

Abstract
Excess body weight is a major risk factor for cardiovascular disease, increasing the risk of hypertension, hyperglycaemia and dyslipidaemia, recognized as the metabolic syndrome. Adipose tissue acts as an endocrine organ by producing various signalling cytokines called adipokines (including leptin, free fatty acids, tumour necrosis factor-α, interleukin-6, C-reactive protein, angiotensinogen and adiponectin). A chronic dysregulation of certain adipokines can have deleterious effects on insulin signalling. Chronic sympathetic overactivity is also known to be present in central obesity, and recent findings demonstrate the consequence of an elevated sympathetic outflow to organs such as the heart, kidneys and blood vessels. Chronic sympathetic nervous system overactivity can also contribute to a further decline of insulin sensitivity, creating a vicious cycle that may contribute to the development of the metabolic syndrome and hypertension. The cause of this overactivity is not clear, but may be driven by certain adipokines. The purpose of this review is to summarize how obesity, notably central or visceral as observed in the metabolic syndrome, leads to adipokine expression contributing to changes in insulin sensitivity and overactivity of the sympathetic nervous system.
Christopher Minson is a Professor of Human Physiology at the University of Oregon. He has a long-standing interest in sympathetic and vascular regulation in humans. Primary areas of research include investigating the effects of sex steroids on sympathetic and vascular function, and understanding neural–vascular interactions in the skin. Current projects include investigating the role of the sympathetic nervous system in obesity and insulin resistance, particularly related to sex steroid regulation of endocrine function in women, and how alterations in normal function may contribute to clinical conditions such as the polycystic ovary syndrome.
 |
Introduction
Obesity is a growing world epidemic resulting in massive increases in medical costs. It has been estimated that medical costs for obese patients is 42% higher than normal weight patients and the annual financial burden from obesity on the medical system in the United States alone is estimated to be as high as $147 billion annually (Finkelstein et al. 2009). In addition to cost, there are a cluster of well known co-morbidities associated with obesity, including but not limited to cardiovascular disease, type 2 diabetes, osteoarthritis and specific cancers (Dixon, 2009). Obesity also contributes to cognitive impairment and increased risk for dementia later in life, independent of co-morbidities such as cardiovascular disease and diabetes (Whitmer et al. 2008).
The purpose of this review is to present an overview of our current understanding of the relationships between obesity, insulin resistance and sympathetic outflow. A review of this topic is timely in that obesity has recently been recognized to contribute to cardiovascular mortality and morbidity through an increased sympathetic drive leading to end organ damage and hypertension (Lambert et al. 2010). Additionally, an elevated sympathetic outflow contributes to the decline of insulin sensitivity and increased mortality from diseases like advanced heart failure (Munhoz et al. 2009) and post-acute thromboembolic cerebrovascular accident (Sander et al. 2001). Examining the different factors that may contribute to increased sympathetic outflow could help in the development of targets for treatment in an effort to reduce the consequences of obesity. This paper will review the effects of increased expression of key cytokines released from adipocytes, termed adipokines, including how they work independently and together to create a state of insulin resistance and chronic sympathetic overactivity.
Obesity and sympathetic overdrive
Increased body fat deposition has been specifically correlated to sympathetic overdrive at rest, with resting levels of muscle sympathetic nerve activity (MSNA) in the obese reported as greater than 50% higher in some studies (Alvarez et al. 2002; Sivenius et al. 2003; Grassi et al. 2004). Elevated levels of MSNA are associated with obesity-induced subclinical organ damage to the heart, blood vessels, and kidneys in young subjects, even in the absence of hypertension (Lambert et al. 2010). To date, there have been four competing hypotheses put forth to explain the aetiology of the metabolic syndrome, and sympathetic overactivity seems to play a fundamental role in all of them (Straznicky et al. 2008). This strongly suggests the sympathetic nervous system (SNS) and factors that contribute to its overactivity in obesity should be considered a target for intervention early in the process of metabolic syndrome development.
Activation of the SNS is not equally targeted to a given body type or a specific organ system. Individuals with central obesity demonstrate augmented sympathetic outflow when compared to non-central adiposity body types (Alvarez et al. 2004). This has been observed via MSNA recordings of post-ganglionic sympathetic nerve fibres (Huggett et al. 2004), urinary noradrenaline (NA) excretion (Lee et al. 2001) and renal NA spillover (Rumantir et al. 1999) even when hypertension is not present, although the presence of hypertension in metabolic syndrome results in a further augmentation of sympathetic activity (Huggett et al. 2004; Grassi et al. 2005). Within this central adiposity phenotype, activation of the SNS is not homogeneous. Specifically, SNS outflow to the skeletal muscle vasculature and the renal system is elevated, whereas activation to the heart is reduced (Vaz et al. 1997). SNS activation to the skin (Grassi et al. 1998a) and splanchnic regions (Vaz et al. 1997) remain unchanged in both forms of obesity. Importantly, the increase in MSNA observed in obese subjects results from an increase in the recruitment of previously silent fibres, and not from an increase in the firing probability of already active sympathetic vasoconstrictor fibres (Lambert et al. 2007). Of note, adrenal adrenaline secretion does not appear to be increased in obese humans (Morgan et al. 1993; Brunner et al. 2002).
The underlying cause of the observed SNS overdrive in obesity is not fully understood, but is likely to be multifactorial and starts as part of a homeostatic mechanism aimed at expending the excess energy as heat rather than additional storage of fat. The accumulation of body fat resulting from a positive energy balance was first shown in animal models to result in SNS activation to a variety of peripheral tissues as a key component of the counter-regulatory response (Landsberg & Young, 1981; Landsberg, 1990; Landsberg & Young, 1993). SNS activation is also seen in humans following overfeeding (Arone et al. 1995; Dulloo & Jacquet, 2001), and is decreased with fasting (Young & Landsberg, 1977). The chronic increase in basal SNA is presumably aimed at stimulating β-adrenergic thermogenesis to prevent further fat storage (Dulloo, 2002), but can also stimulate lipolysis to increase non-esterified free fatty acids (NEFAs), contributing to insulin resistance. Adipose tissue itself can act as an endocrine organ and express various adipokines, which may directly or indirectly increase sympathetic activity. A chronically elevated SNS outflow could in turn impair β-adrenergic signalling, reduce stimulation of metabolism, and evolve into a cycle that contributes to obesity, insulin resistance, and morphological changes in target tissues.
Insulin, insulin resistance and sympathetic outflow
It has been well documented that insulin can augment sympathetic outflow in animals via intracerebroventricular administration (Muntzel et al. 1994; Rahmouni et al. 2004). Sympathetic outflow increases upon the injection of insulin into the third cerebral ventricle of rats (Muntzel et al. 1994), with recent data identifying the arcuate nucleus, via the paraventricular nucleus of the hypothalamus, as the specific site at which insulin acts to increase the sympathetic nervous system and increase sympathetic baroreflex gain (Cassaglia et al. 2011). While very little insulin is produced in the central nervous system, central insulin receptors are found on the hypothalamus (Hopkins & Williams, 1997), and can cause a co-activation of the SNS through transport-mediated uptake across the blood–brain barrier of peripherally secreted insulin. In addition, the arcuate nucleus is unusual in that it contains highly permeable capillaries (Ciofi, 2011), such that insulin may directly activate receptors in this area without a specific transport mechanism (Dampney, 2011). In humans, evidence demonstrates that insulin release in response to a mixed meal (Fagius & Berne, 1994; Cox et al. 1995; Young et al. 2010) or during a hyperinsulinaemic euglycaemic clamp (Anderson et al. 1991; Berne et al. 1992; Vollenweider et al. 1993; Hausberg et al. 1995; Van De Borne et al. 1999) increases MSNA and enhances the arterial baroreflex gain of sympathetic nerve activity (Young et al. 2010). These findings may be taken to suggest hyperinsulinaemia may contribute to the sympathetic overdrive in obesity.
There is a problem with the theory that excess insulin drives the sympathetic overactivity in obesity, however. Specifically, obese subjects do not appear to retain their sensitivity to the stimulatory effects of insulin on the SNS. In lean subjects, MSNA increased 94% in response to euglycaemic hyperinsulinaemia, much greater than the 9% increase observed in age-matched obese subjects (Vollenweider et al. 1994). Similarly, patients with insulin-resistant metabolic syndrome also have demonstrated blunted sympathetic responses to increased plasma insulin following a glucose load (Straznicky et al. 2009a, b), consistent with central insulin resistance in this population. So what factors are causing the SNS to remain elevated in obesity?
There is emerging evidence that certain adipokines expressed in central obesity may contribute to the state of sympathetic overdrive. In addition, certain adipokines when overexpressed may contribute to insulin resistance, ultimately resulting in a hyperinsulinaemic state in concert with greater sympathetic outflow. Insulin resistance is the eventual failure of glucose transporter-4 (GLUT-4) to translocate to the cell membrane to uptake blood glucose into peripheral tissues, predominately skeletal muscle (DeFronzo et al. 1981). It has been demonstrated that the failure is due to alterations in a cellular signalling mechanism, as insulin resistant individuals have normal GLUT-4 levels (Garvey et al. 1998; Shulman, 2000). When GLUT-4 fails to translocate or become activated and uptake blood glucose into peripheral tissues, blood glucose remains elevated. These abnormal blood glucose levels continue to trigger insulin release (Hedeskov, 1980), causing a hyperinsulinaemic state. Many adipokines expressed in both central and peripheral fat cells are implicated in creating this hyperinsulinaemic state through their deleterious effects on cellular insulin signalling, as displayed in Fig. 1.
Figure 1. The cellular effects of adipokines on insulin signalling.
