

Summary
The discovery of drugs that cause the degradation of their target proteins has been largely serendipitous. Here we report that the tert-butyl carbamate-protected arginine (Boc3Arg) moiety provides a general strategy for the design of degradation-inducing inhibitors. The covalent inactivators ethacrynic acid and thiobenzofurazan cause the specific degradation of glutathione-S-transferase in mammalian cells when linked to Boc3Arg. Similarly, the degradation of dihydrofolate reductase is induced when cells are treated with the noncovalent inhibitor trimethoprim linked to Boc3Arg. Degradation is rapid and robust, with 30–80% of these abundant target proteins consumed within 1.3–5 hours. The proteasome is required for Boc3Arg-mediated degradation, but ATP is not necessary and the ubiquitin pathways do not appear to be involved. These results suggest that the Boc3Arg moiety may provide a general strategy to construct inhibitors that induce targeted protein degradation.
Introduction
Several authors have suggested that the ideal drug dissociates slowly from its target protein, so that new target synthesis must occur to restore function (Copeland et al., 2006; Swinney, 2004; Zhang and Monsma, 2009). Such perfection usually requires very high affinity binding or covalent modification, which accounts for the current resurgence of interest in covalent drugs (Singh et al., 2011). Intriguingly, several drugs meet this metric by inducing the degradation of their targets. Selective estrogen receptor down-regulators (SERDs) such as fulvestrant are the best-characterized examples of this phenomenon (Howell, 2006). These compounds induce degradation of the estrogen receptor via the ubiquitin/26S proteasome pathways. In cell culture, the level of estrogen receptor is decreased by 45–83% in 4 hours (Wittmann et al., 2007). Importantly, tamoxifen-resistant breast cancer cells remain sensitive to fulvestrant and other SERDs, illustrating the advantage of this strategy. Similar degradation of the androgen receptor is observed with bicalutamide treatment (Waller et al., 2000), and the success of the retinoic acid/arsenic trioxide combination in acute promyelocytic leukemia therapy derives from its ability to induce the degradation of the oncogenic fusion protein PML-RARA (Nasr et al., 2009). These examples illustrate the therapeutic potential of small molecules that induce protein degradation. In addition, such compounds could have broad utility as reagents in protein knockdown experiments. As yet, a robust, broadly applicable strategy for the design of degradation-inducing small molecule inhibitors has yet to emerge. Given that inhibitor binding generally stabilizes protein structure, the principles of such design are not obvious.
One approach to this problem is ‘proteolysis targeting chimeric molecules’ (PROTACs) developed by Crews and colleagues (Raina and Crews, 2010). PROTACs contain a ligand that recognizes the target protein linked to a ligand that binds to a specific E3 ubiquitin ligase. Degradation of methionine aminopeptidase, androgen receptor, estrogen receptor and the aryl hydrocarbon receptor have been reported (Lee et al., 2007; Rodriguez-Gonzalez et al., 2008; Sakamoto et al., 2001). In many cases, the E3 ligase-targeting ligand is a peptide, which can limit therapeutic use. More importantly, the ubiquitin pathways are extremely complicated and poorly understood (Clague and Urbe, 2010; Finley, 2009). The details of tissue expression, cellular localization and regulation are known in only a tiny fraction of the over 600 putative E3 ligases in humans (Deshaies and Joazeiro, 2009). Such observations suggest that the reliable manipulation of these pathways may prove challenging.
Conditional protein knockdown is an invaluable tool for delineating protein function. In addition to RNAi, several methods have been developed that rely on small molecule ligands (Raina and Crews, 2010). The most common strategy involves expression of fusion proteins that couple the target to an unstable “degron” domain. The degron domain is usually stabilized by the presence of a ligand; removal of the ligand induces degradation (Banaszynski et al., 2006; Dohmen et al., 1994; Iwamoto et al., 2010; Levy et al., 1999; Pratt et al., 2007; Stankunas et al., 2003). These techniques have proven very useful, but are limited by the requirement for the constant presence of ligand to maintain protein levels. Several systems have been developed to address this problem. One method uses a small molecule to localize a target fusion protein directly to the proteasome (Janse et al., 2004). Importantly, the success of this strategy demonstrates that proteasome localization is sufficient to induce degradation. A similar strategy has been used to target protein degradation in bacteria (Davis et al., 2009). In another clever variation, a small molecule has been used to unmask a cryptic degron (Bonger et al., 2011). Recently, a system based on the covalent attachment of hydrophobic tags to dehalogenase fusion proteins has also been reported (Neklesa et al., 2011). However, while these systems can effectively modulate levels of transgenic fusion proteins, they cannot be used to reduce the levels of endogenous proteins.
We have discovered that inhibitors linked to a tert-butyl carbamate-protected arginine (Boc3Arg) moiety induce selective degradation of their target proteins. Remarkably, this process appears to be ATP- and ubiquitin-independent. We propose that Boc3Arg-induced degradation provides a strategy to induce protein degradation for both conditional protein knockdown and chemotherapeutic applications.
