

Abstract
Dysregulated epithelial fluid and electrolyte transport is a common feature of many intestinal disorders. However, molecular mechanisms that regulate epithelial transport processes are still poorly understood, thereby limiting development of new therapeutics. Previously, we showed that epidermal growth factor (EGF) chronically enhances intestinal epithelial secretory function. Here, we investigated a potential role for altered expression or activity of apical Cl− channels in mediating the effects of EGF. Cl− secretion across monolayers of T84 colonic epithelia was measured as changes in short-circuit current. Protein expression/phosphorylation was measured by RT-PCR and Western blotting. Under conditions that specifically isolate apical Ca2+-activated Cl− channel (CaCC) currents, EGF pretreatment (100 ng ml−1 for 15 min) potentiated carbachol (CCh)-induced responses to 173 ± 25% of those in control cells, when measured 24 h later (n= 26; P < 0.01). EGF-induced increases in CaCC currents were abolished by the transmembrane protein 16A (TMEM16A) inhibitor, T16Ainh-A01 (10 μm). Furthermore, TMEM16A mRNA and protein expression was increased by EGF to 256 ± 38% (n= 7; P < 0.01) and 297 ± 46% (n= 9, P < 0.001) of control levels, respectively. In contrast, EGF did not alter CFTR expression or activity. EGF-induced increases in Cl− secretion, CaCC currents and TMEM16A expression were attenuated by a PKCδ inhibitor, rottlerin (20 μm), and a phosphatidylinositol 3-kinase (PI3K) inhibitor, LY290042 (25 μm). Finally, LY290042 inhibited EGF-induced phosphorylation of PKCδ. We conclude that EGF chronically upregulates Ca2+-dependent Cl− conductances and TMEM16A expression in intestinal epithelia by a mechanism involving sequential activation of PI3K and PKCδ. Therapeutic targeting of EGF receptor-dependent signalling pathways may provide new approaches for treatment of epithelial transport disorders.
Key points
Cl− secretion, the predominant driving force for fluid secretion in the intestine, can be dysregulated in conditions of disease, such as cystic fibrosis.
We have previously shown that acute exposure to epidermal growth factor (EGF) chronically upregulates the capacity of colonic epithelial cells to secrete Cl−.
Here, we show that the effects of EGF are mediated by upregulation of the Ca2+-dependent Cl− channel, transmembrane protein 16A (TMEM16A), in the apical membrane of colonic epithelial cells.
EGF-induced TMEM16A expression is mediated by sequential activation of phosphatidylinositol 3-kinase and PKCδ.
These findings are among the first to elucidate molecular mechanisms that regulate TMEM16A expression in epithelial cells and suggest the channel represents a good target for development of new therapeutics for intestinal transport disorders.
Introduction
The secretion of water across intestinal epithelial cells is a vital process that serves to hydrate the luminal contents and enhance mucosal barrier function. Fluid movement across epithelial cells is driven by the active transport of ions, with Cl− secretion being the predominant driving force for fluid secretion. Dysregulated Cl− secretion leads to disturbances in fluid transport and is a common feature of a number of disorders including infectious diseases, inflammatory bowel disease and cystic fibrosis (Keely et al. 2009). Despite the prevalence of such diseases, there is still a lack of therapeutic agents that can specifically and directly modulate epithelial transport processes in their treatment.
The components of the epithelial Cl− secretory pathway have been quite well elucidated and represent good targets for the development of new therapeutics (Barrett & Keely, 2000). The energy for the secretory process is derived from the activity of basolateral Na+,K+-ATPase pumps, which transport Na+ out of the cell in exchange for K+. The activity of the ATPase creates a gradient for Na+ uptake through the Na+–K+–2Cl− cotransporter (NKCC1) along with K+ and Cl−. Since K+ can be recycled through channels in the basolateral membrane, the net activity of these basolateral transporters serves to specifically accumulate Cl− inside the cell so that a gradient for its exit exists when channels in the apical membrane are opened. The best characterized epithelial Cl− channel is the cystic fibrosis transmembrane conductance regulator (CFTR), which opens in response to agonists which increase intracellular cAMP. However, other Cl− channels are also known to exist, including those activated by agents that elevate intracellular Ca2+ levels. Although for many years the molecular identity of epithelial Ca2+-dependent Cl− channels (CaCC) remained elusive, several studies now suggest an important role for the recently identified transmembrane protein 16A (TMEM16A) (Caputo et al. 2008; Schroeder et al. 2008; Yang et al. 2008). This channel has been shown to mediate Ca2+-dependent Cl− conductances in the airways, biliary tract, kidneys and intestines (Ousingsawat et al. 2009; Namkung et al. 2010; Romanenko et al. 2010; Dutta et al. 2011; Tian et al. 2011).
The expression, trafficking and activity of epithelial transport proteins is under tight regulation by an array of hormones, neuroimmune mediators and growth factors (Keely et al. 2009). In particular, epidermal growth factor (EGF) has been shown to be an important regulator of various transport processes in the airways and intestine (Borok et al. 1996; Donowitz et al. 2000; Nielsen et al. 2001; Chung et al. 2002; Xu et al. 2010). Previous studies have also shown that EGF is an important regulator of intestinal secretory function. In the short term, acute exposure of epithelial cells to EGF dampens their ability to evoke responses to secretagogues (Uribe et al. 1996a), an effect which is mediated by rapid inhibition of basolateral K+ channels (Chow et al. 2000). However, our recent studies show that over more prolonged periods of time acute exposure to EGF chronically potentiates epithelial secretory function (O'Mahony et al. 2008). This chronic prosecretory action of EGF involves, at least in part, enhanced expression of NKCC1, which promotes basolateral entry of Cl− into the cells thereby increasing the driving force for its exit across the apical membrane. However, to date there is no information in the literature regarding potential effects of EGF on the expression or activity of the channels that provide the apical exit pathway for Cl− in intestinal epithelial cells. Thus, the current study set out to address this gap in our knowledge by investigating the potential role for EGF in regulating the activity and expression of cAMP and Ca2+-dependent Cl− channels in intestinal epithelial cells.
Methods
Cell culture and treatments
T84 colonic epithelial cells were cultured in Dulbecco's modified Eagle's medium (DMEM)–Ham's F12 nutrient mixture (1:1) supplemented with 5% newborn calf serum (HyClone, Logan, UT, USA) in an atmosphere of 5% CO2 at 37°C. For Ussing chamber/voltage clamp studies, approximately 5 × 105 cells were seeded onto 12 mm Millicel-HA Transwells (Millipore, Bedford, MA, USA). For Western blotting experiments, approximately 106 cells were seeded onto 30 mm Millicel-HA Transwells. Cells were cultured on filters for 10–28 days prior to use. Under these conditions T84 cells develop the polarized, electrically resistant phenotype of native epithelial cells. Prior to treatment with EGF, T84 monolayers were washed in serum-free medium and allowed to equilibrate for 1 h. Unless otherwise noted, cells were treated basolaterally with EGF at a concentration of 100 ng ml−1 for 15 min. EGF-containing medium was then aspirated and cells were washed twice in serum-free medium and maintained at 37°C for 24 h prior to experimentation. When pharmacological inhibitors were employed, T84 cells were treated bilaterally for 30 min with inhibitors (or vehicle) prior to treatment with EGF. After EGF treatment the basolateral solution was removed, cells were washed in serum-free medium and then incubated in the continued presence of inhibitors before experimentation.
