

Abstract
Objective
Objectives of this investigation were not merely to perform a comparative study with original docetaxel, but to define anti‐proliferative and apoptotic effects of novel hydrophilic docetaxel (CQMU‐0519) analogue on A549 lung, SKVO3 ovary and MCF7 breast carcinoma cell lines.
Materials and methods
The materials for the study consist of a completely new docetaxel analogue (CQMU‐0519), synthesized by the Department of Pharmacology, Chongqing Medical University, China, which is completely soluble in water. 50 nm of drug concentration was utilized on all three cell lines where cell population growth was assessed using cell culture kit‐8 and flow cytometry analysis, whereas apoptotic pathways were unveiled by use of annexin‐V FITC, apoptosis DNA ladder, caspases‐3, 6, 8 and 9; in the meanwhile, regulation of Bcl‐2 family members was analysed by western blotting.
Result
The novel docetaxel analogue (CQMU‐0519) suppressed cell proliferation in all three cell lines, inhibition of cell proliferation and cell cycle arrest being more evident in G2/M phase. Also, in both lung and ovarian cell lines, apoptotic levels were higher as measured by the various tests performed, and downregulation of Bcl‐2 and Bcl‐xL with increased expressions of Bad and Bax indicated the intrinsic pathway for apoptosis. Nevertheless, it was found that MCF7 cells, although also manifesting high levels of apoptosis, used the extrinsic pathway instead. Hence, it was shown that novel docetaxel analogue (CQMU‐0519) may have some prospective use in future clinical trials.
Conclusions
Novel hydrophilic docetaxel analogue (CQMU‐0519) inhibited cell proliferation and enhanced the intrinsic apoptotic pathway in lung and ovarian carcinoma cells, whereas it used the extrinsic one in breast adenocarcinoma cells.
Introduction
Docetaxel (Taxotere®) is one of the second generation taxanes – anti‐cancer drugs, mitotic poisons or mitotic inhibitors, currently undergoing profound and meticulous laboratory and clinical investigation. It is derived from a renewable natural source, Taxus baccata, (needles of the European yew) 1. It is slightly more soluble in water than Taxol® 2, but it needs to be solubilized in polysorbate 80 for its commercial formulation, thus potentially bearing associated side effects. Moreover, docetaxel has been known to inhibit cell population growth and induce apoptosis of various cancer cells, and several chemotherapeutic mechanisms have described that its cytotoxicity is related either to its excessive polymerization or depolymerization of microtubules 3, or even association with β‐tubulin 4, 5, where microtubules' mechanism of action is suppressed 6, 7, 8. Thus, there is aberrant mitosis and diminished cell proliferation during subsequent cell cycles. Besides, taxanes have been found to not be cell‐specific with not all concentrations displaying similar effects on microtubule dynamics 6, 7, 8, 9, 10, 11, 12, relating to dissimilar cytotoxic, anti‐proliferative and apoptotic effects on various cancer cell lines, namely breast, lung, ovary, prostate, hepatic, melanocyte and leukaemic 13, 14, 15, 16, 17, 18, 19, 20.
Pioneering studies have shown that docetaxel can involve both extrinsic and intrinsic pathways either singly or in combination 21, 22, 23, where the Bcl‐2 family, including Bcl‐2 (B‐cell lymphoma 2), Bax (Bcl‐2‐associated X protein) and Bcl‐xL (B‐cell lymphoma‐extra large); p53, p21WAF1 and the caspase family are closely involved 24, 25, 26. Although killing malignant cells is the main aim of all anti‐cancer researchers, scientists, cancer physicians and pharmaceutical companies, there are still many hedges and thorns to be overcome to attain successful and irreproachable cancer therapy. Nevertheless, Taxotere® has remained amongst those drugs that can achieve favourable anti‐proliferative and apoptotic results on a whole range of cancer cells both in vivo and in vitro 13, 14, 15, 16, 17, 18, 19, 20. Nowadays, due to its poor oral bioavailability, solubility, drug resistance and numerous side effects, better derivatives such as Abraxane® and docetaxel with several combinations of cisplatin, cyclophosphamide, capecitabine and even radiation 3, 27, 28, 29, 30, 31, are being used in clinical trials and have yielded acceptable outcomes on patients' conditions.
Thus, we have initiated this novel docetaxel analogue (CQMU‐0519), synthesized with the objective of making it more water soluble compared to the original formulation. This study has been performed to highlight the main effects of suppression of cell proliferation and enhancement of cell suicide, using the new drug.
Materials and methods
Reagents used
Original docetaxel (Sigma‐Aldrich, St Louis, MO, USA) was purified, while the novel docetaxel analogue (CQMU‐0519) (Patent numbers: CN 200910104454.2; CN 201010299678.6) was synthesized by the Department of Pharmacology, Chongqing Medical University, China; the analogue is completely soluble in 0.95% normal saline (NS) compared to original docetaxel. Stock solutions of original docetaxel and novel docetaxel analogue (CQMU‐0519) were of 10 000 nm, which were then made up to 25, 50, 100 nm concentrations; due to the poor solubility of Taxotere®, we dissolved the two drugs in 1% methanol and 99% (0.95%) NS to achieve appropriate controls for comparison.
Cell culture
Human A549 lung adenocarcinoma, SKVO3 ovarian and MCF7 breast cancer cell lines were obtained, thanks to Professor Ye Xiu‐Feng, Department of Pathology, Chongqing Medical University, China, and cultured in DMEM‐high glucose medium (Hyclone, Logan, UT, USA) supplemented with 10% foetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin (Hyclone), at 37 °C in a 5% CO2 incubator. Individual cell lines were treated for 6, 12 and 24 h with control (1% methanol & 99% NS) solutions; 25, 50, 100 nm original docetaxel and CQMU‐0519, at final concentrations, were used in cell viability assays at the beginning; however, later in other experiments, cells were cultured at 24‐h incubation with control, 50 nm of original and CQMU‐0519, as better responses were obtained. Thus, our experiments consisted of these three major groups for each cell line.
