

Abstract
Stimuli responsive (or “smart”) polymer brushes represent a non-toxic approach for achieving release of biofouling layers. Thermo-responsive poly(N-isopropylacrylamide) (PNIPAAm) polymer brushes have been shown to modulate bacterial adhesion and release through transition between temperatures above and below the lower critical solution temperature (LCST ~32 °C) of PNIPAAm in water. In this article, we describe a convenient method to synthesize grafted PNIPAAm brushes over large areas for biological studies using a relatively simple and rapid method which allows atom transfer radical polymerization (ATRP) in presence of air using the activator regenerated electron transfer (ARGET) mechanism. PNIPAAm brushes were characterized using X-ray photoelectron spectroscopy, time-of-flight secondary ion mass spectroscopy, Fourier transform infrared spectroscopy, ellipsometry, and contact angle measurements. Our studies demonstrate that uniform, high purity PNIPAAm brushes with controlled and high molecular weight can be easily produced over large areas using ARGET–ATRP. We also report the use of a spinning disk apparatus to systematically and quantitatively study the detachment profiles of bacteria from PNIPAAm surfaces under a range (0–400 dyne/cm2) of shear stresses.
Introduction
Biofilms are layers of organic compounds, microorganisms and their byproducts that form rapidly on synthetic surfaces submerged in non-sterile aqueous environments [1–3]. They are ubiquitous in nature and are often referred to as a type of biofouling, reflecting the reduction of the useful life or efficiency of materials. Biofouling is of concern for its deleterious effects seen in medical devices [4], water reservoirs [5], chemical process industries [5, 6] and on equipment operated in marine environments [7, 8], requiring billions of dollars to be spent annually in mitigation. While heavy-metal based coatings are ancient and effective strategies to control biofouling, there is significant concern regarding their health and environmental impacts that has resulted in restrictions prohibiting their use [9, 10]. Developing environmental friendly non-toxic surfaces to prevent and reduce biofouling has therefore, been an area of research over the last several decades [11].
We have explored both self-assembled monolayers and stimuli-responsive polymers (SRPs) as model non-toxic fouling-release surfaces [12–15]. Such surfaces can undergo significant and reversible chemical or conformational responses to changes in external conditions, such as temperature, pH or light [16, 17]. Poly(N-isopropylacrylamide) (PNIPAAm), perhaps one of the most studied SRPs [13, 15, 18–20], is thermo-responsive and exhibits a phase change in aqueous solutions at its lower critical solution temperature (LCST ~ 32°C) [21]. When PNIPAAm is tethered to a surface, this phase change is reflected as a change in molecular hydration above and below the LCST; below the transition temperature PNIPAAm surfaces are relatively hydrophilic and above the transition temperature they become slightly more hydrophobic. PNIPAAm in various forms (e.g., free, adsorbed, tethered, cross-linked gel) has been used in applications which include drug delivery [22], membrane separations [23], chromatographic separations [24], controlling the adhesion of cells (mammalian [25–27] and bacterial [13, 15]) and proteins [28].
Polymer brushes represent versatile tools for surface modification and functionalization that can dramatically affect properties such as adhesion, lubrication, wettability, friction and biocompatibility [29–31]. These surface properties can be modulated in a reversible manner using end-grafted SRP brushes [16, 32–34]. Surface-initiated “grafting from” approaches are popular and efficient methods that enable control over polymer grafting density, thickness, and polydispersity [35].
Atom transfer radical polymerization (ATRP) is one such method to synthesize polymers with precisely controlled molecular architecture (topology, composition, functionality) [36–39] and for the preparation of various hybrid materials and bioconjugates [40, 41]. ATRP relies on dynamic equilibrium between dormant and active species catalyzed by redox active transition-metal complexes such as copper coordinated to nitrogen based ligands (see Fig. 1a) [42]. As a result of the persistent radical effect [43], the equilibrium is strongly shifted towards the dormant species (kact ≪ kdeact); thus, the radical concentration is low and termination is suppressed. The degree of control in ATRP is affected by the position of the equilibrium (KATRP = kact/kdeact), which, in turn, depends on solvent, temperature, monomer and oxidation state of the Cu species.
Fig. 1.

a Schematic of the ARGET–ATRP reaction. b ARGET–ATRP of PNIPAAm from initiator immobilized on a silicon wafer in presence of air
A disadvantage of ATRP is that the transition metal complexes have to be removed from the reaction mixture, adding expense to purification of the desired resultant polymer [44]. Additionally, special handling procedures are required to remove oxygen from reaction systems prior and during polymerization to avoid oxidation of the reduced copper catalyst [44–46]. To get around these limitations, Matyjaszewski et al. reported an efficient and convenient procedure for ATRP reactions using the activator regenerated electron transfer (ARGET) mechanism. As shown in Fig. 1a, in ARGET–ATRP, Cu(I) complexes are regenerated from oxidatively stable Cu(II) species by the action of reducing agents within the polymerization reaction system [47]. This conversion step does not involve the formation of monomer radicals, therefore, polymers and also, copolymers with complex architectures (e.g., block, random and star copolymers) can be formed from the oxidatively stable catalyst-precursor complexes [48].