Electrophysiological measurements
For measurements of transepithelial Cl− secretion T84 cell monolayers were mounted in Ussing chambers (aperture = 0.6 cm2), voltage-clamped to zero potential difference and monitored for changes in short circuit current (ΔIsc) using an EVC4000 voltage clamp (World Precision Instruments, Sarasota, FL, USA). Under such conditions secretagogue-induced ΔIsc across T84 monolayers are wholly reflective of changes in electrogenic Cl− secretion (Cartwright et al. 1985). Isc measurements were carried out in Ringer's solution containing (in mm): 140 Na+, 5.2 K+, 1.2 Ca2+, 0.8 Mg2+, 119.8 Cl−, 25 HCO3−, 2.4 H2PO42− and 10 glucose. Results were normalized to an area of 1 cm2 and were expressed as ΔIsc (μA cm−2).
Basolateral K+ conductance was analysed by a method described by Kirk & Dawson (1983). T84 cell monolayers in Ussing chambers were apically permeabilized with amphotericin B (50 μm). A K+ gradient (123.2–5.2 mm) was created across the basolateral membrane by addition of a high K+ (123.2 mm) Ringer's solution to the apical reservoir, in which NaCl was replaced with KCl, while the basolateral reservoir contained normal Ringer's solution with a K+ concentration of 5.2 mm. Ouabain (100 μm) was added basolaterally to inhibit the Na+, K+-ATPase pump activity. Under these conditions changes in Isc are wholly reflective of changes in basolateral K+ conductance (IK+).
Na+, K+-ATPase pump activity was measured as previously described (Lam et al. 2003). T84 cell monolayers were mounted in Ussing chambers and bathed bilaterally in low-sodium (25 mm) Ringer's solution, where NaCl was substituted with equimolar N-methyl-d-glucamine (NMDG)-Cl-. Apical membranes were permeabilized with amphotericin (50 μm). Under these conditions (i.e. in the absence of ionic gradients across the permeabilized monolayer) changes in Isc are reflective of electrogenic transport through the Na+, K+-ATPase.
Measurements of cAMP-dependent Cl− currents were carried out using a protocol originally described by Anderson and Welsh and later modified by Hallows and colleagues (Anderson & Welsh, 1991; Hallows et al. 2003). Basolateral membranes were permeabilized with nystatin (100 μg ml−1) and the basolateral Ringer's solution was replaced with a low Cl− solution, containing 25 mm NaCl and 95 mm sodium gluconate, to establish a apical-to-basolateral Cl− concentration gradient. A apical-to-basolateral gradient is employed in these experiments in order to prevent cell swelling that occurs when such experiments are conducted with a basolateral-to-apical gradient. Under these conditions changes in Isc after forskolin (FSK) stimulation reflect CFTR-mediated currents.
Ca2+-activated Cl− channel currents (ICaCC) were measured by a protocol previously described (Schultheiss et al. 2005). Cells were bathed bilaterally in a high K+-containing solution (111.5 mm), which effectively depolarizes the membrane, thereby removing the driving force for K+ movement. Additionally, a basolateral to apical directed Cl− gradient (5.7–113 mm was applied to drive Cl− exit through apical channels. The CFTR inhibitor, CFTRinh172 (10 μm), was added apically in all experiments to block any contribution of CFTR to Isc responses (Ma et al. 2002). Under these conditions changes in Isc stimulated by carbachol (CCh) reflect CaCC-mediated currents. The composition of solutions employed were (in mm): 1.8 Na2HPO4.3H2O, 0.2 NaH2PO4, 25 NaHCO3, 1 MgSO4, 1.25 CaCl2 and 10 glucose. Additionally, the basolateral solution contained 111.5 mm KCl, while the apical solution contained 4.5 mm KCl and 107 mm potassium gluconate. The TMEM16A inhibitor, T16Ainh-A01 (Namkung et al. 2011), was provided as a gift by Prof. Alan Verkman (University of California, San Francisco).
RNA preparation and RT-PCR
After treatment, T84 monolayers were washed (×2) in ice-cold phosphate-buffered saline (PBS) and scraped into ice-cold 1.5 ml Eppendorf tubes. Cell suspensions were centrifuged (130 g; 4 min) and RNA was extracted using RNeasy® Mini Kit (Qiagen, Germany), according to the manufacturer's protocol. RNA was treated with DNA-free reagent (Ambion, Austin, TX, USA) to remove genomic DNA. cDNA was synthesized by reverse transcription using the Improm-II Reverse Transcription kit (Promega, Madison, WI, USA) and the cDNA obtained was used for PCR reactions with previously published primer sets for TMEM16A, GAPDH or 18s rRNA (Carles et al. 2006). The sequence of primers used was as follows: TMEM16A (accession number: NM_018043.5) forward: 5′-CTCCT GGACGAGGTGTATGG-3′, reverse 5′- GAACGCCAC GTAAAAGATGG-3′, GAPDH (NM_002046.3) forward: 5′-TCCCTGAGCTGAACGGGAAG-3′, reverse 5′-GGAGGAGTGGGTGTCGCTGT-3′, and 18s rRNA (NR_003286.1) forward 5′-GTCCCCCAACTTCTTAGAG-3′, reverse 5′-CACCTACGGAAACCTTGTTAC-3′. Primers were obtained from MWG Biotechnology (Ebersberg, Germany) (18s rRNA) or Invitrogen (Carlsbad, CA, USA) (GAPDH, TMEM16A primers). TMEM16A mRNA expression was measured by real-time PCR or semi-quantitive PCR. For real–time PCR analyses, expression was measured over 40 cycles using SYBR-Green (Applied Biosystems, Foster City, CA, USA) as a reporter dye and a 7500 Fast Real-time PCR system (Applied Biosystems). Measurements were done in triplicate. Relative quantity (RQ) of mRNA expression was calculated from ΔΔCt values and control mRNA expression was set as the reference sample. For semi-quantitive PCR, GoTaq® green master mix (Promega) was used in all reactions. The RT-PCR product was analysed by electrophoresis in 2% TAE agarose gel and imaged using a UV light source.
Western blot analysis
After EGF treatment, monolayers were washed (×2) in ice-cold PBS and then lysed for 30 min in lysis buffer (50 mm Tris base, 150 mm NaCl, 1% NP40). Prior to use, the lysis buffer was supplemented with Complete protease inhibitor cocktail (Roche Diagnostics, UK), sodium orthovanadate (100 nm) and PMSF (0.1 mg ml−1). Lysates were scraped into eppendorf tubes and centrifuged at 15,300 g for 10 min at 4°C. The pellets were discarded and after normalizing for protein content, 2× gel loading buffer (50 mm Tris-HCl, 100 mm DTT, 40% glycerol and 4% SDS) was added to the supernatants. After heating to 95°C, samples were separated by SDS-PAGE and transferred to PVDF membranes. Membranes were preblocked with 5% non-fat dry milk (or 5% bovine serum albumin when analysing phosphoproteins) in Tris-buffered saline 0.1% Tween 20 (TBS-T) for 1 h at room temperature after which they were probed overnight at 4°C with antibodies against proteins of interest: TMEM16A (1:5000 dilution; Abcam Inc., Cambridge, MA, USA, cat. no. ab16293), CFTR (1:800 dilution; Millipore, Billerica, MA, USA, cat. no. 05–583), phospho-PKCδ/θ (Ser643/676) (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA, cat. no. 9376) or β-actin (1:10000 dilution; Sigma-Aldrich, St Louis, MO, USA, cat. no. A5316). Membranes were then washed (×5 with TBST) and probed with secondary antibodies conjugated to horseradish peroxidase in 5% blocking buffer. After further washing in TBST, immunoreactive proteins were detected using an enhanced ECL detection kit and exposure of the membrane to X-ray film. Protein levels were quantified by densitometry. Densitometric data were normalized to levels of β-actin in order to control for differences in protein loading between wells, and normalized data were then expressed relative to protein expression in control cells, not treated with EGF.