Results
EA-Boc3Arg induces the degradation of GST-α1 in cell lysates
GST isozymes, especially GST-π, are potential targets for anticancer chemotherapy (Laborde, 2010; Sau et al., 2010). These proteins are well characterized with several readily modified inhibitors such as ethacrynic acid (EA) and thiobenzofurazan (Fur) (Ploemen et al., 1993; Ricci et al., 2005). Therefore we initiated an investigation of protein degradation using C-terminally hemagglutinin (HA)-tagged glutathione-S-transferase α1 (GST-α1) as the target protein. HA-tagged proteins are widely used reagents because horseradish peroxidase coupled anti-HA antibody provides a sensitive assay with a dynamic range of >100. Our plan was to link EA to an arginine residue, reasoning that such an inhibitor would induce protein degradation via the N-end rule pathways (Tasaki and Kwon, 2007). Some indication that the N-end rule pathways can be exploited in this manner is present in the patent literature (Kenten and Roberts, 2001). EA alkylates the active site Cys residue in GST (Figure 1A); the inhibitor’s carboxylate moiety can be readily modified without perturbing this reaction (Figure 1B, (Shi et al., 2006)). One of the compounds produced during the synthetic route was EA-Boc3Arg (Figure 1A), which contains N,N,N-triBoc protected arginine moiety linked to EA via a 1,6-diaminohexane linker. Unfortunately, removal of the Boc groups to produce the unprotected arginine derivative was unsuccessful. Surprisingly, EA-Boc3Arg-modified GST-α1 was readily degraded in HeLa cell lysates (Figure 1C and D). No degradation was observed in the absence of EA-Boc3Arg, with unmodified EA (Figure 1D) or with EA-linker (data not shown). Similar degradation of EA-Boc3Arg-modified GST-α1 was observed in NIH3T3 cell lysates. These results suggested that the Boc3Arg moiety targets the protein for degradation.
Figure 1. Degradation of HA-tagged GST-α1.

(A) Structures of EA and EA-Boc3Arg and the reaction of GST with EA derivatives. (B) EA and EA-Boc3Arg inactivate GST-α1 with similar potency. Purified recombinant GST-α1 (200 nM) was incubated with EA or EA-Boc3Arg and assayed for activity. (C and D) The degradation of EA-Boc3Arg-modified GST-α1. Purified recombinant C-terminally HA-tagged GST-α1 (0.2 mg/ml) was inactivated by EA or EA-Boc3Arg (80 μM) and diluted 50-fold into HeLa cell lysates supplemented with an ATP regenerating system. Samples were analyzed by immunoblotting for the HA tag. Inosine monophosphate dehydrogenase (IMPDH) was used as a loading control. (C) A representative immunoblot. (D) Average of four experiments. Error bars show standard errors. (E) Structure of Fur-Boc3Arg. (F) Degradation of GST-α1 by Fur-Boc3Arg. Purified C-terminally HA-tagged GST-α1 was modified with Fur-Boc3Arg (40 μM) and added to NIH 3T3 cell lysates supplemented with an ATP regenerating system.
Fur-Boc3Arg induces the degradation of GST-α1 in cell lysates
To test the generality of this degradation phenomenon, we synthesized a second Boc3Arg-linked GST inactivator, Fur-Boc3Arg (Figure 1E). Fur also forms a covalent adduct with GST; this molecule can be modified at the indicated positions with retention of activity (Ricci et al., 2005). Fur-Boc3Arg-modified GST-α1 was readily degraded in lysates from NIH 3T3 cells (Figure 1F). These results demonstrate that Boc3Arg-induced degradation does not depend on the nature of the ligand interacting with GST-α1.
TMP-Boc3Arg induces the degradation of eDHFR in cell lysates
To determine if Boc3Arg dependent degradation is a unique property of GST-α1, we turned to the Escherichia coli dihydrofolate reductase/trimethoprim (eDHFR/TMP) system developed by Cornish (Calloway et al., 2007). TMP is a specific inhibitor of eDHFR with much lower affinity for mammalian DHFRs (Kd = 20 pM versus 4 μM for bacterial and mammalian DHFRs, respectively. Importantly, the interaction between eDHFR and TMP is noncovalent, though dissociation is very slow (T1/2 ~20 minutes (Dunn and King, 1980)). TMP can be modified at the 4 position of the B ring with retention of potency and selectivity (Figure 2A; (Calloway et al., 2007)). This strategy has been used to fluorescently label eDHFR fusion proteins in lysates and whole cells (Calloway et al., 2007).
Figure 2. TMP-Boc3Arg induced degradation of eDHFR fusion proteins in cell lysates.

(A) Structures of TMP and derivatives. (B and C) Purified recombinant eDHFR-HA (0.2 mg/ml) was incubated with TMP derivatives (80 μM) and diluted 50-fold into Cos-1 cell lysates supplemented with an ATP regenerating system. (B) Representative immunoblot showing degradation of eDHFR-HA in the presence of TMP and TMP-Boc3Arg. (C) Combined data from 2 experiments after 4 h incubation.
We synthesized a Boc3Arg derivative of TMP using modified published methods (Figure 2A; see Materials and Methods for synthetic procedures; (Calloway et al., 2007; Long et al., 2011)). As above, eDHFR contained a C-terminal HA-tag to facilitate detection. While eDHFR degraded slowly in Cos-1 cell lysates, the eDHFR•TMP complex was stable, as expected (Figure 2B and C). The deprotected compound TMP-Arg also stabilized eDHFR (Figure 2C). In contrast, the DHFR•TMP-Boc3Arg complex was readily degraded. Similar TMP-Boc3Arg-dependent degradation of eDHFR was observed in Hela and NIH3T3 cell lysates (data not shown). Importantly, these observations demonstrated that the Boc3Arg moiety does not need to be covalently attached to the target protein to induce degradation.
