

Highlights
► We report a fully integrated microdevice incorporating a reverse transcription-polymerase chain reaction (RT-PCR) with an immunochromatographic strip (ICS) for visual detection of rapid gene expression of influenza A H1N1 virus on a portable system. ► An assembled five-layered glass–polydimethylsiloxane hybrid microdevice has a functional unit of a pneumatic micropump, a RT-PCR chamber, a microvalve, and an ICS. ► One-step RT-PCR reaction, gold nanoparticle conjugation, and the colorimetric detection on an ICS were performed successively on a portable system to identify the target H1 gene of H1N1 virus.
Keywords: Integrated microdevice, Reverse transcription-polymerase chain reaction, Immunochromatographic strip, Influenza A H1N1 virus, Portable genetic analyzer
Abstract
An integrated microdevice of a reverse transcription-polymerase chain reaction (RT-PCR) reactor and an immunochromatographic strip was constructed for colorimetric detection of gene expression of influenza A virus subtype H1N1. An RT-PCR cocktail, which included Texas Red-labeled primers, dNTP including biotin-labeled dUTP, and RNA templates of influenza A H1N1 virus, was filled in the PCR chamber through the micropump, and the RT-PCR was performed to amplify the target H1 gene (102 bp). The resultant amplicons bearing biotin moieties and Texas Red haptens were subsequently eluted to the immunochromatographic strip, in which they were first conjugated with the gold nanoparticle labeled anti-hapten antibody in the conjugation pad, and then captured on the streptavidin coated test line through the biotin–streptavidin interaction. By observing a violet color in the test line which was derived from the gold nanoparticle, we confirmed the H1N1 target virus. The entire process on the integrated microdevice consisting of a micropump, a 2 μL PCR chamber, and an immunochromatographic strip was carried out on the portable genetic analyzer within 2.5 h, enabling on-site colorimetric pathogen identification with detection sensitivity of 14.1 pg RNA templates.
1. Introduction
Recently, pandemic viral infections, such as novel H1N1 swine origin influenza A (SOIV) (Medina and Garcia-Sastre, 2011, Kash et al., 2006, Vijaykrishna et al., 2010, Panning et al., 2009), acquired immune deficiency syndrome (AIDS) (Kallings, 2008, Gao et al., 1999), foot-and-mouth disease (Yoon et al., 2011), and severe acute respiratory syndrome (SARS) (Smith, 2006, Marra et al., 2003), caused serious human victims and economic loss over the world. To prevent the pandemic and detect fatal pathogens in the early stage, many diagnostic tools have been developed. Microfluidics-based lab-on-a-chip technology (Whitesides, 2006, Chen et al., 2007) has demonstrated its high performance for genetic analysis to address such problems by utilizing the advantages of rapid analysis (Chen et al., 2010a, Chen et al., 2010b), low reagent consumption (Kim et al., 2010), high integration (Hangan et al., 2011), and portability (Liu et al., 2007). Since polymerase chain reaction (PCR) has been considered as a gold standard method for accurate and sensitive genotyping, several types of microdevices have been presented which incorporate a PCR functional unit (Thaitrong et al., 2010, Liu et al., 2011). Easley et al. have reported an integrated microdevice that consists of a solid-phase extraction column for DNA extraction, PCR and capillary electrophoretic (CE) microchannel on a single device to detect B. pertussis from human nasal aspirate (Easley et al., 2006). However, all the processes of the chip operation and fluorescence detection were carried out in the bulky instrumentation which precluded fast and on-site genetic analysis. Beyor et al. showed a lab-on-a-chip system for pathogen detection by combining the functional micro-unit for cell preconcentration, PCR, and CE separation (Beyor et al., 2009). Although they demonstrated high integration and high sensitivity for Escherichia coli O157 identification, the use of laser-induced fluorescence detection requires high-cost and bulky optical laser, and an expertise in genetic analysis is necessary to interpret the CE data. Donhauser et al. presented the stopped PCR combined with chemiluminescence flow-through DNA microarray assay for detecting Escherichia coli O157:H7, Salmonella enterica, and Campylobacter jejuni (Donhauser et al., 2011). They have shown high sensitivity, quantitative analysis, and multiplex detection of pathogenic bacteria, but the PCR step was performed on the off-chip basis which is not suitable for the on-site pathogen detection. Elsholz et al. have proposed an electrical microarray chip for analyzing multiplex PCR products from biowarfare agents (Elsholz et al., 2009). Despite the ability of high-throughput screening and high sensitivity detection of DNA, they also needed a bulky and expensive electrical sensing system, and suffered from a relatively prolonged reaction time. On the other hand, immunochromatographic strip (ICS) technology has been widely adopted for DNA and protein analysis due to its advantages of a user-friendly format, easy interpretation by detecting a colorimetric signal, a short reaction time, a cheap manufacturing, and less environmental influence on the chromatographic separation (Zhang et al., 2006, Xu et al., 2009, Gervais et al., 2011, Peng et al., 2008). Blažková et al. have presented a simple and rapid ICS test for visual detection of PCR products which were obtained by amplifying 16S rRNA templates of Cronobacter spp. (Blažková et al., 2011). Glynou et al. developed a dry-reagent strip biosensor to identify hepatitis C virus through DNA hybridization by using gold nanoparticle (Au NP) labeled probes (Glynou et al., 2003). However, in both cases, the PCR reaction was conducted by a conventional off-chip PCR method which limits high potential of the ICS technology as a point-of-care testing tool. Previous reports presented a PCR microdevice integrated with a lateral flow strip for pathogen detection (Chen et al., 2010a, Chen et al., 2010b, Wang et al., 2006). However, they utilized up-converting phosphor particles as a reporter which needs an IR laser and a fluorescence reader to scan the lateral flow strip to detect the phosphorescence emission.