Cell viability assay
One hundred microlitres of respective cell suspensions (5 × 103 cells/well) were dispensed in triplicate in 96‐well plates and incubated for 24 h; then 10 μl of control, 25, 50 and 100 nm original docetaxel, and 25, 50 and 100 nm CQMU‐0519, were added to individual wells and plates were further incubated for 6, 12 and 24 h. Afterwards, 10 μl cell counting kit‐8 (CCK‐8; Dojindo, Kumamoto, Japan) solution was added to the wells, and plates were incubated for 1 h. Absorbance (optical density‐OD) was read using a universal microplate reader (Bio‐Tek, Winooski, VT, USA) at 450 nm, and a graph of A450 (absorbance at 450 nm wavelength) against concentration was plotted; best incubation period chosen was 24 h. Each point represents mean of collected readings and the procedure was repeated at least three times. Percentage inhibition of proliferation was calculated (mean OD for control, mean OD for each drug)/mean OD for control × 100%.
Cell cycle analysis
Dose of 50 nm was chosen, as it had provided the best anti‐proliferative results in all 3 cancer cell lines (*P < 0.05) (Fig. 1); hence we used this particular concentration in all following experimental trials. 50 nm concentration, being a relatively low and safe level 22, 24, has already resulted in good prognosis in several preclinical studies 17, 32, 33, 34 and a rationale for clinical trials 35, was opted for. Thus, A549, SKVO3 and MCF7 cells 1 × 106 cells/well were allowed to adhere for 24 h in 6‐well plates; then control, 50 nm of original docetaxel and 50 nm CQMU‐0519 were added to each well and incubated for a further period of 24 h. Cells were trypsinized, washed three times in cold phosphate‐buffered saline (PBS), fixed in 70% absolute ice‐cold ethanol, kept at −20 °C overnight then stained in a mixture of propidium (PI) RNase A; using Becton Dickinson flow cytometer, cell cycle analysis was performed (FACScan). This experiment was repeated three times.
Figure 1.

Growth of viable cells was quantified using CCK ‐8 Assay. Data points were expressed as mean of A450 ± SD from three separate experiments for each cell line for 24 h. A549, SKVO3 and MCF7 cells exposed to CQMU‐0519 showed an increase in inhibition of cell proliferation compared to original docetaxel (*P < 0.05).
Light microscopy
Cells of the respective cell lines were grown in 6‐well plates and incubated with control; 50 nm of original docetaxel and CQMU‐0519 concentrations for 24 h; ten high power fields (10 HPF) were visualized, apoptotic bodies were counted and running mean percentage of apoptotic bodies was calculated. Fields were photographed using a Nikon Eclipse inverted microscope (Japan).
Hoechst staining
A549, SKVO3 and MCF7 cell lines were grown on coverslips in 6‐well plates until they reached 70% confluence and respective 50 nm original docetaxel, 50 nm of CQMU‐0519, and control were added for 24 h. Then cells were fixed, stained with 500 μl Hoechst 33258 for 5 min, washed three times in PBS and mounted on slides for viewing on a fluorescence microscope at emission wavelength 460 nm and excitation wavelength 350 nm.
Annexin‐V FITC
Lung adenocarcinoma, ovarian and breast cancer cells were cultured in 6‐well plates at 1 × 106 cells/well for 24 h and incubated with control, 50 nm original docetaxel and 50 nm CQMU‐0519, for a further period of 24 h. Cells were trypsinized, washed three times in warm PBS before annexin‐V FITC staining and analysed by FACScan (Becton Dikinson flow cytometer, CA, USA). Experiments were performed three times.
DNA fragmentation assay
Apoptosis was induced by desired control and 50 nm of both original docetaxel and CQMU‐0519 in all cell lines. Cells were pelleted at 1 × 106, washed in PBS and lysed, and DNA was purified according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). A quantity of 20 μl of each sample was loaded separated on 1.2% agarose gel (Spain) containing 0.5 μg/ml ethidium bromide (Invitrogen), visualized by ultraviolet transillumination (Bio‐Rad, CA, USA) and photographed.
Caspases‐3, ‐6, ‐8 and ‐9 colorimetric activity assays
All three cell lines were grown and incubated with control or respective drugs for 24 h; 2 × 106 cells/ml were trypsinized, washed in cold PBS and lysed, all steps being carried out on ice. Supernatants were collected, immediate Bradford protein concentration assay (Beyotime, Jiangsu, China) was performed for protein concentrations, and supernatants were assayed using commercial kits (Caspase Activity Assay kit; Beyotime) for activities of caspases‐3, ‐6, ‐8 and ‐9 to convert Ac‐DEVD‐PNA (Acetyl‐Asp‐Glu‐Val‐Asp P‐nitroanilide), Ac‐VEID‐PNA (Acetyl‐Val‐Asp‐Glu‐Ilel P‐nitroanilide), Ac‐IETD‐PNA (Acetyl‐Ile‐Glu‐Thr‐Asp P‐nitroanilide) and Ac‐LEHD‐PNA (Acetyl‐Leu‐Glu‐His‐Asp P‐nitroanilide) into the yellow formazan product of PNA (P‐nitroaniline), whose increase of absorbance at 405 nm was used to quantify activity of the caspases. All procedures were performed three times.