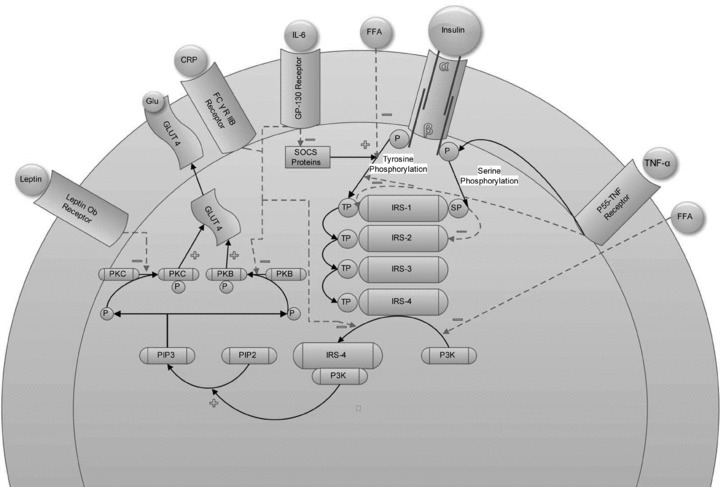
Leptin has been shown to decrease phosphorylation of protein kinase C (PKC) which causes a failure of GLUT-4 translocation which then cannot bring glucose (glu) into the cell. C-reactive protein (CRP) can act in concert with interleukin-6 (IL-6) to decrease protein kinase B (PKB) phosphorylation, which will result in decreased GLUT-4 translocation. IL-6 can also inhibit the activation of insulin receptor substrate (IRS) by phosphatidylinositol 3-kinase (PI3K) and down-regulate the expression of the suppressor of cytokine signalling (SOCS) protein which normally serves a protective role in the effect of cytokines on cellular signalling. It is important to note that IL-6 only demonstrates deleterious effects on insulin signalling when chronically released. Free fatty acids (FFA) can impair one of the first steps in insulin signalling, tyrosine phosphorylation (TP) and impair activation of IRS by PI3K. Tumour necrosis factor-α (TNF-α) can impair insulin signalling by up-regulating serine/threonine phosphorylation, which has been associated with a decrease in GLUT-4 translocation, or by inhibiting phosphorylated tyrosine binding to IRS.
In addition to the effects a dysregulation of adipokines may have on insulin sensitivity resulting from impaired GLUT-4 translocation, chronically elevated SNS activity secondary to increased adipokine expression may reduce the efficiency by which GLUT-4 transports glucose into the cell. Sustained SNS activity on adipose α1-adrenergic receptors can elevate intracellular calcium, which can then inhibit the action of GLUT-4 via activation of phospholipase-Cβ and impaired dephosphorylation of GLUT-4 (McCarty, 2004). When GLUT-4 is phosphorylated, it can still translocate to the cell membrane but its intrinsic activity is decreased, thus impairing the ability of the cell to uptake glucose (Reusch et al. 1991). Evidence for the positive relationship between intracellular calcium and insulin resistance has been demonstrated in rats (Jang et al. 2002) and in humans diagnosed with hyperparathyroidism (Kumar et al. 1994).
In obesity induced hyperinsulinaemia, the sympathetic nervous system may also be co-activated by the hypothalamic–pituitary–adrenal (HPA) axis. Co-activation has been observed by measuring MSNA (Anderson et al. 1991; Berne et al. 1992; Vollenweider et al. 1995), plasma NA (Rowe et al. 1981; O'Hare et al. 1989; Berne et al. 1992; Vollenweider et al. 1995), or NA spillover (Lembo et al. 1992), during direct intracerebroventricular injection of cortisol releasing hormone (Habib et al. 2000). When dexamethasone was used to acutely block cortisol releasing hormone stimulated by stress or hyperinsulinaemia, no increase in MSNA was observed (Stratakis & Chrousos, 1995), providing strong evidence the two brain centres can be co-activated in the presence of hyperinsulinaemia.
Another mechanism that could contribute to SNS hyperactivity is a polymorphism on the gly16 allele of the β2-adrenergic receptor found on pancreatic β-cells. This particular polymorphism is associated with decreased insulin sensitivity, elevated blood pressure and NA levels, and a blunted response to β2-adrenergic agonists (Masuo et al. 2005). Since these particular β2-adrenergic receptors are implicated in augmenting the release of insulin, a decreased sensitivity would require additional sympathetic influence to achieve desired insulin levels. It is not clear whether diminished receptor response causes sympathetic overdrive, or if sympathetic overdrive, secondary to insulin resistance, causes decreased receptor sensitivity. Using either explanation, the end result may be that a decreased sensitivity of β2-adrenergic receptors found on pancreatic β-cells is partially responsible for the sympathetic overdrive (Masuo et al. 2005).
In addition to the well documented neural SNS overactivity at rest, obese subjects demonstrate baroreflex impairments and blunted responses to sympathoexcitatory manoeuvres. Diminished cardiovagal baroreflex responses have been reported in normotensive obese humans in a number of studies (Grassi et al. 1995; Alvarez et al. 2002; Beske et al. 2002), with a further attenuation observed in obese hypertensive patients (Grassi et al. 2000). The sympathetic baroreflex has also been reported to be reduced with obesity (Grassi et al. 1995), although this finding is not ubiquitous (Alvarez et al. 2002). The reason for the discrepancy in findings may be related to the degree of obesity in the studies. In the study by Alvarez et al. (2002), the subjects’ BMI was less than 28 kg m−2 in both the low abdominal visceral fat and high abdominal visceral fat groups versus 40 kg m−2 in the study by Grassi et al. (1995).
The exact cause of changes in baroreflex function with obesity is not entirely clear. In lean humans, insulin is known to increase the sympathetic baroreflex in response to a hyperinsulinaemic–euglycaemic clamp, and in response to a mixed meal (Young et al. 2010). Thus, decreased insulin-mediated sympathetic baroreflex function in the presence of insulin resistance may be expected. However, weight gain and obesity are also associated with increased stiffness of the central arteries in which the baroreceptors lie (Orr et al. 2008), suggesting obesity-related changes in baroreceptor signalling may be a contributing factor to sympathetic overactivity and reduced baroreflex responsiveness. In further support of this notion, it has been demonstrated that obesity is associated with accelerated atherosclerosis in the carotid artery (Lakka et al. 2001), conceivably in part through the actions of adipokines such as leptin, which is independently associated with a thickening of the carotid intima-media (Ciccone et al. 2001). In children, obesity is also associated with a decreased arterial compliance, presumably as an effect of both endothelial dysfunction and structural changes to vessels, and these are possibly early markers of atherogenesis (Tounian et al. 2001). Even in older, overweight humans without insulin resistance or diagnosed metabolic syndrome, carotid intima media thickness was greater than their normal weight peers (Lind et al. 2011). Blood glucose values are also associated with carotid intima-media thickening, even when these blood glucose values lie on the high end of what is considered normal (Thomas et al. 2004). In the Linosa study, an investigation of the metabolic syndrome in an isolated Italian community, insulin resistance correlated negatively with cardiovagal baroreflex gain, and the degree of this baroreflex impairment was associated with increased carotid artery thickness (Lucini et al. 2006). Adult obese Zucker rats also have demonstrated baroreflex-mediated changes in SNS activity similar to humans (Grassi et al. 2000). However, a recent study demonstrated that these changes were due to a central mechanism and not to deficits in the responsiveness of aortic baroreceptors to changes in arterial pressure (Huber & Schreihofer, 2010). Lastly, oxidative stress is known to be increased in obesity (Keaney et al. 2003) and may contribute to cardiovagal dysfunction. Ascorbic acid infusion has been shown to improve cardiovagal baroreflex sensitivity in older adults (Monahan et al. 2004), indicating a link between excess reactive oxygen species and the cardiovagal baroreflex. As discussed in greater detail below, reactive oxygen species may also have a role in sympathetic overactivity and autonomic dysfunction in obesity.
Weight loss through caloric restriction can reduce arterial stiffness in overweight/obese humans (Dengo et al. 2010), and has been demonstrated to increase the sensitivity of the cardiovagal baroreflex (Alvarez et al. 2005) and the sympathetic baroreflex (Grassi et al. 1998b). In a recent study in obese subjects assigned to a hypocaloric diet resulting in weight loss of 9% (Straznicky et al. 2011), MSNA and whole body NA spillover rate were dramatically reduced during active weight loss. However, during weight maintenance following weight loss, MSNA rebounded while NA spillover was preserved, suggesting organ-specific differentiation in SNS adaptation under conditions of negative versus stable energy balance. These intriguing findings demonstrate our knowledge on SNS in obesity, including what factors drive the overactivity and the role of the baroreflexes, is far from complete.
Independent and interactive effects of adipokines on sympathetic regulation
The following are several key cytokines and enzymes released from adipocytes which may alter SNS activity and impair insulin signalling. Among the most prominent in contributing to these observed symptoms in obesity and metabolic syndrome include leptin, non-esterified free fatty acids (NEFAs), tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), C-reactive protein (CRP), angiotensinogen and adiponectin (Fig. 2). A complicated relationship exists between these adipokines that we are just beginning to understand, as many of them work in concert with others to impair insulin signalling, create a state of inflammation, and/or interact to alter sympathetic regulation (Fig. 3).
Figure 2. Specific pathways by which adipokines augment sympathetic outflow in obesity.
