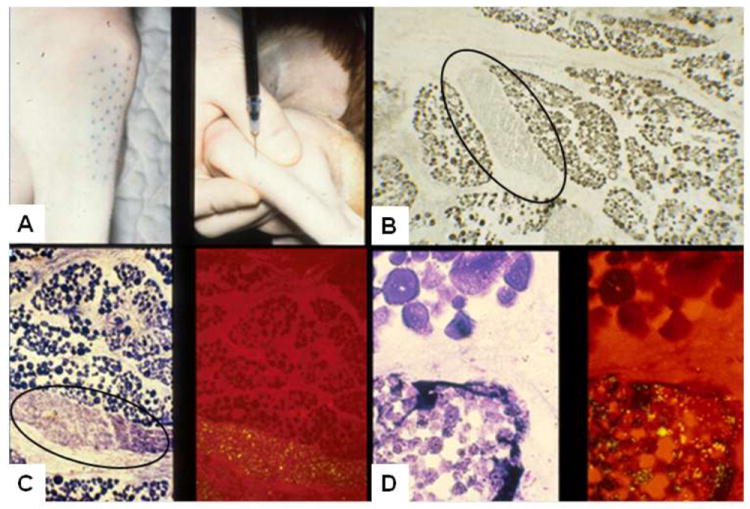
Cells were injected percutaneously (A, right); methylene blue identifies sites of injection (A, left). Large aggregates of cells containing fluorescent microspheres are forming fascicles (circles in B and C). Muscle fibers in these fascicles stain with ATPase (pH 9.4) (B) and toluidine blue (left image in C and D) and contain fluorescent microspheres (right image in C and D) at 24 days post-transplantation.
Bone marrow provides a paradigm for an accessible and pluripotent stem cell population. Despite promising results in mdx mice (Corti et al 2002), bone marrow stem cell (BMSC) transplantation did not restore dystrophin expression in affected dogs (Dell’Agnola et al 2004). While there was successful allogeneic bone marrow engraftment, BMSCs did not contribute either skeletal muscle or muscle precursor cells. Other adult stem cell cell populations may contribute to muscle regeneration. Mesoangioblasts are mesodermal stem cells derived from the vascular wall that contribute to muscle regeneration in both mice and dogs (De Angelis et al 1999; Sampaolesi et al 2006). These cells are multipotent, with capacity to give rise to osteogenic, adipogenic, myogenic, and endothelial lineages (Minasi et al 2002). The Cossu laboratory showed that a clinically-relevant cell mass can be obtained for transplant into the GRMD model and that mesoangioblasts injected in the femoral artery gained access to muscle through downstream capillary beds (Sampaolesi et al 2006). GRMD dogs treated with mesoangioblasts had immunohistochemical and Western blot evidence of dystrophin expression. However, questions have been raised about the outcome parameters used and the role that immunosuppression may have played in improvement (Bretag 2007).
Human immature dental pulp stem cells were transplanted by either arterial or muscular injections into four young GRMD littermate dogs (Kerkis et al 2008). Cell implantation, chimeric fibers, and minimal dystrophin expression were seen. In two recent studies from this same group, muscular implantation was achieved after systemic administration of umbilical cord mesenchymal stromal cells (MSCs; intra-arterial) (Zucconi et al 2011) and human-adipose-derived stromal cells (hASCs; (intravenous) (Vieira et al 2011) in GRMD dogs. Immunosuppression was not used in either study. Donor-recipient pairs were matched for the dog leukocyte antigen (DLA; major histocompatibility complex) in the MSC study but not for hASCs. While dystrophin was evident with hASCs, there was no expression with MSCs. In another study, intraarterial delivery of allogenic muscle stem cells led to long-term dystrophin expression and apparent stabilization of pathologic lesions and clinical function in three GRMD dogs (Rouger et al 2011).
Pharmacological therapies
The GRMD model has been used less commonly for pharmacologic approaches. Perhaps because of the perceived lower risk of such treatments, there has been a tendency to go directly from mdx mouse studies to human trials. In preparation for high-dose and early-treatment regimens of prednisone that might be impractical in human patients, we showed that a dose of 2 mg/kg, orally once daily for 4 months from 2 to 6 months of age, increased extensor muscle force in GRMD dogs, while causing a paradoxical decrease in flexor force (Liu et al 2004). The reduced flexor force probably occurred due to a reduction in early necrosis and a less pronounced regenerative response that can otherwise lead to functional hypertrophy. Treated dogs had remarkable myofiber mineralization, raising questions about potential deleterious side effects. Perhaps, most importantly, this prednisone preclinical trial established the 2 to 4 month time frame as an appropriate window for conducting pharmacologic studies in GRMD dogs.
Using GRMD dogs provided by our group, the Metzger laboratory demonstrated that chronic infusion of membrane-sealing poloxamer reduced myocardial fibrosis, blocked increases of serum cardiac troponin I and brain type natriuretic peptide, and prevented left-ventricular remodeling (Townsend et al 2010). A reduction in GRMD versus mdx mouse cardiac myocyte compliance was associated with a lack of utrophin upregulation in affected dogs, perhaps providing insight for their more severe cardiac phenotype. These results indicated that poloxamer may serve as an effective membrane-stabilizing chemical surrogate in dystrophin/utrophin deficiency.
Se-Jin Lee and colleagues have shown that the myostatin gene (growth/differentiation factor 8) is a key negative regulator of muscle growth (McPherron et al 1997; Lee et al 1999; Lee 2000; McPherron and Lee 2002). Dystrophin-deficient mdx mice in which myostatin is knocked out (Wagner et al 2002) or inhibited postnatally (Wagner et al 2005) have a less severe phenotype. Based, in part, on these results, there has been increasing interest in treatments to inhibit myostatin and thus promote muscle growth in DMD and other muscle wasting disorders. We investigated whether myostatin blockade could be achieved by an AAV-8-canine myostatin propeptide construct in two normal 3-month-old dogs treated via regional limb delivery (Qiao et al 2009). AAV vector DNA and propeptide gene expression were detected by quantitative polymerase chain reaction, Western blotting, and immunofluorescence staining of muscle biopsies. Over expression of the myostatin propeptide resulted in enhanced muscle growth, as evidenced by larger myofibers in multiple muscles and increased muscle volume on MRI. Separately, using four each treated and control 9 to 10-month-old GRMD dogs provided by our group, the Sweeney laboratory studied the muscle effects of liver directed gene transfer of a self-complementary AAV-8 vector expressing a myostatin peptide (Bish et al 2011). Treated GRMD dogs had increased muscle mass as assessed by MRI and confirmed by necropsy, together with hypertrophy of Type IIA fibers and reduced serum creatine kinase and muscle fibrosis. Independently, we have crossbred dystrophin-deficient GRMD dogs with whippets carrying a spontaneous two nucleotide myostatin gene deletion (Moscher et al 2007). A total of four dystrophic, myostatin-heterozygous GRippets and three dystrophic myostatin wild type dogs from two litters have been evaluated at 6 to 8 months of age using various phenotypic tests discussed above. Rather than showing improvement, the dystrophic myostatin-heterozygous GRippets were more severely affected than their dystrophic myostatin-wild type littermates, apparently because disproportionate enlargement and atrophy/hypoplasia of certain muscles led to postural instability and joint contractures (Kornegay JN et al, unpublished).