Western blot analysis
A549, SKVO3 and MCF7 cells were exposed to appropriate control, 50 nm original docetaxel and 50 nm CQMU‐0519, for 24 h. Later, cells were washed in PBS, scraped into Eppendorf tubes and extracts were obtained following the protein extraction method (Beyotime). Protein extractions were determined using BCA protein Assay (Beyotime) and respective proteins 25 μg/lane were loaded on to 12% polyacrylamide gels (SDS–PAGE), except for PARP where 6% SDS–PAGE was used. Proteins were separated by electrophoresis, transferred to PVDF membranes (Millipore, Billerica, MA, USA) and proteins concerned were blocked in 5% skimmed milk for 2 h at room temperature. Primary antibodies against Bcl‐2, Bax (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bcl‐xL, Bad (Beyotime), Bid (BIOSS), PARP (Santa Cruz Biotechnology) and β‐actin (Beyotime) were of 1 μg/ml concentration and were applied overnight at 4 °C. Next day, membranes were washed three times in TBST, secondary antibodies (horseradish peroxidase‐conjugate goat anti‐mouse, and goat anti‐rabbit IgG; Beyotime) were added and incubated for 2 h. Then, membranes were again washed three times in TBST; enhanced chemiluminescence reagents (Pierce Biotechnology, Rockford, IL, USA) were applied and blots were subjected to gel formatter (Bio‐Rad). All experiments were performed at least three times.
Statistical analysis
Quantitative data were expressed as mean ± standard deviation. Statistical analysis was performed by one‐way ANOVA or Student's t‐test using spss 13.0 software package; P‐value of <0.05 was considered statistically significant.
Results
Novel docetaxel analogue inhibited cell population growth in all three cell lines
After incubation with control and 25, 50 and 100 nm of original docetaxel and novel docetaxel analogue (CQMU‐0519) at 50 nm concentration and after 24 h incubation, novel docetaxel analogue (CQMU‐0519) showed that cell proliferation inhibitory effects were evident and significant in A549 lung – 27.90%, SKVO3 – 18.6% ovarian and MCF7 breast – 19.20% cancer cell lines (*P < 0.05) (Fig. 1), with IC50 values of 11.75, 9.52 and 12.67 μg/ml respectively, calculated from concentration dependent curves. IC50 values were calculated only upon concentration, as percentage inhibition of proliferation in all three cell lines for 6 and 12 h did not have significant effects. Best outcome noted was at 24 h, therefore, this was chosen to calculate IC50 values for all three cell lines. Furthermore, although 25, 50 and 100 nm concentrations were used to check the effects of inhibition of cell growth, at 50 nm, the best results were yielded (*P < 0.05) for all three cell lines; hence, this concentration was chosen for subsequent experimental trials.
Novel docetaxel analogue (CQMU‐0519) affects the cell cycle
Phases of the cell cycle were affected by novel docetaxel analogue (CQMU‐0519) (Fig. 2a–c) in all cancer cell lines tested here; this was determined by flow cytometry using PI staining. Proportions of cells in G2/M were much higher (A549 – 43.43%, SKVO3 – 32.13% and MCF7 – 76.93% cancer cell lines) than phases G0/G1 and S, showing mitotic cell cycle arrest in G2/M phase (Table 1), which is relevant to DNA damage checkpoints. Besides, 3.64% apoptotic cells (sub‐G0/G1) in MCF7 cells were also noted (Fig. 2c).
Figure 2.
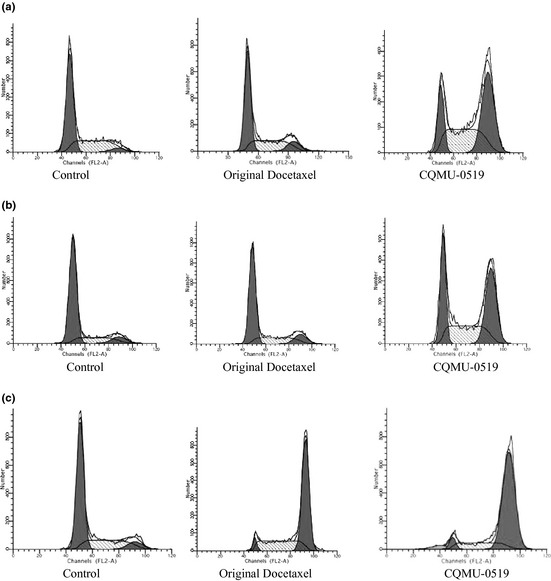
Cell cycle analysis using PI staining. (a) A549, (b) SKVO3 and (c) MCF7. Effects of CQMU‐0519 on cell cycle of all three cell lines was analysed by flow cytometry analysis, using channels (FL2‐A), and different phases G0/G1 (left peak), S and G2/M (right peak) were analysed.
Table 1.
Effects of 50 nm original docetaxel and novel docetaxel analogue (CQMU‐0519) on cell cycle phases G0/G1, S and G2/M in A549, SKVO3 and MCF7 cell lines
| G0/G1 (%) | S (%) | G2/M (%) | |
|---|---|---|---|
| A549 lung control | 62.22 | 33.44 | 4.34 |
| Original docetaxel | 53.07 | 36.24 | 10.69 |
| CQMU‐0519 | 18.46 | 38.12 | 43.42 |
| SKVO3 ovary control | 84.30 | 12.12 | 3.58 |
| Original docetaxel | 57.21 | 18.68 | 24.11 |
| CQMU‐0519 | 28.71 | 39.13 | 32.13 |
| MCF7 breast control | 65.75 | 26.54 | 7.74 |
| Original docetaxel | 4.80 | 31.21 | 64.37 |
| CQMU‐0519 | 3.97 | 18.27 | 76.93 |
Assessment of apoptosis using light microscopy
Apoptotic cells could be seen at ×10 magnification, with chromatin condensation, membrane blebbing and vacuolation (black arrows, Fig. 3a–c). The effects of CQMU‐0519 on the three different cell lines resulted in a running mean percentage of 10 apoptotic bodies visualized per 10 consecutive HPF for A549 cell line, 11.2% for SKVO3 and 11% for MCF7 cancer cell lines which were acceptable 36, 37.