Statistical analysis
All data are expressed as means ± SEM for a series of n experiments. Student's paired t test or one-way analysis of variance (ANOVA) with the Student–Neuman–Keuls post hoc test was used as appropriate. P values ≤ 0.05 were considered to be statistically significant.
Results
EGF chronically and specifically potentiates a Ca2+-dependent Cl− conductance in colonic epithelial cells
We first investigated the possibility that chronic enhancement of epithelial secretory responses by EGF could be mediated by alterations in apical Cl− conductances. Polarized monolayers of T84 cells were acutely treated with EGF (100 ng ml−1 for 15 min) and after allowing them to recover in serum-free medium for 24 h, were mounted in Ussing chambers for measurements of Isc. Responses to cAMP and calcium-dependent secretagogues were then measured under various conditions to isolate specific types of current as described in Methods. As previously reported (O'Mahony et al. 2008), under conditions that measure transepithelial Cl− secretion, transient (15 min) treatment with EGF chronically enhanced Isc responses to both Ca2+ (CCh; 100 μm) and cAMP (FSK; 10 μm)-dependent agonists (Fig. 1A). Control responses to CCh (100 μm) were 94.5 ± 6.7 μA cm−2 compared to 146.9 ± 9.2 μA cm−2 in EGF-pretreated cells (n= 53; P < 0.001), while control responses to FSK were 129.5 ± 4.6 μA cm−2 compared to 158.6 ± 6.1 μA cm−2 in cells treated with EGF (n= 59; P < 0.001).
Figure 1. EGF chronically potentiates Ca2+-dependent Cl− conductances in colonic epithelial cells.

Monolayers of T84 cells were acutely treated with EGF (basolateral; 100 ng ml−1; 15 min) and 24 h later were mounted in Ussing chambers for measurements of transepithelial Cl− secretion or isolated apical Cl− currents. A, representative tracing showing Cl− secretory responses to basolateral addition of CCh and apical addition of FSK in control or EGF-pretreated cells. The right panel shows mean changes in secretory responses to CCh (n= 53) and FSK (n= 59) in control and EGF-pretreated cells. B, under conditions to isolate cAMP-dependent Cl− conductances, EGF pretreatment did not alter ICl(apical) responses to FSK (10 μm; n= 5). C, under conditions to isolate Ca2+-dependent Cl− conductances EGF significantly potentiated ICaCC responses to CCh (100 μm; n= 26). Asterisks denote statistically significant differences from control cells that were not pretreated with EGF. **P < 0.01; ***P < 0.001.
Currents mediated by apical cAMP-dependent Cl− channels (ICl(apical)) were isolated as previously described (Anderson & Welsh, 1991; Hallows et al. 2003). Under these conditions responses to FSK were practically insensitive to the NKCC1 inhibitor, bumetanide, demonstrating effective permeabilization of the basolateral membrane and isolation of apical currents (data not shown). These currents were also found to be inhibited 80 ± 5% (n= 5) by the CFTR inhibitor, CFTRinh172 (10 μm), confirming that they are primarily mediated by CFTR. However, in cells pretreated with EGF, FSK (10 μm)-stimulated (ICl(apical) responses were found to be 97 ± 9% of those in control cells (n= 5), suggesting that potentiation of cAMP-mediated Cl− currents by the growth factor is not due to upregulation of CFTR conductances (Fig. 1B).
CaCC conductances were measured using a protocol previously described by Schultheiss et al. (2005). In this protocol bilateral addition of high K+-containing solutions prevent K+ movement and the imposition of a basolateral to apical Cl− gradient drives Cl− movement through apical channels. CFTRinh172 (10 μm) was also added apically to block CFTR, and on its own reduced CCh-stimulated currents by 59 ± 10% (n= 9). The remaining CCh-stimulated currents were insensitive to inhibition of either NKCC1 or basolateral Ca2+-activated K+ channels, by bumetanide (100 μm) and clotrimazole (10 μm), respectively (data not shown). Under these conditions, which specifically measure CFTR-independent, Ca2+-stimulated Cl− currents, CCh-stimulated an Isc response of 22.1 ± 1.7 μA cm−2 (n= 26). Interestingly, in cells pretreated with EGF, these responses were enhanced to 173 ± 25% of those in controls (n= 26, P < 0.05) (Fig. 1C).
The effects of EGF on the activity of other important components of the Cl− secretory pathway, namely basolateral Na+, K+-ATPase pumps and K+ channels were also investigated. However, the activity of neither of these transport proteins was found to be altered by the growth factor. In EGF-treated cells, CCh-stimulated Na+, K+-ATPase activity and K+ channel currents were 107 ± 13% (n= 5) and 92 ± 14% (n= 8) of those in control cells, respectively.
EGF induces TMEM16A expression in colonic epithelial cells
Since CaCC currents are chronically potentiated by EGF in colonic epithelial cells, we went on to investigate effects of the growth factor on expression of TMEM16A, which has recently been identified as a Ca2+-dependent Cl− conductance in intestinal epithelial cells. Monolayers of T84 cells were treated with EGF (100 ng ml−1; 15 min) and at various times afterwards TMEM16A mRNA levels were measured by real-time PCR. EGF treatment increased TMEM16A mRNA expression with a maximal effect occurring 4 h after treatment, when levels were increased 2.6 ± 0.4-fold compared to those of control cells (n= 4; P < 0.01) (Fig. 2A). In further experiments, Western blot analysis revealed that EGF also increased the abundance of a doublet of bands migrating at approximately 114 kDa, corresponding to the molecular weight of TMEM16A. TMEM16A protein expression was increased by 297 ± 46% compared to control cells when measured 24 h after treatment (n= 9; P < 0.001) (Fig. 2B). In contrast, EGF did not alter cellular expression of CFTR, which, 24 h after treatment with the growth factor, was found to be 105 ± 21% of that in control cells (n= 8) (Fig. 2C). EGF was also without effect on protein expression of either the basolateral K+ channels, KCNN4 and KCNQ1, or the α and β subunits of Na+, K+-ATPase pumps (data not shown). To determine if EGF-induced increases in TMEM16A expression mediate the effects of the growth factor on Ca2+-dependent Cl− currents, we employed a specific blocker of TMEM16A, T16Ainh-A01, as previously described (Namkung et al. 2010). We found that apical pretreatment of the cells with T16Ainh-A01 (10 μm) abolished the potentiating effects of EGF on CCh-stimulated Ca2+-dependent Cl− currents (Fig. 2D). EGF enhanced these currents to 28 ± 6 μA cm−2 compared to control responses of 14 ± 3 μA cm−2 (n= 7, P < 0.01). However, in the presence of T16Ainh-A01, responses to CCh in EGF-pretreated cells were similar to those in controls (10 ± 4 μA cm−2; n= 7).