In this work, we report the ARGET–ATRP of PNIPAAm brushes in ambient environment (open to air) using a surface immobilized initiator, a minute amount of Cu(II)/ligand complex, and a non-toxic reducing agent. Several complementary characterization techniques were used to study the structure, purity and thermo-responsive behavior of PNIPAAm including ellipsometry, X-ray photoelectron spectroscopy (XPS), time-of-flight secondary ion mass spectroscopy (ToF–SIMS), Fourier-transform infrared spectroscopy (FTIR), gel permeation chromatography (GPC), surface wettability measurements and UV–vis spectrophotometry.
To exemplify the utility of ARGET–ATRP in forming SRP brushes for cell adhesion studies, we also describe the use of a spinning disk apparatus, to systematically apply a range of shear forces over a PNIPAAm brush, as a quantitative means to study the detachment profiles of a model marine bacterial species from these surfaces. This process represents an improvement in reproducibility and control of shear force application over previous methods of studying bacterial detachment [15, 20]. When combined with the relatively high-throughput methods for generation of model SRP brushes described above, this technique provides an efficient tool for careful exploration of conditions for, and mechanism of, release of bacteria from SRP surfaces.
Materials and Methods
Materials
N-isopropyl acrylamide (NIPAAm), copper bromide (Cu(II)Br) (98% pure), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, 99 % pure), ascorbic acid (reagent grade, 20–200 mesh), anisole (anhydrous, 99.7 % pure) and tin(II) 2-ethylhexanoate (Sn[EH]2, ~95 % pure) were purchased from Sigma-Aldrich (St. Louis, MO). The NIPAAm monomer was recrystallized twice from a benzene/hexane mixture and then dried under vacuum before use. The ATRP initiator (3-trimethoxysilyl) propyl 2-bromo 2-methylpropionate was purchased from Gelest, Inc. (Morrisville, PA) and stored under dehumidified condition until used. PNIPAAm (molecular weight ~40,000) for spin coating was purchased from Polysciences, Inc. (Warrington, PA).
Bacterial Culture
Cobetia marina (basonym, Halomonas marina) (ATTC 25374) [49–51], was cultured in a chemostat as previously described [15]. Artificial sea water (ASW) used in bacterial detachment studies was composed of 400 mM NaCl, 100 mM MgSO4, 20 mM KCl,10 mM CaCl2 [52]. The measured concentration of the chemostat cultured bacterial suspension was ~107 cells/mL.
Formation of Silane Self-Assembled Monolayers (SAMs)
Silicon wafers (25 mm × 13 mm) were cleaned using HCl (98 %)/MeOH (1:1 by vol) for 30 min followed by H2SO4 (98 %) for 30 min. Wafers were then rinsed with copious amounts of DI water and dried with nitrogen. Cleaned samples were dried under nitrogen and placed in staining jar containing 4 mM of (3-trimethoxysilyl) propyl 2-bromo 2-methylpropionate (ATRP initiator) in anhydrous toluene solution at 25 °C for at least 12 h. The samples were then rinsed three times with toluene and dried in a stream of nitrogen prior to the succeeding polymerization reaction. SAMs for cell detachment studies were prepared in the same manner on circular glass cover slips (25 mm dia, thickness No. 2, VWR).
Polymerization Procedure
A grooved beaker (staining jar) containing the initiator grafted SAMs and a small stir bar was charged with 0.75 M NIPAAm monomer, 0.4 mM Cu(II)Br and 0.7 mM PMDETA dissolved in 30 mL of either (i) anisole for use of Sn[EH]2 as reducing agent or (ii) DI water/MeOH (1:1 by vol) for use of ascorbic acid as reducing agent. Addition of 8 mM reducing agent to this mixture initiated the polymerization reaction. After allowing the reaction to progress at room temperature for various preset polymerization time intervals, the samples were sequentially rinsed with acetone, methanol and distilled water. The samples then underwent Soxhlet extraction at 80 °C in methanol overnight to remove any unreacted monomer and other reagents that might be entrained on the polymer grafted surfaces.
PNIPAAm synthesis in bulk aqueous solution (using ascorbic acid) was performed using the same contents and molar proportions, but by the addition of ATRP initiator to the reaction vessel in lieu of immobilizing it on a surface. After the desired polymerization time, 3 mL of sample was collected using a syringe and quenched with acetone and the precipitated PNIPAAm was collected using rotary evaporation. The precipitate was dissolved in distilled water for standard dialysis purification against methanol after which it was collected again by rotary evaporation and dried in a vacuum desiccator to yield a pure PNIPAAm polymer.
Spin Coated PNIPAAm
Silicon wafers (5 × 5 mm) (University Wafer, MA) were washed for successive 10 min intervals in dichloromethane, acetone, and methanol in an ultrasonic cleaner and dried with nitrogen. A mixture containing 35 mg of PNIPAAm, 5 mL of distilled water and 200 μL 1 N HCl was prepared and 200 μL of this solution was evenly distributed on treated silicon wafer placed on a spin coater (Brewer Science, Inc., Rolla MO) and spun at 2,000 rpm for 60 s [26].