Figure 3.
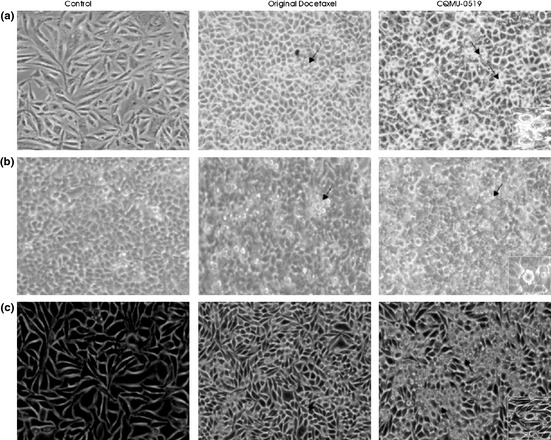
Light microscopy with 100% white balance. (a) A549, (b) SKVO3 and (c) MCF7 cells were treated with our reagents, similar incubation period, and visualized using an inverted light microscope with 100% white balance. Black arrows indicate cells showing nuclear condensation, blebbing and vacuolation (signs of apoptosis).
Assessment of apoptosis by Hoechst staining
Apoptotic figures, blebs and cytoplasmic vacuolation were seen to be significant in all three cell lines exposed to CQMU‐0519; however, fewer apoptotic bodies were noted in SKVO3 cell lines (Fig. 4a–c).
Figure 4.
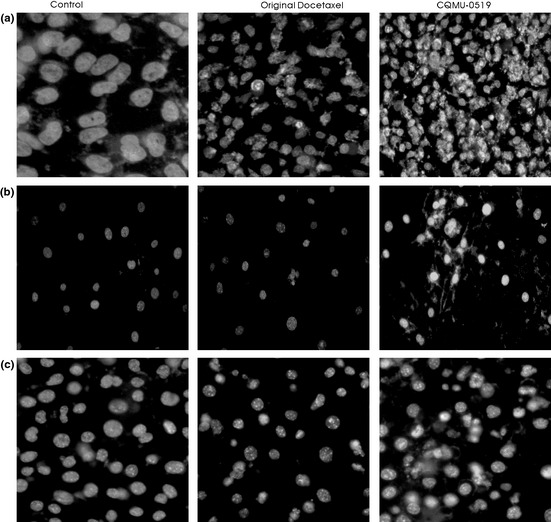
Hoechst 33258 staining. (a) A549, (b) SKVO3 and (c) MCF7 cells treated with docetaxel and CQMU‐0519, similar incubation period, stained with Hoechst 33258 staining.
Determination of apoptosis by annexin‐V FITC assay
It was found that novel docetaxel analogue (CQMU‐0519) caused high levels of apoptosis (early and late): 61.21% in A549, 45.05% in MCF7 and 11.33% in SKVO3 cells, according to flow cytometry using annexin‐V staining (Fig. 5a–c and Table 2). Moreover, A549 cells exhibited 17.37% necrotic cells in response to CQMU‐0519, which was not displayed by breast and ovarian cancer cells.
Figure 5.
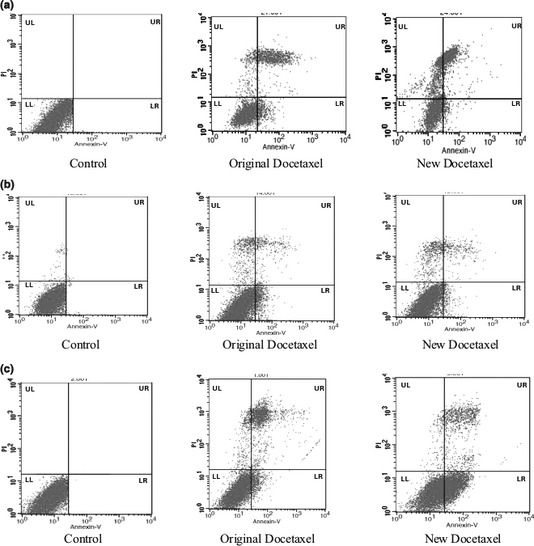
Annexin‐V FITC was used to analyse apoptosis of respective cells. (a) A549, (b) SKVO3 and (c) MCF7 cells, treated with 50 nm of both original and novel docetaxel analogue (CQMU‐0519), for 24 h and analysed using annexin‐V FITC staining. Diagrams representative of three independent experiments. UR, necrotic cells; LL, viable cells; LR, early apoptosis, UL; late apoptosis, in percentages (%).
Table 2.
Percentage of apoptotic cells obtained in A549 lung, SKVO3 ovary and MCF7 breast cancer cells treated with 50 nm original docetaxel and novel docetaxel analogue (CQMU‐0519)
| LL (Live cells %) | LR (Early apoptotic cells %) | UR (Late apoptotic cells %) | UL (Necrotic cells %) | |
|---|---|---|---|---|
| A549 lung control | 98.99 | 1.00 | 0.01 | 0.00 |
| Original docetaxel | 61.90 | 9.31 | 22.27 | 2.92 |
| CQMU‐0519 | 21.43 | 2.10 | 59.11 | 17.37 |
| SKVO3 ovary control | 99.15 | 0.05 | 0.04 | 0.76 |
| Original docetaxel | 86.40 | 2.71 | 3.09 | 7.80 |
| CQMU‐0519 | 83.31 | 7.9 | 3.43 | 5.36 |
| MCF7 breast control | 99.85 | 0.15 | 0.00 | 0.00 |
| Original docetaxel | 73.59 | 2.03 | 19.92 | 4.46 |
| CQMU‐0519 | 53.53 | 35.57 | 9.48 | 1.42 |
Novel docetaxel analogue (CQMU‐0519) induced DNA fragmentation confirming apoptosis
A549 (Fig. 6a) and SKVO3 cells treated with CQMU‐0519, not only displayed DNA fragmentation represented by smears but also DNA ladder formation, although laddering was faint for SKVO3 cells (Fig. 6b); MCF7 cells did displayed no apoptotic DNA fragmentation nor laddering due to their lack of caspase‐3 (Fig. 6c), correlating with our other results (Fig. 6c).