Boc3Arg-induced protein degradation in whole cells
We constructed a variety of GST and eDHFR fusion proteins to determine the efficacy of Boc3Arg-mediated protein degradation in mammalian tissue culture cells. Importantly, TMP-Boc3Arg has no effect on cell viability at concentrations up to 135 μM over the course of these experiments (Figure S1A and B). Likewise, no toxicity was observed when Cos-1 cells were treated with EA-Boc3Arg (100 μM), though HeLa cells displayed some sensitivity (Figure S1C and D). Endogenous GST isozymes are abundant proteins, so we chose to focus our initial efforts on ectopically expressed eDHFR fusion proteins. As noted above, the eDHFR/TMP system is used to selectively label proteins in cells (Calloway et al., 2007), which suggested that the TMP-Boc3Arg would induce the degradation of eDHFR fusion proteins in whole cells.
Boc3Arg-initiated protein degradation was monitored using a modified global protein stability (GPS) assay (Yen et al., 2008). GFP-fusion proteins are co-expressed with Discosoma sp. red fluorescent protein (RFP) in a bicistronic construct under control of a CMV promoter, with GFP-fusion protein translation occurring at the internal ribosome binding site. Thus a single mRNA is responsible for the production of both RFP and GFP-fusion protein, so that both proteins are produced in a constant ratio in all transfected cells (Yen et al., 2008). Changes in the relative intensities of red and green fluorescence are measured by flow cytometry, providing a facile and quantitative measure of the protein stability in living cells and real time. This assay has been used to monitor the stability of ~8000 human proteins to identify substrates of the proteasome and E3 ligases (Yen and Elledge, 2008; Yen et al., 2008).
In the present application, an eDHFR-HA-GFP fusion protein is expressed with translation occurring at the internal ribosome binding site. The ratio of GFP/RFP does not change in the presence of TMP (Figure 3A). In contrast, the GFP/RFP ratio decreases in the presence of TMP-Boc3Arg (Figure 3B,C). We confirmed the degradation of eDHFR-HA-GFP with both anti-HA and anti-GFP blotting (Figure 3D). Approximately 30–40% of the GFP signal was lost over 5 hours. Similar decreases in GFP/RFP ratios were observed in MCF-7 cells. These experiments demonstrate that Boc3Arg-induced degradation is sufficiently robust to compete with CMV-driven protein expression.
Figure 3. Degradation of eDHFR fusion proteins in whole cells.

(A and B) Global protein stability assay. HeLa cells co-express RFP and eDHFR-HA-GFP from a bicistronic construct. Red and green fluorescence was measured by flow cytometry. (A) TMP (80 μM). (B) TMP-Boc3Arg (80 μM). (C) Quantitation of four independent GPS experiments. (D) Degradation of eDHFR-GFP was confirmed by anti-HA and anti-GFP immunoblotting. Supported by Figure S1.
In the context of a whole cell, depletion of target protein levels will be a function of the rate of protein synthesis, the rate of degradation and the rate of uptake of Boc3Arg-ligand. To further gauge the contribution of new protein synthesis to the extent of protein knockdown, we monitored TMP-Boc3Arg-induced degradation in the presence of cycloheximide, which blocks the synthesis of new proteins. Under these conditions, eDHFR-HA-GFP levels are reduced to ~10% within 3 hours in cells treated with TMP-Boc3Arg (Figure 4). No degradation was observed when cells were treated with DMSO alone or with TMP, demonstrating that degradation is induced by the Boc3Arg degron. Given that eDHFR-HA-GFP expression is driven by a CMV promoter, and that GFP is a very stable protein, these observations suggest that Boc3Arg-induced degradation will be sufficiently efficient to knockdown most proteins.
Figure 4. Degradation of eDHFR fusion proteins in cycloheximide treated cells.

(A) Experiment as in Figure 3, but HeLa cells were incubated with 0.2 mg/mL cycloheximide for 20 minutes prior to TMP-Boc3Arg treatment. The rightmost panel is an independent blot. (B) Quantitation of three independent experiments.
EA-Boc3Arg induced degradation of endogenous GST-π
Encouraged by the above results, we investigated whether EA-Boc3Arg could cause the degradation of endogenous GST-π. This protein is abundant; its concentration can be as high as 50 μM in cancer cells (Ricci et al., 2005). Moreover, alkylators induce the expression of GST (Hu et al., 2006a; Thimmulappa et al., 2002), so this is another demanding test of the efficacy of Boc3Arg-mediated degradation. Cos-1 cells were incubated with EA or EA-Boc3Arg for 3 hours. EA-treatment increased the level of endogenous GST-π by 30% relative to the DMSO control (Figure 5A). In contrast, EA-Boc3Arg treatment reduced the levels of endogenous GST-π by 30% relative to the DMSO control and by 50% relative to EA treatment (Figure 5A). GST-π is a bona fide anticancer target (Raj et al., 2011; Sau et al., 2010), so this experiment provides proof of concept with a pharmaceutical relevant protein.
Figure 5. EA-Boc3Arg-induced degradation of endogenous and ectopic GST in whole cells.