In this work, we report a fully integrated microdevice incorporating a micro-RT-PCR with an ICS for visual detection of rapid gene expression of influenza A H1N1 virus on a portable system. In particular, we employed a Au NP as a signal label that facilitate colorimetric detection with naked eyes. An assembled five-layered glass–polydimethylsiloxane (PDMS) hybrid microdevice has a functional unit of a pneumatic micropump, a RT-PCR chamber, a microvalve, and an ICS. One step RT-PCR reaction, Au NP conjugation, and the colorimetric detection on an ICS were performed successively on a portable system to identify the target H1 gene of H1N1 virus. The combination of the RT-PCR microdevice with an ICS technology enables us to accomplish the genetic analysis of H1N1 virus with high speed and convenience.
2. Experimental
2.1. Structure and fabrication of the ICS
The ICS consists of a buffer loading pad, a conjugate pad, a nitrocellulose membrane with a test and control line, and an absorbent pad as shown in Fig. 1A. The conjugate area was immobilized with Au NP labeled anti-hapten antibodies, and the test and control line were coated with streptavidin proteins and anti-mouse IgG, respectively (Zhang et al., 2006, Glynou et al., 2003). The target H1 gene was amplified from the H1N1 viral RNA by using a Texas Red labeled primer and biotin–dUTP, and then the Texas Red and biotin labeled RT-PCR amplicons were absorbed in the conjugate pad. The Texas Red dyes were linked with Au NP labeled antibodies, and a 40 μL of the running buffer (1 × PBS (pH 7.6), 1% BSA, 0.5% PEG, 1% sucrose, 0.1% Tween 20, 0.1% sodium azide) was added in the buffer loading zone to move the Au NP linked DNA conjugates to the nitrocellulose membrane. The Au NP linked amplicons passed through the test line where the streptavidin–biotin interaction occurs, while the excess of Au NP labeled anti-hapten antibody was immobilized with the anti-mouse IgG in the control line (Fig. 1B–D).
Fig. 1.
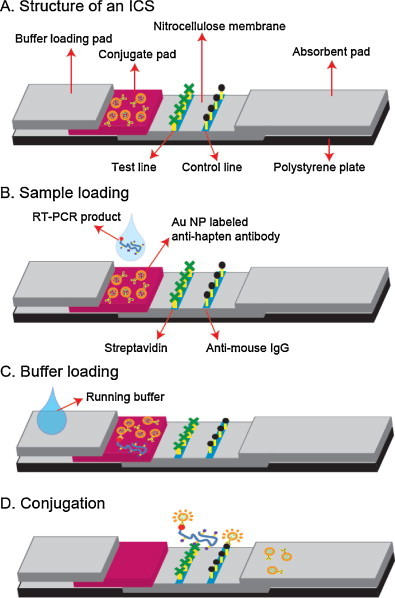
(A) The structure of an ICS. The reaction mechanism on the ICS: (B) The RT-PCR product bearing the biotin and Texas Red hapten was loaded in the conjugate pad to be linked with Au NP labeled anti-hapten antibodies. (C) The Au NP labeled RT-PCR amplicons were moved to the nitrocellulose membrane by adding a buffer in the buffer loading pad. (D) The product was captured by streptavidin on the test line, while the excess of the Au NP labeled anti-hapten antibodies were immobilized on the anti-mouse IgG in the control line.