Figure 6.
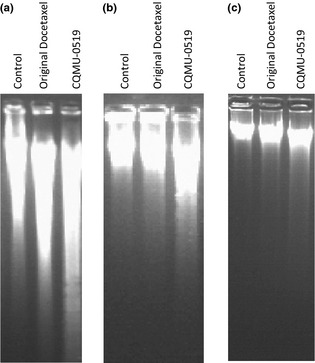
DNA fragmentation assay was performed on all three cell lines. (a) A549, (b) SKVO3 and (c) MCF7 cells. DNA gel electrophoresis of control (lane 1), original docetaxel‐treated (lane 2), novel docetaxel analogue (CQMU‐0519) (lane 3). DNA from all three cell lines was treated with 50 nm respectively for 24 h and was extracted (as described in the Materials and methods section) in 1.2% agarose.
Activation of caspases
In both A549 and SKVO3 cells, novel docetaxel analogue (CQMU‐0519) displayed an increase in effector caspases‐3 and 6, and in initiator caspases‐8, 9 (*P < 0.05) (Fig. 7a,b), suggesting activation of both extrinsic and intrinsic pathways for apoptosis. Nevertheless, it was found that in MCF7 breast cancer cells, there was no activation of caspases‐3 and 9 just an increase in effector caspase‐6 and initiator caspase‐8 (*P < 0.05), indicating that apoptosis in MCF7 breast cancer cells could be effected by the extrinsic pathway only (Fig. 7c), sustaining our findings during DNA fragmentation.
Figure 7.
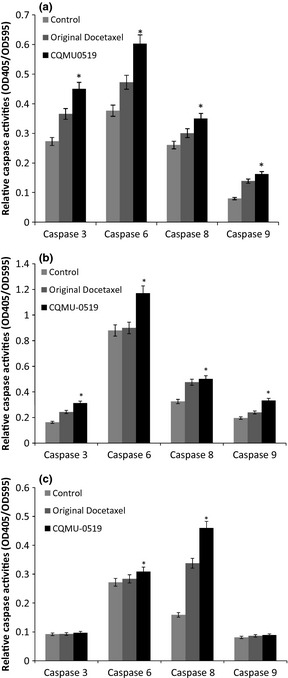
Role of capases in apoptosis. Role of caspases in apoptosis in (a) A549, (b) SKVO3 and (c) MCF7 cells, induced by both original docetaxel and novel docetaxel analogue (CQMU‐0519) – and each cell line had its own control. Colorimetric assessment of activation of caspases‐3, ‐6, ‐8, ‐9 was performed; graphs are mean values (±SD) of three independent experiments, where A549 and SKVO3 cell lines displayed increases of all four caspases (*P < 0.05) but MCF7 cells had activation of caspases 6 and 8 only (*P < 0.05).
Regulation of Bcl‐2 family members and PARP cleavage
Protein expression of Bcl‐2 and Bcl‐xL, being anti‐apoptotic regulators, were reduced in both A549 and SKVO3 cells, by the original docetaxel as well as by the original docetaxel analogue; moreover, pro‐apoptotic protein regulators Bax, tBid (truncated/cleaved) and Bad expressions were elevated compared to control (*P < 0.05) (Fig. 8a–d), indicating that apoptosis was executed through the intrinsic pathway rather than through the extrinsic one, in those two cell lines. Nevertheless, in the MCF7 breast adenocarcinoma cell line, there was no upregulation of Bax, Bid or Bad expression, and there was no change in expression of Bcl‐2 or Bcl‐xL. This illustrates that apoptosis probably occurred using the extrinsic pathway, not through an intrinsic one (Fig. 8e,f); moreover, in all three cell lines, it was clearly seen that PARP (Poly (ADP‐ribose) polymerase)‐116 kDa was cleaved to 85 kDa, a significant event in apoptosis (Fig. 8a,c,e) being related to Bcl‐2 family activity as well as to caspases.
Figure 8.
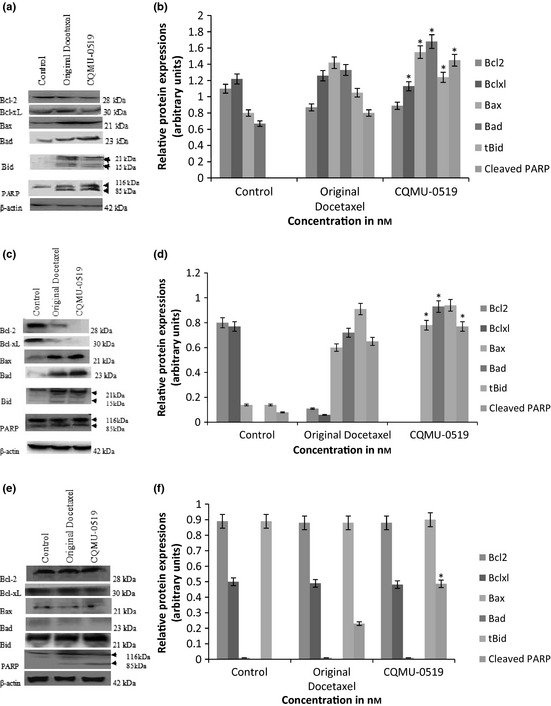
Western blot analysis outlining effects of original docetaxel and novel docetaxel analogue (CQMU‐0519) on Bcl‐2 family of apoptosis regulatory proteins. Densitometric analysis revealed upregulation of Bax, Bad, tBid (15 kDa) with lower expressions of Bcl‐2 and Bcl‐xL in A549 and SKVO3 cells (a–d) (*P < 0.05). However, there was no increase in target levels of these proteins in treated MCF7 cells (e–f). However, PARP (85 kDa) was cleaved in all three cell lines (a–f) (*P < 0.05). β‐actin was used as an internal loading control.