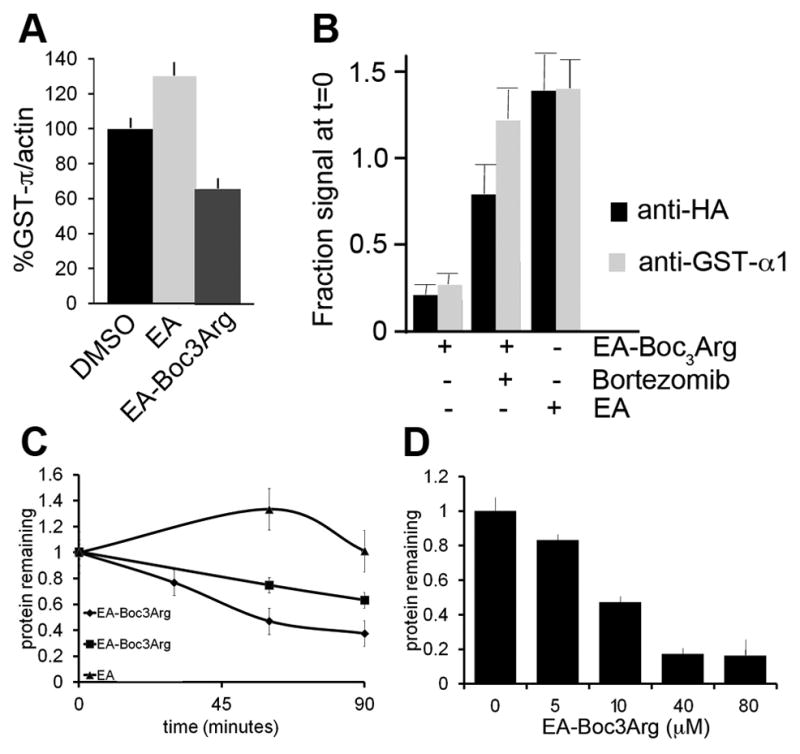
(A) Degradation of endogenous GST-π control when Cos-1 cells are treated with EA and EA-Boc3Arg for 3 hrs (N = 4). (B) Degradation of eDHFR-HA-GST-α1 in HeLa cells. Cells expressing eDHFR-HA-GST-α1 were treated with EA-Boc3Arg (80 μM) over 1.8 hours. Protein was measured by both anti-GST and anti-HA immunoblotting (N≥3). (C) HeLa cells expressing eDHFR-HA-GST-α1 were treated with: triangles EA (80 μM); squares EA-Boc3Arg (8 μM); diamonds EA-Boc3Arg (80 μM). At the indicated time points, the eDHFR-HA-GST-α1 was quantitated and normalized to GAPDH (D) HeLa cells expressing eDHFR-HA-GST-α1 were treated with a range of concentrations of EA-Boc3Arg or just DMSO. After 1.2 hours, the amount of eDHFR-HA-GST-α1 was quantitated. Supported by Figure S2.
EA-Boc3Arg induced degradation of ectopic proteins in whole cells
Compounds such as EA are known to amplify the expression of GST-π (Higgins and Hayes, 2011), so it is possible that the rate of GST-π synthesis was increasing over the course of the above experiment. Therefore we ectopically expressed GST-α1-fusion proteins under control of a CMV promoter to ensure that the target protein would be expressed at a constant rate. Gratifyingly, treatment with EA-Boc3Arg caused the rapid and robust degradation of eDHFR-HA-GST-α1. Fusion protein levels decreased by ~70% within 2 hours (Figure 5B and C). As observed with GST-π, eDHFR-HA-GST-α1 levels increased when cells were treated with EA alone, suggesting that the unmodified inhibitor stabilized the protein. Degradation was dose dependent, with 10 μM causing ~50% degradation in 1.2 h (Figure 5D). Preincubation of cells with EA protected eDHFR-HA-GST-α1 against degradation, confirming the specificity of EA-Boc3Arg (Figure S2).
The eDHFR-HA-GST-α1 fusion protein allows the direct comparison of degradation induced by EA-Boc3Arg and TMP-Boc3Arg. While 80 μM EA-Boc3Arg induces nearly complete degradation of the fusion protein in under 2 hours, only ~25% degradation is observed in 5 h when cells are treated with the same concentration of TMP-Boc3Arg (data not shown). Though it is possible that the superior efficacy of EA-Boc3Arg derives from better cellular uptake, these results may also indicate that a covalent attachment between the target protein and ligand is the preferred design strategy.
Degradation requires the proteasome, but not ubiquitylation
The ubiquitin-proteasome system is the major route for intracellular protein degradation (Xie, 2010). SERDs-induced degradation of the estrogen receptor utilizes these pathways (Howell, 2006), and the degradation of dehalogenase fusion proteins with hydrophobic tagging also has been shown to require the proteasome (Neklesa et al., 2011). Therefore we investigated whether Boc3Arg-induced degradation also utilized the ubiquitin-proteasome system. The specific proteasome inhibitor bortezomib blocks the Boc3Arg-mediated degradation of eDHFR and endogenous GST-α1-GFP in whole cells (Figures 4A and 5A). Lactacystin, a second specific proteasome inhibitor, blocks Boc3Arg-induced degradation in Cos-1 cell lysates (Figure 6A), as does the less specific inhibitor MG132 (Figure 6D). However, we failed to observe the accumulation of higher molecular weight forms of any of the proteins in the presence of proteasome inhibitors, even when deubiquitylating enzyme inhibitors were included with proteasome inhibitors. The experiment with ubiquitin aldehyde addition is shown Figure 6C; similar results were obtained with IU1, an inhibitor of USP14 (not shown; (Lee et al., 2010)). ATP is required both for ubiquitylation and for the degradation of ubiquitylated proteins by the 26S proteasome, but omission of the ATP regenerating system did not inhibit Boc3Arg-induced degradation (Figure 6B). Although it can often be difficult to detect ubiquitylation (Jariel-Encontre et al., 2008), nevertheless these observations suggest that the Boc3Arg group induces degradation of the target protein by a mechanism that is independent of the ubiquitin pathways.