The calpain and ubiquitin-proteasome systems are involved in muscle degradation. Inhibition of involved enzymes has been proposed as a treatment for DMD, based largely on evidence that enzyme activities are elevated in the mdx mouse and that blockade can improve their phenotype (Spencer et al 1995; Badalamente and Stracher 2000; Bonuccelli et al 2007). We evaluated the efficacy of a novel calpain inhibitor in 6-week-old GRMD dogs treated daily with either active compound (n=9) or placebo (n=7) for 8 weeks (Childers et al, in press). Several tests were performed at baseline and every two weeks over the course of therapy. Calpain inhibition did not improve tibiotarsal joint angle, increase tibiotarsal extension force, or reduce eccentric contraction decrement; furthermore, there was no improvement in lean muscle mass or muscle histopathologic changes. Thus, consistent with findings from a recent mdx mouse study (Selsby et al 2010), we were unable to demonstrate efficacy for calpain inhibition in the GRMD model. Separately, in collaboration with the Willis laboratory, we have measured mRNA expression of ubiquitin-proteasome and calpain system components and calpain 1 and 2 and proteasome activities in several GRMD muscles and the heart at 6 months of age (Wadosky et al 2011). Less than half of the muscles tested had increases in proteasome activity and only half had increased calpain activity. Moreover, numerous components of the ubiquitin-proteasome system were significantly decreased in the heart. These results illustrate that ubiquitin-proteasome and calpain system expression varies among skeletal muscles and the heart, suggesting that pharmacological inhibition could lead to unexpected consequences. mRNA studies.
Potential role for modifier genes in DMD and GRMD
Symptoms in DMD patients vary even though most completely lack dystrophin, regardless of the specific gene mutation (Beggs et al 1991; Winnand et al 1993). Microarray gene expression profiling in DMD and dystrophin-deficient animals has provided a platform to study the role of modifier genes in phenotypic variation. Initial studies in both the mdx mouse (Porter et al 2002) and DMD (Chen et al 2000; Haslett et al 2003) have generally shown up-regulation of genes associated with inflammation and the regenerative response. With the advent of canine microarrays (Holzwarth et al 2005), analogous studies have been done in dogs with various diseases (Clements et al 2007). However, to our knowledge, expression arrays have not been evaluated in GRMD. As discussed above, we have collected functional data from GRMD dogs, both in the context of natural history and preclinical treatment studies. We also have muscle biopsy samples from many of these dogs. Importantly, these samples have been collected systematically from the same muscles at similar ages. Thus, by correlating expression array and functional data from these dogs, we should gain insight into the role that modifier genes play in phenotypic variation at both the individual dog and muscle level. In an ongoing study, we have performed 72 microarray profiles on the cranial sartorius, long digital extensor, and vastus lateralis muscles of eight GRMD and four normal dogs at 4-9 weeks and 6 months of age to evaluate mRNA expression on a genome-wide level. So as to determine genes that could protect against the dystrophic phenotype, we identified specific transcripts of the dystrophin-glycoprotein complex that were associated with cranial sartorius size at 6 months of age. Follow-up immunofluoresent microscopy revealed expression of these proteins in the perimembranous region in all three muscles despite the lack of dystrophin. We were interested in identifying additional genes that could be driving muscle hypertrophy. Using quantitative PCR to confirm the array studies, targeted genes involved with muscle growth were confirmed to be differentially expressed within each muscle. In conclusion, expression profiling of multiple skeletal muscles revealed unique patterns of transcript expression in each muscle, suggesting specific molecular signatures in response to the primary molecular defect (Figure 10). Therapies that target these genes could be beneficial in DMD.
Figure 10. Heat map of unsupervised hierarchical clustering showing differential transcript expression between GRMD and normal profiles.
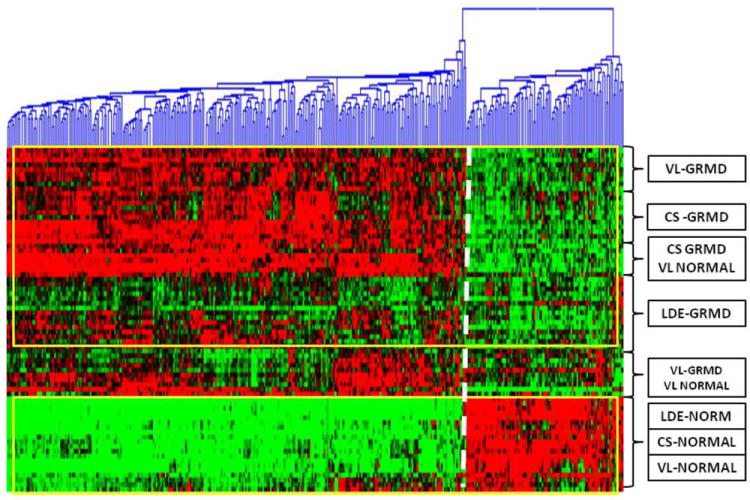
The heat map was prepared from 72 mRNA profiles from the cranial sartorius (CS), long digital extensor (LDE), and vastus lateralis (VL) muscles of eight GRMD and four normal dogs at 4-9 weeks and 6 months of age. Four hundred transcripts that were differentially regulated between GRMD CS profiles versus normal CS profiles (p < 0.0001; 200 transcripts) and GRMD LDE profiles versus normal LDE profiles (p < 0.0001; 200 transcripts) were clustered against the 72 mRNA profiles. Two main clusters are outlined in yellow. The top yellow cluster shows up-regulated genes (red color) for GRMD VL, CS and LDE and some normal VL profiles on the left and down-regulated transcripts (green) to the right of the white dotted lines. The bottom yellow cluster shows the opposite for all normal profiles. The profiles in between the yellow outlined clusters (VL GRMD and VL normal box) show a similar clustering pattern as the GRMD CS and normal VL profile box within the top yellow cluster. Note that there is some heterogeneity within GRMD profile expression. The heat map was prepared in HCE 3.5 Power Analysis; http://bioinformatics.cnmcresearch.org.
Conclusion
Unlike the dystrophin-deficient mdx mouse, which remains relatively normal clinically, GRMD dogs develop progressive, fatal disease strikingly similar to the human condition. To better utilize the GRMD model in therapeutic trials, we and others have developed various phenotypic tests to objectively characterize disease progression. The availability of these tests has set the stage for use of GRMD dogs in preclinical trials. Indeed, as outlined in this review, the GRMD model has been used in a range of genetic, cellular, and pharmacologic studies, setting the stage for clinical trials in DMD.