Cell Adhesion Assay Using Spinning Disk Apparatus
A custom built spinning disk apparatus similar to that used by Nathan and Garcia [53] was used to measure the adhesion strength of marine bacteria attached to PNIPAAm brushes. The spinning disk consists of a cylindrical chamber that holds artificial sea water; the sample is introduced into the liquid after being immobilized, via vacuum, on a sample holder at the end of a shaft. The shaft in turn is driven by a DC motor with its speed controlled by an optical sensor. The detailed working principle has been described elsewhere [53].
Samples of ARGET–ATRP grafted PNIPAAm using ascorbic acid reducing agent on glass cover slips were pre-equilibrated to 37 °C in a 30 mL suspension of C. marina collected from a chemostat. After incubation for 15 min, the sample was removed from the suspension and affixed to the sample holder of the spinning disk apparatus. The sample was accelerated for 30 s to reach the preset rotational speed, held at constant speed for 29 min and decelerated back to zero over 30 s (total spin cycle: 30 min). Five identical sample replicates were prepared; each spun using the spinning disk at different rotational speeds (1,200, 2,000, 3,600 and 4,000 rpm) in separate experiments using cells freshly collected from a chemostat culture. Each of the spun samples was rinsed once gently with d-H2O water to remove loosely attached cells and salts, dried under a low-pressure stream of nitrogen and examined under a phase contrast optical microscope (Zeiss, Carl Zeiss Microimaging, Inc., USA) through a sampling template. The template helps in capturing each image at a known radial distance from center of the sample. Using a 40× objective and the template, at least five images were captured at various known radial distances from the center using a CCD camera (Axiocam, Carl Zeiss, Inc., USA). The data transfer to computer was done using Axiovision software and later ImageJ image processing software (NIH [54]) was used for counting the cell densities.
Analysis of Polymers
See supplementary information for details on standard surface and polymer characterization techniques including ellipsometry, X-ray photoelectron spectroscopy (XPS), time of flight-secondary ion mass spectroscopy (ToF–SIMS), Fourier transform infrared (FTIR) spectroscopy, gel permeation chromatography (GPC), UV–Vis spectrophotometry, and surface wettability measurements.
Results and Discussion
The reaction schematic for the surface polymerization of PNIPAAm is shown in Fig. 1. ARGET–ATRP was carried out using two different reducing agents (tin(II) ethylhexanoate and ascorbic acid). In order to compensate for competitive complexation of the low amount of added metal catalyst, an excess in ligand (PMDETA) concentration over that typically required for conventional ATRP is recommended [47].
Figure 2a presents PNIPAAm dry brush thickness as a function of polymerization time for reactions carried out using tin-ethylhexanoate (Sn[EH]2) as a reducing agent in anisole. The film thickness is linearly correlated (R2 = 0.98) with polymerization time over at least 6 h with a growth rate that is similar to that obtained by conventional ATRP [55]. Figure 2b illustrates the polymer brush thickness versus polymerization time using ascorbic acid as reducing agent in water/methanol (1:1 v/v), which also showed a linear correlation but only over the first 6 min (R2 = 0.95). The increase in brush thickness obtained using ascorbate as a reducing agent was nearly two orders of magnitude faster than that obtained using Sn[EH]2. This difference is because of the fact that the ATRP polymerization reactions are faster in aqueous solution in comparison to organic solvents [56]. Also, the rate of polymerization in ATRP depends on the ratio of the concentration of activator (Cu(I)) and deactivator (Cu(II)) [43, 48]. Ascorbic acid, being a stronger reducing agent promotes faster conversion of Cu(II) to Cu(I) species which might result in faster activation and thus leads to a faster rate of polymerization. On the other hand, Sn[EH]2, a mild reducing agent with relatively low solubility in anisole, maintains a lower concentration of Cu(I) radical leading to a relatively low reaction rate. These results suggests that, within proper experimental windows, use of ascorbic acid as a reducing agent in surface initiated ARGET–ATRP can very quickly yield well defined polymer brushes of PNIPAAm with dry thickness at least up to 90 nm.
Fig. 2.

Ellipsometric thickness of grafted PNIPAAm brush measured in dry state for different polymerization times by (a) use of tin 2-ethylhexanoate as the reducing agent, and (b) use of ascorbic as the reducing agent. The standard deviation for each measurement is within the size of the data point
To further examine the role of ascorbic acid as a reducing agent in ARGET–ATRP, we performed GPC analysis to examine the molecular weight distribution of PNIPAAm obtained from polymerization carried out in bulk solution (Table 1). The data in Table 1 indicate that there is an increase in molecular weight of the polymer with reaction time along with slight increase in the polydispersity index (PDI). However, all the polydispersity values remain less than 1.5 indicating a good degree of consistency in polymer chain length. At polymerization times greater than 10 min, the rate of increase in PNIPAAm molecular weights decreased and the PDI increased (PDI = 1.4 with reaction time of 12 min). The GPC data shown in Table 1 indicate that ARGET–ATRP using ascorbic acid provides good control over the PNIPAAm polymer molecular weight up to approximately 150,000 g mol−1.
Table 1.