Figure 6. Mechanism of Boc3Arg-induced degradation.

(A) Effect of lactacystin (50 μM) on the TMP-Boc3Arg-induced degradation of eDHFR in Cos-1 cell lysates supplemented with an ATP regeneration system. (B) Degradation of GST-α1 (0.2 mg/ml) was modified by EA-Fur-Boc3Arg (40 μM) then diluted 50-fold into NIH 3T3 cell lysates. closed diamonds, standard assay buffer containing ATP regenerating system; closed squares, ATP regenerating system omitted. (C) Effect of lactacystin (50 μM) and ubiquitin aldehyde (20 μM) on the degradation of eDHFR•TMP-Boc3Arg in Cos-1 lysates. (D) Effect of MG132 (100 μM) on the EA-Boc3Arg-induced degradation of GST-α1 in Cos-1 cell lysates supplemented with an ATP regeneration system.
Discussion
The Boc3Arg moiety provides a general strategy for the design of degradation-inducing inhibitors
The use of small molecules to stabilize misfolded proteins is a promising approach for the treatment of several inherited diseases (Bernier et al., 2004; Ringe and Petsko, 2009). Such chemical/pharmacological chaperones exploit the common ability of inhibitor binding to stabilize protein structure. In contrast, the experiments described above demonstrate that the Boc3Arg moiety provides a general strategy for the construction of small molecules that induce protein degradation. Importantly, a covalent interaction between the inhibitor and target is not required, which suggests that the Boc3Arg-induced degradation design may be broadly applicable. EA is a diuretic, GST-π has an essential role in the development of colon cancer (Dang et al., 2005), TMP is a widely used antibiotic and DHFR is an important target for cancer, immunosuppressive and antimicrobial chemotherapy, so this work demonstrates proof of concept with therapeutically relevant inhibitors and targets.
Boc3Arg-induced degradation can be rapid and robust- our work demonstrates that as much as 30–80% of an abundant protein can be consumed in 2–5 hours. Haploinsufficiency is a frequent cause of autosomal dominant disorders, which suggests that such levels of protein knockdown should be sufficient to observe a phenotype. Further, similar levels of estrogen receptor knockdown are observed when tissue culture cells are treated with SERDS (Wittmann et al., 2007), demonstrating Boc3Arg-induced degradation should be sufficient to make a pharmacological impact. Boc3Arg-induced protein knockdown is more rapid than RNAi, which typically requires several days to deplete the target mRNA. Of course, the extent of protein knockdown will depend on the rate of synthesis and the intrinsic structural stability of the protein target, but these issues also determine the efficacy of RNAi.
The structure-activity profile of Boc3Arg-induced degradation has not been defined, and our work towards this end is ongoing. The Boc3Arg and linker together have a molecular weight approaching 600 Da, with polar surface area of more than 150 Å2. While the linker requirements are likely to be target specific, more effective degrons may be obtained with an optimization program that seeks to reduce the size of the Boc3Arg moiety. The properties of the recognition ligand must also be defined. We expect that Boc3Arg-induced degradation will have a bell-shaped dependence on the affinity of the recognition ligand. If this interaction is too weak, the association will not be long enough for the target to transit to the proteasome. If the interaction is too strong, the target will not unfold and enter the proteolytic chamber. It seems reasonable to expect that more effective examples of inhibitor-mediated degradation will be realized.
The pharmacological potential of Boc3Arg-mediated degradation
Drugs that induce protein degradation are likely to have different pharmacological effects than those that simply obstruct a binding site. For example, inhibition of the kinase activity of the Bcr-Abl kinase is not sufficient to block activation of its downstream signaling pathways (Hu et al., 2006b); obviously such kinase-independent signaling would be eliminated if Bcr-Abl was degraded. Treatment of botulinum toxin (BoNT) intoxication highlights another advantage of this strategy. BoNT has a half-life of 90 days, so patients must be kept on respirators for many months (Foran et al., 2003); a small molecule inhibitor of BoNT would also have to be administered for many months, but an agent that could induce the degradation of BoNT would require a much reduced treatment schedule. The degradation of aberrant proteins also provides a novel and much-needed strategy for treating many hereditary diseases. For example, mutations in IMPDH1 cause retinitis pigmentosa, an inherited blindness resulting from the degeneration of rod photoreceptors. IMPDH1 knockout mice display only a mild retinopathy (Aherne et al., 2004), suggesting that the removal of the mutant protein will prevent disease.
The knockdown of GST-π illustrates the potential of inhibitor mediated protein degradation. EA increased the level of GST-π relative to the DMSO-treated control. In contrast, EA-Boc3Arg decreased the levels of GST-π, so that only 50% of the protein is present relative to EA-treated cells. In both cases, the active site of GST-π is blocked, disrupting phase II detoxification and protein glutathionylation reactions. However, GST-π has additional functions that appear to be independent of the active site (Tew and Townsend, 2011), including a ‘ligandin’ activity that binds large planar aromatic anions outside the active site. This ligandin activity may serve to prevent the generation of reactive oxygen species. GST-π also forms a complex with c-Jun N-terminal kinase 1 to negatively regulate MAP kinase signaling. Lastly, GST-π forms higher level oligomers under conditions of cellular stress, though the functional consequences of oligomerization are not known. These processes are likely to be regulated by the levels of GST-π, which suggests that the pharmacology of EA and EA-Boc3Arg treatment will be distinct.
