

Abstract

The receptor tyrosine kinase AXL has emerged in recent years as an potential oncology target due to its overexpression in several types of cancers coupled with its ability to promote tumor growth and metastasis. To identify small molecule inhibitors of AXL, we built a homology model of its catalytic domain to virtually screen and identify scaffolds displaying an affinity for AXL. Further computational and structure-based design resulted in the synthesis of a series of 2,4,5-trisubstitued pyrimidines, which demonstrated potent inhibition of AXL in vitro (IC50 = 19 nM) and strongly inhibited the growth of several pancreatic cell lines.
Keywords: AXL kinase inhibitors, anticancer, structure−activity relationship
AXL is a receptor tyrosine kinase (RTK) involved in the growth, differentiation, survival, and motility of many different cell types.1 It belongs to the TAM family of RTKs that also consists of MER and TYRO3. This family of kinases is similar to other RTKs in that they have an intracellular kinase domain and span the cell membrane to an extracellular domain. In contrast, the extracellular portion of AXL (and TAM family members) is particularly unique in that it has properties and structural elements similar to cell adhesion molecules that may participate in cell-to-cell contacts that are not fully understood.2 In addition to this, it is clear that AXL binds to a soluble ligand called GAS6 (growth arrest-specific 6) that activates downstream signaling networks responsible for its biological effects. GAS6 binding to AXL results in receptor dimerization and AXL autophosphorylation.3 This activation results in downstream signaling to the PI3K/AKT,4 MAPK,5 and NF-κB6 pathways.
AXL has been reported to be overexpressed in numerous tumor types.1 It was first discovered in chronic myelogenous leukemia (CML),7 although more recent reports primarily associate AXL expression with solid tumors, such as breast, brain, lung, pancreatic, prostate, esophageal, and renal tumors. Target validation studies using genetic tools (RNAi) or therapeutic agents (antibodies and small molecules) demonstrate that inhibiting AXL function in cell culture and animal models has favorable antitumorigenic effects.8 For example, an AXL-targeted monoclonal antibody was shown to decrease xenograft tumor growth, inhibit angiogenesis, and sensitize cancer cells to standard-of-care therapies.9 Similarly, several small molecule AXL inhibitors (Figure 1), particularly R428, were shown to inhibit angiogenesis and to synergize with cisplatin in reducing liver metastases.10 Finally, using shRNA, it was shown that knockdown of AXL expression decreased pancreatic cancer cell growth in soft agar, inhibited cell invasion and migration, and downregulated the expression of epithelial-mesenchymal transition (EMT)-associated genes.11 Taken together, these studies suggest that AXL is a potential therapeutic target for the treatment of several tumor types.
Figure 1.

Reported selective AXL kinase inhibitors.
Our initial approach to rationally design AXL kinase inhibitors relied on in silico scaffold-based flexible docking carried out with the ICM docking algorithm12 as implemented in the ICM-Pro program (v3.7-1g, Molsoft, LLC) followed by the synthesis of potential leads. The structure of the catalytic domain of AXL remaining unsolved, we first built a homology model of the active site to conduct docking experiments. Three closely related proteins with solved X-ray crystal structures were used as templates: MER, c-Met, and IGF-1R. Initial cross-docking experiments utilizing the homology model of AXL with an in-house collection of compounds led to the identification of novel 2,4,6-trisubstituted pyrimidine 2,4-diamines 1 and 2 (shown in Figure 2) as potential leads with AXL inhibitory activity of 33 and 47 μM in a biochemical assay.
Figure 2.