Molecular weight and polydispersity index of PNIPAAm synthesized in solution via ARGET–ATRP using ascorbic acid for different polymerization time periods
| Polymerization time (min) | Mn (g mol−1) | PDI |
|---|---|---|
| 0.5 | 14,052 | 1.0353 |
| 1 | 39,116 | 1.0828 |
| 2 | 63,211 | 1.1573 |
| 4 | 98,015 | 1.1701 |
| 8 | 141,225 | 1.2569 |
| 12 | 159,205 | 1.4105 |
The significant increase in PDI value of polymer synthesized in solution at longer polymerization times (>10 min) is mirrored by the observed decrease in measured brush growth rate at higher polymerization times and may be due to one or more factors, including those that lead to accumulation of dead chains by termination reactions at longer polymerization times. Matyjaszewksi and coworkers [57] explained that free monomer present in the solvent can coordinate with Cu(1)/PMDETA complex, and the reactivity of the coordinated monomers can be altered. In our case, the same situation could be occurring; due to the use of a strong reducing agent (ascorbic acid), longer polymerization time results in higher concentration of Cu(1)/PMDETA in the reaction mixture and could lead to complexation with free monomer and thereby, alter the polymer growth and may led to the increased PDI value observed at longer polymerization time. Further, one of the proposed limitations in the synthesis of high molecular weight polymers by ATRP is the side reactions that Cu(II) species can undergo during the polymerization reaction (e.g., the oxidation of polystyryl radical to a carbocation by the Cu(II) species during the ATRP of polystyrene [58]). Since ARGET–ATRP can be successfully carried out with small amounts of Cu(II) species, further optimization may make it feasible to synthesize PNIPAAm with much higher molecular weight (>150,000 g mol−1) and low polydispersity, both in solution and on surfaces.
XPS was used to examine the ATRP initiators immobilized on silicon substrates (i.e., the SAMs) and the PNIPAAm brushes. As analyzed from survey spectra, the surfaces consisted of carbon, oxygen, silicon and bromine for the initiator SAMs (data not shown). The measured relative elemental composition (5.27 ± 0.6 (C/Br), 1.2 ± 0.2 (C/O)) of the SAM samples was consistent with the elemental composition of the initiator silane (7 (C/Br), 1.4 (C/O)), thereby confirming the immobilization of the initiator silane. The survey spectra of PNIPAAm brushes (Fig. S1) synthesized using Sn[EH]2 and ascorbic acid showed characteristic signals attributed to carbon, nitrogen and oxygen. Brushes synthesized using the Sn[EH]2 reducing agent also showed an additional peak due to tin (Sn 3d) at a binding energy (BE) of 504.25 eV (Table 2). Elemental compositions measured for the brushes synthesized using ascorbic acid were consistent with the structure of PNIPAAm.
Table 2.
XPS elemental compositions of PNIPAAm surface grafted via ARGET–ATRP using Sn[EH]2 (I) and ascorbic acid (II)
| Atom | Theoretical | Experimental (I) | Experimental (II) |
|---|---|---|---|
| C | 75 | 77 ± 0.6 | 76.4 ± 0.2 |
| N | 12.5 | 10.4 ± 0.5 | 12 ± 0.2 |
| O | 12.5 | 9.5 ± 0.8 | 11.6 ± 0.2 |
| Sn (I) | 0 | 3.1 ± 0.4 | 0 |
| C–C/C–H | 66.7 | 70.2 ± 8.2 | 64.5 ± 4.8 |
| C–N | 16.7 | 11.9 ± 1.4 | 18.8 ± 3.8 |
| N–C=O | 16.7 | 17.9 ± 7.1 | 16.7 ± 1.2 |
High resolution carbon (C1s) spectra of PNIPAAm grafted brushes were resolved by curve-fitting into three component peaks (Fig. 3a, b): (1) a peak with a BE at 285.0 eV attributable to the aliphatic hydrocarbon (labeled C–H/C–C); (2) a peak with a BE at 286.5 eV corresponding to the –CH– unit adjacent to the –NH– group (labeled C–N); and (3) peak with a BE at 287.9 eV attributable to the amide group (labeled N–C=O). Similar spectra and binding environments (C–H, C–N and N–C=O) were observed for PNIPAAm brushes synthesized using either Sn[EH]2 or ascorbic acid reducing agents, and both spectra of both types of samples closely match those typically obtained from spin coated PNIPAAm surfaces [26]. Thus, XPS spectra of samples prepared by ARGET–ATRP confirm the synthesis of PNIPAAm.
Fig. 3.

High resolution XPS C1 s spectra of PNIPAAm grafted surfaces synthesized using (a) tin(II) 2-ethylhexanoate and (b) and ascorbic acid
As mentioned above, XPS analysis indicated the presence of tin on PNIPAAm samples synthesized using Sn[EH]2 as a reducing agent (Table 2). The presence of organotin constituents on surfaces is undesirable and may be problematic for studies of cell/surface interactions or other biological applications. Even small amounts of organotin compounds can be highly toxic [9, 10]. Hence, we have avoided the use of Sn[EH]2 as a reducing agent for ARGET–ATRP of polymer brushes used for marine bacterial adhesion studies (see below). Though PNIPAAm samples synthesized using ascorbic acid reducing agent showed no detectable impurities (e.g., Cu) by XPS analysis, we further used ToF–SIMS as a sensitive probe for impurities in the synthesized PNIPAAm polymer brushes. ToF–SIMS is a highly sensitive surface analysis technique that can detect elements present on a surface the part per billion range [59].