The ICS was made of a buffer loading pad (3.6 mm × 14 mm), a conjugate pad (3.6 mm × 10 mm), a nitrocellulose membrane (3.6 mm × 25 mm × 8 μm), an absorbent pad (3.6 mm × 19 mm) and a polystyrene plate, which were purchased from Millipore. The nitrocellulose membrane was attached on the middle of the polystyrene plate and then the absorbent pad was adhesive on the nitrocellulose membrane at the right side. The buffer loading pad and conjugate pad were attached on the nitrocellulose membrane at the left side. The embedment of the nanoparticle, streptavidin and antibodies on the ICS was carried out by modifying the process published in the previous report (Zhang et al., 2006). Briefly, colloidal Au NPs (avg. diameter:71.5 nm) were synthesized by adding 0.7 mL of trisodium citrate (1%, w/v) to 100 mL of a chloroauric acid solution (0.01%, w/v) and heating at 90 °C followed by boiling for 5 min. Purified anti-hapten antibody (1 mg) was mixed with 25 mL of the colloidal Au NP solution at pH 8.4. The Au NP conjugated anti-hapten antibody was sprayed onto a conjugate pad. The streptavidin (2.5 mg/mL) and anti-mouse IgG (0.4 mg/mL) were jetted linearly onto the nitrocellulose membrane using a dispensing platform to form a test and control line (0.1 μL/mm2). The prepared nitrocellulose membrane, the conjugate and the buffer loading pad, and the absorbent pad were assembled with an adhesive card.
2.2. Design of an ICS integrated RT-PCR microdevice
The integrated RT-PCR-ICS microdevice is composed of five layers as shown in Fig. 2A: an ICS, a four-point resistance temperature detector (RTD) wafer, a 4 inch glass channel wafer in which a PCR chamber and microvalves (Beyor et al., 2008, Grover et al., 2006, Zhang et al., 2009) were fabricated on the bottom and the top respectively, a monolithic PDMS membrane, and a glass manifold. Schematics of the assembled microdevice are shown in Fig. 2B. The pneumatic micropump was used to load the RT-PCR cocktail from the sample reservoir to the PCR chamber through a hole. For thermal cycling, a film heater (Minco, Minnesota, USA) covered the PCR chamber at the bottom for heating and a fan, placed above the microdevice, was used for fast cooling. Electrical circuits for temperature control were similar to the previous method (Lagally et al., 2004, Toriello et al., 2006). A constant 10 mA current was applied to the RTD through the outer electrodes, and the resultant voltage was sensed in the inner electrodes. The signal was processed using an active low-pass filter at 5 Hz and then transferred to the DAQ board. A proportional/integral/differential (PID) module within the in-house LabVIEW program outputs through the DAQ board to control the heater power supply. The PCR chamber was connected with two exits through the PDMS microvalve: one is for the waste outlet and the other is for the product outlet. The ICS was attached on the bottom of the RTD wafer, and the product outlet and buffer inlet were positioned onto the conjugate pad and buffer loading pad, respectively.
Fig. 2.
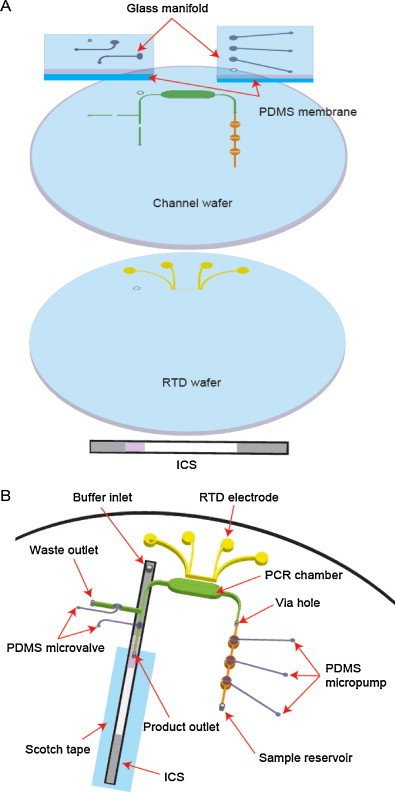
(A) Exploded view of five layers for constructing an integrated RT-PCR-ICS microdevice: from bottom to top, an ICS, a RTD wafer, a channel wafer, a monolithic PDMS membrane, and a glass manifold. (B) Schematics of an integrated RT-PCR-ICS microdevice: a pneumatic micropump (orange), a PCR chamber (green), a microfabricated RTD (yellow), a PDMS microvalve (violet), and an ICS (grey). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
2.3. Microfabrication
The microvalves, a PCR chamber, and microfluidic channels of a channel wafer were fabricated on a 4 in. glass wafer. A 1.1 mm thick borofloat glass was coated with 200 nm amorphous silicon on both sides, and was primed with hexamethyldisilazane (HMDS). Photoresist (Shipley 1818, Marlborough, MA) was spin-coated at 2500 rpm and soft-baked at 120 °C for 90 s. The channel pattern was transferred to the coated substrate via UV exposure and developed using Microposit developer (1:1 with deionized water, Shipley). The developed area of amorphous silicon was removed using SF6 reactive ion etching (RIE) and was immersed in a 49% HF solution for isotropic wet etching. The remaining photoresist was stripped using acetone with sonication, and the amorphous silicon was removed with SF6 RIE. To fabricate a RTD electrode, 20 nm Ti and 200 nm Pt were coated on a borofloat glass wafer, and the RTD pattern was covered with a photoresist. A wet etching with an aqua regia (HCl:HNO3 = 3:1) removed the background of Ti/Pt, and the RTD electrode was finally obtained after cleaning the remaining photoresist with acetone. The channel wafer and the RTD wafer were thermally bonded in a vacuum furnace at 680 °C for 6 h. The RTD electrode was positioned close to the PCR chamber for accurate temperature measurement. The glass manifold was fabricated by using the same procedure as above and was diced with an automatic dicing saw machine (Disco Corp.). To form the microvalve, a monolithic PDMS membrane was pretreated in a UV-ozone cleaner for 1 min, and then sandwiched between the bonded wafer and the glass manifold. An ICS was attached to the bottom of the RTD wafer by using a scotch tape. The digital image of the integrated RT-PCR-ICS microdevice is shown in Fig. 3A.