Discussion
Several studies involving different cancer cell lines treated with docetaxel have demonstrated a whole spectrum of cell proliferation inhibition, induction of apoptosis and inhibition of angiogenesis 38. Nevertheless, although docetaxel has yielded some very good outcomes in both experimental and clinical trials 3, 27, 28, 29, 30, its poor solubility, associated with numerous side effects 2, has been a major hurdle in cancer therapy and there are numerous ongoing studies to produce better taxanes in terms of solubility, mechanism of action and fewer side effects, for the overall benefit of patients. Moreover, that apoptosis is the main explored mechanism for killing cancer cells by these mitotic inhibitors, it has been investigated over the past number of decades 39 and is still being thoroughly investigated. It has been found to be closely associated with microtubule depolymerization preventing cell cycle completion and consequently leading to programmed cell death. Furthermore, it has been found that docetaxel and its derivatives disrupt microtubules ultimately leading to DNA damage, affecting the intrinsic pathway of cell death. The mitochondrial components released at that time, triggered the activation of both pro‐ and anti‐apoptotic members of the Bcl‐2 family (22,23) and mitochondrial involvement significant 18 of G2/M arrest. Besides, single docetaxel concentration, either low or high, induces better and more acceptable pro‐apoptotic responses compared to taxol 22, 24, 36, 37, 38, 39, but several studies have demonstrated that docetaxel in combination with other drugs has better response levels and fewer side effects, with prolongation of overall surviving time, hence representing an advantage in the optimal treatment of cancers.
In our study, 50 nm concentration of the novel docetaxel analogue (CQMU‐0519) and an incubation time of 24 h were preferred having yielded better effects on cell population growth inhibition (Fig. 1) and this concentration was considered to be a relatively low and safe one 22, 24 to result in good preclinical outcomes 32, 33, 34, 35. Original docetaxel was purified, and as it is already known to be poorly soluble in water, we used 1% methanol and 99% normal saline as solvent for it; however, the novel docetaxel analogue (CQMU‐0519), synthesized by our college of pharmacy, is completely soluble in water and hence represents an important characteristic for clinical trials in the sense that there may be fewer side effects. Our control was set up to rule out harmful or apoptotic effects of its own, on the A549 lung adenocarcinoma, SKVO3 ovarian and MCF7 breast epithelial cancer cell lines, in comparison to DMSO and Cremophor EL, known to affect cytotoxicity of taxanes. Our main aim was to define anti‐proliferative and apoptotic effects of CQMU‐0519 on these three cancer cell lines; however, it was noted that CQMU‐0519 had better results for inhibition of cell proliferation and cell cycle arrest, compared to its predecessor. A549, SKVO3 and MCF7 cells were treated with 25, 50 and 100 nm of respective drugs for 24 h; it was seen that cell population growth was successfully inhibited by CQMU‐0519 (Fig. 1), where A549 displayed highest percentage of inhibition. This was further confirmed by flow cytometry results, which showed that CQMU‐0519 induced cell cycle arrest at the G2/M phase, accounting for depolymerization of microtubules 3, 6, 7, which was appreciated in all three cancer cell lines (Fig. 2a–c, Table 1). Nevertheless, MCF7 had a hypodiploid sub G0/G1 peak, which indicates onset of DNA fragmentation, which could have occurred earlier, or due to loss of PI staining 39. Moreover, during annexin‐V FITC staining, more cells in all three cell lines had undergone early and late apoptotic phases during the 24 h (Fig. 5a–c, Table 2), where it became higher in A549 and MCF7 than in SKVO3 cells. Morphological apoptotic changes, including chromatin clumping, membrane blebbing and general vacuolation, were mostly seen after CQMU‐0519 (Fig. 3a–c) correlating with the running mean. In both breast and ovarian cancer cell lines, percentage of necrotic cells was not countable; however, A549 cells exhibited 17.37% necrotic cells in response to CQMU‐0519, showing that perhaps apoptosis in these cells may not be the only pathway for cancer cell death (Fig. 5a). DNA cleavage was further assessed by DNA laddering patterns; although DNA smears were evident in A549 and SKVO3 cells treated with CQMU‐0519, DNA ladder patterns were weak 40; this could signify some degree of necrosis or chromatin fragmentation occurring randomly, not at characteristic inter‐nucleosomal sites 39, or that exposure time to the drug affected only a fraction of the cell population 39. Nevertheless, MCF7 cells showed no appreciable DNA smear nor visible ladder (Fig. 6c). This is significant as human MCF7 cells present a good model for investigation, due to their lack of the caspase 3 gene(47 bp within exon3); thus, these cells do not exhibit any DNA fragmentation visible by DNA ladder patterning 41, 42. This was confirmed by caspase‐3 activity assay, which showed no activation of the gene (Fig. 7c); expression of caspase‐9, being an initiator of the caspase intrinsic pathway, was not raised 42. Caspases‐8 and ‐6 expressions were increased (*P < 0.05), showing that apoptosis might have been effected through the extrinsic pathway at this particular concentration, or that there may have been a further cellular mechanism that compensated for the function of caspase‐3 on caspase‐6 activation 43. To confirm this, we tested for the Bcl‐2 family by western blot analysis, where basal levels of