Representative ToF–SIMS positive-ion spectra (0–200 m/z) (of five replicates) of a grafted PNIPAAm brush formed using ascorbate and of commercially available PNIPAAm spin-coated on silicon wafer are shown in Fig. 4. Both spectra show peaks corresponding to characteristic PNIPAAm molecular fragments [25] such as NH4+ (m/z = 18.0371), C2H3+ (27.0229), C3H7+ (43.0550), C3H3O+ (55.0190), C5H7+ (67.0613) and C3H8NO+ (74.0638). By comparison of the SIMS spectra Fig. 4a, b we observe that PNIPAAm brushes synthesized via ARGET–ATRP yielded more fragments with relatively large mass (e.g., 58.0681, 114.0923) than the spin coated PNIPAAm sample, which yielded more smaller mass fragments (e.g., 18.0371, 27.0229). The high intensity peaks of monomer (C6H12NO+, 114.0923) and isopropyl (C3H8N+, 58.0681) fragments strongly suggest that NIPAAm structural units of the polymer are present on the brush surface without cross-linking. Positive-ion ToF–SIMS spectra obtained showed no peaks that are characteristic to Cu2+ (62.9277). Negative-ion ToF–SIMS spectra were also obtained from five sample replicates (spectra not shown) and did not show peaks (e.g., m/z 71 (C3H3O2−) or 72 (C2O3−)) characteristic of ascorbic acid, suggesting that negligible amounts of ascorbic acid remained entrained in the polymer brushes.
Fig. 4.

Time-of-flight secondary ion mass spectroscopy positive ion spectra of (a) ARGET–ATRP grafted PNIPAAm brush formed using ascorbic acid as a reducing agent, and (b) spin coated PNIPAAm on silicon substrates
Since ToF–SIMS generally yields hundreds of peaks in the 0–200 m/z range, for detailed spectral interpretation and comparison, principal component analysis (PCA) was used to identify related variables and focusing on the differences between the spectra [60, 61]. ToF–SIMS positive-ion spectra from five replicates of each sample type (SAM, spin coated PNIPAAm and ARGET–ATRP grafted PNIPAAm) were compared using PCA. The results of this PCA analysis (Fig. S2) are presented in detail in Supplementary Information. In summary, the PCA results obtained by comparison of ToF–SIMS spectra obtained from five replicates of spin-coated and five replicates of ARGET–ATRP PNIPAAm samples reveal that mass fragments from spin-coated PNIPAAm were randomly distributed in the scores plot and comprised mostly smaller mass fragments. These results suggest that polymer chain conformation and/or orientation are inconsistent within the spin coated samples leading to inconsistent chain fragmentation. On the other hand, most of ARGET–ATRP PNIPAAm fragments identified were of higher mass and closely clustered in the scores plots indicating that polymer molecules on the surface undergo consistent fragmentation likely due to their more consistent molecular conformation and/or orientation.
The FTIR absorbance spectrum (Fig. S3a) of PNIPAAm brush grafted on silicon wafer (brush thickness ~25 nm) using ARGET–ATRP with ascorbic acid reducing agent also confirms the presence of NIPAAm structural units [62]. The spectrum obtained is similar to the FTIR spectra previously obtained for PNIPAAm brushes [63] and peaks due to carbonyl (at 1,650 cm−1) and amide (at 1,460 cm−1) groups confirm the presence of grafted PNIPAAm [63]. Taken together, all the surface characterization results indicate that we were able to successfully synthesize consistent, reproducible, high purity PNIPAAm grafted brushes over large surface areas using ascorbic acid mediated ARGET–ATRP in ambient environments at a fast polymerization rate.
To examine the thermo-responsive behavior of PNIPAAm brushes synthesized using ARGET–ATRP, contact angle (θ) measurements were taken using a captive-bubble of air under water [64]. The average θ of the grafted PNIPAAm surfaces (dry thickness 15 nm) taken over five replicates was 28 ± 0.2° at 22 °C and 47 ± 0.9° at 45 °C. (The thermo-responsive behavior of PNIPAAm synthesized in solution was examined using turbidity measurements and showed a drastic increase in its absorbance at ~32 °C, the LCST; Fig. S3b). The significant difference observed in contact angle is due to the different extent of hydration of PNIPAAm brush above and below the temperature corresponding to the LCST and thus, verifies the thermo responsive behavior of PNIPAAm grafted surfaces synthesized using ARGET–ATRP.
The thermo-responsive behavior of PNIPAAm surfaces can be used to reversibly regulate the adhesion and detachment of bacterial and mammalian cells. We have previously shown [12, 13, 15, 20] that fouling bacteria can be detached from surface grafted PNIPAAm when they are placed under shear while simultaneously inducing a temperature-mediated polymer transition; the shear rates applied in previous studies were, however, only qualitatively similar and not rigorously controlled [12]. In some of the previous studies, PNIPAAm brushes were synthesized using conventional ATRP, which limits sample preparation throughput in comparison to the ARGET–ATRP methods described here. Hence, use of ARGET–ATRP synthesis coupled with a carefully controlled shear stress device can significantly provide a faster and accurate means for quantitatively studying the combined effects of polymer brush dynamics and shear forces on bacterial detachment.