Mechanism of Boc3Arg-induced degradation
The three tert-butyl groups of Boc3Arg create a large hydrophobic surface that may cause for degradation in a similar manner to the hydrophobic tagging of dehalogenase fusion proteins recently reported by Crews and colleagues (Neklesa et al., 2011). The exposure of hydrophobic surfaces is also believed to be the underlying cause of SERD-induced degradation of the estrogen receptor, oxidized protein degradation and unfolded protein degradation (Breusing and Grune, 2008; Howell, 2006). While the characterization of these processes is far from complete, it is already clear that multiple pathways are at work. SERD-induced degradation involves additional estrogen receptor cofactors such as SRC3/AIB1 and requires polyubiquitylation and the 26S proteasome (Callige and Richard-Foy, 2006). Further, each SERD exposes a distinct hydrophobic surface (Wittmann et al., 2007; Wu et al., 2005), suggesting that different sets of cofactors/ubiquitylation enzymes could be utilized in each case. The degradation of oxidatively damaged proteins is similarly complex. The ubiquitin pathways are needed when damage occurs to newly synthesized proteins (Medicherla and Goldberg, 2008), while the 20S proteasome degrades the majority of oxidized proteins in an ubiquitin- and ATP-independent process (Jung et al., 2009; Shringarpure et al., 2001). Unfolded proteins are also substrates for the 20S proteasome (Asher et al., 2006; Breusing and Grune, 2008). In addition, protein cofactors such as HSP90 can act in concert with the 20S proteasome to promote degradation (Whittier et al., 2004).
Boc3Arg-mediated degradation requires the proteasome, but appears to be ubiquitin- and ATP-independent. These observations suggest that the 20S proteasome may be involved. The 20S proteasome accounts for approximately 40% of the total proteasome pool in cells (Tanahashi et al., 2000), so a large proteolytic capacity is available for ubiquitin-independent degradation. Proteasome inhibitors also block the hydrophobic tagging/dehalogenase degradation system (Neklesa et al., 2011), though it is not yet known if this is a ubiquitin- and ATP-independent process. Enzyme•inhibitor complexes are generally more stable than unliganded enzymes, which makes degradation all the more curious. One possibility is that the Boc3Arg moiety might enter the proteasome and drag the rest of the protein into the proteolytic chamber. While such a process is easily envisioned with a covalently attached inhibitor, it is difficult to understand how this mechanism could work with noncovalent interaction. Another possibility is that the Boc3Arg group intercalates into the target protein, exposing a hydrophobic surface that then interacts with the 20S proteasome, or perhaps Boc3Arg interacts with other protein factors such as HSP90. Which of these mechanisms is operable has important implications for the optimization of Boc3Arg-induced degradation. How the Boc3Arg moiety targets proteins to the proteasome remains to be elucidated.
Significance
Drugs such as fulvestrant cause the degradation of their target proteins. Such compounds generally display superior pharmacodynamic properties relative to analogs that do not induce degradation. However, the discovery of compounds with this property has been largely serendipitous and a robust general strategy for the design of small molecules that cause protein degradation has yet to emerge. Given that inhibitor binding generally stabilizes protein structure, the principles of such design are not obvious. We have discovered that Boc3Arg-linked inhibitors induce degradation of their target proteins in mammalian tissue culture cells. Robust degradation was observed with the Boc3Arg moiety in the context of three different inhibitor scaffolds and with three different target proteins, including an abundant endogenous protein. Importantly, the inhibitors can be covalent or noncovalent. Thus Boc3Arg provides a general strategy for the design of small molecules that induce protein degradation that will be useful in the design of drugs and chemical tools.
Experimental procedures
Materials
All reagents were of the highest commercial grade. Monoclonal rat anti-hemagglutinin (HA) conjugated to horseradish peroxidase (anti-HA-HRP, clone 3F10) was from Roche Diagnostics (Indianapolis, IN). Mouse monoclonal anti-GAPDH conjugated to HRP (anti-GAPDH-HRP), mouse anti-tubulin, rabbit anti-actin and MG132 were from Sigma Aldrich (St. Louis, MO). Rabbit anti-human IMPDH and secondary antibody HRP conjugates were purchased from Abcam (Cambridge, MA). Rabbit anti-GST-α1 and anti-GST-π were from Oxford Biomedical (Oxford, UK). Rabbit anti-GFP was from Chemicon (Billerica, MA). Purified 20S proteasome from HeLa, immunoproteasome from mouse spleen, lactacystin and IU1 were from Boston Biochem (Cambridge, MA). Bortezomib was from Sellek Chemicals LLC (Boston, MA). We thank Charles Morrow (Wake Forest University) for the GST-α1 expression plasmid pOXO4-α1 and the Elledge laboratory for the PMSCV-vector containing the DSRED_IRES_eGFP casette. Recombinant GST-α1 and eDHFR proteins were purified by affinity chromatography as described in Supporting Materials. Compound synthesis and characterization is also described in Supporting Materials.
Cell culture
For lysate experiments, cells were grown to confluence in DMEM with 10% FBS, then cultured without FBS for 2 days. Lysates were prepared in buffer A (300 mM phosphate, pH 7.8, 200 mM sucrose, 42 mM MgCl2, 10 mM NaCl). Protein content was determined using the Bradford assay with IgG as a standard (BioRad, Hercules, CA).