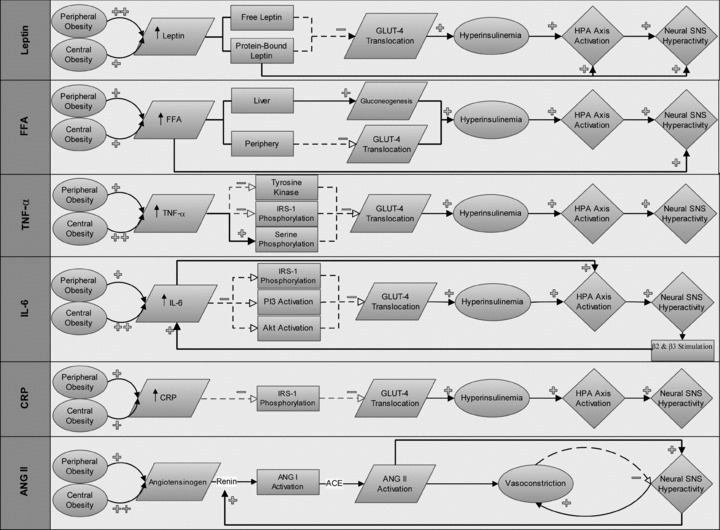
Leptin is preferentially expressed in peripheral fat cells and both free and protein-bound leptin can impair glucose transporter-4 (GLUT-4) which creates a hyperinsulinaemic state and results in a co-activation of the sympathetic nervous system (SNS). Leptin can also act through a central mechanism. Free fatty acids (FFA) are equally expressed in central and peripheral obesity and in addition to causing a decrease in GLUT-4 translocation, can increase blood sugar and ultimately insulin levels through up-regulating gluconeogenesis in the liver. There is also evidence FFA can stimulate the SNS by a central mechanism. TNF-α is preferentially expressed in central obesity and through interacting with several key steps of insulin transduction, can cause a decrease in GLUT-4 translocation and ultimately increase blood glucose levels. Interleukin-6 (IL-6) is preferentially expressed in central obesity and stimulates neural SNS hyperactivity through effects on insulin signalling, direct HPA activation and in positive feedback fashion with β2- and β3-adrenoceptors on adipocytes. C-reactive protein (CRP) acts by impairing insulin signalling causing the resultant hyperinsulinaemia and co-activation of the neural SNS. Angiotensinogen expression is up-regulated in obesity which, in concert with the observed increase in renal SNS and resultant renin release, may act through mass action to cause a vasoconstriction and activation of the SNS by angiotensin II (ANG II) formation.
Figure 3. Co-expression and mechanisms of adipokines in obesity.
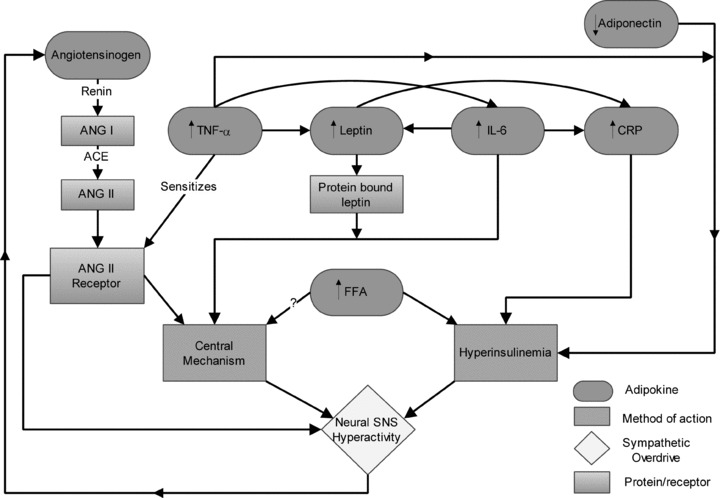
In obesity, key adipokines which can result in sympathetic overdrive are dysregulated (angiotensinogen, tumour necrosis factor-α, adiponectin, leptin, free fatty acids, interleukin-6 and C reactive protein) and can act as endocrine and paracrine hormones to up-regulate expression of other adipokines. In the case of TNF-α they can sensitize angiotensin II receptors which, when activated, can cause sympathetic outflow through a central mechanism. These adipokines then work to increase sympathetic outflow either through their effects on insulin sensitivity, or through central cardiovascular control centres directly.
Leptin
Leptin is a 16 kDa protein produced principally in adipocytes in proportion to the amount of fat mass, although it has also been shown to be released from the brain (Esler et al. 1998). Production of leptin from adipocytes acts in a negative feedback loop to suppress appetite and prevent weight gain, as postulated in the lipostatic theory of body-weight set point (Friedman, 2000). Leptin infusion has been observed to increase sympathetic outflow in a dose-dependent manner in Sprague–Dawley rats (Haynes et al. 1997a,b), similar to the pattern of sympathetic activation in human obesity. Some notable exceptions in humans are that adrenaline secretion rates are normal and sympathetic activity to the heart is not increased (Vaz et al. 1997; Esler et al. 2006), whereas these changes were observed with leptin in rats.
Leptin concentration may act centrally to impact SNS overactivity. Two key sites associated with autonomic regulation have been identified that have leptin receptors. Leptin receptors in the hypothalamic arcuate nucleus (Grill, 2006) appear to be responsible for regulating glucose homeostasis and body weight (Coppari et al. 2005), and may also act to signal SNS activity. There are also leptin receptors on the nucleus tractus solitarii (NTS; Mark et al. 2009), a site where leptin can cause direct SNS activation. Evidence for this exists in the fact that leptin is found in two forms: protein-bound leptin and unbound leptin. Protein-bound leptin can cross the blood–brain barrier (Brabant et al. 2000). Tank et al. (2003) reported that unbound leptin is not associated with MSNA, as opposed to protein bound leptin. Chronic leptin stimulation may be expected to result in brain leptin resistance. However, it has been suggested that brain leptin resistance may be selective, such that the effect on appetite is inhibited, whereas that on sympathetic activity is preserved (Mark et al. 1999). More recently, leptin has been shown in an animal model to act through cardiovascular control centres in the brain to specifically target sympathetic outflow to the kidney (Mark et al. 2009). Further, microinjection of leptin into the NTS has been shown to impair baroreflex control of heart rate (Arnold et al. 2009).
It has been demonstrated that leptin levels are more closely linked in humans to peripheral than central fat stores (Russell et al. 1998; Van Harmelen et al. 1998). Thus, the association of leptin to SNS activity is more controversial than in animal models. Some studies show a positive but not strong relationship between leptin and measures of whole body and regional sympathetic activity (Snitker et al. 1997; Monroe et al. 2000), while others do not (Narkiewicz et al. 2001; Alvarez et al. 2002). Alvarez et al. (2002) studied non-obese men and men with subcutaneous obesity but did not find a correlation between leptin and MSNA, despite the group with subcutaneous obesity having a 2.6-fold higher plasma leptin concentration. This underscores the concept that leptin may only contribute to sympathetic outflow in visceral obesity. Additionally, there may be an interplay between leptin and other adipokines associated with central obesity that ultimately drive the SNS overactivity. Leptin itself exerts a paracrine effect on fat cells, and its expression and secretion by fat cells can be induced by IL-6 and inhibited by TNF-α (Lau et al. 2005).
Non-esterified fatty acids
In humans, circulating levels of NEFAs are increased in obesity and augmented levels are inversely correlated with insulin sensitivity (Bruce et al. 1994; Perseghin et al. 1997). NEFAs act on all aspects of glucose homeostasis, from uptake at peripheral tissue to hepatic production and disposal. Elevated NEFA levels inhibit glucose uptake into peripheral tissues by impairing phosphatidylinositol kinase (PI3-kinase) activation and IRS-1 phosphorylation following insulin stimulation, which ultimately may impair insulin-mediated GLUT-4 translocation (Kruszynska et al. 2002). NEFA disrupting insulin signalling may also be due to impaired PI3-kinase activation due to excess protein kinase C (Griffin et al. 1999). Further supporting this notion is the evidence that PI3-kinase activation is lost as a result of a high fat diet in mice (Zierath et al. 1997).
As NEFA can cross the blood–brain barrier (Lam et al. 2005), it has been found to cause a central activation of MSNA in lean subjects (Florian & Pawelczyk, 2009) and reduce cardiovagal baroreflex in lean and obese subjects (Gadegbeku, 2002). NEFA also stimulates plasminogen activator inhibitor-1, which may also contribute to the association between increased plasma NEFA in obesity and the augmentation of MSNA (Kohler & Grant, 2000). However, this concept has been the subject of conflicting results, with another study demonstrating no significant effect of NEFA on MSNA or sympathetic baroreflex sensitivity (Monahan et al. 2007). Furthermore, whole body and renal noradrenaline spillover did not change during NEFA infusion (Grekin, 2005).
Tumour necrosis factor-α
TNF-α is a protein that acts as a cytokine with a primary role in stimulating the acute phase of inflammation. It also has roles in inducing apoptosis, and inhibiting viral replication and tumorigenesis. TNF-α is capable of crossing the blood–brain barrier (Banks et al. 1995) and may exert its effects in this capacity as well as peripherally and integratively with other adipokines. In obesity, it has been shown in both the animal (Hofmann et al. 1994; Hotamisligil et al. 1994) and human model (Hotamisligil et al. 1995; Kern et al. 1995) that TNF-α is over-expressed when compared with lean subjects. TNF-α has a positive relationship with waist-to-hip ratio but not BMI, suggesting a preferential expression in central adiposity (Tsigos et al. 1999). TNF-α will also stimulate lipolysis (Feingold et al. 1992), which will increase NEFA levels. These augmented NEFA levels and resultant impairment of insulin signalling can cause a hyperinsulinaemic state and possibly a co-activation of the SNS as previously discussed. TNF-α is also responsible for signalling increased IL-6 production (Berne et al. 1992), further underscoring the interplay between adipokines in SNS hyperactivity associated with obesity.
Interleukin-6
IL-6 is a protein that acts in the acute-phase reaction of the inflammation model and is considered to have pro-inflammatory and anti-inflammatory effects. IL-6 is produced from smooth muscle and helper T cells and as an adipokine. It is the interleukin associated with direct action on the hypothalamus by crossing the blood–brain barrier (Banks et al. 1995) to induce fever, and may cause a co-activation of the SNS through its effects on the HPA axis. IL-6 also releases acute phase proteins into the blood, including CRP. Paradoxically, IL-6 can also be released from muscular contraction, as appearance of IL-6 has been observed following exercise (Febbraio & Pedersen, 2005) and in this context can augment insulin sensitivity (Petersen & Pedersen, 2005).