To illustrate such a controlled study, we used a custom-built spinning disk device that can reproducibly generate well-defined and systematically varied range of laminar shear flow conditions across a single and reasonably sized (1 sq. inch) sample, the details of which have been described elsewhere [53, 65]. The applied shear stress τ, on the sample surface in the spinning disk apparatus varies with the radial position as given by:
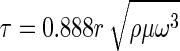 |
1 |
where r (m) is the radial distance from the center of the sample, ρ (kg m−3) is the density of artificial sea water (ASW), μ (kg m−1 s−1) viscosity of ASW and ω (s−1) is the angular frequency of the disk. The fraction of adherent cells at any given radial position is calculated by normalization of measured cell densities to the cell density at the center of the sample disk (i.e. r = 0) where the effective shear stress (τ) is zero.
Our previous work demonstrated that C. marina attaches more readily to hydrophobic surfaces than to hydrophilic surfaces and that it can be detached upon transition of PNIPAAm to a more hydrophilic state [12, 20]. We therefore examined the release of C. marina (attached at 37 °C) under different shear rates below the LCST of PNIPAAm. Figure 5 shows the effect of shear rate on the fraction of remaining C. marina cells on PNIPAAm brushes (15 nm dry thickness) at temperatures above and below the LCST of PNIPAAm in ASW. At 22 and 4 °C, i.e., below the LCST of PNIPAAm, a shear rate of at least 9,980 s−1 (corresponding to a shear stress of 100 dyne/cm2) was required for any detachment to occur, after which a linear cell detachment profile was observed until a shear rate of ~40,000 s−1 (shear stress = 401 dyne/cm2). A small fraction (~0.1) of cells remained adhered even under large shear rates (>40,000 s−1). On the other hand, for the spinning disk bacterial detachment experiment conducted at 37 °C, there was only 40 % release. These results indicate that the thermo-responsive behavior PNIPAAm brushes has a significant effect on the detachment of bacteria under shear, and up to 90 % of cell release can be attained by changing the solution temperature from 37 to 22 °C. The ability to directly correlate a measurable shear stress to bacterial attachment and release represents an important step forward in our understanding between PNIPAAm and bacterial cells. However, there are many other factors to be considered that can quantitatively effect release of cells from PNIPAAm grafted substrates such the time period of cell attachment [15], grafting density and brush thickness [19, 66]. In an upcoming publication, we will report on the effects of such parameters to more precisely understand the mechanism of bacterial detachment from PNIPAAm grafted surfaces.
Fig. 5.

Fraction of adherent C. marina cells on PNIPAAm grafted glass coverslip substrate as a function of shear rate applied at different temperature (inverted filled triangles 37 °C, filled circles 25 °C, filled diamonds 4 °C) using the spinning disk device using spinning disk device. Errors bars are within the size of the data point symbols
Conclusions
In this report, we demonstrate an ARGET–ATRP method of synthesizing thermally-responsive PNIPAAm brushes that can provide a simple, fast and convenient means of producing uniform and controlled polymer brushes over large surface areas in ambient environments using very low concentrations of copper catalyst (~100 ppm). In this process, the copper (II) complex formed during polymer chain elongation is continuously reduced to copper (I) complex in the presence of ascorbic acid or tin(II) 2-ethylhexanoate. Surface grafting of PNIPAAm using tin(II) 2-ethylhexanoate resulted in the entrainment of trace amounts of tin that may be toxic to micro-organisms or other biological systems. In contrast, ascorbic acid, a highly soluble reducing agent, was not detected in the PNIPAAm brushes. A spinning disk apparatus can be used as an efficient device to analyze the effect of fluid shear forces on cell detachment quantitatively. PNIPAAm brushes below its LCST showed a non-linear cell detachment profile for adhered C. marina bacteria, with a needed critical shear force (50% release) of ~250 dyne/cm2.