2,4,6-Trisubstituted pyrimidine hits 1 and 2 derived from in silico scaffold-based design and 2,4,5-trisubstituted pyrimidine leads 3 and 4 chosen for optimization.
However, their binding modes suggest strong steric clashes between the bulky substituents in the 6-position of the pyrimidine core and the hinge, gate keeper, and other residues present in the hydrophobic pocket of AXL. This observation prompted us to focus on compounds featuring pyrimidine cores with small hydrophobic substituents that can be accommodated by the Leu620 gate keeper residue of AXL. This led to the identification of 5-methylpyrimidines 3 and 4, which exhibited IC50 values against AXL in the low micromolar range (2–6 μM) and were chosen as leads for optimization. Both of these series were prepared according to Scheme 1.
Scheme 1. Preparation of 2,4-Substituted: 5-Alkylpyrimidines.
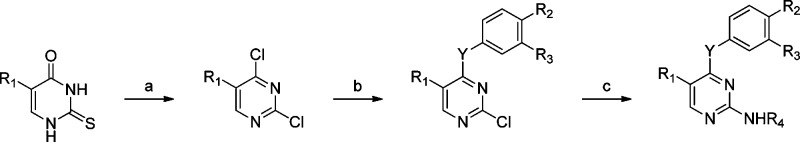
Reagents and conditions: (a) Oxalyl chloride, DMF, dioxane, reflux, 12 h for R1 = cyclopropyl. (b) R2R3PhB(OH)2, Pd(OAc)2, PPh3, aqueous Na2CO3, THF, reflux, 12 h or MW, 150 °C, 30 min for Y = bond; R2R3PhOH, K2CO3, acetone, reflux, 12 h for Y = O; R2R3PhNH2, DIPEA, IPA, reflux, 12 h or MW, 150 °C, 30 min for Y = NH. (c) R4NH2, Pd2(dba)3, Xantphos, K2CO3, tBuOH, reflux, 12 h.
A further 2-fold improvement in activity was achieved with 5 (IC50 = 0.73 μM) in which a methylene linker was introduced between the 2-aryl substituent and the piperazine heterocycle, enhancing the interaction between the N-methylpiperazine moiety and the solvent-accessible region of the kinase. A further extension of the linker to a methoxyethyl group (along with replacing the piperazine with a piperidine, compound 6) resulted in decreased activity. Replacing the N-methylpiperazine with a morpholine ring led to a dramatic decrease in the activity of the inhibitor (IC50 > 10 μM), presumably due to the lack of desolvation. Similar observations were made when the 2-aryl ring was replaced by an electron-rich pyridine; however, replacing the methylene linker with N-methylamine and the N-methylpiperazine with N-methylpiperidine led to a modest improvement in potency (7, IC50 = 0.42 μM). We expected the in vivo stability of this moiety to be less than that of 5, however, prompting us to select the methylene-linked aryl piperazine for further compounds.
The study of the binding mode of 5 with the homology model of AXL revealed the critical importance of the substituent in the 4-position of the pyrimidine ring. This moiety was found to extend deep inside the hydrophobic cavity of the kinase, suggesting that stronger binding could be achieved by introducing various electron-withdrawing groups on the 4-aryl group.
Thus, eight additional compounds were prepared featuring various electron-deficient 4-aryl substituents. Mild improvements in potency were observed when a chlorine atom was installed in the meta position (8, IC50 = 0.44 μM) or a gemdimethylcyano group in the para position (9, IC50 = 0.29 μM). The effect of the linkage between the pyrimidine core and its 4-substituent was also investigated, but replacing the C–C bond of 5 with either an ether (10) or an amine (11) linkage resulted in decreased potency (IC50 = 1.4 μM and >10 μM, respectively). In all cases, the biochemical activities (summarized in Table 1) were found to complement the calculated binding energies, confirming the validity of our AXL homology model.
Table 1. Structure–Activity Relationship and Docking Parameters for 5-Alkylpyrimidine-Based Inhibitors.