In obesity, up to 30% of circulating IL-6 is secreted from adipocytes (Mohamed-Ali et al. 2001) and is preferentially secreted from visceral adipocytes at a rate 2–3 times that of subcutaneous adipocytes (Lang & Dobrescu, 1989). Rather than an over-expression of IL-6 from adipocytes, it has been demonstrated that IL-6 production is linked to the increasing number of adipocytes in obesity (Bastard et al. 1999). In the human and rat model, stimulation of β2- and β3-adrenoceptors on adipocytes results in increased production of IL-6 (Mohamed-Ali et al. 1997), which could conceivably act in a positive feedback loop with the SNS, potentially contributing to the observed elevations in obesity.
An increased production of IL-6 may drive SNS activity by acting centrally. In IL-6 knockout mice, there is a blunted heart rate and energy expenditure response to stress and exposure to cold, respectively (Wernstedt et al. 2006). With central administration of IL-6, an enhancement of uncoupling protein activity and an increase in oxygen consumption is seen, suggesting a direct link to SNS activation (Wallenius et al. 2002). Evidence for IL-6 activating the HPA axis is provided by the appearance of adrenocorticotrophic releasing hormone (Mastorakos et al. 1994) and corticosterone (Lenczowski et al. 1999) following IL-6 administration. This co-activation is normally considered a part of the inflammation process (Chrousos, 1995), but may partially explain the effects of increased IL-6 production in obesity and the correlated increase in sympathetic outflow. In this case, the SNS activation may be an indication of the chronic inflammatory state observed in central obesity.
IL-6 can also affect insulin sensitivity. Plasma IL-6 levels are significantly and inversely proportional to insulin sensitivity (Kern et al. 2001). Mechanistically this may be attributed to the hypothesis that cytokines such as IL-6 can induce cellular protein expression, through a specific family of proteins called ‘suppressors of cytokine signalling’ (SOCSs). SOCSs normally act to suppress the effects of cytokines on insulin transduction steps such as IRS-1 phosphorylation, PI3 activation or protein kinase B activation (Senn et al. 2003). Inhibition of any one of these steps will cause a failure of GLUT-4 translocation and result in a hyperinsulinaemic state (see Fig. 1). These SOCS proteins are normally expressed in a pulsatile fashion. However, in obesity and the resultant chronic elevation of IL-6, SOCS proteins are inhibited.
Conflicting with the role of IL-6 in down-regulating insulin transduction is the paradoxical finding that increasing IL-6 levels can also be correlated to the postexercise period and observed heightened insulin sensitivity. Presumably, IL-6 is being released due to muscle contraction as a myokine. It has been demonstrated that in this capacity, IL-6 can enhance the action of insulin (Petersen et al. 2005), promote glucose uptake into cells (Carey et al. 2008) and drive fatty acid oxidation in skeletal muscle (Kelly et al. 2004). This action is proposed to occur by IL-6 activating AMP kinase to promote glucose uptake and fatty acid metabolism, but is not fully understood at this time. The difference in observations may be explained in that the sustained IL-6 release in obesity and its interaction with other adipokines results in SNS activation, whereas with exercise there is a transient release of IL-6 which is expressed in the absence of other adipokines. IL-6 acts with TNF-α to increase the synthesis of C-reactive protein (CRP) (Yudkin et al. 1999), and it may be that CRP, under the control of IL-6 expressed from adipocytes, contributes to insulin resistance and SNS overactivity indirectly.
C-reactive protein
CRP is synthesized mainly in the liver. It is rapidly synthesized in response to trauma, infection and inflammation, and reduced quickly when these symptoms are ameliorated. In obesity, CRP has been shown to be chronically elevated and the two are correlated in a dose-dependent fashion, with no preferential expression from visceral or subcutaneous adipocytes (Ridker et al. 2003). Although CRP has not been reported to directly stimulate sympathetic outflow, the use of β-blockers lowers CRP, and there is a strong correlation between CRP and IL-6 levels in developing insulin resistance (Festa et al. 2002; Freeman et al. 2002). Leptin (Chen et al. 2006), IL-6 and TNF-α (Yudkin et al. 1999) can induce CRP expression, again highlighting the interplay between adipokines in obesity. CRP is also responsible for increased expression of the angiotensin I receptor in vascular smooth muscle, indicating that CRP may indirectly contribute to SNS outflow through the up-regulation of the angiotensin II pathway (Wang et al. 2003), as discussed below.
Angiotensinogen
Both the SNS and the renin–angiotensin–aldosterone system (RAS) are activated in obesity, and both systems can upregulate the action of the other. Sympathetic nerves stimulate renin release and the formation of angiotensin II, and angiotensin II can alter SNS outflow directly (Matsukawa et al. 1991), or can augment SNS responsiveness by potentiating release of noradrenaline from sympathetic nerve terminals and amplification of α1-receptor mediated vasoconstriction. Part of this bidirectional augmentation may be through the actions of angiotensinogen, the production of which is elevated in obesity (Umemura et al. 1997; Van Harmelen et al. 2000), more so in central adiposity than peripheral. An increase in angiotensinogen could allow for greater synthesis of angiotensin II by a mass action shift with increased availability of angiotensinogen (Umemura et al. 1997) and augmented renin release secondary to higher renal sympathetic nerve traffic.
Increased MSNA might also be explained by the over-expression of TNF-α in central obesity, which sensitizes the effects of angiotensin II (Zera et al. 2008). Obese women have been shown to express angiotensin II receptors in adipocytes at twice the rate of normal weight women, further implicating the RAS system in the transduction of sympathetic outflow (Gorzelniak et al. 2002). Angiotensin II has also been shown to induce CRP expression in human aortic endothelial cells (Han et al. 2010). Taken together, these findings indicate the RAS is not only implicated in the observed sympathetic overdrive in obesity, but may provide a mechanism through which sympathetic overactivity leads to chronic hypertension. Lending support to this concept, when angiotensin II was inhibited for three months in obese, hypertensive humans, MSNA activity was reduced by 21% (Grassi et al. 2003). However, this was attributed to the concomitant improvement in insulin sensitivity rather than a direct effect on SNS outflow. In lean insulin-sensitive hypertensives, angiotensin II receptor blocker did not alter SNS activity (Struck et al. 2002).
Adiponectin
Adiponectin is a protein highly expressed in adipocytes (Maeda et al. 1996). It has been shown that adiponectin improves insulin sensitivity and glucose tolerance (Yamauchi et al. 2002), and is reduced in obese humans (Arita et al. 1999). Levels of adiponectin are inversely proportionate to circulating leptin concentration and insulin sensitivity (Matsubara et al. 2002). Furthermore, adiponectin levels are sex specific, favouring production in women (Nishizawa et al. 2002). Adiponectin is also attenuated in lean humans with type 2 diabetes (Hotta et al. 2000), suggesting a closer association with metabolic dysfunction rather than as an adipokine.
Administering adiponectin to overweight mice consuming a high fat, high sugar diet was associated with a decrease in NEFAs and plasma glucose (Fruebis et al. 2001), suggestive of an improvement in insulin transduction. There does not seem to be any direct, central mechanism by which adiponectin alters SNA, and thus any relationship seems to be in response to peripheral changes. Specifically, adiponectin acts to augment hepatic but not muscle insulin signalling.
Reactive oxygen species
Reactive oxygen species (ROS) are known to have important roles in cell signalling but can participate in the dysregulation of autonomic balance and control of blood pressure in humans. ROS production is normally countered by enzymatic and non-enzymatic reducing agents to prevent the associated negative effects, but in a disease state excessive ROS may be generated. Germane to this review, ROS are capable of modifying central activation of sympathetic nervous activity (Campese et al. 2004; Han et al. 2005), and may serve as an important link between adipokine expression and SNS overactivity. As an example, autonomic dysfunction in obesity may be related to angiotensin II as a source of ROS, as links have now been established between angiotensin II and superoxide signalling in the CNS relating to the regulation of SNS activity (Zimmerman et al. 2002; Campese et al. 2005; Gao et al. 2005; Han et al. 2005). In support of this concept, direct injections of superoxide dismutase, an enzyme capable of reducing ROS, into the rostral ventrolateral medulla of a porcine model reduces sympathetic nerve activity (Zanzinger & Czachurski, 2000). Likewise, direct injections of a superoxide dismutase mimetic into the medulla of rats produced the same quieting of the sympathetic nervous system, and these effects were partially independent of nitric oxide production in these vascular beds (Campese et al. 2004).
ROS production is noted to be elevated in central adipose tissue (Cancello et al. 2006), possibly resulting from increased infiltration of macrophages. Macrophage-induced oxidative stress may be influenced by dysregulation of other adipokines as well, creating a positive feedback loop with SNS activity. For example, adiponectin has been noted to have an inhibitory effect on cell adhesion molecules and down-regulation could conceivably promote monocyte infiltration (Kawanami et al. 2004). The production of ROS resulting from dysregulation of adipokines is an interesting possible link between chronic inflammation and SNS overactivity in obesity. Future studies should be directed at investigating these potentially important relationships.
Summary
There is growing evidence that suggests links between an unfavourable expression of adipokines to insulin resistance and an increase in neural SNS activation. In addition to sympathetic overdrive in central obesity, there is an observed blunted response of the sympathetic nervous system to excitatory manoeuvres. A better understanding of the relationships between adipokines, insulin resistance, and sympathetic activity at rest and in response to stressors is necessary if we are to develop targeted strategies to improve health and decrease risks associated with obesity.