Electronic supplementary material
REFERENCES
- 1.Callow JA and Callow ME, Prog Mol Subcell Biol 42, 141 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Beer D and Stoodley P, The prokaryotes (vol 1, 3rd edn). A handbook on the biology of bacteria: symbiotic associations, biotechnology applied microbiology (Springer, Singapore, 2006). [Google Scholar]
- 3.Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M and Marrie TJ, Ann Rev Microbiol 41, 435 (1987) 10.1146/annurev.mi.41.100187.002251. [DOI] [PubMed] [Google Scholar]
- 4.Bryers JD, Biotechnol Bioeng 100, 1 (2008) 10.1002/bit.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flemming HC, Appl Microbiol Biotechnol 59, 629 (2002) 10.1007/s00253-002-1066-9. [DOI] [PubMed] [Google Scholar]
- 6.Bott TR (1999) In: Keevil CW, Godfree A, Holt D, Dow C (eds) Biofilms in the aquatic environment. Royal Society of Chemistry, London
- 7.Flemming HC, Murthy PS, Venkatesan R and Cooksey K, Biofilms: marine and industrial biofouling (Springer, New York, 2009). [Google Scholar]
- 8.Callow ME and Callow JA, Biologist (London) 49, 1 (2002). [PubMed] [Google Scholar]
- 9.Ozretic B, Petrovic S and Ozretic MK, Chemosphere 37, 1109 (1998) 10.1016/S0045-6535(98)00109-X. [DOI] [Google Scholar]
- 10.Baul TSB, Rynjah W, Singh KS, Pellerito C, D’Agati P and Pellerito L, Appl Organometal Chem 19, 1189 (2005) 10.1002/aoc.971. [DOI] [Google Scholar]
- 11.Maréchal JP and Hellio C, Int J Mol Sci 10, 4623 (2009) 10.3390/ijms10114623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ista LK, Mendez S and Lopez GP, Biofouling 26, 111 (2010) 10.1080/08927010903383455. [DOI] [PubMed] [Google Scholar]
- 13.Ista LK, Mendez S, Balamurugan SS, Balamurugan S, Rao VGR, Lopez GP (2009) In: Provder T, Baghdachi J (eds) Smart coatings II. ACS Symposium Series
- 14.Balamurugan S, Ista LK, Yan J, Lopez GP, Fick J, Himmelhaus M and Grunze M, J Am Chem Soc 127, 14548 (2005) 10.1021/ja054156g. [DOI] [PubMed] [Google Scholar]
- 15.Ista LK, Perez-Luna VH and Lopez GP, Appl Environ Microbiol 65, 1603 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik Z, Zauscher S, Luzinov I and Minko S, Nat Mater 9, 101 (2010) 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 17.Alercon CH, Pennadam S and Alexander C, Chem Soc Rev 34, 276 (2004) 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 18.Cooperstein MA and Canavan HE, Langmuir 26, 7695 (2010) 10.1021/la902587p. [DOI] [PubMed] [Google Scholar]
- 19.Akiyama Y, Kikuchi A, Yamato M and Okano T, Langmuir 20, 5506 (2004) 10.1021/la036139f. [DOI] [PubMed] [Google Scholar]
- 20.Ista LK and Lopez GP, J Ind Microbiol Biotechnol 20, 121 (1998) 10.1038/sj.jim.2900490. [DOI] [Google Scholar]
- 21.Schild HG, Prog Polym Sci 17, 163 (1992) 10.1016/0079-6700(92)90023-R. [DOI] [Google Scholar]
- 22.Nakayama M and Okano T, React Funct Polym 71, 235 (2011) 10.1016/j.reactfunctpolym.2010.08.006. [DOI] [Google Scholar]
- 23.Rao GVR, Krug ME, Balamurugan S, Xu HF, Xu Q and Lopez GP, Chem Mater 14, 5075 (2002) 10.1021/cm020627b. [DOI] [Google Scholar]
- 24.Kanazawa H, Sunamoto T, Ayano E, Matsushima Y, Kikuchi A and Okano T, Anal Sci 18, 45 (2002) 10.2116/analsci.18.45. [DOI] [PubMed] [Google Scholar]
- 25.Reed JA, Love SA, Lucero AE, Haynes CL and Canavan HE, Langmuir 28, 2281 (2012) 10.1021/la300735u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reed JA, Lucero AE, Hu S, Ista LK, Bore MT, Lopez GP and Canavan HE, ACS Appl Mater Interfaces 2, 1048 (2010) 10.1021/am900821t. [DOI] [PubMed] [Google Scholar]
- 27.Okano T, Kikuchi A, Sakurai Y, Takei Y and Ogata N, J Contr Rel 36, 125 (1995) 10.1016/0168-3659(95)00054-C. [DOI] [Google Scholar]
- 28.Huber DL, Maginell RP, Samara MA, Kim BI and Bunker BC, Science 301, 352 (2003) 10.1126/science.1080759. [DOI] [PubMed] [Google Scholar]
- 29.Bhat RR, Tomlinson MR, Wu T and Genzer J, Adv Polym Sci 198, 51 (2006) 10.1007/12_060. [DOI] [Google Scholar]
- 30.Kato K, Uchida E, Kang ET, Uyama Y and Ikada Y, Prog Polym Sci 28, 209 (2003) 10.1016/S0079-6700(02)00032-1. [DOI] [Google Scholar]
- 31.Zhao B and Brittain WJ, Prog Polym Sci 25, 677 (2000) 10.1016/S0079-6700(00)00012-5. [DOI] [Google Scholar]
- 32.Luzinov I, Minko S and Tsukruk V, Soft Matter 4, 714 (2008) 10.1039/b718999k. [DOI] [PubMed] [Google Scholar]
- 33.Minko S, Polym Rev 46, 397 (2006). [Google Scholar]