However, the slow incremental nature of our progress prompted us to focus our efforts toward optimizing the 5-position of the pyrimidine core, which interacts with the Leu620 gate keeper of AXL. It was hypothesized that this hydrophobic mildly sized amino acid could interact more favorably with larger more hydrophobic substituents than a methyl group. To this intent, a 5-cyclopropyl analogue of 5 was prepared but surprisingly exhibited poor activity (12, IC50 = 3.4 μM). Our attempts at production of an aziridine analogue of 5 were unsuccessful, and we resorted to scaffold hopping to explore novel pharmacophores. This led to the identification of scaffolds featuring sulfonyl groups positioned within the distance required for Mg2+ coordination (2.69 Å) and exhibiting high binding energies. We investigated different positions for the sulfonyl moiety, resulting in the synthesis of 13, a compound featuring a 5-chloropyrimidine core and an amine-linked N-dimethylbenzenesulfonamide moiety at the 4-position, which extends in the hydrophobic site; 13 displayed a potent activity against AXL with an IC50 of 0.027 μM. Studies of the bound complex reveal that the pyrimidine nitrogen of 13 is involved in a hydrogen-binding interaction with the Met623 residue in the hinge region of the kinase. As shown in Figure 3, the hydrophobic electron-withdrawing 5-Cl atom interacts favorably with the Leu620 gate keeper while the electron-deficient 4-aryl moiety and the N-dimethylsulfonamide are involved in charge–charge interactions with the lipophilic “roof” of the β-sheet (GXGXG) and the DFG loop of AXL, respectively. An additional strong interaction is formed between a Mg2+ ion, critical for AXL activity, and the electron-rich oxygen atoms of the polarizable sulfonamide moiety, possibly explaining the considerable improvement in potency observed with 6.
Figure 3.

Structure of the bound complex formed between 13 and the homology model of the ATP binding site of AXL kinase.
The importance of the electron-deficient 4-aryl group was confirmed by synthesizing an analogue of 13 in which the aryl ring is replaced by a piperazine ring directly attached to the pyrimidine core through one of its nitrogen atoms and retaining a N-dimethylsulfonamide on the other. As expected, this compound displayed poor affinity for AXL with an IC50 in the micromolar range. The role of the sulfonamide proved more ambiguous when it was found that substitution with a N-dimethylamide had little effect on binding (14, IC50 = 0.088 μM). This was rationalized based on the similar geometry of the bound complexes, the methyl groups being involved in hydrophobic van der Walls interactions with the DFG loop, and the polarizable oxygen atom coordinating the Mg2+ ion.
On the basis of the potency of 13 in biochemical assays, its activity in cell-based studies was evaluated. It showed extremely potent activity in cell viability assays with an IC50 of 6 nM against the pancreatic cancer cell line PSN-1 (Table 2). More importantly, 13 was evaluated for its ability to block GAS6-mediated activation of AXL in pancreatic cancer cells. PSN-1 cells were serum-starved and then stimulated with GAS6 in the presence of various concentrations of 13. Lysates from these treatments were generated and evaluated for phospho-AKT (S473) using the Meso Scale Discovery platform and phospho-AXL using a standard ELISA. Compound 13 showed a concentration-dependent decrease in phospho-AKT and phospho-AXL levels with an EC50 of 305 and 222 nM, respectively (Figure 4).
Table 2. Structure–Activity Relationship and Binding Energy Terms for Amide- and Sulfonamide-Containing Inhibitors.

Figure 4.

Pancreactic cancer cells (PSN1) were serum-starved followed by GAS6 (3 μg/mL) stimulation in the presence of the indicated concentrations of 13. Cells were harvested, and lysates from the cells were analyzed for phospho-Akt (Ser473) and phospho-Axl. Compound 13 demonstrated a concentration-dependent effect of lowering these cell signaling pathways.
These data demonstrate that 13 inhibits the intended molecular target (AXL kinase) with nanomolar potency in cultured cancer cells. However, the disconnect between the IC50 in the cell viability assay and the EC50 in the phospho-AKT/AXL assays led us to believe that the strong antiproliferative activity of 13 was primarily driven by a target other than AXL.
To identify other kinase targets of 13, profiling studies were conducted to evaluate its selectivity toward AXL. Not surprisingly, the pyrimidine scaffold, which is a very common motif among protein kinase inhibitors, imparted 13 with a broad selectivity profile: When tested at 200 nM against a panel of 75 kinases, 13 displayed over 50% inhibition for 11 of them (Figure 5).
Figure 5.