Perspectives
Just as clinical diagnoses of visceral obesity, hypertension, dyslipidaemia and glucose intolerance have evolved to be recognized as functionally related in the metabolic syndrome, recognition that sympathetic overdrive may be a common thread to these pathologies is evolving. Designing strategies to treat the neural sympathetic overdrive may result in improved outcomes in certain patient groups. As an example, Schlaich et al. (2011) recently performed bilateral sympathetic renal denervation by endovascular radiofrequency in obese women with polycystic ovary syndrome, a disease characterized by SNS activation, insulin resistance, and blood pressure elevation. In these patients, baseline SNA was elevated ∼3-fold from normal values. Renal nerve ablation resulted in a reduction in overall SNA (measured by whole body NA spillover and MSNA), a moderate reduction in blood pressure and significant improvement in insulin sensitivity. This suggests selectively targeting SNA overdrive may have profound improvements on insulin resistance and possibly comorbidities associated with the metabolic syndrome. However, there remains much to learn about the impact of chronic SNA overactivity in humans, including some basic information on sex differences, the influences of the sex steroids, the role of inflammation, and how the well-known age-related increase in SNA and body fat might contribute to morbidity. An integrative approach to our thinking is necessary to understand how sympathetic overdrive may be a key underlying component to the constellation of diseases resulting from the metabolic syndrome, and may even have a causal role in insulin resistance. Integrative physiological research should continue to test hypotheses and interventions which establish novel therapies in circumventing or ameliorating the cyclical relationship of neural sympathetic overdrive, insulin resistance and visceral obesity. Improving our understanding of the integrative effects of certain adipokines and how they may alter the regulation of sympathetic tone and contribute to insulin resistance is paramount in this regard.
Acknowledgments
Christopher T. Minson is supported by NIH HL081671.
References
- Alvarez GE, Ballard TP, Beske SD, Davy KP. Subcutaneous obesity is not associated with sympathetic neural activation. Am J Physiol Heart Circ Physiol. 2004;287:H414–418. doi: 10.1152/ajpheart.01046.2003. [DOI] [PubMed] [Google Scholar]
- Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation. 2002;106:2533–2536. doi: 10.1161/01.cir.0000041244.79165.25. [DOI] [PubMed] [Google Scholar]
- Alvarez GE, Davy BM, Ballard TP, Beske SD, Davy KP. Weight loss increases cardiovagal baroreflex function in obese young and older men. Am J Physiol Endocrinol Metab. 2005;289:E665–669. doi: 10.1152/ajpendo.00487.2004. [DOI] [PubMed] [Google Scholar]
- Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest. 1991;87:2246–2252. doi: 10.1172/JCI115260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- Arnold AC, Shaltout HA, Gallagher PE, Diz DI. Leptin impairs cardiovagal baroreflex function at the level of the solitary tract nucleus. Hypertension. 2009;54:1001–1008. doi: 10.1161/HYPERTENSIONAHA.109.138065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arone LJ, Mackintosh R, Rosenbaum M, Leibel RL, Hirsch J. Autonomic nervous system activity in weight gain and weight loss. Am J Physiol Regul Integr Comp Physiol. 1995;269:R222–225. doi: 10.1152/ajpregu.1995.269.1.R222. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation. 1995;2:241–248. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation. 1999;99:2221–2222. [PubMed] [Google Scholar]
- Berne C, Fagius J, Pollare T, Hjemdahl P. The sympathetic response to euglycaemic hyperinsulinaemia. Evidence from microelectrode nerve recordings in healthy subjects. Diabetologia. 1992;35:873–879. doi: 10.1007/BF00399935. [DOI] [PubMed] [Google Scholar]
- Beske SD, Alvarez GE, Ballard TP, Davy KP. Reduced cardiovagal baroreflex gain in visceral obesity: implications for the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2002;282:H630–635. doi: 10.1152/ajpheart.00642.2001. [DOI] [PubMed] [Google Scholar]
- Brabant G, Horn R, von zur Mühlen A, Mayr B, Wurster U, Heidenreich F, Schnabel D, Grüters-Kieslich A, Zimmermann-Belsing T, Feldt-Rasmussen U. Free and protein bound leptin are distinct and independently controlled factors in energy regulation. Diabetologia. 2000;43:438–442. doi: 10.1007/s001250051326. [DOI] [PubMed] [Google Scholar]
- Bruce R, Godsland I, Walton C, Crook D, Wynn V. Associations between insulin sensitivity, and free fatty acid and triglyceride metabolism independent of uncomplicated obesity. Metabolism. 1994;43:1275–1281. doi: 10.1016/0026-0495(94)90222-4. [DOI] [PubMed] [Google Scholar]
- Brunner EJ, Hemingway H, Walker BR, Page M, Clarke P, Juneja M, Shipley MJ, Kumari M, Andrew R, Seckl JR, Papadopoulos A, Checkley S, Rumley A, Lowe GD, Stansfeld SA, Marmot MG. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: nested case-control study. Circulation. 2002;106:2659–2665. doi: 10.1161/01.cir.0000038364.26310.bd. [DOI] [PubMed] [Google Scholar]
- Campese VM, Shaohua Y, Huiquin Z. Oxidative stress mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2005;46:533–539. doi: 10.1161/01.HYP.0000179088.57586.26. [DOI] [PubMed] [Google Scholar]
- Campese VM, Ye S, Zhong H, Yanamadala V, Ye Z, Chiu J. Reactive oxygen species stimulate central and peripheral sympathetic nervous system activity. Am J Physiol Heart Circ Physiol. 2004;287:H695–703. doi: 10.1152/ajpheart.00619.2003. [DOI] [PubMed] [Google Scholar]
- Cancello R, Tordjman J, Poitou C, Guilhem G, Bouillot JL, Hugol D, Coussieu C, Basdevant A, Bar HenA, Bedossa P, Guerre-Millo M, Clement K. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes. 2006;55:1554–1561. doi: 10.2337/db06-0133. [DOI] [PubMed] [Google Scholar]
- Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK, Febbraio MA. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes. 2008;55:2688–2697. doi: 10.2337/db05-1404. [DOI] [PubMed] [Google Scholar]
- Cassaglia P, Hermes S, Aicher S, Brooks V. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol. 2011;589:1643–1662. doi: 10.1113/jphysiol.2011.205575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Li F, Li J, Cai H, Strom S, Bisello A, Kelley DE, Friedman-Einat M, Skibinski GA, McCrory MA, Szalai AJ, Zhao AZ. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat Med. 2006;12:425–432. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- Ciccone M, Vettor R, Pannacciulli N, Minenna A, Bellacicco M, Rizzon P, Giorgino R, De Pergola G. Plasma leptin is independently associated with the intima-media thickness of the common carotid artery. Int J Obes Relat Metab Disord. 2001;25:805–810. doi: 10.1038/sj.ijo.0801623. [DOI] [PubMed] [Google Scholar]
- Ciofi P. The arcuate nucleus as a circumventricular organ in the mouse. Neurosci Lett. 2011;487:187–190. doi: 10.1016/j.neulet.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Lowell BB, Elmquist JK. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Cox HS, Kaye DM, Thompson JM, Turner AG, Jennings GL, Itsiopoulos C, Esler MD. Regional sympathetic nervous activation after a large meal in humans. Clin Sci (Lond) 1995;89:145–154. doi: 10.1042/cs0890145. [DOI] [PubMed] [Google Scholar]
- Dampney RA. Arcuate nucleus – a gateway for insulin's action on sympathetic activity. J Physiol. 2011;589:2109–2110. doi: 10.1113/jphysiol.2011.208579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- Dengo AL, Dennis EA, Orr JS, Marinik EL, Ehrlich E, Davy BM, Davy KP. Arterial destiffening with weight loss in overweight and obese middle-aged and older adults. Hypertension. 2010;55:855–861. doi: 10.1161/HYPERTENSIONAHA.109.147850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JB. The effect of obesity on health outcomes. Mol Cell Endocrinol. 2009;316:104–108. doi: 10.1016/j.mce.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Dulloo AG. Biomedicine. A sympathetic defense against obesity. Science. 2002;297:780–781. doi: 10.1126/science.1074923. [DOI] [PubMed] [Google Scholar]
- Dulloo AG, Jacquet J. An adipose-specific control of thermogenesis in body weight regulation. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S22–29. doi: 10.1038/sj.ijo.0801907. [DOI] [PubMed] [Google Scholar]
- Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension. 2006;48:787–796. doi: 10.1161/01.HYP.0000242642.42177.49. [DOI] [PubMed] [Google Scholar]
- Esler M, Vaz M, Collier G, Nestel P, Jennings G, Kaye D, Seals D, Lambert G. Leptin in human plasma is derived in part from the brain, and cleared by the kidneys. Lancet. 1998;351:879. doi: 10.1016/S0140-6736(05)70289-0. [DOI] [PubMed] [Google Scholar]
- Fagius J, Berne C. Increase in muscle nerve sympathetic activity in humans after food intake. Clin Sci (Lond) 1994;86:159–167. doi: 10.1042/cs0860159. [DOI] [PubMed] [Google Scholar]
- Febbraio MA, Pedersen BK. Contraction-induced myokine production and release: is skeletal muscle an endocrine organ? Exerc Sport Sci Rev. 2005;33:114–119. doi: 10.1097/00003677-200507000-00003. [DOI] [PubMed] [Google Scholar]
