

Abstract
Commercially available (2-fluoro)allyl chloride serves as an efficient allyl donor in highly enantioselective iridium catalyzed carbonyl (2-fluoro)allylations from the alcohol or aldehyde oxidation level via transfer hydrogenation. Diastereoselective Crabtree hydrogenation of the resulting homoallylic alcohols provides syn-3-fluoro-1-alcohols.
Organofluorine compounds represent over 20% of approved pharmaceutical agents and 30–40% of commercially available agrochemicals.1 The importance of organofluorine compounds, along with the fact that approximately 80% of the small molecule drugs entering the market are estimated to contain one or more chiral centers,2 have driven development of enantioselective methods for the preparation of fluorinated compounds.3 Under the conditions of C–C bond forming transfer hydrogenation,4,5 we recently reported an enantioselective iridium catalyzed carbonyl (α-trifluoromethyl)allylation from the alcohol or aldehyde oxidation level.5k Given the commercial availability of (2-fluoro)allyl chloride, corresponding carbonyl (2-fluoro)-allylations were considered. Remarkably, despite decades of work on enantioselective carbonyl allylation,6 enantioselective carbonyl (2-fluoro)allylations have not been reported. Here, under the conditions of iridium catalyzed transfer hydrogenation, we report the first enantioselective (2-fluoro)allylations, which are achieved with equal facility from the alcohol or aldehyde oxidation level. These adducts participate in diastereoselective Crabtree hydrogenation,7 enabling the synthesis of syn-3-fluoro-1-alcohols via consecutive C–C and C–H bond forming hydrogenations (Scheme 1).
Scheme 1.

Synthesis of syn-3-fluoro-1-alcohols via consecutive C–C and C–H bond forming hydrogenations.
Olefin coordination is a prerequisite to the ionization of allylic leaving groups by low valent transition metals. Consequently, in iridium catalyzed carbonyl allylations employing allylic carboxylates, allyl donors that incorporate monosubstituted olefins are generally required, as the stability of late transition metal-olefin π-complex decreases with increasing degree of olefin substitution.8 In recently established catalytic C–C couplings of methallyl chloride,5l it was found that use of a more reactive leaving group in the form of chloride compensates for the shorter lifetime associated with more highly substituted olefin-iridium complexes. To probe the expansion of substrate scope potentially availed by this effect, and given the aforementioned significance of organofluorine compounds, a study on the use of (2-fluoro)allyl chloride as an allyl donor was undertaken (Scheme 2).
Scheme 2.

More reactive chloride leaving groups compensate for decreased stability of π-complex.
Studies began with a preliminary screen of (2-fluoro)allyl chloride and alcohol 2g using the chromatographically isolated cyclometallated iridium π-allyl complex of 4-cyano-3-nitrobenzoic acid and BIPHEP under conditions optimized for the reaction of methallyl chloride.5l Although the product 4g was obtained in good isolated yield, competing defluorination to form 5g was observed. Attempts were made to attenuate this side reaction. Variation of solvent, concentration and base provided no improvement beyond the initially applied conditions, which involve THF (1.0 M) andK3PO4 (100 mol%). Reaction temperature had a more significant impact. For allylic and benzylic alcohols, which dehydrogenate and, hence, couple at lower temperature, reactions conducted at 40 °C were optimal in terms of maximizing product formation and minimizing defluorination. For aliphatic alcohols, the optimal reaction temperature was determined to be 60 °C.
To assess whether these trends in reactivity translate to enantioselective processes, the chiral complex Ir-Cat-I (Scheme 3), which is isolated by conventional silica gel chromatography, was prepared and assayed in the coupling of (2-fluoro)allyl chloride to alcohols 2a–2i. To our delight, aliphatic alcohols 2a–2c, allylic alcohols 2d–2f and benzylic alcohols 2g–2i participate in highly enantioselective C–C coupling to furnish the corresponding products of (2-fluoro)allylation 4a–4i. In general, the products of (2-fluoro)allylation 4a–4i may be separated from the defluorination products 5a–5i by silica gel chromatography. However, to ensure an accurate evaluation of the product distribution, 4a–4i and 5a–5i were isolated as mixtures (Table 1). An equivalent set of adducts 4a–4i may be generated from aldehydes 3a–3i using isopropanol as the terminal reductant under otherwise identical conditions. Comparable isolated yields and enantioselectivities are observed. Thus, carbonyl (2-fluoro)allylation may be accomplished from the alcohol or aldehyde oxidation level (Table 1).
Scheme 3.
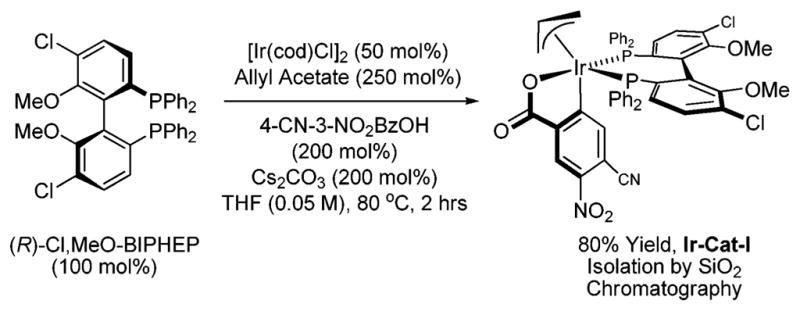
Preparation of iridium complex Ir-Cat-I.
Table 1.
Enantioselective iridium catalyzed (2-fluoro)allylation from the alcohol or aldehyde oxidation levela