Figure 2. EGF induces TMEM16A expression in colonic epithelial cells.

A, monolayers of T84 cells were treated with EGF (basolateral; 100 ng ml−1; 15 min) and at various times (2, 4, 6 and 24 h) afterwards TMEM16A mRNA expression was measured by real-time PCR. B–C, CFTR and TMEM16A protein expression was investigated 24 h after EGF pretreatment by Western blotting. CFTR protein migrated as a band with a molecular weight of 170 kDa and TMEM16A as a doublet of bands at 114 kDa. EGF pretreatment significantly increased TMEM16A mRNA (n= 4) (A) and protein expression (n= 9) (B), without altering levels of CFTR protein expression (n= 8) (C). D, cells were pretreated with EGF (100 ng ml−1; 15 min) and after 24 h were mounted in Ussing chambers under conditions for measuring Ca2+-dependent Cl− conductances. Cells were then apically treated with the TMEM16A blocker, T16Ainh-A01 (10 μm), and ICaCC responses to CCh (100 μm) were measured. T16Ainh-A01 abolished the potentiating effects of EGF on CCh-induced ICaCC responses (n= 7). *P < 0.05 **P < 0.01; ***P < 0.001.
Protein kinase C (PKC)δ mediates EGF-induced increases in Ca2+-dependent Cl− conductances
PKC is a well-established effector of epidermal growth factor receptor (EGFR) activation and is an important regulator of epithelial transport (Chow et al. 2000; Del Castillo et al. 2005; Broughman et al. 2006; Lau et al. 2010). Thus, we investigated if PKC might have a role to play in mediating enhanced secretory responses to EGF in colonic epithelial cells. To answer this, cells were pretreated for 30 min with PKC inhibitors before treatment with EGF (100 ng ml−1; 24 h). We found that when cells were pretreated with a general PKC inhibitor, GF109203X (5 μm), EGF-induced potentiation of Cl− secretion was abolished. Responses to CCh in cells treated with EGF in presence of GF109203X were 107 ± 13% of controls, compared to 206 ± 31% in those treated with EGF alone (n= 6; P < 0.01) (Fig. 3A). To further characterize the role of PKC in mediating effects of EGF, pharmacological inhibitors that block the activity of specific PKC isoforms were employed. Although EGF has been previously shown to activate PKCɛ in T84 cells (Chow et al. 2000; Del Castillo et al. 2005), an established inhibitor of this PKC isoform, known as PKCɛ translocation inhibitor (Meyer et al. 2009), was found to be without effect on EGF-induced potentiation of secretory responses (Fig. 3B). In EGF-pretreated cells CCh-stimulated responses were 192 ± 37% of controls (n= 5; P < 0.05) compared to 198 ± 30% (n= 5; P < 0.05) in the presence of the PKCɛ translocation inhibitor. Similarly, a PKCα and β inhibitor, Gö6976 (1 μm), was found to be without effect, suggesting these isoforms are not involved (Fig. 3C). This is supported by the observation that EGF treatment did not alter levels of PKCα phosphorylation at any time after treatment (n= 3) (Fig. 3E). We next employed an inhibitor of PKCδ, rottlerin, to investigate the role of this isoform in mediating responses to EGF. Rottlerin (20 μm), at a concentration known to differentiate between PKCδ and other PKC isoforms (Gschwendt et al. 1994), abolished the potentiating effect of EGF on transepithelial Cl− secretion induced by CCh (Fig. 3D). CCh-induced responses after EGF treatment alone were 191 ± 63% of control responses (n= 4, P < 0.01) compared to 121 ± 19% in rottlerin-treated cells (n= 4). In addition, EGF was found to induce phosphorylation of PKCδ with a maximal response of 171 ± 22% compared to unstimulated cells, occurring 1 h after treatment (n= 3 − 10; P < 0.001; Fig. 3E).
Figure 3. PKCδ mediates EGF-induced increases in Ca2+-dependent Cl− secretion.
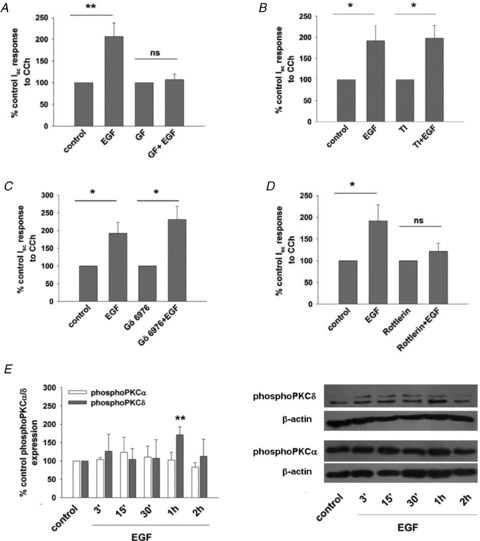
A–D, monolayers of T84 cells were treated with EGF (basolateral; 100 ng ml−1; 15 min) alone or in presence of a general PKC inhibitor, GF109203X (GF) (5 μm; n= 6) (A), a PKCɛ translocation inhibitor (TI) (200 μm; n= 5) (B), PKCα/β inhibitor, Gö6976 (1 μm; n= 6) (C), or a PKCδ inhibitor, rottlerin (20 μm; n= 4) (D). All pharmacological inhibitors were added bilaterally for 30 min before EGF treatment. After 24 h monolayers were mounted in Ussing chambers and Cl− secretory responses to basolateral CCh (100 μm) were measured. Results are expressed as mean ± SEM percentage control Isc responses to CCh in cells not treated with EGF. E, monolayers of T84 cells were treated with basolateral EGF (100 ng ml−1; 15 min) and at various times afterwards PKCα or PKCδ phosphorylation was measured by Western blotting. Densitometric analysis of several experiments is shown with results expressed as mean ± SEM percentage control PKCα or PKCδ expression in untreated cells. Since Western blots for PKCδ revealed 2 closely migrating bands that may represent splice variants of the protein, both of these bands were included in densitometric analyses. EGF stimulated phosphorylation of PKCδ (n= 3 − 10) but not PKCα (n= 3). Representative blots are shown in the right panel. *P < 0.05; **P < 0.01.
Having shown that PKCδ mediates EGF-induced increases in transepithelial Cl− secretion, we next investigated if the growth factor also regulates Ca2+-dependent Cl− conductances in the apical membrane. Under experimental conditions that specifically isolate these currents, pretreatment with rottlerin (20 μm) also inhibited potentiation of CCh-induced responses by EGF. In cells treated with EGF alone, responses to CCh were 179 ± 26% of controls (n= 4; P < 0.01), compared to 115 ± 51% in cells treated with the growth factor in the presence of rottlerin (n= 4) (Fig. 4A). Further experiments revealed that rottlerin also abolished the effects of EGF on TMEM16A protein expression (Fig. 4B). Rottlerin did not alter basal TMEM16A expression.