Bcl‐2, Bcl‐xL, Bax, tBid and Bad remained unaltered (Fig. 8e–f) 44. Such different findings from our results may be due to differences in variants of MCF7 cells 45; moreover, several studies have shown that there are various differences in apoptotic responses, illustrated by differences in DNA fragmentation and expression of Bcl‐2 family proteins, depending on these variants 42, 45, 46, 47. In addition, in all 3 cancer cell lines, there was cleavage of poly (ADP‐ribose) polymerase (PARP) (Fig. 8a–f), known to be a mediator of cell death initiated by the caspases, although some lines of study have indicated it to be dispensable for apoptosis 48 and has been found to be an early event preceding DNA fragmentation and cell suicide 49. In our experimentals, it could be suggested that caspase‐8 activated caspase‐6, which in turn led to PARP cleavage and apoptosis, hence, supporting our findings that the apoptotic pathway for MCF7 breast adenocarcinoma treated with 50 nm CQMU‐0519 for 24 h, is associated with the extrinsic caspase apoptotic pathway only. On the other hand, A549 and SKVO3 cell lines both showed increase in all 4 caspase activities (caspase‐8 & 9 initiators, caspase‐3 & 6 effectors) at this particular concentration and incubation period, where it could be that apoptosis did not occur solely by the intrinsic pathway, but also occurs extrinsically; this could not be ruled out. Nevertheless, cleavage of full length Bid (21 kDa) into its truncated form (tBid – 15 kDa) illustrated that activation of caspase 8 could have led to this, as it is the bridge between the extrinsic apoptosis pathway, allowing convergence into the intrinsic mechanism. This is where tBid is known to have a close relationship with Bax intra‐mitochondrially 50, 51, contributing to the mitochondrial pathway of programmed cell death. In some studies 26, it has been shown that docetaxel can induce apoptosis without activation of caspase 8 and Bid. However, our further western blotting results clearly showed that Bcl‐2 and Bcl‐xL were downregulated in the cell lines, with increase in expressions of Bax and Bad (Fig. 8a–d). These two are pro‐apoptotic regulatory proteins and it is crucial to test for them in particular, as they reflect mitochondrial integrity; any loss of such leads to release of apoptosis‐inducing inter‐membrane proteins into the cytosol, activating caspases‐2 and 9 41. The latter in turn initiates the caspase cascade, involving downstream executors caspases‐3 and 6, cleaving PARP into its 85 kDa active fragment. This consequently leads to apoptosis and formation of apoptosomes. Therefore, in both our lung and ovarian cell lines, apoptosis is brought about by the intrinsic pathway not the extrinsic one.
In conclusion, our novel docetaxel analogue (CQMU‐0519) has, as anticipated, anti‐apoptotic effects on all three of our cancer cell lines, although the mechanism by which it takes place maybe variable 52. In addition, it was seen that at 50 nm concentration for 24 h, apoptotic responses and pathways were the same for both original docetaxel and novel docetaxel analogue (CQMU‐0519). Our results have forecast that under these optimal conditions, CQMU‐0519 may evince superior effects in A549 lung, SKVO3 ovarian, MCF7 breast cancer cells, where there has been inhibition of cell proliferation with cell cycle delay at G2/M phases along with induction of apoptosis. This also may be also associated with some levels of necrosis in lung adenocarcinoma that also results in cell death. Further studies, however, are needed to confirm this statement. It was also seen that in A549 and SKVO3 cells, there was activation of the caspase intrinsic pathway towards apoptosis, while MCF7 cells displayed the extrinsic pathway. The novel docetaxel analogue (CQMU‐0519) was revealed to elicit apoptotic responses, however, further thorough experimental trials are needed before it could be introduced into clinical trials.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the Major State Science and Technology Special Project of Chongqing Innovative Medicine Incubation Base; Platform for New Drug Design and Screening (2010ZX9401‐306‐1‐1), and by the National Nature Science Foundation of China (NSFC:30870946).
We would also like to thank Mo Na, Yi Yang, Gie Qong Wang and Yong Tao Sheng for their valuable help during this experiment.
References
- 1. Bissery MC, Guénard D, Guéritte‐Voegelein F, Lavelle F (1991) Experimental antitumor activity of taxotere (RP56976, NSC 628503), a taxol analogue. Cancer Res. 51, 4845–4852. [PubMed] [Google Scholar]
- 2. Hennenfent KL, Govindan R (2006) Novel formulations of taxanes: a review. Old wine in a new bottle? Ann. Oncol. 17, 735–749. [DOI] [PubMed] [Google Scholar]
- 3. Calderoni A, Cerny T (2001) Taxanes in lung cancer: a review with focus on the European experience. Crit. Rev. Oncol. Hematol. 38, 105–127. [DOI] [PubMed] [Google Scholar]
- 4. Rao S, He L, Chakravarty S, Ojima I, Orr GA, Horwitz SB (1999) Characterization of the Taxol binding site on the microtubule. Identification of Arg282 in β‐tubulin as the site of photoincorporation of a 7‐benzophenone analogue of taxol. J. Biol. Chem. 274, 37990–37994. [DOI] [PubMed] [Google Scholar]
- 5. Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E (2001) The binding conformation of Taxol in b‐tubulin: a model based on electron crystallographic density. Proc. Natl. Acad. Sci. USA 98, 5312–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yvon AMC, Wadsworth P, Jordan MA (1999) Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol. Biol. Cell 10, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jordan MA, Wilson L (1998) Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr. Opin. Cell Biol. 10, 123–130. [DOI] [PubMed] [Google Scholar]