References
- Ambrósio CE, Valadares MC, Zucconi E, Cabral R, Pearson PL, Gaiad TP, Canovas M, Vainzof M, Miglino MA, Zatz M. Ringo, a Golden Retriever Muscular Dystrophy (GRMD) dog with absent dystrophin but normal strength. Neuromuscul Disord. 2008;18:892–893. doi: 10.1016/j.nmd.2008.06.385. [DOI] [PubMed] [Google Scholar]
- Ambrósio CE, Fadel L, Gaiad TP, Martins DS, Araujo KP, Zucconi E, Brolio MP, Giglio RF, Morini AC, Jazedje T, Froes TR, Feitosa ML, Valadares MC, Beltrao-Braga PC, Meirelles FV, Miglino MA. Identification of three distinguishable phenotypes in golden retriever muscular dystrophy. Genet Mol Res. 2009;8:389–396. doi: 10.4238/vol8-2gmr581. [DOI] [PubMed] [Google Scholar]
- Badalamente MA, Stracher A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve. 2000;23:106–111. doi: 10.1002/(sici)1097-4598(200001)23:1<106::aid-mus14>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Baltzer WI, Calise DV, Levine JM, Shelton GD, Edwards JF, Steiner JM. Dystrophin-deficient muscular dystrophy in a Weimaraner. J Am Anim Hosp Assoc. 2007;43:227–232. doi: 10.5326/0430227. [DOI] [PubMed] [Google Scholar]
- Barthélémy I, Barrey E, Thibaud J-L, Uriarte A, Voit T, Blot S, Hogrel J-V. Gait analysis using accelerometry in dystrophin-deficient dogs. Neuromuscul Disord. 2009;19:788–96. doi: 10.1016/j.nmd.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Barthélémy I, Barrey E, Aguilar P, Uriarte A, Le Chevoir M, Thibaud JL, Voit T, Blot S, Hogrel JY. Longitudinal ambulatory measurements of gait abnormality in dystrophin-deficient dogs. BMC Musculoskelet Disord. 2011;12:75. doi: 10.1186/1471-2474-12-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett RJ, Stockinger S, Denis MM, Bartlett WT, Inverardi L, Le TT, Man NT, Morris GE, Bogan DJ, Metcalf-Bogan J, Kornegay JN. In vivo targeted repair of a point mutation in the canine dystrophin gene by a chimeric RNA/DNA oligonucleotide. Nat Biotech. 2000;18:615–622. doi: 10.1038/76448. [DOI] [PubMed] [Google Scholar]
- Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86:45–48. doi: 10.1007/BF00205170. [DOI] [PubMed] [Google Scholar]
- Beggs AH, Hoffman EP, Snyder JR, Arahata K, Specht L, Shapiro F, Angelini C, Sugita H, Kunkel LM. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: Dystrophin gene and protein studies. Am J Hum Genet. 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- Bish LT, Sleeper MM, Forbes SC, Morine KJ, Reynolds C, Singletary GE, Trafny D, Bogan J, Kornegay JN, Walter GA, Sweeney HL. Long-term systemic myostatin inhibition via liver-targeted gene transfer in golden retriever muscular dystrophy. Hum Gene Ther. 2011 Jul 25; doi: 10.1089/hum.2011.102. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy KN, Roche BM, Schwartz DS, Nakayama T, Hamlin RL. Evaluation of the six-minute walk test in dogs. Am J Vet Res. 2004;65:311–313. doi: 10.2460/ajvr.2004.65.311. [DOI] [PubMed] [Google Scholar]
- Bonuccelli G, Sotgia F, Capozza F, Gazzerro E, Minetti C, Lisanti MP. Localized treatment with a novel FDA-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx mice. Cell Cycle. 2007;6:1242–1248. doi: 10.4161/cc.6.10.4182. [DOI] [PubMed] [Google Scholar]
- Bretag AH. Stem cell treatment of dystrophic dogs. Nature. 2007;450:7173–E23. doi: 10.1038/nature06437. discussion E23-5. [DOI] [PubMed] [Google Scholar]
- Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP, Province MA. Clinical Investigation in Duchenne dystrophy: 2. Determination of the “power” of therapeutic trials based on the natural history. Muscle Nerve. 1983;6:91–103. doi: 10.1002/mus.880060204. [DOI] [PubMed] [Google Scholar]
- Brumitt JW, Essman SC, Kornegay JN, Graham P, Weber WJ, Berry CR. Radiographic features of Golden Retriever muscular dystrophy. Vet Radiol Ultrasound. 2006;47:574–580. doi: 10.1111/j.1740-8261.2006.00188.x. [DOI] [PubMed] [Google Scholar]
- Buchthal F, Rosenfalck P. Electrophysiologic aspects of myopathy with particular reference to progressive muscular dystrophy. In: Bourne GH, Golarz MN, editors. Muscular Dystrophy in Man and Animals. Hafner Publishing Company; New York, NY: 1963. pp. 193–243. [Google Scholar]
- Bulfield G, Siller WG, Wight PAL, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey CT, Nussbaum RL, Cohan LC, Pollack L. Sporadic occurrence of Duchenne Muscular Dystrophy: evidence for new mutation. Clinical Genetics. 1980;18:329–341. doi: 10.1111/j.1399-0004.1980.tb02293.x. [DOI] [PubMed] [Google Scholar]
- Cerletti M, Negri T, Cozzi F, Colpo R, Andreetta F, Croci D, Davies KE, Cornelio F, Pozza O, Karpati G, Gilbert R, Mora M. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Therapy. 2003;10:750–757. doi: 10.1038/sj.gt.3301941. [DOI] [PubMed] [Google Scholar]
- Chakkalakal JV, Thompson J, Parks RJ, Jasmin BJ. Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB J. 2005;19:880–891. doi: 10.1096/fj.04-1956rev. [DOI] [PubMed] [Google Scholar]
- Chen Y-W, Zhao P, Borup R, Hoffman EP. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151:1321–1336. doi: 10.1083/jcb.151.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connuck DM, Sleeper LA, Colan SD, Cox GR, Towbin JA, Lowe AM, Wilkinson JD, Orav EJ, Cuniberti L, Salbert BA, Lipshultz SE. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: A comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2008;15:998–1005. doi: 10.1016/j.ahj.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox GR, Kunkel LM. Dystrophies and heart disease. Curr Opin Cardiol. 1997;12:329–343. [PubMed] [Google Scholar]
- Chetboul V, Carlos C, Blot S, Thibaud JL, Escriou C, Tissier R, Retortillo JL, Pouchelon J-L. Tissue Doppler assessment of diastolic and systolic alterations of radial and longitudinal left ventricular motions in golden retrievers during the preclinical phase of cardiomyopathy associated with muscular dystrophy. Am J Vet Res. 2004;65:1335–1341. doi: 10.2460/ajvr.2004.65.1335. [DOI] [PubMed] [Google Scholar]
- Childers MK, Kornegay JN, Aoki R, Otaviani L, Bogan DJ, Petroski G. Evaluating motor endplate-targeted injections of botulinum toxin type A in a canine model. Muscle Nerve. 1998;21:653–655. doi: 10.1002/(sici)1097-4598(199805)21:5<653::aid-mus15>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Childers MK, Okamura CS, Bogan DJ, Bogan JR, Sullivan MJ, Kornegay JN. Myofiber injury and regeneration in a canine homologue of Duchenne muscular dystrophy. Am J Phys Med Rehab. 2001;80:175–181. doi: 10.1097/00002060-200103000-00004. [DOI] [PubMed] [Google Scholar]