Figure 4. PKCδ mediates EGF-induced increases in Ca2+-dependent Cl− conductances and TMEM16A expression.

A, monolayers of T84 cells were acutely treated with basolateral EGF (100 ng ml−1; 15 min) alone or in presence of the PKCδ inhibitor, rottlerin (20 μm; bilateral addition). Cells were pretreated with rottlerin for 30 min before addition of EGF. After 24 h cell monolayers were mounted in Ussing chambers under conditions to isolate Ca2+-dependent Cl− conductances. Data are expressed as mean ± SEM percentage control ICaCC responses to basolateral CCh (100 μm). Rottlerin reversed EGF potentiation of CCh-stimulated CaCC conductances (n= 4). B, monolayers of T84 cells were acutely treated with EGF alone or in the presence of rottlerin (20 μm). Cells were then lysed and TMEM16A expression measured by Western blotting. The lower panel shows densitometric analysis of 5 experiments with data expressed as mean ± SEM percentage TMEM16A expression in cells not treated with EGF. Rottlerin abolished EGF-induced increases in TMEM16A expression. **P < 0.01.
Phosphatidylinositol 3-kinase (PI3K) mediates EGF-induced potentiation of Ca2+-dependent Cl− conductances
PI3K is a well-described effector protein that is activated downstream of the EGFR and upstream of PKC isoforms in a number of systems, including colonic epithelial cells (Ecay et al. 2000; Bertelsen et al. 2004; Tuo et al. 2009; Hoda et al. 2010). We therefore sought to investigate if PI3K might also be involved in the chronic actions of EGF on colonic epithelial secretion. To test this, T84 cells were pretreated with the PI3K inhibitor LY294002 (25 μm) for 30 min before treatment with EGF (100 ng ml−1; 15 min). After 24 h CCh-stimulated transepithelial Cl− secretory responses or apical Cl− conductances were measured. As shown in Fig. 5A, PI3K inhibition significantly attenuated the enhancement of CCh-stimulated transepithelial secretory responses brought about by EGF. In cells treated with EGF, transepithelial Cl− secretion in response to CCh was enhanced 188 ± 30% of that in control cells (n= 6; P < 0.01), while pretreatment with LY290042 reduced these responses to 134 ± 15% of controls (n= 6), which were not significantly different from control responses. Next, we investigated if PI3K inhibition also alters EGF-induced potentiation of Ca2+-dependent Cl− conductances. In EGF treated cells Isc responses to CCh were significantly increased to 30 ± 6 μA cm−2 compared to 14 ± 3 μA cm−2 in controls (n= 7; P < 0.05) (Fig. 5B). However, responses in cells treated with EGF in the presence of LY290042 were similar (16 ± 4 μA cm−2) to those in cells treated with LY290042 alone (21 ± 4 μA cm−2) (n= 7). Furthermore, inhibition of PI3K was also found to prevent the effects of EGF on both TMEM16A mRNA and protein expression (Fig. 5C and D).
Figure 5. PI3K mediates EGF-induced increases in Ca2+-dependent Cl− conductances, TMEM16A expression and PKCδ activation.

A and B, monolayers of T84 cells were acutely treated with basolateral EGF (100 ng ml−1; 15 min) alone or in the presence of the PI3K inhibitor, LY294002 (LY) (25 μm; bilateral addition). Cells were pretreated with LY290042 for 30 min before addition of EGF. After 24 h cell monolayers were mounted in Ussing chambers for measurements of Cl− secretion (A) or Ca2+-dependent Cl− conductances (ICaCC) stimulated by basolateral CCh (100 μm) (B). Data are expressed as mean ± SEM percentage control Isc responses to CCh in cells not treated with EGF (A) or Isc response to CCh (B). C, TMEM16A mRNA expression was measured by RT-PCR 4 h after EGF treatment in the absence or presence of LY290042 (n= 4). D, TMEM16A protein expression was measured by Western blotting 24 h after EGF treatment in the absence or presence of LY290042 (n= 5). E, PKCδ phosphorylation was measured 1 h after EGF treatment in the absence or presence of LY290042 (n= 5). *P < 0.05; **P < 0.01.
Finally, to determine if EGF-induced PI3K activation occurs upstream of PKCδ, the effects of LY294002 on PKCδ activation were examined. In cells treated with EGF alone, phosphorylation of PKCδ was increased 167 ± 22% (n= 5; P < 0.05) (Fig. 5E), while in cells pretreated with LY290042 this response was reduced to 120 ± 22% of controls.
Discussion
Fluid and electrolyte secretion in the intestine are critical processes for normal digestive function and in maintaining appropriate hydration of the mucosal surfaces. The physiological importance of fluid secretion is underlined in pathological conditions such as cystic fibrosis and diarrhoeal diseases, where under- or overexpression of Cl− secretion can lead to constipation, obstruction, diarrhoea or maldigestion (Keely et al. 2009). There is currently a lack of safe and specific therapeutic approaches for epithelial transport disorders, which is largely due to our incomplete understanding of the regulatory mechanisms involved. Growth factors, such as EGF, are emerging as important regulators of intestinal transport function in the short and long term and as such they, and the signalling pathways they activate, present good targets for the development of new treatments for epithelial transport disorders. Previous studies in cultured human colonic epithelial cells and in mouse colonic tissue have shown that acute exposure to EGF inhibits subsequent epithelial responses to secretagogues (Uribe et al. 1996a; Schultheiss & Deiner, 2005; McCole & Barrett, 2009), although the mechanisms involved appear to be species dependent since similar antisecretory effects were not observed in rats (Schultheiss & Diener, 2005). Furthermore, we have recently shown that over more prolonged periods of time EGF enhances secretory function (O'Mahony et al. 2008). This latter effect involves upregulation of NKCC1, which constitutes the basolateral entry pathway for Cl−. Our current studies sought to further elucidate molecular mechanisms underlying chronic potentiation of secretion by EGF and reveal that the growth factor chronically and specifically upregulates TMEM16A, a Ca2+-dependent channel that constitutes an apical exit pathway for Cl− in epithelial cells.
While CFTR has long been known to mediate Cl− secretion in response to cAMP-dependent secretagogues, the identity, and even the existence, of specific Ca2+-dependent Cl− conductances in intestinal epithelia has been a subject of controversy. Several early studies failed to detect CaCCs in differentiated intestinal epithelia (Berschneider et al. 1988; Anderson & Welsh, 1991), leading investigators to believe that CFTR also mediates secretory responses to Ca2+-dependent agonists, with the proposed model being that hyperpolarization of the basolateral membrane due to efflux of K+ provided the electrical driving force for Cl− secretion through a pool of constitutively open apical CFTR channels. This hypothesis is supported by our current findings where we found that under conditions for isolating Ca2+-activated Cl− currents in T84 cells, subsequent Isc responses were inhibited 60% by the CFTR blocker, CFTRinh172. However, a large body of evidence now exists to support the idea that intestinal epithelia also express a distinct Ca2+-dependent Cl− conductance. Several studies have shown that two Cl− conductances are present in the apical membrane of cultured colonic epithelial cells and animal tissues, one activated by cAMP and the other by Ca2+ (Vaandrager et al. 1991; Valverde et al. 1993; McEwan et al. 1994; Merlin et al. 1998; Hennig et al. 2008; Tradtrantip et al. 2010). Moreover, silencing of CFTR in T84 cells by antisense oligodeoxynucleotides decreases cAMP-stimulated Cl− currents without altering Ca2+-dependent Cl− secretion (Wagner et al. 1992). Using an established experimental approach to isolate Ca2+-dependent and CFTR-independent Cl− conductances (Hennig et al. 2008), our studies also support the existence of a functional Ca2+-dependent Cl− channel in T84 cells. Furthermore, our data show that acute treatment with EGF chronically potentiates this conductance. This effect of EGF is specific since the growth factor did not alter cAMP-dependent currents mediated by CFTR. These data are the first to demonstrate a role for EGF in regulating Cl− channel conductances in intestinal epithelia and develop our understanding of how EGF can regulate epithelial secretory function in the long term. In addition to enhancing uptake of Cl− through increased NKCC1 expression, EGF simultaneously upregulates the exit pathway for Cl− in the apical membrane, but only to agonists that stimulate Ca2+-dependent secretion. These findings may account for our observations that transepithelial secretory responses to Ca2+-dependent agonists are more sensitive to potentiation by EGF than those elicited by cAMP.