In vitro degradation assays
Lysates were validated for proteolytic activity using a fluorescence-based proteasome assay (Sigma Aldrich, St. Louis, MO). Addition of MG132 (100 μM) or lactacystin (50 μM) inhibited this reaction, demonstrating the presence of active proteasomes. An optimized concentration (1 mg/mL) lysate was used in degradation assays. Unless otherwise stated, the lysate was supplemented with: 10 mM ATP, 63 mM MgCl2, 100 mM creatine phosphate and 0.01 mg/ml creatine kinase. The target enzyme (0.2 mg/mL, 8.3 μM) was preincubated with the respective inhibitor (typically 80 μM) for 20 minutes at 37°C in 50 mM Tris-Cl buffer, pH 8.0. Then inhibitor-enzyme complex was diluted 50-fold into lysate, overlaid with Chill-out wax® (Biorad) and incubated at 37°C. Aliquots (15 μL) were added to 10 μL of 5X SDS loading buffer at the appropriate time intervals. Protein was visualized with the appropriate primary and secondary antibodies using ECL Plus (GE, Bucks, UK). Densitometry was carried out using IMAGE-J V 1.44p.
In cell degradation assays
Cells were grown to approximately 60% confluence, then transfected with the required plasmid using Transit2020® (except HeLa for which Transit HeLaMonster® was used) (Mirus, Pittsburg, PA). Fresh media containing compound was added to initiate the assay. Cells were incubated at 37°C and harvested by trypsinization and lysis at the appropriate time intervals. For Fluorescence Activated Cell Sorting (FACS), cells were collected by centrifugation, then resuspended in FACS buffer (1% FBS in PBS). FACS was carried out on a Becton Dickinson FACScalibur instrument. Details of analysis are included in Supporting Information.
Supplementary Material
Highlights.
Two Boc3Arg-linked covalent inhibitors induce the degradation of GST proteins in cells
A Boc3Arg-linked noncovalent inhibitor induces the degradation of DHFR in cells
30–80% degradation of the target protein is observed within 1.3–5 hours
Proteasome inhibitors block targeted degradation, but ubiquitylation is not required
Acknowledgments
The authors thank Craig Stropkay and Prof. Ruibao Ren for assistance with FACS and for the use of their fluorescence microscope and Dr. B. Chris Hoefler for useful and insightful discussions including suggesting the CD experiments and Dr. Iain S. MacPherson for invaluable help with cloning. This work was supported by NIH GM54403 (LH) and AI075466 (LH). MJCL is supported by a Howard Hughes Medical Instititute International Student Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aherne A, Kennan A, Kenna PF, McNally N, Lloyd DG, Alberts IL, Kiang AS, Humphries MM, Ayuso C, Engel PC, et al. On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa. Hum Mol Genet. 2004;13:641–650. doi: 10.1093/hmg/ddh061. [DOI] [PubMed] [Google Scholar]
- Asher G, Reuven N, Shaul Y. 20S proteasomes and protein degradation “by default”. Bioessays. 2006;28:844–849. doi: 10.1002/bies.20447. [DOI] [PubMed] [Google Scholar]
- Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15:222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Bonger KM, Chen LC, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nature chemical biology. 2011;7:531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breusing N, Grune T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol Chem. 2008;389:203–209. doi: 10.1515/BC.2008.029. [DOI] [PubMed] [Google Scholar]
- Callige M, Richard-Foy H. Ligand-induced estrogen receptor alpha degradation by the proteasome: new actors? Nucl Recept Signal. 2006;4:e004. doi: 10.1621/nrs.04004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calloway NT, Choob M, Sanz A, Sheetz MP, Miller LW, Cornish VW. Optimized fluorescent trimethoprim derivatives for in vivo protein labeling. Chembiochem. 2007;8:767–774. doi: 10.1002/cbic.200600414. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Urbe S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Dang DT, Chen F, Kohli M, Rago C, Cummins JM, Dang LH. Glutathione S-transferase pi1 promotes tumorigenicity in HCT116 human colon cancer cells. Cancer research. 2005;65:9485–9494. doi: 10.1158/0008-5472.CAN-05-1930. [DOI] [PubMed] [Google Scholar]
- Davis JH, Baker TA, Sauer RT. Engineering synthetic adaptors and substrates for controlled ClpXP degradation. J Biol Chem. 2009;284:21848–21855. doi: 10.1074/jbc.M109.017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Dohmen RJ, Wu P, Varshavsky A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Dunn SM, King RW. Kinetics of ternary complex formation between dihydrofolate reductase, coenzyme, and inhibitors. Biochemistry. 1980;19:766–773. doi: 10.1021/bi00545a024. [DOI] [PubMed] [Google Scholar]
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran PG, Mohammed N, Lisk GO, Nagwaney S, Lawrence GW, Johnson E, Smith L, Aoki KR, Dolly JO. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J Biol Chem. 2003;278:1363–1371. doi: 10.1074/jbc.M209821200. [DOI] [PubMed] [Google Scholar]
- Higgins LG, Hayes JD. Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev. 2011;43:92–137. doi: 10.3109/03602532.2011.567391. [DOI] [PubMed] [Google Scholar]
- Howell A. Fulvestrant (‘Faslodex’): current and future role in breast cancer management. Crit Rev Oncol Hematol. 2006;57:265–273. doi: 10.1016/j.critrevonc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, Lin W, Reddy B, Chan JY, Kong AN. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 2006a;243:170–192. doi: 10.1016/j.canlet.2005.11.050. [DOI] [PubMed] [Google Scholar]
- Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006b;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Bjorklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol. 2010;17:981–988. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse DM, Crosas B, Finley D, Church GM. Localization to the proteasome is sufficient for degradation. J Biol Chem. 2004;279:21415–21420. doi: 10.1074/jbc.M402954200. [DOI] [PubMed] [Google Scholar]