Compound 13 was profiled against a panel of 40 kinases at 200 nM using Invitrogen's SelectScreen kinase profiling service. Significant activity (>80%) was seen against 11 of the kinases including the three TAM family members (AXL, MER, and TYRO3) as well as Aurora A, JAK2, ALK, and ABL1.
A strong activity against related TAM family members MER and TYRO3 was observed as well as against other Ser/Thr and Tyr kinases such as Aurora A and B, JAK2, ALK, ABL1, and CHEK1. The IC50 of 13 against the Auroras was determined to be 3.0 and 12.4 nM, respectively, likely explaining the strong antiproliferative activity observed in cell-based studies. To further explore this, cell cycle analyses were performed on cells treated with 13, and consistent with Aurora A and B inhibition, treated cells showed a significant increase in the G2/M population of cells and also an accumulation of cells with ≥4N DNA, which is indicative of Aurora B inhibition and endoreduplication (Figure 6).
Figure 6.

Pancreatic cancer cells (PSN-1) were treated with 30 nM 13 for 24 h. Cells were collected, stained with propidium iodide, and analyzed by flow cytometry. Consistent with Aurora A and Aurora B inhibition, there was a significant increase in the G2/M (4N) portion of cells in the treated group. Furthermore, there was also the emergence of an 8N peak, which is indicative of endoreduplication and Aurora B inhibition.
The differences between the ATP binding pockets of AXL and the Auroras are minute. The gate keeper residues are identical, the DFG loop of both kinases is the “in” conformation, and changes in the solvent binding region of 13 lead to loss of activity. Thus, our efforts at optimizing the selectivity initially focused on the residue flanking the gate keeper: hydrophobic Pro for AXL and acidic Glu for Auroras. We expected a more lipophilic electron-withdrawing moiety in the 5-position of the pyrimidine core to enhance AXL binding while destabilizing the hinge region of the Auroras. To this intent, we prepared 5-Me, 5-F, and 5-CF3 analogues of 13 or 14, but in all cases, the AXL activity was lost, presumably due to a steric clash with the Leu620 gate keeper residue and loss of one hydrogen bond in the hinge.
Our next approach exploited the differences in the hydrophobic pockets of the kinases. A favorable hydrophobic interaction exists in the case of AXL only between the 4-aryl group and the Leu542 residue in the roof of the β-sheet forming the hydrophobic pocket. Strengthening this interaction can be achieved by enhancing the hydrophobic contacts between the inhibitor and the DGF “in” loop of the kinase, inducing a slight conformational change, which reduces the distance between the 4-aryl substituent and the roof of the hydrophobic pocket residues. To this intent, the N-dimethyl groups of 13 and 14 were replaced by more lipophilic pyrrolidinyl or N-cyclopropyl groups (18–20; see Table 2). This resulted in moderate AXL binding improvement in all cases, but the Aurora activity did not decrease. The addition of electron-withdrawing atoms on the 4-aryl ring was also studied (21–25; synthesis shown in Scheme 2): F was well tolerated but did not improve activity or selectivity, and Cl resulted in loss of AXL activity. Methylation of the amine linker also resulted in loss of activity, presumably as a result of increased conformational strain. When the aryl ring was directly connected to the pyrimidine core (15, 16, 26, and 27), potent AXL inhibitors were obtained, but again, they exhibited strong aurora activity. Finally, meta-substituted analogues of 13 featuring both NH and C–C bound 4-aryl groups were prepared, but decreased activities were observed, presumably due to steric clashes arising from the change in orientation of the aryl group in the hydrophobic pocket.
Scheme 2. Preparation of Sulfonamide-Based Inhibitors.