The discovery of TMEM16A as a Ca2+-dependent Cl− conductance represented a great step forward in the field of epithelial physiology (Caputo et al. 2008; Schroeder et al. 2008; Yang et al. 2008). The channel is expressed in a wide array of tissues including epithelia from the airways, kidneys and the intestine where it has been shown to mediate Ca2+-dependent Cl− secretion in animal models and cultured cells (Galietta, 2009). However, the extent to which TMEM16A mediates Ca2+-dependent secretory responses in the intestine in vivo is not yet clear. While in postnatal TMEM16A knockout mice Ca2+-dependent Cl− secretion is abolished in the intestine, in cultured colonic epithelia it has been reported that, although the channel is present and functional, it mediates only a minor component of Ca2+-dependent secretory responses (Ousingsawat et al. 2009; Tradtrantip et al. 2010; Namkung et al. 2011). However, like all transport proteins, it is likely that expression levels of TMEM16A are tightly regulated in health and disease and a greater understanding of the mechanisms involved could lead to new ways to manipulate this protein in treatment of intestinal transport disorders. Here, we have shown that in conjunction with its effects on Ca2+-dependent Cl− secretion, EGF also upregulates expression of TMEM16A in colonic epithelial cells. This effect was specific, since consistent with its lack of effect on cAMP-dependent Cl− conductances, the growth factor did not alter expression of CFTR. Furthermore, that TMEM16A mediates increased apical Cl− currents to Ca2+-dependent agonists was evidenced by our observations that T16Ainh-A01, a selective blocker of TMEM16A, abolished the effects of EGF on CCh-stimulated ICaCC responses. Interestingly, we found that T16Ainh-A01 slightly, but not significantly, reduces CCh-induced responses in T84 cells that were not pretreated with EGF. This confirms previous observations that TMEM16A is normally a minor component of Ca2+-dependent secretion in T84 cells (Namkung et al. 2011), and suggests that expression of the channel must first be induced before it can play a significant role.
The current studies are the first to describe intracellular signalling mechanisms that regulate the expression of TMEM16A in epithelial cells. Through the use of biochemical and pharmacological approaches we demonstrate that the effects of EGF on both TMEM16A expression and Ca2+-dependent Cl− conductances involve activation of PKCδ. Interestingly, it has been previously shown in rabbits that the age-dependent expression of PKCδ in colonic epithelial cells correlates with their ability to secrete Cl− in response to Ca2+-dependent secretagogues (Kanchanapoo et al. 2007). This supports the idea that PKCδ is necessary for the expression of a transport protein involved in the Ca2+-dependent secretory pathway. Our current findings are also intriguing in light of previous studies which show that the acute antisecretory effects of EGF are mediated by a different PKC isoform, PKCɛ, which rapidly inhibits the activity of basolateral K+ channels (Chow et al. 2000). Furthermore, in more recent studies, both PKCδ and PKCɛ have been shown to be part of a signalling mechanism that regulates NKCC1 endocytosis at the basolateral membrane (Tang et al. 2010). While we found no evidence of a role for PKCɛ in mediating chronic actions of EGF on epithelial secretory function, it is interesting to note that both PKCɛ and PKCδ activation occur downstream of PI3K, an enzyme that rapidly associates with the EGF receptor after ligand binding (Uribe et al. 1996b). Thus, although it is initially a common PI3K-mediated signalling mechanism that regulates both the short and long-term actions of EGF on epithelial secretory capacity, these effects are ultimately determined by differential actions of different PKC isoforms. Indeed, it has long been known that PKC isoforms exert complex pro- and antisecretory actions on intestinal epithelial cells (Kachintorn et al. 1992) but there still is much work to be done to fully elucidate their roles in regulating the activity, trafficking and expression of transport proteins.
Although our data show that EGF chronically upregulates Ca2+-dependent Cl− secretion in intestinal epithelial cells, the physiological significance of these findings remains to be determined. While some studies suggest that Ca2+-dependent secretion is either absent or only a minor contributor to secretory responses in normal adult humans, our findings suggest that this prosecretory pathway may become more relevant in conditions of intestinal injury or stress. For example, EGF is considered as a luminal surveillance peptide that does not normally have access to its receptor on the basolateral membrane (Playford & Wright, 1996). However, should epithelial barrier function become compromised, for example due to sloughing off of epithelial cells as digested food moves through the lumen, then EGF can gain access to its basolateral receptors to bring about an integrated response. One component of this response may be to enhance epithelial secretion in order to lubricate the mucosa, thereby promoting barrier function. A similar scenario can be envisioned in the pathological setting of bowel inflammation where enhanced mucosal levels of the EGFR ligand, TGF-α, are known to occur (Myhre et al. 2004). In addition to promoting restitution, we would expect that the growth factor would also induce long-term increases in secretory capacity which would serve to augment barrier function and also to flush the intestine of cellular debris, inflammatory mediators and other noxious substances. Our findings may also have relevance for the future development of treatments for cystic fibrosis. There has long been great interest in targeting Ca2+-dependent Cl− conductances as a means to bypass defective CFTR in therapy of this disease. However, such approaches may have limited efficacy in the intestine due to low levels of Ca2+-dependent Cl− conductances being present. Thus, pharmaceutical agents that have the capability to upregulate intestinal expression of TMEM16A are likely to be of therapeutic benefit.
In summary, we have shown that acute stimulation with the growth factor EGF chronically upregulates Ca2+-dependent Cl− conductances and TMEM16A expression in intestinal epithelial cells through a mechanism involving sequential activation of PI3K and PKCδ. These findings provide valuable new insights into the molecular mechanisms by which intestinal secretory function may be regulated in health and disease. In future work we aim to further elucidate transcriptional and post-transcriptional mechanisms by which EGF exerts its effects on epithelial transport protein function with the hope that this will lead to the identification of new targets for treatment of intestinal transport disorders.
Acknowledgments
This work was supported by a Principal Investigator Award from Science Foundation Ireland to S.J.K. We gratefully acknowledge Prof. Alan Verkman and Dr Zhen Yao for providing us with the TMEM16A inhibitor, T16Ainh-A01.