- Feingold KR, Doerrler W, Dinarello CA, Fiers W, Grunfeld C. Stimulation of lipolysis in cultured fat cells by tumor necrosis factor, interleukin-1, and the interferons is blocked by inhibition of prostaglandin synthesis. Endocrinology. 1992;130:10–16. doi: 10.1210/endo.130.1.1370149. [DOI] [PubMed] [Google Scholar]
- Festa A, D'Agostino R, Tracy RP, Haffner SM. Insulin Resistance Atherosclerosis Study. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002;51:1131–1137. doi: 10.2337/diabetes.51.4.1131. [DOI] [PubMed] [Google Scholar]
- Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff. 2009;28:w822–831. doi: 10.1377/hlthaff.28.5.w822. [DOI] [PubMed] [Google Scholar]
- Florian JP, Pawelczyk JA. Non-esterified fatty acids increase arterial pressure via central sympathetic activation in humans. Clin Sci. 2009;118:61–69. doi: 10.1042/CS20090063. [DOI] [PubMed] [Google Scholar]
- Freeman DJ, Norrie J, Caslake MJ, Gaw A, Ford I, Lowe GD, O'Reilly DS, Packard CJ, Sattar N. C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes. 2002;51:1596–1600. doi: 10.2337/diabetes.51.5.1596. [DOI] [PubMed] [Google Scholar]
- Friedman JM. Obesity in the new millennium. Nature. 2000;404:632–634. doi: 10.1038/35007504. [DOI] [PubMed] [Google Scholar]
- Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadegbeku CA, Dhandayuthapani A, Sadler ZE, Egan BM. Raising lipids acutely reduces baroreflex sensitivity. Am J Hypertens. 2002;15(6):479–485. doi: 10.1016/s0895-7061(02)02275-6. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Simvastatin therapy normalizes sympathetic neural control in experimental heart failure: roles of angiotensin II type 1 receptors and NAD(P)H oxidase. Circulation. 2005;112:1763–1770. doi: 10.1161/CIRCULATIONAHA.105.552174. [DOI] [PubMed] [Google Scholar]
- Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorzelniak K, Engeli S, Janke J, Luft FC, Sharma AM. Hormonal regulation of the human adipose-tissue renin-angiotensin system: relationship to obesity and hypertension. J Hypertens. 2002;20:965–973. doi: 10.1097/00004872-200205000-00032. [DOI] [PubMed] [Google Scholar]
- Grassi G, Colombo M, Seravalle G, Spaziani D, Mancia G. Dissociation between muscle and skin sympathetic nerve activity in essential hypertension, obesity, and congestive heart failure. Hypertension. 1998a;31:64–67. doi: 10.1161/01.hyp.31.1.64. [DOI] [PubMed] [Google Scholar]
- Grassi G, Dell'Oro R, Facchini A, Quarti TrevanoF, Bolla GB, Mancia G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens. 2004;22:2363–2369. doi: 10.1097/00004872-200412000-00019. [DOI] [PubMed] [Google Scholar]
- Grassi G, Dell'Oro R, Quarti-Trevano F, Scopelliti F, Seravalle G, Paleari F, Gamba PL, Mancia G. Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia. 2005;48:1359–1365. doi: 10.1007/s00125-005-1798-z. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Cattaneo BM, Lanfranchi A, Colombo M, Giannattasio C, Brunani A, Cavagnini F, Mancia G. Sympathetic activation in obese normotensive subjects. Hypertension. 1995;25:560–563. doi: 10.1161/01.hyp.25.4.560. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Colombo M, Bolla G, Cattaneo BM, Cavagnini F, Mancia G. Body weight reduction, sympathetic nerve traffic, and arterial baroreflex in obese normotensive humans. Circulation. 1998b;97:2037–2042. doi: 10.1161/01.cir.97.20.2037. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Dell'Oro R, Trevano FQ, Bombelli M, Scopelliti F, Facchini A, Mancia G. CROSS Study. Comparative effects of candesartan and hydrochlorothiazide on blood pressure, insulin sensitivity, and sympathetic drive in obese hypertensive individuals: results of the CROSS study. J Hypertens. 2003;9:1761–1769. doi: 10.1097/00004872-200309000-00027. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Dell'Oro TurriC, Bolla GB, Mancia G. Adrenergic and reflex abnormalities in obesity-related hypertension. Hypertension. 2000;36:538–542. doi: 10.1161/01.hyp.36.4.538. [DOI] [PubMed] [Google Scholar]
- Grekin RJ, Ngarmukos CO, Williams DM, Supiano MA. Renal norepinephrine spillover during infusion of nonesterified fatty acids. Am J Hypertens. 2005;18(3):422–426. doi: 10.1016/j.amjhyper.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- Grill HJ. Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity. 2006;14:216S–221S. doi: 10.1038/oby.2006.312. [DOI] [PubMed] [Google Scholar]
- Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL, Atkinson AJ, Schulkin J, Contoreggi C, Chrousos GP, McCann SM, Suomi SJ, Higley JD, Gold PW. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A. 2000;97:6079–6084. doi: 10.1073/pnas.97.11.6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Liu J, Liu X, Li M. Angiotensin II induces C-reactive protein expression through ERK1/2 and JNK signaling in human aortic endothelial cells. Atherosclerosis. 2010;212:206–212. doi: 10.1016/j.atherosclerosis.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Han Y, Zhang Y, Wang HJ, Gao XY, Wang W, Zhu GQ. Reactive oxygen species in paraventricular nucleus modulates cardiac sympathetic afferent reflex in rats. Brain Res. 2005;1058:82–90. doi: 10.1016/j.brainres.2005.07.055. [DOI] [PubMed] [Google Scholar]
- Hausberg M, Mark AL, Hoffman RP, Sinkey CA, Anderson EA. Dissociation of sympathoexcitatory and vasodilator actions of modestly elevated plasma insulin levels. J Hypertens. 1995;13:1015–1021. doi: 10.1097/00004872-199509000-00012. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997a;100:270–278. doi: 10.1172/JCI119532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WG, Sivitz WI, Morgan DA, Walsh SA, Mark AL. Sympathetic and cardiorenal actions of leptin. Hypertension. 1997b;30:619–623. doi: 10.1161/01.hyp.30.3.619. [DOI] [PubMed] [Google Scholar]
- Hedeskov CJ. Mechanism of glucose-induced insulin secretion. Physiol Rev. 1980;60:442–509. doi: 10.1152/physrev.1980.60.2.442. [DOI] [PubMed] [Google Scholar]
- Hofmann C, Lorenz K, Braithwaite SS, Colca JR, Palazuk BJ, Hotamisligil GS, Spiegelman BM. Altered gene expression for tumor necrosis factor-α and its receptors during drug and dietary modulation of insulin resistance. Endocrinology. 1994;134:264–270. doi: 10.1210/endo.134.1.8275942. [DOI] [PubMed] [Google Scholar]
- Hopkins DF, Williams G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet Med. 1997;14:1044–1050. doi: 10.1002/(SICI)1096-9136(199712)14:12<1044::AID-DIA508>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-α. J Clin Invest. 1994;94:1543–1549. doi: 10.1172/JCI117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, Nishida M, Kihara S, Sakai N, Nakajima T, Hasegawa K, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Hanafusa T, Matsuzawa Y. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- Huber DA, Schreihofer AM. Attenuated baroreflex control of sympathetic nerve activity in obese Zucker rats by central mechanisms. J Physiol. 2010;588:1515–1525. doi: 10.1113/jphysiol.2009.186387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggett RJ, Burns J, Mackintosh AF, Mary DA. Sympathetic neural activation in nondiabetic metabolic syndrome and its further augmentation by hypertension. Hypertension. 2004;44:847–852. doi: 10.1161/01.HYP.0000147893.08533.d8. [DOI] [PubMed] [Google Scholar]
- Jang YJ, Ryu HJ, Choi YO, Kim C, Leem CH, Park CS. Improvement of insulin sensitivity by chelation of intracellular Ca2+ in high-fat-fed rats. Metabolism. 2002;51:912–918. doi: 10.1053/meta.2002.33351. [DOI] [PubMed] [Google Scholar]
- Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Imai Y, Manabe I, Utsunomiya K, Nagai R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: a new insight into adipocytokine endothelial cell interactions. Biochem Biophys Res Commun. 2004;314:415–419. doi: 10.1016/j.bbrc.2003.12.104. [DOI] [PubMed] [Google Scholar]
- Keaney JF, Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- Kelly M, Keller C, Avilucea PR, Keller P, Luo Z, Xiang X, Giralt M, Hidalgo J, Saha AK, Pedersen BK, Ruderman NB. AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem Biophys Res Commun. 2004;320:449–454. doi: 10.1016/j.bbrc.2004.05.188. [DOI] [PubMed] [Google Scholar]
- Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2001;280:E745–751. doi: 10.1152/ajpendo.2001.280.5.E745. [DOI] [PubMed] [Google Scholar]
- Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95:2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–1801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, Olefsky JM. Fatty acid-induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab. 2002;87:226–234. doi: 10.1210/jcem.87.1.8187. [DOI] [PubMed] [Google Scholar]
- Kumar S, Olukoga AO, Gordon C, Mawer EB, France M, Hosker JP, Davies M, Boulton AJ. Impaired glucose tolerance and insulin insensitivity in primary hyperparathyroidism. Clin Endocrinol (Oxf) 1994;40:47–53. doi: 10.1111/j.1365-2265.1994.tb02442.x. [DOI] [PubMed] [Google Scholar]
- Lakka TA, Lakka HM, Salonen R, Kaplan GA, Salonen JT. Abdominal obesity is associated with accelerated progression of carotid atherosclerosis in men. Arteriosclerosis. 2001;154:497–504. doi: 10.1016/s0021-9150(00)00514-1. [DOI] [PubMed] [Google Scholar]
- Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci. 2005;8:579–584. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- Lambert E, Sari CI, Dawood T, Nguyen J, McGrane M, Eikelis N, Chopra R, Wong C, Chatzivlastou K, Head G, Straznicky N, Esler M, Schlaich M, Lambert G. Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension. 2010;56:351–358. doi: 10.1161/HYPERTENSIONAHA.110.155663. [DOI] [PubMed] [Google Scholar]
- Lambert E, Straznicky N, Schlaich M, Esler M, Dawood T, Hotchkin E, Lambert G. Differing pattern of sympathoexcitation in normal-weight and obesity-related hypertension. Hypertension. 2007;50:862–868. doi: 10.1161/HYPERTENSIONAHA.107.094649. [DOI] [PubMed] [Google Scholar]
- Landsberg L. Insulin resistance, energy balance and sympathetic nervous system activity. Clin Exp Hypertens A. 1990;12:817–830. doi: 10.3109/10641969009073502. [DOI] [PubMed] [Google Scholar]
- Landsberg L, Young JB. Diet-induced changes in sympathoadrenal activity: implications for thermogenesis. Life Sci. 1981;28:1801–1819. doi: 10.1016/0024-3205(81)90352-0. [DOI] [PubMed] [Google Scholar]
- Landsberg L, Young JB. Sympathoadrenal activity and obesity: physiological rationale for the use of adrenergic thermogenic drugs. Int J Obes Relat Metab Disord. 1993;17(Suppl 1):S29–34. [PubMed] [Google Scholar]
- Lang CH, Dobrescu C. Sepsis-induced changes in in vivo insulin action in diabetic rats. Am J Physiol Endocrinol Metab. 1989;3:E301–308. doi: 10.1152/ajpendo.1989.257.3.E301. [DOI] [PubMed] [Google Scholar]
- Lau DCW, Dhillon G, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atherosclerosis. Am J Physiol Heart Circ Physiol. 2005;288:H2031–H2041. doi: 10.1152/ajpheart.01058.2004. [DOI] [PubMed] [Google Scholar]
- Lee ZS, Critchley JA, Tomlinson B, Young RP, Thomas GN, Cockram CS, Chan TY, Chan JC. Urinary epinephrine and norepinephrine interrelations with obesity, insulin, and the metabolic syndrome in Hong Kong Chinese. Metabolism. 2001;50:135–143. doi: 10.1053/meta.2001.19502. [DOI] [PubMed] [Google Scholar]
- Lembo G, Napoli R, Capaldo B, Rendina V, Laccarino G, Volpe M, Trimarco B, Saccà L. Abnormal sympathetic overactivity evoked by insulin in the skeletal muscle of patients with essential hypertension. J Clin Invest. 1992;90:24–29. doi: 10.1172/JCI115842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenczowski MJ, Bluthé RM, Roth J, Rees GS, Rushforth DA, van Dam AM, Tilders FJ, Dantzer R, Rothwell NJ, Luheshi GN. Central administration of rat IL-6 induces HPA activation and fever but not sickness behavior in rats. Am J Physiol Regul Integr Comp Physiol. 1999;276:R652–658. doi: 10.1152/ajpregu.1999.276.3.R652. [DOI] [PubMed] [Google Scholar]
- Lind L, Siegbahn A, Ingelsson E, Sundstrom J, Arnlov J. A detailed cardiovascular characterization of obesity without the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2011;31:e27–34. doi: 10.1161/ATVBAHA.110.221572. [DOI] [PubMed] [Google Scholar]
- Lucini D, Cusumano G, Bellia A, Kozakova M, Difede G, Lauro R, Pagani M. Is reduced baroreflex gain a component of the metabolic syndrome? Insights from the LINOSA study. J Hypertens. 2006;24:361–370. doi: 10.1097/01.hjh.0000202817.02836.9c. [DOI] [PubMed] [Google Scholar]
- Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose most abundant gene transcript 1) Biochem Biophys Res Commun. 1996;221:286–289. doi: 10.1006/bbrc.1996.0587. [DOI] [PubMed] [Google Scholar]
- Mark AL, Agassandian K, Morgan DA, Liu X, Cassell MD, Rahmouni K. Leptin signaling in the nucleus tractus solitarii increases sympathetic nerve activity to the kidney. Hypertension. 2009;53:375–380. doi: 10.1161/HYPERTENSIONAHA.108.124255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark AL, Correia M, Morgan DA, Shaffer RA, Haynes WG. State-of-the-art-lecture: Obesity-induced hypertension: new concepts from the emerging biology of obesity. Hypertension. 1999;33:537–541. doi: 10.1161/01.hyp.33.1.537. [DOI] [PubMed] [Google Scholar]
- Mastorakos G, Weber JS, Magiakou MA, Gunn H, Chrousos GP. Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion. J Clin Endocrinol Metab. 1994;79:934–939. doi: 10.1210/jcem.79.4.7962300. [DOI] [PubMed] [Google Scholar]
- Masuo K, Katsuya T, Fu Y. β2-Adrenoceptor polymorphisms relate to insulin resistance and sympathetic overactivity as early markers of metabolic disease in nonobese, normotensive individuals. Am J Hypertens. 2005;18:1009–1014. doi: 10.1016/j.amjhyper.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Matsubara M, Maruoka S, Katayose S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur J Endocrinol. 2002;147:173–180. doi: 10.1530/eje.0.1470173. [DOI] [PubMed] [Google Scholar]
- Matsukawa T, Gotoh E, Minamisawa K, Kihara M, Ueda S, Shionoiri H, Ishii M. Effects of intravenous infusions of angiotensin II on muscle sympathetic nerve activity in humans. Am J Physiol Regul Integr Comp Physiol. 1991;261:R690–696. doi: 10.1152/ajpregu.1991.261.3.R690. [DOI] [PubMed] [Google Scholar]
- McCarty MF. Elevated sympathetic activity may promote insulin resistance syndrome by activating alpha-1 adrenergic receptors on adipocytes. Med Hypotheses. 2004;62:830–838. doi: 10.1016/j.mehy.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Mohamed-Ali V, Flower L, Sethi J, Hotamisligil G, Gray R, Humphries SE, York DA, Pinkney J. beta-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab. 2001;86:5864–5869. doi: 10.1210/jcem.86.12.8104. [DOI] [PubMed] [Google Scholar]
- Mohamed-Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS, Klein S, Coppack SW. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-α, in vivo. J Clin Endocrinol Metab. 1997;82:4196–4200. doi: 10.1210/jcem.82.12.4450. [DOI] [PubMed] [Google Scholar]
- Monahan KD, Dyckman DJ, Ray CA. Effect of acute hyperlipidemia on autonomic and cardiovascular control in humans. J Appl Physiol. 2007;103:162–169. doi: 10.1152/japplphysiol.00167.2007. [DOI] [PubMed] [Google Scholar]
- Monahan KD, Eskurza I, Seals DR. Ascorbic acid increases cardiovagal baroreflex sensitivity in healthy older men. Am J Physiol Heart Circ Physiol. 2004;286:H2113–2117. doi: 10.1152/ajpheart.01054.2003. [DOI] [PubMed] [Google Scholar]
- Monroe MB, Van Pelt RE, Schiller BC, Seals DR, Jones PP. Relation of leptin and insulin to adiposity associated elevations in sympathetic activity with age in humans. Int J Obes Relat Metab Disord. 2000;24:1183–1187. doi: 10.1038/sj.ijo.0801364. [DOI] [PubMed] [Google Scholar]
- Morgan DA, Balon TW, Ginsberg BH, Mark AL. Nonuniform regional sympathetic nerve responses to hyperinsulinemia in rats. Am J Physiol Regul Integr Comp Physiol. 1993;264:R423–427. doi: 10.1152/ajpregu.1993.264.2.R423. [DOI] [PubMed] [Google Scholar]
- Munhoz RT, Negrão CE, Barretto AC, Ochiai ME, Cardoso JN, Morgado PC, Del Carlo CH, Ramires JA. Microneurography and venous occlusion plethysmography in heart failure: correlation with prognosis. Arq Bras Cardiol. 2009;92:46–53. doi: 10.1590/s0066-782x2009000100008. [DOI] [PubMed] [Google Scholar]
- Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol. 1994;267:R1350–1355. doi: 10.1152/ajpregu.1994.267.5.R1350. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Kato M, Phillips BG, Pesek CA, Choe I, Winnicki M, Palatini P, Sivitz WI, Somers VK. Leptin interacts with heart rate but not sympathetic nerve traffic in healthy male subjects. J Hypertens. 2001;19:1089–1094. doi: 10.1097/00004872-200106000-00014. [DOI] [PubMed] [Google Scholar]
- Nishizawa H, Shimomura I, Kishida K, Maeda N, Kuriyama H, Nagaretani H, Matsuda M, Kondo H, Furuyama N, Kihara S, Nakamura T, Tochino Y, Funahashi T, Matsuzawa Y. Androgens decrease plasma adiponectin, an insulin-sensitizing adipocyte-derived protein. Diabetes. 2002;51:2734–2741. doi: 10.2337/diabetes.51.9.2734. [DOI] [PubMed] [Google Scholar]
- O'Hare JA, Minaker KL, Meneilly GS, Rowe JW, Pallotta JA, Young JB. Effect of insulin on plasma norepinephrine and 3,4-dihydroxyphenylalanine in obese men. Metabolism. 1989;38:322–329. doi: 10.1016/0026-0495(89)90118-2. [DOI] [PubMed] [Google Scholar]
- Orr JS, Gentile CL, Davy BM, Davy KP. Large artery stiffening with weight gain in humans: role of visceral fat accumulation. Hypertension. 2008;51:1519–1524. doi: 10.1161/HYPERTENSIONAHA.108.112946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46:1001–1009. doi: 10.2337/diab.46.6.1001. [DOI] [PubMed] [Google Scholar]
- Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. 2005;98:1154–1162. doi: 10.1152/japplphysiol.00164.2004. [DOI] [PubMed] [Google Scholar]
- Petersen EW, Carey AL, Sacchetti M, Steinberg GR, Macaulay SL, Febbraio MA, Pedersen BK. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am J Physiol Endocrinol Metab. 2005;288:E155–162. doi: 10.1152/ajpendo.00257.2004. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Morgan DA, Morgan GM, Liu X, Sigmund CD, Mark AL, Haynes WG. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest. 2004;114:652–658. doi: 10.1172/JCI21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch JE, Begum N, Sussman KE, Draznin B. Regulation of GLUT-4 phosphorylation by intracellular calcium in adipocytes. Endocrinology. 1991;129:3269–3273. doi: 10.1210/endo-129-6-3269. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation. 2003;107:391–397. doi: 10.1161/01.cir.0000055014.62083.05. [DOI] [PubMed] [Google Scholar]