- Childers MK, Okamura CS, Bogan DJ, Bogan JR, Petroski GF, McDonald K, Kornegay JN. Eccentric contraction injury in dystrophic canine muscle. Arch Phys Med Rehabil. 2002;83:1572–1578. doi: 10.1053/apmr.2002.35109. [DOI] [PubMed] [Google Scholar]
- Childers MK, Grange RW, Kornegay JN. In vivo canine muscle function assay. J Vis Exp. 2011 Apr 5;(50) doi: 10.3791/2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers MK, Bogan JR, Bogan DJ, Greiner H, Staley J, Holder M, Grange RW, Kornegay JN. Chronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of Duchenne muscular dystrophy. Front Integr Physiol. 2011 doi: 10.3389/fphar.2011.00089. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011 Aug 13;378(9791):595–605. doi: 10.1016/S0140-6736(11)60756-3. Epub 2011 Jul 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements DN, Carter SD, Innes JF, Ollier WER, Day PJR. Gene expression profiling of normal and ruptured canine anterior cruciate ligaments. Osteoarthritis Cartilage. 2007;16:195–203. doi: 10.1016/j.joca.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Cooper BJ, Winand WN, Stedman H, Valentine BA, Hoffman EP, Kunkel LM, Scott M-O, Fischbeck KH, Kornegay JN, Avery RJ, Williams JR, Schmickel RD, Sylvester JE. The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature. 1988;334:154–156. doi: 10.1038/334154a0. [DOI] [PubMed] [Google Scholar]
- Corti S, Strazzer S, Del Bo R, Salani S, Bossolasco P, Fortunato F, Locatelli F, Soligo D, Moggio M, Ciscato P, Prelle A, Borsotti C, Bresolin N, Scarlato G, Comi GP. A subpopulation of murine bone marrow cells fully differentiates along the myogenic pathway and participates in muscle repair in the mdx dystrophic mouse. Exp Cell Res. 2002;277:74–85. doi: 10.1006/excr.2002.5543. [DOI] [PubMed] [Google Scholar]
- Cowan J, Macdessi J, Start A, Morgan G. Incidence of Duchenne muscular dystrophy in New South Wales and the Australian Capital Territory. J Med Genet. 1980;17:245–249. doi: 10.1136/jmg.17.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros D, Harnden P, Pellissier JF, Serratrice G. Muscle hypertrophy in Duchenne muscular dystrophy. A pathological and morphometric study. J Neurol. 1989;236:43–47. doi: 10.1007/BF00314217. [DOI] [PubMed] [Google Scholar]
- Dell’Agnola C, Want Z, Storb R, Tapscott SJ, Kuhr CS, Hauschka SD, Lee RS, Sale GE, Zellmer E, Gisburne S, Bogan J, Kornegay JN, Cooper BJ, Gooley TA, Little M-T. Hematopoietic stem cell transplantation does not restore dystrophin expression in Duchenne muscular dystrophy dogs. Blood. 2004;104:4311–4318. doi: 10.1182/blood-2004-06-2247. [DOI] [PubMed] [Google Scholar]
- De Angelis L, Berghella L, Coletta M, Lattanzi L, Zanchi M, Cusella-De Angelis MG, Ponzetto C, Cossu G. Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J Cell Biol. 1999;147:869–78. doi: 10.1083/jcb.147.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, Hart KJ, Tuohy T, White S, den Dunnen JT, Weiss RB, Flanigan KM. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005;134:295–298. doi: 10.1002/ajmg.a.30617. [DOI] [PubMed] [Google Scholar]
- de Lima AR, Nyengaard JR, Jorge AAL, Balieiro JCC, Peixoto C, Fioretto ET, Ambrósio CE, Miglino MA, Zatz M, Ribeiro AACM. Muscular dystrophy-related quantitative and chemical changes in adenohypophysis GH-cells in golden retrievers. Growth Horm IGF Res. 2007;2007:489–491. doi: 10.1016/j.ghir.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Desmedt JE, Borenstein S. Regeneration in Duchenne muscular dystrophy. Electromyographic evidence. Arch Neurol. 1976;33:642–650. doi: 10.1001/archneur.1976.00500090048010. [DOI] [PubMed] [Google Scholar]
- Devaux J-Y, Cabane L, Esler M, Flaouters H, Duboc D. Non-invasive evaluation of the cardiac function in golden retriever dogs by radionuclide angiography. Neuromusc Disord. 1993;3:429–432. doi: 10.1016/0960-8966(93)90090-7. [DOI] [PubMed] [Google Scholar]
- Edwards RHT, Jones DA, Newham DJ, Chapman SJ. Role of mechanical damage in the pathogenesis of proximal myopathy in man. Lancet. 1984;1:548–552. doi: 10.1016/s0140-6736(84)90941-3. [DOI] [PubMed] [Google Scholar]
- Emeryk-Szajewska B, Kopeć J. Electromyographic pattern in Duchenne and Becker muscular dystrophy. Part I: Electromyographic pattern in subsequent stages of muscle lesion in Duchenne muscular dystrophy. Electromyogr Clin Neurophysiol. 2008;48:265–277. [PubMed] [Google Scholar]
- Engel AG, Ozawa E. Dystrophinopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology. Basic and Clinical. McGraw-Hill; New York: 2004. pp. 961–1025. [Google Scholar]
- Fan Y, Maley M, Beilharz M, Grounds M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve. 1996;19:853–860. doi: 10.1002/(SICI)1097-4598(199607)19:7<853::AID-MUS7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Fan Z, Kocis K, Valley R, Howard JF, Chopra M, An H, Lin W, Muenzer J, Powers W. Safety and feasibility of high-pressure transvenous limb perfusion with 0.9% saline in human muscular dystrophy. Mol Ther. 2011 Jul 19; doi: 10.1038/mt.2011.137. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlini A, Sewry C, Melis MA, Mateddu A, Muntoni F. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromus Disord. 1999;9:339–346. doi: 10.1016/s0960-8966(99)00015-2. [DOI] [PubMed] [Google Scholar]
- Fine DM, Shin J-H, Yue Y, Volkmann D, Leach SB, Smith BF, McIntosh M, Duan D. Age-matched comparison reveals early electrocardiography and echocardiography changes in dystrophin-deficient dogs. Neuromuscul Disord. 2011 Jul;21(7):453–61. doi: 10.1016/j.nmd.2011.03.010. Epub 2011 May 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis JM. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil. 1999;20:605–625. doi: 10.1023/a:1005545325254. [DOI] [PubMed] [Google Scholar]
- Goyenvalle A, Seto JT, Davies KE, Chamberlain J. Therapeutic approaches to muscular dystrophy. Hum Mol Genet. 2011;20(R1):R69–78. doi: 10.1093/hmg/ddr105. Epub 2011 Mar 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griggs RC, Moxley RT, 3rd, Mendell JR, Fenichel GM, Brooke MH, Miller PJ, Mandel S, Florence J, Schierbecker J, Kaiser KK, King W, Pandya S, Robison J, Signore L. Randomized, double-blind trial of maziindol in Duchenne dystrophy. Muscle Nerve. 1990;13:1169–1173. doi: 10.1002/mus.880131212. [DOI] [PubMed] [Google Scholar]