Reagents and conditions: (a) XPhB(OH)2, Pd(OAc)2, PPh3, aqueous Na2CO3, THF, reflux, 12 h or MW, 150 °C, 30 min for Y = bond; XPhNH2 or XPhNHMe DIPEA, IPA, reflux, 12 h or MW, 150 °C, 30 min for Y = NH or NMe. (b) Chlorosulfonic acid, room temperature or 90 °C, 1–12 h. (c) R2R3NH, DIPEA, IPA, room temperature, 1 h. (d) R4NH2, Pd2(dba)3, Xantphos, K2CO3, tBuOH, reflux, 12 h.
In conclusion, we have prepared a series of potent AXL kinase inhibitors featuring a 5-Cl pyrimidine core carrying an arylsulfonamide moiety in its 4-position. The pyrimidine scaffold was chosen based on the results on in silico screening of fragments and in-house compounds against a homology model of AXL kinase. Several compounds displayed low nanomolar activity against AXL in biochemical assays and potently inhibited the growth of PSN1 cells. However, cell cycle studies revealed that the compounds induced strong G2/M arrest by potently inhibiting Aurora A and B as well as several other kinases. Synthetic efforts are underway to improve the selectivity profile of our inhibitors while retaining potency.
Supporting Information Available
Full experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
We acknowledge support from the Huntsman Cancer Foundation and NCI grant number P30 CA042014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Linger R. M.; Keating A. K.; Earp H. S.; Graham D. K. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 2010, 14101073–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno J.; Mansukhani A.; Basilico C. A putative receptor tyrosine kinase with unique structural topology. Oncogene 1991, 6101909–1913. [PubMed] [Google Scholar]
- Stitt T. N.; Conn G.; Gore M.; Lai C.; Bruno J.; Radziejewski C.; Mattsson K.; Fisher J.; Gies D. R.; Jones P. F.; Masiakowski P.; Ryan T. E.; Tobkes N. J.; Chen D. H.; DiStefano P. S.; Long G. L.; Basilico C.; Goldfarb M. P.; Lemke G.; Glass D. J.; Yancopoulos G. D. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 804661–670. [DOI] [PubMed] [Google Scholar]
- Sawabu T.; Seno H.; Kawashima T.; Fukuda A.; Uenoyama Y.; Kawada M.; Kanda N.; Sekikawa A.; Fukui H.; Yanagita M.; Yoshibayashi H.; Satoh S.; Sakai Y.; Nakano T.; Chiba T. Growth arrest-specific gene 6 and Axl signaling enhances gastric cancer cell survival via Akt pathway. Mol. Carcinog. 2007, 462155–164. [DOI] [PubMed] [Google Scholar]
- Fridell Y. W.; Jin Y.; Quilliam L. A.; Burchert A.; McCloskey P.; Spizz G.; Varnum B.; Der C.; Liu E. T. Differential activation of the Ras extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell. Biol. 1996, 161135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarchi F.; Verardo R.; Varnum B.; Brancolini C.; Schneider C. Gas6 anti-apoptotic signaling requires NF-kappa B activation. J. Biol. Chem. 2001, 2763431738–31744. [DOI] [PubMed] [Google Scholar]
- Liu E.; Hjelle B.; Bishop J. M. Transforming genes in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. U.S.A. 1988, 8561952–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Ye X.; Tan C.; Hongo J. A.; Zha J.; Liu J.; Kallop D.; Ludlam M. J.; Pei L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28393442–3455. [DOI] [PubMed] [Google Scholar]
- Ye X.; Li Y.; Stawicki S.; Couto S.; Eastham-Anderson J.; Kallop D.; Weimer R.; Wu Y.; Pei L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29385254–5264. [DOI] [PubMed] [Google Scholar]
- Holland S. J.; Pan A.; Franci C.; Hu Y.; Chang B.; Li W.; Duan M.; Torneros A.; Yu J.; Heckrodt T. J.; Zhang J.; Ding P.; Apatira A.; Chua J.; Brandt R.; Pine P.; Goff D.; Singh R.; Payan D. G.; Hitoshi Y. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 7041544–1554. [DOI] [PubMed] [Google Scholar]
- Koorstra J. B.; Karikari C. A.; Feldmann G.; Bisht S.; Rojas P. L.; Offerhaus G. J.; Alvarez H.; Maitra A. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol. Ther. 2009, 87618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abagyan R.; Totrov M.; Kuznetsov D. ICM—A new method for protein modeling and design—Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.