| ||||
|---|---|---|---|---|
| Entry | Product | [o] Level | Y [%] 4a (5a) | ee [%] |
| 1 |
4a |
Alcohol | 76 (10) | 99b |
| Aldehyde | 61 (5) | 99b | ||
| 2 |
4b |
Alcohol | 65 (6) | 98b |
| Aldehyde | 55 (6) | 98b | ||
| 3 |
4c |
Alcohol | 89 (5) | 99b |
| Aldehyde | 65 (6) | 98b | ||
| 4 |
4d |
Alcohol | 76 (4) | 98c |
| Aldehyde | 74 (3) | 98c | ||
| 5 |
 4e |
Alcohol | 80 (5) | 98c |
| Aldehyde | 93 (4) | 98c | ||
| 6 |
4f |
Alcohol | 75 (4) | 95c |
| Aldehyde | 74 (3) | 96c | ||
| 7 |
 4g |
Alcohol | 86 (7) | 99c |
| Aldehyde | 89 (5) | 99c | ||
| 8 |
 4h |
Alcohol | 81 (3) | 99c |
| Aldehyde | 88 (5) | 99c | ||
| 9 |
 4i |
Alcohol | 65 (5) | 99c |
| Aldehyde | 73 (5) | 99c | ||
Products 4 and 5 are isolated as mixtures, but were separated for the purpose of characterization. See Supporting Information for further details.
60 °C.
40 °C.
With adducts 4a–4i in hand, methods for diastereoselective hydrogenation were explored. Reduction of 4g occurs efficiently using palladium on carbon, however, the saturated product 6g forms as a 1 : 1 mixture of diastereomers. In contrast, using the Crabtree catalyst,7 hydrogenation of 4g at 25 °C provides 6g as a 6 : 1 mixture of diastereomers favouring the syn-diastereomer. Under these conditions, vinyl fluorides 4a–c and 4g were converted to the syn-3-fluoro-1-alcohols 6a–c and 6g, respectively (Scheme 4). In these experiments, it was found that diastereoselectivity improved with lower temperature, lower loadings of the Crabtree catalyst and higher dilution. Fortuitously, the syn-3-fluoro-1-alcohol 6g is crystalline, allowing relative and absolute stereochemical assignment via single crystal X-ray diffraction analysis† by the anomalous dispersion method. On this basis, the absolute stereochemistry of (2-fluoro)allylation products 4a–4i is assigned.
Scheme 4.