- Gussoni E, Blau HM, Kunkel LM. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat Med. 1997;3:970–977. doi: 10.1038/nm0997-970. [DOI] [PubMed] [Google Scholar]
- Haslett JN, Sanoudou D, Kho AT, Han M, Bennett RR, Kohane KS, Beggs AH, Kunkel LM. Gene expression profiling of Duchenne muscular dystrophy skeletal muscle. Neurogenetics. 2003;4:163–171. doi: 10.1007/s10048-003-0148-x. [DOI] [PubMed] [Google Scholar]
- Hilton P, Stalberg E, Trontelg J, Mihelin M. Causes of the increased fiber density in muscular dystrophies studied with single fiber EMG during electrical stimulation. Muscle Nerve. 1985;8:383–388. doi: 10.1002/mus.880080507. [DOI] [PubMed] [Google Scholar]
- Hoogerwaard EM, van der Wouw PA, Wilde AAM, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, van Essen AJ, Leschot NJ, de Visser M. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1999;9:347–351. doi: 10.1016/s0960-8966(99)00018-8. [DOI] [PubMed] [Google Scholar]
- Holloway SM, Wilcox DE, Wilcox A, Dean JCS, Berg JN, Goudie DR, Denvir MA, Porteous MEM. Life expectancy and death from cardiomyopathy amongst carriers of Duchenne and Becker muscular dystrophy in Scotland. Heart. 2008;94:633–636. doi: 10.1136/hrt.2007.125948. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Holzwarth JA, Middleton RP, Roberts M, Mansourian R, Raymond F, Hannah SS. The development of a high-density canine microarray. J Hered. 2005;96:817–820. doi: 10.1093/jhered/esi130. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Howard JF, Wheeler SJ, Kornegay JN. Stimulated single fibre electromyography in normal dogs. J Sm Anim Pract. 1993;34:271–276. [Google Scholar]
- Howell JM, Fletcher S, O’Hara A, Johnsen RD, Lloyd F, Kakulas BA. Direct dystrophin and reporter gene transfer into dog muscle in vivo. Muscle Nerve. 1998a;21:159–165. doi: 10.1002/(sici)1097-4598(199802)21:2<159::aid-mus2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Howell JM, Lochmuller H, O’Hara A, Fletcher S, Kakulas BA, Massie B, Nalbantoglu J, Karpati G. High-level dystrophin expression after adenovirus mediated dystrophin minigene transfer to skeletal muscle of dystrophic dogs: prolongation of expression with imunosuppression. Hum Gene Ther. 1998b;9:629–634. doi: 10.1089/hum.1998.9.5-629. [DOI] [PubMed] [Google Scholar]
- Jaegger G, Marcellin-Little DJ, Levine D. Reliability of goniometry in Labrador retrievers. Am J Vet Res. 2002;63:979–986. doi: 10.2460/ajvr.2002.63.979. [DOI] [PubMed] [Google Scholar]
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernback SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- Jones BR, Brennan S, Mooney CT, Callahan JJ, McAllister H, Guo LT, Martin PT, Engvall E, Shelton GD. Muscular dystrophy with truncated dystrophin in a family of Japanese Spitz dogs. J Neurol Sci. 2004;217:143–149. doi: 10.1016/j.jns.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Kane AM, DeFrancesco TC, Boyle MC, Malarkey DE, Ritchey JW, Atkins CE, Cullen JM, Kornegay JN, Keene BW. Cardiac structure and function in female carriers of a canine model of Duchenne muscular dystrophy. doi: 10.1016/j.rvsc.2012.09.027. submitted. [DOI] [PubMed] [Google Scholar]
- Karpati G, Ajdukovic D, Arnold D, Gledhill RB, Guttmann R, Holland P, Koch PA, Shoubridge E, Spence D, Vanasse M, Watters GV, Abrahamowicz M, Duff C, Worton RG. Myoblast transfer in Duchenne muscular dystrophy. Ann Neurol. 1993;34:8–17. doi: 10.1002/ana.410340105. [DOI] [PubMed] [Google Scholar]
- Kellman P, Hernando D, Shah S, Hoyt RF, Kotin RM, Keene BW, Kornegay JN, Aletras AH, Arai AE. Myocardial fibro-fatty infiltration in Duchenne muscular dystrophy canine model detected using multi-echo Dixon method of water and fat separation imaging. International Society of Magnetic Resonance Medicine (ISMRM) 2009 [Google Scholar]
- Kerkis I, Ambrósio CE, Kerkis A, Martins DS, Zucconi E, Fonseca SAS, Cabral RM, Maranduba CMC, Gaiad TP, Morini AC, Vieira NM, Brolio MP, Sant’ Anna OA, Miglino MA, Zatz M. Early transplantation of human immature dental pulp stem cells from baby teeth to golden retriever muscular dystrophy (GRMD) dogs: Local or systemic? J Transl Med. 2008;6:35. doi: 10.1186/1479-5876-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowler C, Wheeler SJ. Neospora caninum infection in three dogs. J Sm Anim Pract. 1995;36:172–177. doi: 10.1111/j.1748-5827.1995.tb02875.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Nakamura A, Hasegawa D, Fujita M, Orima H, Takeda S. Evaluation of dystrophic dog pathology by fat-suppressed T2-weighted imaging. Muscle Nerve. 2009;40:815–826. doi: 10.1002/mus.21384. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Tuler SM, Miller DM, Levesque DC. Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve. 1988;11:1056–1064. doi: 10.1002/mus.880111008. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Prattis SM, Bogan DJ, Sharp NJH, Bartlett RJ, Alameddine HS, Dykstra MJ. Results of myoblast transplantation in a canine model of muscle injury. In: Kakulas BA, Howell JMc, Roses AD, editors. Duchenne Muscular Dystrophy. Animal Models and Genetic Manipulation. Raven Press; 1992. pp. 203–212. [Google Scholar]
- Kornegay JN, Sharp NJ, Bogan DJ, Van Camp SD, Metcalf JR, Schueler RO. Contraction tension and kinetics of the peroneus longus muscle in golden retriever muscular dystrophy. J Neurol Sci. 1994a;123:100–107. doi: 10.1016/0022-510x(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Sharp NJ, Schueler RO, Betts CW. Tibiotarsal joint contracture in dogs with golden retriever muscular dystrophy. Lab Anim Sci. 1994b;44:331–333. [PubMed] [Google Scholar]