Glossary
Abbreviations
- CCh
carbachol
- CFTR
cystic fibrosis transmembrane conductance regulator
- EGF
epidermal growth factor
- FSK
forskolin
- ICaCC
Ca2+-dependent Cl− channel conductance
- Isc
short-circuit current
- NKCC
Na+–K+–2Cl− co-transporter
- PI3K
phosphatidylinositol 3-kinase
- PKC
protein kinase C
- TMEM16A
transmembrane protein 16A
Author contributions
These experiments were conducted in the Molecular Medicine Laboratories, Royal College of Surgeons in Ireland. Both M.M. and S.K. contributed to (i) conception and design of experiments, (ii) collection analysis and interpretation of data and (iii) drafting and reviewing the article for important intellectual content. Both authors approve the manuscript in its submitted form.
References
- Anderson MP, Welsh MJ. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc Natl Acad Sci U S A. 1991;88:6003–6007. doi: 10.1073/pnas.88.14.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- Berschneider HM, Knowles MR, Azizkhan RG, Boucher RC, Tobey NA, Orlando RC, Powell DW. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988;2:2625–2629. doi: 10.1096/fasebj.2.10.2838365. [DOI] [PubMed] [Google Scholar]
- Bertelsen LS, Barrett KE, Keely SJ. Gs protein-coupled receptor agonists induce transactivation of the epidermal growth factor receptor in T84 cells: implications for epithelial secretory responses. J Biol Chem. 2004;279:6271–6279. doi: 10.1074/jbc.M311612200. [DOI] [PubMed] [Google Scholar]
- Borok Z, Hami A, Danto SI, Lubman RL, Kim KJ, Crandall ED. Effects of EGF on alveolar epithelial junctional permeability and active sodium transport. Am J Physiol Lung Cell Mol Physiol. 1996;270:L559–565. doi: 10.1152/ajplung.1996.270.4.L559. [DOI] [PubMed] [Google Scholar]
- Broughman JR, Sun L, Umar S, Scott J, Sellin JH, Morris AP. Chronic PKC-β activation in HT-29 Cl.19a colonocytes prevents cAMP-mediated ion secretion by inhibiting apical membrane current generation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G318–330. doi: 10.1152/ajpgi.00355.2005. [DOI] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Carles A, Millon R, Cromer A, Ganguli G, Lemaire F, Young J, Wasylyk C, Muller D, Schultz I, Rabouel Y, Dembele D, Zhao C, Marchal P, Ducray C, Bracco L, Abecassis J, Poch O, Wasylyk B. Head and neck squamous cell carcinoma transcriptome analysis by comprehensive validated differential display. Oncogene. 2006;25:1821–1831. doi: 10.1038/sj.onc.1209203. [DOI] [PubMed] [Google Scholar]
- Cartwright CA, McRoberts JA, Mandel KG, Dharmsathaphorn K. Synergistic action of cyclic adenosine monophosphate- and calcium-mediated chloride secretion in a colonic epithelial cell line. J Clin Invest. 1985;76:1837–1842. doi: 10.1172/JCI112176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JY, Uribe JM, Barrett KE. A role for protein kinase Cɛ in the inhibitory effect of epidermal growth factor on calcium-stimulated chloride secretion in human colonic epithelial cells. J Biol Chem. 2000;275:21169–21176. doi: 10.1074/jbc.M002160200. [DOI] [PubMed] [Google Scholar]
- Chung BM, Wallace LE, Hardin JA, Gall DG. The effect of epidermal growth factor on the distribution of SGLT-1 in rabbit jejunum. Can J Physiol Pharmacol. 2002;80:872–878. doi: 10.1139/y02-115. [DOI] [PubMed] [Google Scholar]
- Del Castillo IC, Fedor-Chaiken M, Song JC, Starlinger V, Yoo J, Matlin KS, Matthews JB. Dynamic regulation of Na+-K+-2Cl− cotransporter surface expression by PKCɛ in Cl− secretory epithelia. Am J Physiol Cell Physiol. 2005;289:C1332–1342. doi: 10.1152/ajpcell.00580.2004. [DOI] [PubMed] [Google Scholar]
- Donowitz M, Janecki A, Akhter S, Cavet ME, Sanchez F, Lamprecht G, Zizak M, Kwon WL, Khurana S, Yun CH, Tse CM. Short-term regulation of NHE3 by EGF and protein kinase C but not protein kinase A involves vesicle trafficking in epithelial cells and fibroblasts. Ann N Y Acad Sci. 2000;915:30–42. doi: 10.1111/j.1749-6632.2000.tb05221.x. [DOI] [PubMed] [Google Scholar]
- Dutta AK, Khimji AK, Kresge C, Bugde A, Dougherty M, Esser V, Ueno Y, Glaser SS, Alpini G, Rockey DC, Feranchak AP. Identification and functional characterization of TMEM16A, a Ca2+-activated Cl− channel activated by extracellular nucleotides, in biliary epithelium. J Biol Chem. 2011;286:766–776. doi: 10.1074/jbc.M110.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecay TW, Dickson JL, Conner TD. Wortmannin inhibition of forskolin-stimulated chloride secretion by T84 cells. Biochim Biophys Acta. 2000;1467:54–64. doi: 10.1016/s0005-2736(00)00204-2. [DOI] [PubMed] [Google Scholar]
- Galietta LJ. The TMEM16 protein family: a new class of chloride channels? Biophys J. 2009;97:3047–3053. doi: 10.1016/j.bpj.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK. Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am J Physiol Cell Physiol. 2003;284:C1297–1308. doi: 10.1152/ajpcell.00227.2002. [DOI] [PubMed] [Google Scholar]
- Hennig B, Schultheiss G, Kunzelmann K, Diener M. Ca2+-induced Cl− efflux at rat distal colonic epithelium. J Membr Biol. 2008;221:61–72. doi: 10.1007/s00232-007-9078-0. [DOI] [PubMed] [Google Scholar]
- Hoda MR, Scharl M, Keely SJ, McCole DF, Barrett KE. Apical leptin induces chloride secretion by intestinal epithelial cells and in a rat model of acute chemotherapy-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G714–721. doi: 10.1152/ajpgi.00320.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachintorn U, Vongkovit P, Vajanaphanich M, Dinh S, Barrett KE, Dharmsathaphorn K. Dual effects of a phorbol ester on calcium-dependent chloride secretion by T84 epithelial cells. Am J Physiol Cell Physiol. 1992;262:C15–22. doi: 10.1152/ajpcell.1992.262.1.C15. [DOI] [PubMed] [Google Scholar]
- Kanchanapoo J, Ao M, Prasad R, Moore C, Kay C, Piyachaturawat P, Rao MC. Role of protein kinase C-δ in the age-dependent secretagogue action of bile acids in mammalian colon. Am J Physiol Cell Physiol. 2007;293:C1851–1861. doi: 10.1152/ajpcell.00194.2007. [DOI] [PubMed] [Google Scholar]
- Keely S, Montrose M, Barrett K. Secretion and absorption: small intestine and colon. In: Yamada T, Kalloo AN, Alpers D, Kaplowitz CO, Powell D, editors. Textbook of Gastroenterology. 5th edn. Philadelphia: Lippencott-Raven; 2009. pp. 330–367. [Google Scholar]