Fig. 3.

(A) Digital image of the integrated RT-PCR-ICS microdevice on the portable genetic analyzer. (B) Schematics of the portable genetic analyzer which consists of power supplies, a diaphragm pump, solenoid valves, and an electronic control board. (C) Thermal cycling profile with cooling ramping rate of 5.7 °C/s and heating ramping rate of 4.4 °C/s.
2.4. Portable genetic analyzer
To perform the RT-PCR reaction and colorimetric immunochromatographic detection on a portable system, high-voltage power supplies (AA12-P4, Ultravolt, USA), a diaphragm pump (60615, Thomas, USA), solenoid valves (GV010E1, KOGANEI, Japan), and an electronic control board (PCE-e, Nanoscope Systems Inc., Korea) were miniaturized to be packed into the case (21 cm × 24 cm × 10 cm). The portable genetic analyzer (Fig. 3B) communicated with a PC by using a RS-232 protocol and the command from a PC was distributed and calculated in the FGA based control board. Control board regulated the constant current, the change of the voltage across the RTD sensor, and the heater (Blazej et al., 2006, Liu et al., 2007). The sampling frequency of the voltage was 2 MHz and every 512 samples were averaged on the control board. The diaphragm pump and solenoid valves supplied pressure (4.3 psi) and vacuum (−5.8 psi) through a pneumatic line tube to function the PDMS microvalves. The three serial microvalves operated for pneumatic pulsatile on-chip pumping by a LabVIEW program. An external cooling fan was equipped above the integrated microdevice for rapid cooling ramping rate during the PCR thermal cycling, and the thermal cycling temperature was controlled with a PID module. A representative PCR thermal cycling profile is shown in Fig. 3C. All the operation of a microvalve, micropump, and RT-PCR was automatically performed through an in-house LabVIEW program.
2.5. Colorimetric detection of the H1 gene expression of influenza A H1N1 virus on a chip
Prior to introducing a RT-PCR cocktail, the microfluidic channels and the PCR chamber were thoroughly cleaned by a piranha solution (H2SO4:H2O2 = 3:1) at 80 °C for 30 min. Then, 1 M sodium hydroxide and hydrochloric acid were successively injected to remove any impurities in the inner surface of the chip, followed by vigorous washing with deionized water. A 1.45 μg/μL bovine serum albumin (BSA) solution was injected and incubated for 30 min to prevent non-specific interaction of biomolecules on the surface. For one-step RT-PCR reaction, the PCR cocktail was prepared by using Qiagen OneStep RT-PCR kit. In a 10 μL reaction volume, 2 μL of 5× reaction buffer (Tris–HCl, KCl, (NH4)2SO4, 12.5 mM MgCl2, DTT, pH 8.7), 1 μL of 5 mM dNTP mixed with biotin–dUTP (16.4 pmol/μL), 0.4 μL of forward primer (5′-Texas Red-GTG CTA TAA ACA CCA GCC TYC CA-3′; 0.8 pmol/μL) and reverse primer (5′-Texas Red-CGG GAT ATT CCT TAA TCC TGT RGC-3′; 0.8 pmol/μL), Qiagen OneStep enzyme mix (1.6 μL), water (3.6 μL), and 1.0 μL of RNA templates of influenza A H1N1 virus. The concentration of biotin–dUTP was controlled so that most of the biotin–dUTP could be captured in the streptavidin in the test line. The primer design was generated from the primer 3 program and the specificity for amplifying the target H1 gene was confirmed in the capillary electrophoresis by showing only the H1 amplicon peak in the electropherogram without non-specific peak.