- Kornegay JN, Bogan DJ, Bogan JR, Childers MK, Cundiff DD, Petroski GF, Schueler RO. Contraction force generated by tibiotarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci. 1999;166:115–121. doi: 10.1016/s0022-510x(99)00118-5. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Cundiff DD, Bogan DJ, Bogan JR, Okamura CS. The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul Disord. 2003;13:493–500. doi: 10.1016/s0960-8966(03)00025-7. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H, Wang B, Qiao C, Howard JF, Jr, Xiao X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol Ther. 2010;18:1501–1508. doi: 10.1038/mt.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Grange RW. Golden retriever muscular dystrophy (GRMD): Developing and maintaining a colony and physiological functional measurements. In: Duan D, editor. Muscle Gene Therapy: Methods and Protocols. Methods in Molecular Biology. Vol. 709. Humana Press; New York: 2011. pp. 105–123. [DOI] [PubMed] [Google Scholar]
- Lamminen AE. Magnetic resonance imaging of primary skeletal muscle diseases: patterns of distribution and severity of involvement. Br J Radiol. 1990;63:946–950. doi: 10.1259/0007-1285-63-756-946. [DOI] [PubMed] [Google Scholar]
- Lee S-J, McPherron AC. Myostatin and the control of skeletal muscle mass. Curr Opin Genet Dev. 1999;9:604–607. doi: 10.1016/s0959-437x(99)00004-0. [DOI] [PubMed] [Google Scholar]
- Lee S-J. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- Li J, Bogan J, Chen C, Bogan D, Wang B, Yuan Z, Qiao C, Kornegay JN, Xiao X. Hydrodynamic limb vein injection of AAV9 results in regional and systemic long-term expression of minidystrophin in young adult GRMD dogs. Mol Ther. 2009;17(S1):S278. [Google Scholar]
- Liu GC, Jong Y-J, Chiang C-H, Jaw T-S. Duchenne muscular dystrophy: MR grading system with functional correlation. Radiology. 1993;186:475–480. doi: 10.1148/radiology.186.2.8421754. [DOI] [PubMed] [Google Scholar]
- Liu JMK, Okamura CS, Bogan DJ, Bogan JR, Childers MK, Kornegay JN. Effects of prednisone in canine muscular dystrophy. Muscle Nerve. 2004;30:767–773. doi: 10.1002/mus.20154. [DOI] [PubMed] [Google Scholar]
- Lovitt S, Moore SL, Marden FA. The use of MRI in the evaluation of myopathy. Clin Neurophysiol. 2006;117:486–495. doi: 10.1016/j.clinph.2005.10.010. [DOI] [PubMed] [Google Scholar]
- McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL, Atkinson L, Reha A, Hirawat S, Miller LL. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. 2010;41:500–510. doi: 10.1002/mus.21544. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee S-J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lee S-J. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest. 2002;109:595–601. doi: 10.1172/JCI13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Hart K, Klamut H, Thomas N, Bodrug S, Burghes A, Bobrow M, Harper P, Thompson M, Ray P, Worton R. Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy. Science. 1988;242:755–759. doi: 10.1126/science.3055295. [DOI] [PubMed] [Google Scholar]
- Marden FA, Connolly AM, Siegel MJ, Rubin DA. Compositional analysis of muscle in boys with Duchenne muscular dystrophy using MR imaging. Skeletal Radiol. 2005;34:140–148. doi: 10.1007/s00256-004-0825-3. [DOI] [PubMed] [Google Scholar]
- Marsh AP, Eggebeen JD, Kornegay JN, Markert CD, Childers MK. Kinematics of gait in golden retriever muscular dystrophy. Neuromuscul Disord. 2010;20:16–20. doi: 10.1016/j.nmd.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Mioreelli M, Mostacciuolo ML, Fasoli G, Angelini C, Volta SD. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation. 1996;94:3168–3175. doi: 10.1161/01.cir.94.12.3168. [DOI] [PubMed] [Google Scholar]
- Meola G. Advanced microscopic and histochemical techniques: diagnostic tools in the molecular era of myology. Eur J Histochem. 2005;49:93–96. [PubMed] [Google Scholar]
- Miller RG, Sharma KR, Pavlath GK, Gussoni E, Mynhier M, Yu P, Lanctot AM, Greco CM, Steinman L, Blau H. Myoblast transplantation in Duchenne muscular dystrophy: The San Francisco Study. Muscle Nerve. 1997;20:469–478. doi: 10.1002/(sici)1097-4598(199704)20:4<469::aid-mus10>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, Caprioli A, Sirabella D, Baiocchi M, De Maria R, Boratto R, Jaffredo T, Broccoli V, Bianco P, Cossu G. The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development. 2002;129:2773–83. doi: 10.1242/dev.129.11.2773. [DOI] [PubMed] [Google Scholar]
- Miyazato LG, Moraes JRE, Baretta DC, Kornegay JN. Muscular dystrophy in dogs: Does the crossing of different breeds influence disease phenotype? Vet Pathol. 2011;48:655–662. doi: 10.1177/0300985810387070. [DOI] [PubMed] [Google Scholar]
- Moens P, Baatsen PHWW, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Mus Res Cell Motil. 1993;14:446–451. doi: 10.1007/BF00121296. [DOI] [PubMed] [Google Scholar]
- Moise NS, Valentine BA, Brown CA, Erb HN, Beck KA, Cooper BJ, Gilmour RF. Duchenne’s cardiomyopathy in a canine model: Electrocardiographic and echocardiographic studies. J Am Coll Cardio. 1991;17:812–820. doi: 10.1016/s0735-1097(10)80202-5. [DOI] [PubMed] [Google Scholar]
- Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:779–786. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F. Cardiomyopathy in muscular dystrophies. Curr Opin Neurol. 2003;16:577–583. doi: 10.1097/01.wco.0000093100.34793.81. [DOI] [PubMed] [Google Scholar]
- Nakagaki K, Gebhard DH, Bogan DJ, Kornegay JN. A quantitative comparison of NCAM-positive muscle cells from normal and dystrophic dogs. Muscle Nerve. 1994;17(Suppl 1):S193. [Google Scholar]
- Nakagaki K, Gebhard DH, Bogan DJ, Kornegay JN. Selection of canine myogenic cells with panning. Muscle Nerve. 1994;17(Suppl 1):S260. [Google Scholar]
- Nicholson HL, Osmotherly PG, Smith BA, McGowan CM. Determinants of passive hip range of motion in adult Greyhounds. Aust Vet J. 2007;85:217–221. doi: 10.1111/j.1751-0813.2007.00145.x. [DOI] [PubMed] [Google Scholar]
- Nigro G, Comi LI, Politano L, Limongelli FM, Nigro V, Rimini ML, Giugliano MAM, Petretta VR, Passamano L, Restucci B, Fattore L, Tebloev K, Comi L, De Luca F, Raia P, Esposito M. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve. 1995;18:283–291. doi: 10.1002/mus.880180304. [DOI] [PubMed] [Google Scholar]
- Nguyen F, Cherel Y, Guigand L, Goubault-Leroux I, Wyers M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J Comp Path. 2002;126:100–108. doi: 10.1053/jcpa.2001.0526. [DOI] [PubMed] [Google Scholar]
- Nandedkar SD, Barkhaus PE, Charles A. Multi-motor unit action potential analysis (MMA) Muscle Nerve. 1995;10:1155–1166. doi: 10.1002/mus.880181012. [DOI] [PubMed] [Google Scholar]