- 34.Luzinov I, Minko S and Tsukruk VV, Prog Polym Sci 29, 635 (2004) 10.1016/j.progpolymsci.2004.03.001. [DOI] [Google Scholar]
- 35.Granville AM and Brittain WJ, Polymer brushes: synthesis, characterization, applications, edited by Advincula RC, Brittian WJ, Caster KC and Ruhe J (Wiley-VCH Verlag GmbH & Co. KG&A, Weinheim, 2005). [Google Scholar]
- 36.Gao H, Tsarevsky NV and Matyjaszewski K, Macromolecules 38, 5995 (2005) 10.1021/ma0503099. [DOI] [Google Scholar]
- 37.Davis KA, Matyjaszewski K (2002) Adv Polym Sci. doi:10.1007/3-540-45806-9
- 38.Matyjaszewski K and Xia JH, J Chem Rev 101, 2921 (2001) 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 39.Coessens V, Pintauer T and Matyjaszewski K, Prog Polym Sci 26, 337 (2001) 10.1016/S0079-6700(01)00003-X. [DOI] [Google Scholar]
- 40.Chen G, Huynh D, Felgner PL and Guan ZJ, J Am Chem Soc 128, 4298 (2006) 10.1021/ja0573864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Licciardi M, Tang Y, Billingham NC, Armes SP and Lewis AL, Biomacromolecules 6, 1085 (2005) 10.1021/bm049271i. [DOI] [PubMed] [Google Scholar]
- 42.Wang JS and Matyjaszewski K, J Am Chem Soc 117, 5614 (1995) 10.1021/ja00125a035. [DOI] [Google Scholar]
- 43.Tang W, Tsarevsky NV and Matyjaszewski K, J Am Chem Soc 128, 1598 (2006) 10.1021/ja0558591. [DOI] [PubMed] [Google Scholar]
- 44.Shen Y, Tang H and Ding S, Prog Polym Sci 29, 1053 (2004) 10.1016/j.progpolymsci.2004.08.002. [DOI] [Google Scholar]
- 45.Honigfort ME and Brittain WJ, Macromolecules 36, 3111 (2003) 10.1021/ma021641c. [DOI] [Google Scholar]
- 46.Hong SC and Matyjaszewski K, Macromolecules 2001, 7590 (2002). [Google Scholar]
- 47.Matyjaszewski K, Dong H, Jakubowski W, Pietrasik J and Kusumo A, Langmuir 23, 4528 (2007) 10.1021/la063402e. [DOI] [PubMed] [Google Scholar]
- 48.Tomislav P and Matyjaszewski K, Chem Soc Rev 37, 1087 (2008) 10.1039/b714578k. [DOI] [PubMed] [Google Scholar]
- 49.Baumann L, Bowditch RD and Baumann P, Int J Syst Bacteriol 33, 793 (1983) 10.1099/00207713-33-4-793. [DOI] [Google Scholar]
- 50.Dobson SJ and Franzmann PD, Int J Syst Bacteriol 46, 550 (1966) 10.1099/00207713-46-2-550. [DOI] [Google Scholar]
- 51.Shea C, Lovelace LJ and Smith-Somerville HE, J Ind Microbiol 15, 290 (1995) 10.1007/BF01569982. [DOI] [Google Scholar]
- 52.Kersters K (1992) In: Balows A, Truper HG, Dworkin, Harder W, Schliefer KH (eds) The Prokaryotes—a handbook on the biology of bacteria, ecophysiology, isolation, identification, application, 2nd edn. Springer, New York
- 53.Nathan DG, Garcia AJ (2007) In: Amanda SC (ed) Adhesion protein protocols, 2nd ed., Springer, New Jersey
- 54.Abramoff MD, Magalhaes PJ and Ram SJ, Biophoton Int 11, 36 (2004). [Google Scholar]
- 55.Plunkett KN, Zhu X, Moore JS and Leckband DE, Langmuir 22, 4259 (2006) 10.1021/la0531502. [DOI] [PubMed] [Google Scholar]
- 56.Jianding Y and Narain R, J Phys Chem B 113, 676 (2009) 10.1021/jp808905s. [DOI] [PubMed] [Google Scholar]
- 57.Braunecker WA, Pintauer T, Tsarevsky NV, Kickelbick G and Matyjaszewski K, J Organometal Chem 690, 916 (2005) 10.1016/j.jorganchem.2004.10.035. [DOI] [Google Scholar]
- 58.Lutz JF and Matyjaszewski K, J Polym Sci 43, 897 (2005) 10.1002/polb.20382. [DOI] [Google Scholar]
- 59.Benninghoven A, Angewandte Chemie Int (in English) 33, 1023 (1994) 10.1002/anie.199410231. [DOI] [Google Scholar]
- 60.Jackson JE, J Qual Technol 12, 201 (1980). [Google Scholar]
- 61.Wold S, Esbensen K and Geladi P, Chemometr Intell Lab Syst 2, 37 (1987) 10.1016/0169-7439(87)80084-9. [DOI] [Google Scholar]
- 62.Gordon AJ and Ford RA, The chemist’s companion: a handbook of practical data, techniques, and references (Wiley, New York, 1972). [Google Scholar]
- 63.Kaholek M, Lee WK, LaMattina B, Caster KC and Zauscher S, Polymer brushes: synthesis, characterization, applications, edited by Advincula RC, Brittian WJ, Caster KC and Ruhe J (Wiley-VCH Verlag GmbH & Co. KG&A, Weinheim, 2005). [Google Scholar]
- 64.Drelick J and Miller JD, A systematic comparison of sessile-drop and captive-bubble contact angle methods (SME Annual Meeting, Denver, 1995). [Google Scholar]
- 65.Levich VG, Physicochemical hydrodynamics (Prentice Hall, Englewood Cliffs, 1962). [Google Scholar]
- 66.Yim H, Kent MS, Mendez S, Lopez GP, Satija S and Seo Y, Macromolecules 39, 3420 (2006) 10.1021/ma0520949. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.