For a limit of detection (LOD) test, the RNA template was serially diluted to prepare the concentration of 7.1, 0.71, 0.071, and 0.0071 ng/μL. A 5 μL of RT-PCR cocktail in the sample reservoir was moved to the waste outlet by the PDMS micropump to fill in the PCR chamber which took about 3 min. After closing the microvalves adjacent to the PCR chamber, RT reaction was performed by maintaining the temperature at 42 °C for 30 min. Then, PCR thermal cycling was carried out by an initial activation step at 95 °C for 5 min, 35 amplification cycles of 94 °C for 30 s, 55 °C for 60 s, 72 °C for 60 s, followed by a final extension step at 72 °C for 10 min. The entire RT-PCR reaction was finished in 2 h and 10 min. After RT-PCR reaction, the resultant amplicons were shifted to the product outlet by using a pneumatic pump actuation. The PCR product was absorbed on the conjugate pad of the ICS, and the colorimetric detection on the ICS was completed following the described process in the section of structure and fabrication of the ICS.
3. Results and discussion
3.1. Reaction mechanism on the ICS
The RT-PCR amplicons contained the two moieties of biotin and Texas Red. Biotin was inserted to the DNA strand during the PCR reaction by using the biotin-labeled dUTP, and Texas Red, serving as a hapten, was labeled at the both ends of the RT-PCR amplicons. The ICS consists of the buffer loading zone, the conjugate area, the nitrocellulose membrane which incorporates a test line with streptavidin proteins and a control line with anti-mouse IgG, and an absorbent pad on the polystyrene plate. Once the PCR products were loaded in the conjugate pad, the running buffer was added in the buffer loading pad to make the product move toward the absorbent pad by capillary force. The RT-PCR product was conjugated with Au NP labeled antibodies through the hapten–antibody interaction, and subsequently the biotin of the product was linked with the streptavidins on the test line. Since Au NPs exhibit higher absorption coefficients and scattering cross-sections than the conventional dyes by several orders of magnitude due to strong surface plasmon resonance, Au NP was widely utilized as a reporter for colorimetric detection of biomolecules (Jian and Huang, 2011, Elghanian et al., 1997). Therefore, the presence of target amplicons can be easily recognized by observing the violet color in the test line without any assistance of optical instruments such as IR laser and laser-induced fluorescence detector. The excess of the Au NP labeled antibodies flowed to the control line to be captured on the immobilized anti-mouse IgG. While the control line always showed a violet color as a positive control due to the interaction between the Au NP labeled anti-hapten antibody and the anti-mouse IgG, the test line revealed the same color only when the Au NP labeled amplicons were present. Therefore, the simple colorimetric detection in the test line enables us to identify the target gene expression of influenza A H1N1 virus.
3.2. Portable ICS integrated RT-PCR microsystem
The ICS integrated RT-PCR microdevice in Fig. 3A was fabricated by combining five layers of an ICS, a RTD wafer on which a Ti/Pt electrode was patterned, a channel wafer in which the PCR chamber and the microfluidic channel were fabricated on the bottom while a pneumatic micropump and two microvalves were fabricated on the top side, a PDMS membrane and a glass manifold. The assembled microdevice has three functional units such as a pneumatic microvalve and pulsatile micropump for fluidic control, a RT-PCR reaction, and an ICS based colorimetric detection. The glass channel–PDMS–glass manifold hybrid functions as a pneumatic microvalve, and three serial microvalves work as a pulsatile micropump by opening and closing the valve in consecutive order with 300 ms delay which drives 5 μL of the RT-PCR cocktail in the sample reservoir to the PCR chamber in 3 min without bubble generation. The volume of the PCR chamber was fixed with 2 μL since it turned out that such a relatively large volume was necessary to give a strong signal on an ICS. For rapid thermal cycling of the RT-PCR, the cooling fan was equipped over the integrated microdevice, and the thermal cycling proceeded with the cooling and heating ramping rate of 5.7 °C/s and 4.4 °C/s, respectively. Measured temperature was matched with the set temperature within ±0.5 °C variation through the PID controller. The generated RT-PCR amplicons were loaded to the ICS through the pneumatic micropump. Upon addition of the running buffer, the color change in the test and control lines was observed in 3 min. The disposable ICS which was adherent to the microdevice by a scotch tape was replaced, and the PCR chamber and microfluidic channels were washed with a piranha solution to remove any residual DNA molecules for the next use. The ICS based colorimetric detection allows us to perform the genetic analysis without need for any complicated analytical instrumentation, to interpret the results with ease, and to generate the genetic information with high speed. For portable genetic analysis, a miniaturized hardware including power supplies, a pneumatic pump, solenoid valves, and a control board was constructed, and all the RT-PCR thermal cycling, microvalve actuation, and cooling fan operation were run automatically through the in-house LabVIEW program. Therefore, our portable integrated RT-PCR genetic analyzer could provide an advanced platform for point-of-care DNA analysis.