- Partridge TA, Grounds M, Sloper JC. Evidence of fusion between host and donor myoblasts in skeletal muscle grafts. Nature. 1978;273:306–308. doi: 10.1038/273306a0. [DOI] [PubMed] [Google Scholar]
- Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 1989;337:176–179. doi: 10.1038/337176a0. [DOI] [PubMed] [Google Scholar]
- Partridge T. Myoblast transplantation. Neuromuscul Disord. 2002;12(Suppl 1):S3–6. doi: 10.1016/s0960-8966(02)00076-7. [DOI] [PubMed] [Google Scholar]
- Phillips BA, Cala LA, Thickbroom GW, Melsom A, Zilko PJ, Mastaglia FL. Patterns of muscle involvement in inclusion body myositis: clinical and magnetic resonance imaging study. Muscle Nerve. 2001;24:1526–1534. doi: 10.1002/mus.1178. [DOI] [PubMed] [Google Scholar]
- Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–272. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- Prattis SM, Gebhart DH, Dickson G, Watt DJ, Kornegay JN. Magnetic affinity cell sorting (MACS) separation and flow cytometric characterization of neural cell adhesion molecule-positive, cultured myogenic cells from normal and dystrophic dogs. Exp Cell Res. 1993;208:453–464. doi: 10.1006/excr.1993.1267. [DOI] [PubMed] [Google Scholar]
- Prior TW, Bridgeman SJ. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Molec Diag. 2005;7:317–326. doi: 10.1016/S1525-1578(10)60560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Li J, Zheng H, Bogan J, Li J, Yuan Z, Zhang C, Bogan D, Kornegay J, Xiao X. Hydrodynamic limb vein injection of AAV8 canine myostatin propeptide gene in normal dogs enhances muscle growth. Hum Gene Ther. 2009;20:1–10. doi: 10.1089/hum.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideau Y. Treatment of orthopedic deformity during the ambulation stage of Duchenne muscular dystrophy. In: Serratrice G, editor. Neuromuscular Diseases. Raven Press; New York: 1984. pp. 557–564. [Google Scholar]
- Rouger K, Larcher T, Dubreil L, Deschamps JY, Le Guiner C, Jouvion G, Delorme B, Lieubeau B, Carlus M, Fornasari B, Theret M, Orlando P, Ledevin M, Zuber C, Leroux I, Deleau S, Guigand L, Testault I, Le Rumeur E, Fiszman M, Chérel Y. Systemic delivery of allogenic muscle stem (MuStem) cells Induces Long-Term Muscle repair and clinical efficacy in Duchenne muscular dystrophy dogs. Am J Pathol. 2011 Sep 13; doi: 10.1016/j.ajpath.2011.07.022. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S. Cardiac dysfunction with Becker muscular dystrophy. Am Heart J. 1996;132:642–647. doi: 10.1016/s0002-8703(96)90250-1. [DOI] [PubMed] [Google Scholar]
- Salimena MC, Lagrota-Candido J, Quírico-Santos T. Gender dimorphism influences extracellular matrix expression and regeneration of muscular tissue in mdx dystrophic mice. Histochem Cell Biol. 2004;122:535–544. doi: 10.1007/s00418-004-0707-8. [DOI] [PubMed] [Google Scholar]
- Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthelemy I, Perani L, Mantero S, Guttinger M, Pansarasa O, Rinaldi C, Cusella De Angelis MG, Torrente Y, Bordingnon C, Bottinelli R, Cossu G. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–579. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- Sanders DB, Stalberg EV. AAEM minimonogaph #25: Single-fiber electromyography. Muscle Nerve. 1996;19:1069–1083. doi: 10.1002/(SICI)1097-4598(199609)19:9<1069::AID-MUS1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Schatzberg SJ, Olby NJ, Breen M, Anderson LVB, Langford CF, Dickens HF, Wilton SD, Zeiss SD, Binns MM, Kornegay JN, Morris GE, Sharp NJH. Molecular analysis of a spontaneous dystrophin “knockout” dog. Neuromuscul Disord. 1999;9:289–295. doi: 10.1016/s0960-8966(99)00011-5. [DOI] [PubMed] [Google Scholar]
- Selsby J, Pendrak K, Zadel M, Tian Z, Pham J, Carver T, Acosta P, Barton E, Sweeney HL. Leupeptin-based inhibitors do not improve the mdx phenotype. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1192–1201. doi: 10.1152/ajpregu.00586.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NJH, Kornegay JN, Van Camp SD, Herbstreith MH, Secore SL, Kettle S, Hung W-Y, Constantinou CD, Dykstra MJ, Roses AD, Bartlett RJ. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics. 1991;13:115–121. doi: 10.1016/0888-7543(92)90210-j. [DOI] [PubMed] [Google Scholar]
- Shimatsu Y, Katagiri K, Furuta T, Nakura M, Tanioka Y, Yuasa K, Tomohiro M, Kornegay JN, Nonaka I, Takeda S. Canine X-linked muscular dystrophy in Japan (CXMDJ) Exp Anim. 2003;52:93–97. doi: 10.1538/expanim.52.93. [DOI] [PubMed] [Google Scholar]
- Shimatsu Y, Yoshimura M, Yuasa K, Urasawa N, Tomohiro M, Nakura M, Tanigawa M, Nakamura A, Takeda S. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJ. Acta Myol. 2005;24:145–154. [PubMed] [Google Scholar]
- Siegel IM, Miller JE, Ray RD. Subcutaneous lower limb tenotomy in the treatment of pseudohypertrophic muscular dystrophy. J Bone Joint Surg. 1968;50A:1437–1443. [PubMed] [Google Scholar]
- Skuk D, Paradis M, Goulet M, Tremblay JP. Ischemic central necrosis in pockets of transplanted myoblasts in nonhuman primates: implications for cell-transplantation strategies. Transplantation. 2007;84:1307–1315. doi: 10.1097/01.tp.0000288322.94252.22. [DOI] [PubMed] [Google Scholar]
- Smith BF, Kornegay JN, Duan D. Independent canine models of Duchenne muscular dystrophy due to intronic insertions of repetitive DNA. Mol Ther. 2007;15(Suppl 1):S51. [Google Scholar]
- Smith BF, Yue Y, Woods PR, Kornegay JN, Shin J-H, Williams RR, Duan D. An intronic LINE-1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne-like muscular dystrophy in the corgi breed. Lab Invest. 2011;91:216–231. doi: 10.1038/labinvest.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe GM, Hodgetts SI, Grounds MD. Problems and solutions in myoblast transfer therapy. J Cell Mol Med. 2001;5:33–47. doi: 10.1111/j.1582-4934.2001.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer MJ, Croall DE, Tidball JG. Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem. 1995;270:10909–10914. doi: 10.1074/jbc.270.18.10909. [DOI] [PubMed] [Google Scholar]