- Kirk K, Dawson D. Basolateral potassium channel in turtle colon. Evidence for single-file ion flow. J Gen Physiol. 1983;82:297–329. doi: 10.1085/jgp.82.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam R, App E, Nahirney D, Szkotak A, Vieira-Coelho M, King M, Duszyk M. Regulation of Cl− secretion by α2-adrenergic receptors in mouse colonic epithelium. J Physiol. 2003;548:475–484. doi: 10.1113/jphysiol.2002.036806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Lytle C, Straus DS, DeFea KA. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol Cell Physiol. 2010;300:C113–123. doi: 10.1152/ajpcell.00162.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCole DF, Barrett KE. Decoding epithelial signals: critical role for the epidermal growth factor receptor in controlling intestinal transport function. Acta Physiol (Oxf) 2009;195:149–159. doi: 10.1111/j.1748-1716.2008.01929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan GT, Hirst BH, Simmons NL. Carbachol stimulates Cl− secretion via activation of two distinct apical Cl− pathways in cultured human T84 intestinal epithelial monolayers. Biochim Biophys Acta. 1994;1220:241–247. doi: 10.1016/0167-4889(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Merlin D, Jiang L, Strohmeier GR, Nusrat A, Alper SL, Lencer WI, Madara JL. Distinct Ca2+- and cAMP-dependent anion conductances in the apical membrane of polarized T84 cells. Am J Physiol Cell Physiol. 1998;275:C484–495. doi: 10.1152/ajpcell.1998.275.2.C484. [DOI] [PubMed] [Google Scholar]
- Meyer KD, Zhang H, Zhang L. Prenatal cocaine exposure abolished ischemic preconditioning-induced protection in adult male rat hearts: role of PKCɛ. Am J Physiol Heart Circ Physiol. 2009;296:H1566–1576. doi: 10.1152/ajpheart.00898.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre GM, Toruner M, Abraham S, Egan LJ. Metalloprotease disintegrin-mediated ectodomain shedding of EGFR ligands promotes intestinal epithelial restitution. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1213–1219. doi: 10.1152/ajpgi.00149.2004. [DOI] [PubMed] [Google Scholar]
- Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 2010;24:4178–4186. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CU, Amstrup J, Steffansen B, Frokjaer S, Brodin B. Epidermal growth factor inhibits glycylsarcosine transport and hPepT1 expression in a human intestinal cell line. Am J Physiol Gastrointest Liver Physiol. 2001;281:G191–199. doi: 10.1152/ajpgi.2001.281.1.G191. [DOI] [PubMed] [Google Scholar]
- O'Mahony F, Toumi F, Mroz MS, Ferguson G, Keely SJ. Induction of Na+/K+/2Cl− cotransporter expression mediates chronic potentiation of intestinal epithelial Cl− secretion by EGF. Am J Physiol Cell Physiol. 2008;294:C1362–1370. doi: 10.1152/ajpcell.00256.2007. [DOI] [PubMed] [Google Scholar]
- Ousingsawat J, Martins JR, Schreiber R, Rock JR, Harfe BD, Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J Biol Chem. 2009;284:28698–28703. doi: 10.1074/jbc.M109.012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Playford RJ, Wright NA. Why is epidermal growth factor present in the gut lumen? Gut. 1996;38:303–305. doi: 10.1136/gut.38.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko VG, Catalan MA, Brown DA, Putzier I, Hartzell HC, Marmorstein AD, Gonzalez-Begne M, Rock JR, Harfe BD, Melvin JE. Tmem16A encodes the Ca2+-activated Cl− channel in mouse submandibular salivary gland acinar cells. J Biol Chem. 2010;285:12990–13001. doi: 10.1074/jbc.M109.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheiss G, Siefjediers A, Diener M. Muscarinic receptor stimulation activates a Ca2+-dependent Cl− conductance in rat distal colon. J Membr Biol. 2005;204:117–127. doi: 10.1007/s00232-005-0757-4. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Bouyer P, Mykoniatis A, Buschmann M, Matlin KS, Matthews JB. Activated PKCδ and PKCɛ inhibit epithelial chloride secretion response to cAMP via inducing internalization of the Na+-K+-2Cl− cotransporter NKCC1. J Biol Chem. 2010;285:34072–34085. doi: 10.1074/jbc.M110.137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Kongsuphol P, Hug M, Ousingsawat J, Witzgall R, Schreiber R, Kunzelmann K. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J. 2011;25:1058–1068. doi: 10.1096/fj.10-166884. [DOI] [PubMed] [Google Scholar]
- Tradtrantip L, Namkung W, Verkman AS. Crofelemer, an antisecretory antidiarrheal proanthocyanidin oligomer extracted from Croton lechleri, targets two distinct intestinal chloride channels. Mol Pharmacol. 2010;77:69–78. doi: 10.1124/mol.109.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo B, Wen G, Zhang Y, Liu X, Wang X, Dong H. Involvement of phosphatidylinositol 3-kinase in cAMP- and cGMP-induced duodenal epithelial CFTR activation in mice. Am J Physiol Cell Physiol. 2009;297:C503–515. doi: 10.1152/ajpcell.00460.2008. [DOI] [PubMed] [Google Scholar]
- Uribe JM, Gelbmann CM, Traynor-Kaplan AE, Barrett KE. Epidermal growth factor inhibits Ca2+-dependent Cl− transport in T84 human colonic epithelial cells. Am J Physiol Cell Physiol. 1996a;271:C914–922. doi: 10.1152/ajpcell.1996.271.3.C914. [DOI] [PubMed] [Google Scholar]
- Uribe JM, Keely SJ, Traynor-Kaplan AE, Barrett KE. Phosphatidylinositol 3-kinase mediates the inhibitory effect of epidermal growth factor on calcium-dependent chloride secretion. J Biol Chem. 1996b;271:26588–26595. doi: 10.1074/jbc.271.43.26588. [DOI] [PubMed] [Google Scholar]
- Vaandrager AB, Bajnath R, Groot JA, Bot AG, De Jonge HR. Ca2+ and cAMP activate different chloride efflux pathways in HT-29.cl19A colonic epithelial cell line. Am J Physiol Gastrointest Liver Physiol. 1991;261:G958–965. doi: 10.1152/ajpgi.1991.261.6.G958. [DOI] [PubMed] [Google Scholar]
- Valverde MA, O'Brien JA, Sepulveda FV, Ratcliff R, Evans MJ, Colledge WH. Inactivation of the murine cftr gene abolishes cAMP-mediated but not Ca2+-mediated secretagogue-induced volume decrease in small-intestinal crypts. Pflugers Arch. 1993;425:434–438. doi: 10.1007/BF00374869. [DOI] [PubMed] [Google Scholar]
- Wagner JA, McDonald TV, Nghiem PT, Lowe AW, Schulman H, Gruenert DC, Stryer L, Gardner P. Antisense oligodeoxynucleotides to the cystic fibrosis transmembrane conductance regulator inhibit cAMP-activated but not calcium-activated chloride currents. Proc Natl Acad Sci U S A. 1992;89:6785–6789. doi: 10.1073/pnas.89.15.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhang B, Li J, Chen H, Tooley J, Ghishan FK. Epidermal growth factor inhibits intestinal NHE8 expression via reducing its basal transcription. Am J Physiol Cell Physiol. 2010;299:C51–57. doi: 10.1152/ajpcell.00081.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]