3.3. Optimization of an ICS based colorimetric detection
To obtain a strong colorimetric signal in the ICS, we optimized the concentration and the structure of biotin–dUTP, since the number of biotin in the amplicons and the spacer length between biotin and dUTP eventually influence the conjugation efficiency with the streptavidin in the test line. Therefore, we performed the RT-PCR reaction by tuning the ratio of dTTP:biotin–dUTP as 1:0, 2:1, 4:1 and 8:1. Simultaneously, the number of carbon between the dUTP and the biotin was controlled by 11, 16, and 18 to investigate how the spacer length reduces steric hindrance for streptavidin to capture the biotin-labeled amplicons. The RT-PCR reaction and the ICS based colorimetric detection were performed on the off-chip basis. As the spacer length increased from 11 to 18 (Fig. 4A–C), the band signal more clearly appeared, meaning the longer distance of biotin from the backbone of the amplicons makes the conjugation efficiency higher. In all the cases of Fig. 4A–C, the amplicons synthesized by adding dTTP:biotin–dUTP with 2:1 ratio revealed the most distinct band signal. Although the PCR efficiency would be decreased by the presence of biotin–dUTP and the shorter spacer in the biotin–dUTP would be better substrate for the polymerase mediated incorporation into the growing DNA molecule, the biotin–dUTP with longer spacers and high quantity produced the most improved colorimetric signal by the favorable streptavidin–biotin complex formation. Therefore, we further conducted the on-chip RT-PCR reaction for visual identification of influenza A H1N1 virus on the ICS by using the biotin–dUTP with 18 carbon linker, and 2:1 ratio of dTTP:biotin–dUTP in the RT-PCR cocktail.
Fig. 4.
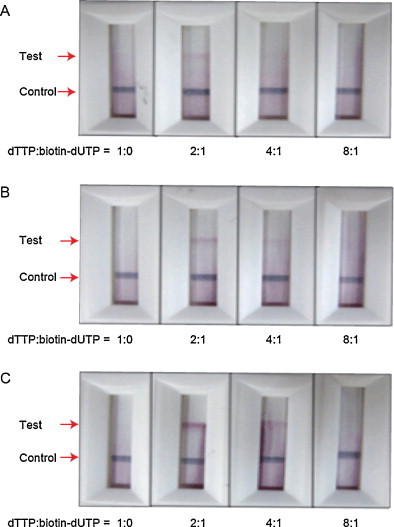
Optimization of an ICS based colorimetric detection by tuning the length of the spacer between biotin and dUTP and the concentration ratio of dTTP and biotin–dUTP. The carbon number of the spacer was (A) 11, (B) 16, and (C) 18. The ratio of dTTP:biotin–dUTP was 1:0, 2:1, 4:1 and 8:1.
3.4. Detection sensitivity comparison
To demonstrate the colorimetric detection sensitivity of the ICS, we compared the intensity of the signal band on the ICS with that of the agarose gel electrophoresis. The target H1 gene (102 bp) was amplified by using one-step RT-PCR reaction on the off-chip basis, and the serially diluted influenza A H1N1 viral RNA templates (1720, 172, 17.2, 1.72 and 0.172 pg) were employed. After RT-PCR reaction, 1 μL of the RT-PCR product was loaded on the ICS (Fig. 5 , top panel) and agarose gel (Fig. 5, bottom panel). While the ICS revealed the violet color signal in the test line even with 17.2 fg of the viral RNA sample, 1.72 pg of RNA was necessary to produce a dim band in the gel electrophoresis, indicating the 100-fold improved detection sensitivity of ICS than the agarose gel. Moreover, the total analysis time of the ICS was within 10 min without any analytical instruments, but the analysis of agarose gel electrophoresis generally takes more than 1 h from the sample loading to UV illumination.
Fig. 5.
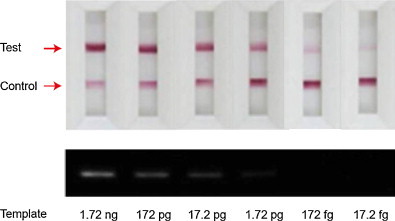
Detection sensitivity comparsion between the ICS (top panel) and the agarose gel electrophoresis (bottom panel) by using the serially diluted viral RNA templates from 1.72 ng to 17.2 fg. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.5. On-chip RT-PCR reaction and ICS based colorimetric detection on the portable genetic analyzer
We performed the gene expression analysis of influenza A H1N1 virus on the integrated microdevice and the miniaturized portable genetic analyzer by using the serially diluted RNA template with 1.41, 0.141, and 0.0141 ng. Fig. 6 shows the resultant ICS data with a negative control experiment. The control line as well as the test line displayed a clear violet color, and the signal intensities were gradually decreased from 1.41 ng to 0.0141 ng of templates from left to right in Fig. 6 0.0141 ng of RNA templates produced a dim band, whereas 0.00141 ng templates resulted in too weak band to be detected. The negative control experiment in which the RNA template was omitted in the RT-PCR cocktail did not give any colorimetric signal in the test line, while the control line had a violet color. Therefore, we could confirm the presence of influenza A H1N1 virus by the colorimetric detection on the portable integrated RT-PCR-ICS microdevice with LOD of 0.0141 ng of RNA templates.