- Tegler CJ, Grange RW, Bogan DJ, Markert CD, Case D, Kornegay JN, Childers MK. Eccentric contractions induce rapid isometric torque drop in dystrophic-deficient dogs. Muscle Nerve. 2010;42:130–132. doi: 10.1002/mus.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibaud JL, Monnet A, Dertoldi D, Barthelemy I, Blot S, Carlier PG. Characterization of dystrophic muscle in golden retriever muscular dystrophy dogs by nuclear magnetic resonance imaging. Neuromuscul Disord. 2007;17:575–584. doi: 10.1016/j.nmd.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1050. doi: 10.1172/JCI41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay JP, Malouin F, Roy R, Huard J, Bouchard JP, Satoh A, Richards CL. Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplant. 1993;2:99–112. doi: 10.1177/096368979300200203. [DOI] [PubMed] [Google Scholar]
- Valentine BA, Cooper BJ, de Lahunta A, O’Quinn R, Blue JT. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci. 1988;88:69–81. doi: 10.1016/0022-510x(88)90206-7. [DOI] [PubMed] [Google Scholar]
- Valentine BA, Cummings JF, Cooper BJ. Development of Duchenne-type cardiomyopathy. Morphologic studies in a canine model. Am J Pathol. 1989a;135:671–678. [PMC free article] [PubMed] [Google Scholar]
- Valentine BA, Kornegay JN, Cooper BJ. Clinical electromyography studies of canine X-linked muscular dystrophy. Am J Vet Res. 1989b;50:2145–2147. [PubMed] [Google Scholar]
- Valentine BA, Cooper BJ, Cummings JF, de Lahunta A. Canine X-linked muscular dystrophy: morphologic lesions. J Neurol Sci. 1990;97:1–23. doi: 10.1016/0022-510x(90)90095-5. [DOI] [PubMed] [Google Scholar]
- Vieira NM, Valadares M, Zucconi E, Secco M, Bueno CR, Brandalise V, Assoni A, Gomes J, Landini V, Andrade T, Caetano HV, Vainzof M, Zatz M. Human Adipose-Derived Mesenchymal Stromal cells injected systemically into GRMD dogs without immunosuppression are able to reach the host muscle and express human dystrophin. Cell Transplant. 2011 Oct 14; doi: 10.3727/096368911X. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Vignos PJ, Jr, Archibald KC. Maintenance of ambulation in childhood muscular dystrophy. J Chron Dis. 1960;12:273–290. doi: 10.1016/0021-9681(60)90105-3. [DOI] [PubMed] [Google Scholar]
- Vignos PJ, Jr, Spencer GE, Jr, Archibald JC. Management of muscular dystrophy of childhood. JAMA. 1963;184:89–96. doi: 10.1001/jama.1963.03700150043007. [DOI] [PubMed] [Google Scholar]
- Wadosky KM, Li L, Rodríguez JE, Min J, Bogan D, Gonzalez J, Kornegay JN, Willis M. Regulation of the calpain and ubiquitin proteasome systems in a canine model of Duchenne muscular dystrophy. Muscle Nerve. 2011 Apr 4; doi: 10.1002/mus.22125. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, McPherron AC, Winik N, Lee S-J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA. 2005;102:2519–24. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A, Stanley R, Chandler K, Marks SL, Muntoni F, Shelton GD, Piercy RJ. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PloS one. 2010;5(no. 1) doi: 10.1371/journal.pone.0008647. Article ID e8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li J, Fu FH, Xiao X. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthop Res. 2009;27:421–426. doi: 10.1002/jor.20781. [DOI] [PubMed] [Google Scholar]
- Wang J, Fan Z, Kornegay JN, Styner MA. MRI-based quantification of Duchenne muscular dystrophy in a canine model. Proc SPIE. 2011;7965:0G1–9. [Google Scholar]
- Wang Z, Allen JM, Riddell SR, Gregorevic P, Storb R, Tapscott SJ, Chamberlain JS, Kuhr CS. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum Gene Ther. 2007a;18:18–26. doi: 10.1089/hum.2006.093. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kuhr CS, Allen JM, Blankenship M, Gregorevic P, Chamberlain JS, Tapscott SJ, Storb R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007b;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- Wang Z, Storb R, Lee D, Kushmerick MJ, Chu B, Berger C, Arnett A, Allen J, Chamberlain JS, Riddell SR, Tapscott SJ. Immune responses to AAV in canine muscle monitored by cellular assays and noninvasive imaging. Mol Ther. 2010;18:617–24. doi: 10.1038/mt.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winand N, Pradham D, Cooper B. Molecular Mechanism of Neuromuscular Disease. Muscular Dystrophy Association; Tucson, Ariz, USA: 1994. Molecular characterization of severe Duchenne-type muscular dystrophy in a family of Rottwiler dogs. [Google Scholar]
- Winnard AV, KIein CJ, Coovert DD, Prior T, Papp A, Snyder P, Bulman DE, Ray PN, McAndrew P, King W, Moxley RT, Mendel JR, Burghes AHM. Characterization of translational frame exception patients in Duchenne/Becker muscular dystrophy. Hum Mol Genet. 1993;2:737–744. doi: 10.1093/hmg/2.6.737. [DOI] [PubMed] [Google Scholar]
- Wulff K, Herrmann FH, Wapenaar MC, Wehner M. Deletion screening in patients with Duchenne muscular dystrophy. J Neurol. 1989;236:470–473. doi: 10.1007/BF00328509. [DOI] [PubMed] [Google Scholar]
- Yokota T, Lu QL, Partridge T, Kobayashi M, Nakamura A, Takeda S, Hoffman E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol. 2009;65:667–76. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa K, Yoshimura M, Urasawa N, Ohshima S, Howell JM, Nakamura A, Hijikata T, Miyagoe-Suzuki Y, Takeda S. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 2007;14:1249–1260. doi: 10.1038/sj.gt.3302984. [DOI] [PubMed] [Google Scholar]
- Yue Y, Ghosh A, Long C, Bostick B, Smith BF, Kornegay JN, Duan D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16:1944–1952. doi: 10.1038/mt.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yugeta N, Urasawa N, Fujii Y, Yoshimura M, Yuasa K, Wada MR, Nakura M, Shimatsu Y, Tomohiro M, Takahashi A, Machida N, Wakao Y, Nakaura A, Takeda S. Cardiac involvement in Beagle-based canine x-linked muscular dystrophy in Japan (CXMDJ): electrocardiographic, echocardiographic, and morphologic studies. BMC Cardiovasc Disord. 2006;6:47–59. doi: 10.1186/1471-2261-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Zatz M, Betti RT, Levy JA. Benign Duchenne muscular dystrophy in a patient with growth hormone deficiency. Am J Med Genet. 1981;10:301–304. doi: 10.1002/ajmg.1320100313. [DOI] [PubMed] [Google Scholar]
- Zucconi E, Vieira NM, Bueno CR, Jr, Secco M, Jazedje T, Costa Valadares M, Fussae Suzuki M, Bartolini P, Vainzof M, Zatz M. Preclinical studies with umbilical cord mesenchymal stromal cells in different animal models for muscular dystrophy. J Biomed Biotechnol. 2011:715251. doi: 10.1155/2011/715251. Epub 2011 Jul 15. [DOI] [PMC free article] [PubMed] [Google Scholar]