- Rowe JW, Young JB, Minaker KL, Stevens AL, Pallotta J, Landsberg L. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes. 1981;30:219–225. doi: 10.2337/diab.30.3.219. [DOI] [PubMed] [Google Scholar]
- Rumantir MS, Vaz M, Jennings GL, Collier G, Kaye DM, Seals DR, Wiesner GH, Brunner-La Rocca HP, Esler MD. Neural mechanisms in human obesity-related hypertension. J Hypertens. 1999;17:1125–1133. doi: 10.1097/00004872-199917080-00012. [DOI] [PubMed] [Google Scholar]
- Russell CD, Petersen RN, Rao SP, Ricci MR, Prasad A, Zhang Y, Brolin RE, Fried SK. Leptin expression in adipose tissue from obese humans: depot-specific regulation by insulin and dexamethasone. Am J Physiol Endocrinol Metab. 1998;275:E507–E515. doi: 10.1152/ajpendo.1998.275.3.E507. [DOI] [PubMed] [Google Scholar]
- Sander D, Winbeck K, Klingelhöfer J, Etgen T, Conrad B. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology. 2001;57:833–838. doi: 10.1212/wnl.57.5.833. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Straznicky N, Grima M, Ika-Sari C, Dawood T, Mahfoud F, Lambert E, Chopra R, Socratous F, Hennebry S, Eikelis N, Bohm M, Krum H, Lambert G, Esler MD, Sobotka PA. Renal denervation: a potential new treatment modality for polycystic ovary syndrome? J Hypertens. 2011;29:991–996. doi: 10.1097/HJH.0b013e328344db3a. [DOI] [PubMed] [Google Scholar]
- Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, Mooney RA. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. 2003;278:13740–13746. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivenius K, Niskanen L, Laakso M, Uusitupa M. A deletion in the α2B-adrenergic receptor gene and autonomic nervous function in central obesity. Obes Res. 2003;11:962–970. doi: 10.1038/oby.2003.133. [DOI] [PubMed] [Google Scholar]
- Snitker S, Pratley RE, Nicolson M, Tataranni PA, Ravussin E. Relationship between muscle sympathetic nerve activity and plasma leptin concentration. Obes Res. 1997;5:338–340. doi: 10.1002/j.1550-8528.1997.tb00561.x. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Chrousos GP. Neuroendocrinology and pathophysiology of the stress system. Ann N Y Acad Sci. 1995;771:1–18. doi: 10.1111/j.1749-6632.1995.tb44666.x. [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Eikelis N, Lambert EA, Esler MD. Mediators of sympathetic activation in metabolic syndrome obesity. Curr Hypertens Rep. 2008;10:440–447. doi: 10.1007/s11906-008-0083-1. [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Grima MT, Eikelis N, Nestel PJ, Dawood T, Schlaich MP, Chopra R, Masuo K, Esler MD, Sari CI, Lambert GW, Lambert EA. The effects of weight loss versus weight loss maintenance on sympathetic nervous system activity and metabolic syndrome components. J Clin Endocrinol Metab. 2011;96:E503–508. doi: 10.1210/jc.2010-2204. [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Lambert GW, Masuo K, Dawood T, Eikelis N, Nestel PJ, McGrane MT, Mariani JA, Socratous F, Chopra R, Esler MD, Schlaich MP, Lambert EA. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am J Clin Nutr. 2009a;89:27–36. doi: 10.3945/ajcn.2008.26299. [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Lambert GW, McGrane MT, Masuo K, Dawood T, Nestel PJ, Eikelis N, Schlaich MP, Esler MD, Socratous F, Chopra R, Lambert EA. Weight loss may reverse blunted sympathetic neural responsiveness to glucose ingestion in obese subjects with metabolic syndrome. Diabetes. 2009b;58:1126–1132. doi: 10.2337/db08-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struck J, Muck P, Trubger D, Handrock R, Weidinger G, Dendorfer A, Dodt C. Effects of selective angiotensin II receptor blockade on sympathetic nerve activity in primary hypertensive subjects. J Hypertens. 2002;20:1143–1149. doi: 10.1097/00004872-200206000-00026. [DOI] [PubMed] [Google Scholar]
- Tank J, Jordan J, Diedrich A, Schroeder C, Furlan R, Sharma AM, Luft FC, Brabant G. Bound leptin and sympathetic outflow in nonobese men. J Clin Endocrinol Metab. 2003;88:4955–4959. doi: 10.1210/jc.2003-030371. [DOI] [PubMed] [Google Scholar]
- Thomas GN, Chook P, Qiao M, Huang XS, Leong HC, Celermajer DS, Woo KS. Deleterious impact of “high normal” glucose levels and other metabolic syndrome components on arterial endothelial function and intima-media thickness in apparently healthy Chinese subjects: the CATHAY study. Arterioscler Thromb Vasc Biol. 2004;24:739–743. doi: 10.1161/01.ATV.0000118015.26978.07. [DOI] [PubMed] [Google Scholar]
- Tounian P, Aggoun Y, Dubern B, Varille V, Guy-Grand B, Sidi D, Girardet JP, Bonnet D. Presence of increased stiffness of the common carotid artery and endothelial dysfunction in severely obese children: a prospective study. Lancet. 2001;358:1400–1404. doi: 10.1016/S0140-6736(01)06525-4. [DOI] [PubMed] [Google Scholar]
- Tsigos C, Kyrou I, Chala E, Tsapogas P, Stavridis JC, Raptis SA, Katsilambros N. Circulating tumor necrosis factor α concentrations are higher in abdominal versus peripheral obesity. Metabolism. 1999;48:1332–1335. doi: 10.1016/s0026-0495(99)90277-9. [DOI] [PubMed] [Google Scholar]
- Umemura S, Nyui N, Tamura K, Hibi K, Yamaguchi S, Nakamaru M, Ishigami T, Yabana M, Kihara M, Inoue S, Ishii M. Plasma angiotensinogen concentrations in obese patients. Am J Hypertens. 1997;6:629–633. doi: 10.1016/s0895-7061(97)00053-8. [DOI] [PubMed] [Google Scholar]
- Van De Borne P, Hausberg M, Hoffman RP, Mark AL, Anderson EA. Hyperinsulinemia produces cardiac vagal withdrawal and nonuniform sympathetic activation in normal subjects. Am J Physiol Regul Integr Comp Physiol. 1999;276:R178–183. doi: 10.1152/ajpregu.1999.276.1.R178. [DOI] [PubMed] [Google Scholar]
- Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obes Res. 2000;8:337–341. doi: 10.1038/oby.2000.40. [DOI] [PubMed] [Google Scholar]
- Van Harmelen V, Reynisdottir S, Eriksson P, Thorne A, Hoffstedt J, Lonnqvist F, Arner P. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes. 1998;47:913–917. doi: 10.2337/diabetes.47.6.913. [DOI] [PubMed] [Google Scholar]
- Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation. 1997;96:3423–3429. doi: 10.1161/01.cir.96.10.3423. [DOI] [PubMed] [Google Scholar]
- Vollenweider L, Tappy L, Owlya R, Jéquier E, Nicod P, Scherrer U. Insulin-induced sympathetic activation and vasodilation in skeletal muscle. Effects of insulin resistance in lean subjects. Diabetes. 1995;44:641–645. doi: 10.2337/diab.44.6.641. [DOI] [PubMed] [Google Scholar]
- Vollenweider P, Randin D, Tappy L, Jequier E, Nicod P, Scherrer U. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Invest. 1994;93:2365–2371. doi: 10.1172/JCI117242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollenweider P, Tappy L, Randin D, Schneiter P, Jéquier E, Nicod P, Scherrer U. Differential effects of hyperinsulinemia and carbohydrate metabolism on sympathetic nerve activity and muscle blood flow in humans. J Clin Invest. 1993;92:147–154. doi: 10.1172/JCI116542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenius V, Wallenius K, Ahrén B, Rudling M, Carlsten H, Dickson SL, Ohlsson C, Jansson JO. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–79. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- Wang CH, Li SH, Weisel RD, Fedak PW, Dumont AS, Szmitko P, Li RK, Mickle DA, Verma S. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation. 2003;107:1783–1790. doi: 10.1161/01.CIR.0000061916.95736.E5. [DOI] [PubMed] [Google Scholar]
- Wernstedt I, Edgley A, Berndtsson A, Fäldt J, Bergström G, Wallenius V, Jansson JO. Reduced stress- and cold-induced increase in energy expenditure in interleukin-6-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;291:R551–557. doi: 10.1152/ajpregu.00514.2005. [DOI] [PubMed] [Google Scholar]
- Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71:1057–1064. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Oike Y, Kamon J, Waki H, Komeda K, Tsuchida A, Date Y, Li MX, Miki H, Akanuma Y, Nagai R, Kimura S, Saheki T, Nakazato M, Naitoh T, Yamamura K, Kadowaki T. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat Genet. 2002;30:221–226. doi: 10.1038/ng829. [DOI] [PubMed] [Google Scholar]
- Young CN, Deo SH, Chaudhary K, Thyfault JP, Fadel PJ. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. J Physiol. 2010;588:3593–3603. doi: 10.1113/jphysiol.2010.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JB, Landsberg L. Suppression of sympathetic nervous system during fasting. Science. 1977;196:1473–1475. doi: 10.1126/science.867049. [DOI] [PubMed] [Google Scholar]
- Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol. 1999;19:972–978. doi: 10.1161/01.atv.19.4.972. [DOI] [PubMed] [Google Scholar]
- Zanzinger J, Czachurski J. Chronic oxidative stress in the RVLM modulates sympathetic control of circulation in pigs. Pflugers Arch. 2000;439:489–494. doi: 10.1007/s004249900204. [DOI] [PubMed] [Google Scholar]
- Zera T, Ufnal M, Szczepanska-Sadowska E. Central TNF-α elevates blood pressure and sensitizes to central pressor action of angiotensin II in the infarcted rats. J Physiol Pharmacol. 2008;59:117–121. [PubMed] [Google Scholar]
- Zierath JR, Houseknecht KL, Gnudi L, Kahn BB. High-fat feeding impairs insulin-stimulated GLUT4 recruitment via an early insulin-signaling defect. Diabetes. 1997;46:215–223. doi: 10.2337/diab.46.2.215. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]