Fig. 6.
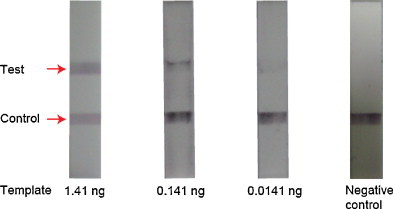
On-chip RT-PCR reaction and ICS based colorimetric detection on the portable genetic analyzer for H1N1 virus identification by using RNA templates of 1.41 ng, 0.141 ng, 0.0141 ng and 0 ng from left to right. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
4. Conclusions
We demonstrated a success of the genetic analysis of influenza A H1N1 virus by using a portable integrated RT-PCR-ICS microdevice within 2.5 h. The use of an ICS as a colorimetric detection method enables us to perform user-friendly interpretation of the results to identify the target virus by naked eyes, thereby eliminating any complicated analytical instrumentation. Once the functional unit for sample pretreatment is further integrated, our advanced microsystem would be adequate for rapid point-of-care genetic analysis in the fields of biomedical diagnostics, food safety testing, and environmental pollutant monitoring.
Acknowledgments
This research was supported by Technology Development Program for Agriculture and Forestry, Ministry for Agriculture, Forestry and Fisheries, Republic of Korea, and by the R&D Program of MKE/KEIT (10035638). We thank Drs. Byung Seon Chun and InCheon Song (Nanoscope Systems, Inc., Daejeon, Korea) for developing the portable system.
References
- Beyor N., Seo T.S., Liu P., Mathies R.A. Biomed. Microdevices. 2008;10:909–917. doi: 10.1007/s10544-008-9206-3. [DOI] [PubMed] [Google Scholar]
- Beyor N., Yi L., Seo T.S., Mathies R.A. Anal. Chem. 2009;81:3523–3528. doi: 10.1021/ac900060r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazej R.G., Kumaresan P., Mathies R.A. Proc. Natl. Acad. Sci. U.S.A. 2006;103:7240–7245. doi: 10.1073/pnas.0602476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blažková M., Javurková B., Fuka L., Rauch P. Biosens. Bioelectron. 2011;26:2828–2834. doi: 10.1016/j.bios.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Chen D., Mauk M., Qiu X., Liu C., Kim J., Pamprasad S., Ongagna S., Abrams W.R., Malamud D., Corstjens P.L.A.M., Bau H.H. Biomed. Microdevices. 2010;12:705–723. doi: 10.1007/s10544-010-9423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Manz A., Day P.J.R. Lab Chip. 2007;7:1413–1423. doi: 10.1039/b708362a. [DOI] [PubMed] [Google Scholar]
- Chen Y., Choi J.Y., Choi S.J., Seo T.S. Electrophoresis. 2010;31:2974–2980. doi: 10.1002/elps.201000270. [DOI] [PubMed] [Google Scholar]
- Donhauser S.C., Niessner R., Seidel M. Anal. Chem. 2011;83:3153–3160. doi: 10.1021/ac2002214. [DOI] [PubMed] [Google Scholar]
- Easley C.J., Karlinsey J.M., Bienvenue J.M., Legendre L.A., Roper M.G., Feldman S.H., Hughes M.A., Hewlett E.L., Merkel T.J., Ferrance J.P., Landers J.P. Proc. Natl. Acad. Sci. U.S.A. 2006;103:19272–19277. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elghanian R., Storhoff J.J., Mucic R.C., Letsinger R.L., Mirkin C.A. Science. 1997;277:1078–1081. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- Elsholz B., Nitsche A., Achenbach J., Ellerbork H., Blohm L., Albers J., Pauli G., Hintsche R., Wörl R. Biosens. Bioelectron. 2009;24:1737–1743. doi: 10.1016/j.bios.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Gao F., Bailes E., Roberson D.L., Chen Y., Rodenburg C.M., Michael S.F., Cummins L.B., Arthur L.O., Peeters M., Shaw G.M., Sharp P.M., Hahn B.H. Nature. 1999;397:436–442. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- Gervais L., Rooij N.D., Delamarche E. Adv. Mater. 2011;23:H151–H176. doi: 10.1002/adma.201100464. [DOI] [PubMed] [Google Scholar]
- Glynou K., Ioannou P.C., Christopoulos T.K., Syriopoulou V. Anal. Chem. 2003;75:4155–4160. doi: 10.1021/ac034256+. [DOI] [PubMed] [Google Scholar]