- Jariel-Encontre I, Bossis G, Piechaczyk M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim Biophys Acta. 2008;1786:153–177. doi: 10.1016/j.bbcan.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Kenten JH, Roberts SF. In: Controlling protein levels in eukaryotic organisms. States U, editor. Proteinix, Inc; 2001. [Google Scholar]
- Laborde E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell death and differentiation. 2010;17:1373–1380. doi: 10.1038/cdd.2010.80. [DOI] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Puppala D, Choi EY, Swanson H, Kim KB. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chembiochem. 2007;8:2058–2062. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- Levy F, Johnston JA, Varshavsky A. Analysis of a conditional degradation signal in yeast and mammalian cells. Eur J Biochem. 1999;259:244–252. doi: 10.1046/j.1432-1327.1999.00024.x. [DOI] [PubMed] [Google Scholar]
- Long MJ, Pan Y, Lin HC, Hedstrom L, Xu B. Cell compatible trimethoprim-decorated iron oxide nanoparticles bind dihydrofolate reductase for magnetically modulating focal adhesion of Mammalian cells. Journal of the American Chemical Society. 2011;133:10006–10009. doi: 10.1021/ja202767g. [DOI] [PubMed] [Google Scholar]
- Medicherla B, Goldberg AL. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J Cell Biol. 2008;182:663–673. doi: 10.1083/jcb.200803022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr R, Lallemand-Breitenbach V, Zhu J, Guillemin MC, de The H. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin Cancer Res. 2009;15:6321–6326. doi: 10.1158/1078-0432.CCR-09-0209. [DOI] [PubMed] [Google Scholar]
- Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nature chemical biology. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploemen JH, van Ommen B, Bogaards JJ, van Bladeren PJ. Ethacrynic acid and its glutathione conjugate as inhibitors of glutathione S-transferases. Xenobiotica. 1993;23:913–923. doi: 10.3109/00498259309059418. [DOI] [PubMed] [Google Scholar]
- Pratt MR, Schwartz EC, Muir TW. Small-molecule-mediated rescue of protein function by an inducible proteolytic shunt. Proc Natl Acad Sci U S A. 2007;104:11209–11214. doi: 10.1073/pnas.0700816104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K, Crews CM. Chemical inducers of targeted protein degradation. J Biol Chem. 2010;285:11057–11060. doi: 10.1074/jbc.R109.078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ricci G, De Maria F, Antonini G, Turella P, Bullo A, Stella L, Filomeni G, Federici G, Caccuri AM. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. The Journal of biological chemistry. 2005;280:26397–26405. doi: 10.1074/jbc.M503295200. [DOI] [PubMed] [Google Scholar]
- Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8:80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, Sakamoto KM. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sau A, Pellizzari Tregno F, Valentino F, Federici G, Caccuri AM. Glutathione transferases and development of new principles to overcome drug resistance. Archives of biochemistry and biophysics. 2010;500:116–122. doi: 10.1016/j.abb.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Shi B, Stevenson R, Campopiano DJ, Greaney MF. Discovery of glutathione S-transferase inhibitors using dynamic combinatorial chemistry. Journal of the American Chemical Society. 2006;128:8459–8467. doi: 10.1021/ja058049y. [DOI] [PubMed] [Google Scholar]
- Shringarpure R, Grune T, Davies KJ. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci. 2001;58:1442–1450. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nature reviews Drug discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Bayle JH, Gestwicki JE, Lin YM, Wandless TJ, Crabtree GR. Conditional protein alleles using knockin mice and a chemical inducer of dimerization. Mol Cell. 2003;12:1615–1624. doi: 10.1016/s1097-2765(03)00491-x. [DOI] [PubMed] [Google Scholar]
- Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004;3:801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. The Journal of biological chemistry. 2000;275:14336–14345. doi: 10.1074/jbc.275.19.14336. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Kwon YT. The mammalian N-end rule pathway: new insights into its components and physiological roles. Trends Biochem Sci. 2007;32:520–528. doi: 10.1016/j.tibs.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Tew KD, Townsend DM. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metab Rev. 2011;43:179–193. doi: 10.3109/03602532.2011.552912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer research. 2002;62:5196–5203. [PubMed] [Google Scholar]
- Waller AS, Sharrard RM, Berthon P, Maitland NJ. Androgen receptor localisation and turnover in human prostate epithelium treated with the antiandrogen, casodex. J Mol Endocrinol. 2000;24:339–351. doi: 10.1677/jme.0.0240339. [DOI] [PubMed] [Google Scholar]
- Whittier JE, Xiong Y, Rechsteiner MC, Squier TC. Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. The Journal of biological chemistry. 2004;279:46135–46142. doi: 10.1074/jbc.M406048200. [DOI] [PubMed] [Google Scholar]
- Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67:9549–9560. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Xie Y. Structure, assembly and homeostatic regulation of the 26S proteasome. J Mol Cell Biol. 2010;2:308–317. doi: 10.1093/jmcb/mjq030. [DOI] [PubMed] [Google Scholar]
- Yen HC, Elledge SJ. Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science. 2008;322:923–929. doi: 10.1126/science.1160462. [DOI] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- Zhang R, Monsma F. The importance of drug-target residence time. Curr Opin Drug Discov Devel. 2009;12:488–496. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.