Synthesis of syn-3-fluoro-1-alcohols 6a–c and 6g via Crabtree hydrogenation of vinyl fluorides 4a–c and 4g, respectively.
In summary, using commercially available (2-fluoro)allyl chloride, direct enantioselective iridium catalyzed C–H (2-fluoro)-allylation of primary alcohols 2a–2i is achieved. Corresponding aldehydes 3a–3i participate in carbonyl (2-fluoro)allylation to furnish an identical set of adducts 4a–4i in the presence of isopropanol under otherwise identical conditions. Diastereo-selective Crabtree hydrogenation of the resulting vinyl fluoride containing homoallylic alcohols 4a–c and 4g provides syn-3-fluoro-1-alcohols 6a–c and 6g, respectively. Thus, using consecutive C–C and C–H bond forming hydrogenations, primary alcohols are converted to chiral fluorine containing building blocks in the absence of stoichiometric metallic reagents or stoichiometric organic byproducts.
Supplementary Material
Acknowledgments
Acknowledgement is made to the Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445) and the University of Texas at Austin, Center for Green Chemistry and Catalysis for partial support of this research. The Higher Education Commission of Pakistan is acknowledged for graduate student fellowship support (AH).
Footnotes
Electronic supplementary information (ESI) available: Characterization data for all new compounds (1H NMR, 13C NMR, IR, HRMS, [α]). Absolute stereochemical assignment of 6g by single crystal X-ray diffraction. CCDC 867899. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc31743e
Notes and references
- 1.(a) Thayer AM. Chem Eng News. 2006;84:15. [Google Scholar]; (b) Mueller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Thayer AM. Chem Eng News. 2007;85:11. [Google Scholar]
- 2.(a) Carey JS, Laffan D, Thomson C, Williams MT. Org Biomol Chem. 2006;4:2337. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]; (b) Farina V, Reeves JT, Senanayake CH, Song JJ. Chem Rev. 2006;106:2734. doi: 10.1021/cr040700c. [DOI] [PubMed] [Google Scholar]
- 3.For selected reviews on enantioselective methods for the preparation of organofluorine compounds, see: Ma JA, Cahard D. Chem Rev. 2004;104:6119. doi: 10.1021/cr030143e.Brunet VA, O’Hagan D. Angew Chem, Int Ed. 2008;47:1179. doi: 10.1002/anie.200704700.Bobbio C, Gouverneur V. Org Biomol Chem. 2006;4:2065. doi: 10.1039/b603163c.Audouard C, Ma JA, Cahard D. Adv Org Synth. 2006;2:431.Ma JA, Cahard D. Chem Rev. 2008;108:1–43. doi: 10.1021/cr800221v.Cao LL, Gao BL, Ma ST, Liu ZP. Curr Org Chem. 2010;14:889.Kang YK, Kim DY. Curr Org Chem. 2010;14:917.Cahard D, Xu X, Couve-Bonnaire S, Pannecoucke X. Chem Soc Rev. 2010;39:558. doi: 10.1039/b909566g.Nie J, Guo HC, Cahard D, Ma JA. Chem Rev. 2011;111:455. doi: 10.1021/cr100166a.
- 4.For selected reviews on C–C bond forming hydrogenation and transfer hydrogenation, see: Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim Acta. 2008;41:95.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem, Int Ed. 2008;48:34. doi: 10.1002/anie.200802938.Han SB, Kim IS, Krische MJ. Chem Commun. 2009:7278. doi: 10.1039/b917243m.Bower JF, Krische MJ. Top Organomet Chem. 34:107. doi: 10.1007/978-3-642-15334-1_5.Hassan A, Krische MJ. Org Process Res Dev. 2011;15:1236. doi: 10.1021/op200195m.
- 5.For enantioselective carbonyl allylation via iridium catalyzed C–C bond forming transfer hydrogenation, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem, Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648.Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760. doi: 10.1021/ja9097675.Zhang YJ, Yang JH, Kim SH, Krische MJ. J Am Chem Soc. 2010;132:4562. doi: 10.1021/ja100949e.Han SB, Gao X, Krische MJ. J Am Chem Soc. 2010;132:9153. doi: 10.1021/ja103299f.Hassan A, Zbieg JR, Krische MJ. Angew Chem, Int Ed. 2011;50:3493. doi: 10.1002/anie.201100646.Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350. doi: 10.1021/jo200068q.Gao X, Zhang YJ, Krische MJ. Angew Chem, Int Ed. 2011;50:4173. doi: 10.1002/anie.201008296.Hassan A, Townsend IA, Krische MJ. Chem Commun. 2011;47:10028. doi: 10.1039/c1cc14392a.
- 6.For selected reviews on enantioselective carbonyl allylation, see: Ramachandran PV. Aldrichim Acta. 2002;35:23.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.Lachance H, Hall DG. Org React. 2008;73:1.Yus M, Gonzáles-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774. doi: 10.1021/cr1004474.
- 7.Crabtree RH, Felkin H, Morris GE. J Organomet Chem. 1977;141:205.For a review, see: Crabtree RH. Acc Chem Res. 1979;12:331.
- 8.(a) Cramer R. J Am Chem Soc. 1967;89:4621. [Google Scholar]; (b) Jesse AC, Cordfunke EHP, Ouweltjes W. Thermochim Acta. 1979;30:293. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.