- Grover W.H., Ivester R.H.C., Jensen E.C., Mathies R.A. Lab Chip. 2006;6:623–631. doi: 10.1039/b518362f. [DOI] [PubMed] [Google Scholar]
- Hangan K.A., Reedy C.R., Uchimoto M.L., Basu D., Engel D.A., Landers J.P. Lab Chip. 2011;11:957–961. doi: 10.1039/c0lc00136h. [DOI] [PubMed] [Google Scholar]
- Jian J., Huang C. Chem. Eur. J. 2011;17:2374–2380. doi: 10.1002/chem.201002825. [DOI] [PubMed] [Google Scholar]
- Kallings L.O.J. Intern. Med. 2008;263:218–243. doi: 10.1111/j.1365-2796.2007.01910.x. [DOI] [PubMed] [Google Scholar]
- Kash J.C., Tumpey T.M., Proll S.C., Carter C., Perwitasari O., Thomas M.J., Basler C.F., Palese P., Taubenberger J.K., Garcia-Saster A., Swayne D.E., Katze M.G. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.J., Shin G.W., Choi S.J., Hwang H.S., Jung G.Y., Seo T.S. Electrophoresis. 2010;31:1108–1115. doi: 10.1002/elps.200900651. [DOI] [PubMed] [Google Scholar]
- Lagally E.T., Scherer J.R., Blazej R.G., Toriello N.M., Diep B.A., Ramchandani M., Sensabaugh G.F., Riley L.W., Mathies R.A. Anal. Chem. 2004;76:3162–3170. doi: 10.1021/ac035310p. [DOI] [PubMed] [Google Scholar]
- Liu P., Li X., Greenspoon S.A., Scherer J.R., Mathies R.A. Lab Chip. 2011;11:1041–1048. doi: 10.1039/c0lc00533a. [DOI] [PubMed] [Google Scholar]
- Liu P., Seo T.S., Beyor N., Shin K., Scherer J.R., Mathies R.A. Anal. Chem. 2007;79:1881–1889. doi: 10.1021/ac061961k. [DOI] [PubMed] [Google Scholar]
- Marra M.A. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Medina R.A., Garcia-Sastre A. Nat. Rev. Microbiol. 2011;9:590–603. doi: 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panning M., eickmann M., Landt O., Monazahian M., Olschlager S., Baumgarte S., Reischl U., Wenzel J.J., Niller H.H., Gunther S., Hollmann1 B., Huzly1 D., Drexler J.F., Helmer A., Becker S., Matz B., Eis-Hubinger A.M., Drosten C. Eur. Surveill. 2009;14(36):19329–19334. [PubMed] [Google Scholar]
- Peng F., Wang Z., Zhang S., Wu R., Hu S., Li Z., Wang X., Bi D. Clin. Vaccine Immunol. 2008;15:569–574. doi: 10.1128/CVI.00273-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.D. Soc. Sci. Med. 2006;63:3113–3123. doi: 10.1016/j.socscimed.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaitrong N., Liu P., Briese T., Lipkin W.I., Chiesl T.N., Higa Y., Mathies R.A. Anal. Chem. 2010;82:10102–10109. doi: 10.1021/ac1020744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello N.M., Liu C.N., Mathies R.A. Anal. Chem. 2006;78:7997–8003. doi: 10.1021/ac061058k. [DOI] [PubMed] [Google Scholar]
- Vijaykrishna D., Poon L.L.M., Zhu H.C., Ma S.K., Li O.T.W., Cheung C.L., Smith G.J.K., Peiris J.S.M., Guan Y. Science. 2010;328:1529. doi: 10.1126/science.1189132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Chen Z., Corstjens P.L.A.M., Mauk M.G., Bau H.H. Lab Chip. 2006;6:46–53. doi: 10.1039/b511494b. [DOI] [PubMed] [Google Scholar]
- Whitesides G.M. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- Xu H., Mao X., Zeng Q., Wang S., Kawde A., Liu G. Anal. Chem. 2009;81:669–675. doi: 10.1021/ac8020592. [DOI] [PubMed] [Google Scholar]
- Yoon S.H., Park W., King D.P., Kim H. Mol. Cells. 2011;31:413–421. doi: 10.1007/s10059-011-0249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G., Gua J., Wang X., Yang J., Yang Y., Li Q., Li X., Deng R., Xiao Z., Yang J., Xing G., Zhao D. Vet. Parasitol. 2006;137:286–293. doi: 10.1016/j.vetpar.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Zhang W., Lin S., Wang C., Hu J., Li C., Zhuang Z., Zhou Y., Mathies R.A., Yang C.J. Lab Chip. 2009;9:3088–3094. doi: 10.1039/b907254c. [DOI] [PubMed] [Google Scholar]