- 8. Derry WB, Wilson L, Jordan MA (1995) Substoichiometric binding of taxol suppresses microtubule dynamics. Biochemistry 34, 2203–2211. [DOI] [PubMed] [Google Scholar]
- 9. Schiff PB, Fant J, Horwitz SB (1979) Promotion of microtubule assembly in vitro by taxol. Nature 277, 665–667. [DOI] [PubMed] [Google Scholar]
- 10. Schiff PB, Horwitz SB (1980) Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 77, 1561–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jordan MA, Toso RJ, Thrower D, Wilson L (1993) Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 90, 9552–9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L (1996) Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 56, 816–825. [PubMed] [Google Scholar]
- 13. Geng CX, Zeng ZC, Wang JY (2003) Docetaxel inhibits SMMC‐7721 human hepatocellular carcinoma cells growth and induces apoptosis. World J. Gastroenterol. 9, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mhaidat NM, Zhang XD, ChenJiang C, Hersey P (2007) Docetaxel‐induced apoptosis of human melanoma is mediated by activation of c‐Jun NH2‐terminal kinase and inhibited by the mitogen‐activated protein kinase extracellular signal‐regulated kinase 1/2 pathway. Clin. Cancer Res. 13, 1308–1314. [DOI] [PubMed] [Google Scholar]
- 15. Kang J, Wu X, Wang Z, Ran H, Xu C, Wu J et al (2010) Antitumor effect of docetaxel‐loaded lipid microbubbles combined with ultrasound‐targeted microbubble activation on VX2 rabbit liver tumors. J. Ultrasound Med. 29, 61–70. [DOI] [PubMed] [Google Scholar]
- 16. Ichite N, Chougule MB, Jackson T, Fulzele SV, Safe S, Singh M (2009) Enhancement of docetaxel anticancer activity by a novel diindolylmethane compound in human non‐small cell lung cancer. Clin. Cancer Res. 15, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahaffey CM, Davies AM, Lara PN Jr, Pryde B, Holland W, Mack PC et al (2007) Schedule dependent apoptosis in K‐ras mutant non‐small‐cell lung cancer cell lines treated with docetaxel and erlotinib: rationale for pharmacodynamic separation. Clin. Lung Cancer 8, 548–553. [DOI] [PubMed] [Google Scholar]
- 18. Nehmé A, Varadarajan P, Sellakumar G, Gerhold M, Niedner H, Zhang Q et al (2001) Modulation of docetaxel‐induced apoptosis and cell cycle arrest by all‐trans retinoic acid in prostate cancer cells. Br. J. Cancer 84, 1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang QW, Lu HL, Song CC, Liu H, Xu CG (2005) Radiosensitivity of human colon cancer cell enhanced by immunoliposomal docetaxel. World J. Gastroenterol. 11, 4003–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferlini C, Di Stefano M, Marone M, Gaggini C, Ferrandina G, Riva A et al (1998) The synergistic anti‐tumour activity of ICI 182,780 in combination with docetaxel is mediated by P‐glycoprotein inhibition. Endocr. Relat. Cancer 5, 315–324. [Google Scholar]
- 21. Choe MS, Chen Z, Klass CM, Zhang X, Shin DM (2007) Enhancement of docetaxel‐induced cytotoxicity by blocking epidermal growth factor receptor and cyclooxygenase‐2 pathways in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 13, 3015–3023. [DOI] [PubMed] [Google Scholar]
- 22. Wang Z, Goulet R III, Stanton KJ (2005) Differential effect of anti‐apoptotic genes Bcl‐xL and c‐FLIP on sensitivity of MCF‐7 breast cancer cells to paclitaxel and docetaxel. Anticancer Res. 25, 2367–2380. [PubMed] [Google Scholar]
- 23. Hwang JJ, Kim YS, Kim MJ (2010) Histone deacetylase inhibitor potentiates anticancer effect of docetaxel via modulation of Bcl‐2 family proteins and tubulin in hormone refractory prostate cancer cells. J. Urol. 184, 2557–2564. [DOI] [PubMed] [Google Scholar]
- 24. Chang JT, Chang GC, Ko JL, Liao HY, Liu HJ, Chen CC et al (2006) Induction of tubulin by docetaxel is associated with p53 status in human non small cell lung cancer cell lines. Int. J. Cancer 118, 317–325. [DOI] [PubMed] [Google Scholar]
- 25. Mediavilla‐Varela M, Pacheco FJ, Almaguel F, Perez J, Sahakian E, Daniels TR et al (2009) Docetaxel‐induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer. doi: 10.1186/1476-4598-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mhaidat NM, Wang Y, Kiejda KA, Zhang XD, Hersey P (2007) Docetaxel induced apoptosis in melanoma cells is dependent on activation of caspase‐2. Mol. Cancer Ther. 6, 752–761. [DOI] [PubMed] [Google Scholar]
- 27. Katopodis O, Polyzos A, Kentepozidis N, Giassas S, Rovithi M, Bozionelou V et al (2010) Second line chemotherapy with capecitabine (Xeloda) and docetaxel (Taxotere) in previously treated, unresectable adenocarcinoma of pancreas: the final results of a phase II trial. Cancer Chemother. Pharmacol. doi: 10.1007/s00280-010-1329-6. [DOI] [PubMed] [Google Scholar]
- 28. Mäenpaä JU (2003) Docetaxel: promising and novel combinations in ovarian cancer. Br. J. Cancer 89, S29–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wenzel C, Steger GG (2006) Adjuvant treatment of breast cancer with Taxanes. Breast Care 1, 171–175. [Google Scholar]
- 30. Pectasides D, Pectasides M, Farmakis D, Kostopoulou V, Nikolaou M, Gaglia A et al (2005) Comparison of docetaxel and docetaxel–irinotecan combination as second‐line chemotherapy in advanced non‐small‐cell lung cancer: a randomized phase II trial. Ann. Oncol. 16, 294–299. [DOI] [PubMed] [Google Scholar]
- 31. Nabell L, Spencer S (2003) Docetaxel with concurrent radiotherapy in head and neck cancer. Semin. Oncol. 30, 89–93. [DOI] [PubMed] [Google Scholar]
- 32. Huang TSW, Northrup S (2004) The cytotoxicity of combined paricalcitol and docetaxel in head and neck squamous cell carcinomas. J. Clin. Oncol. (Meeting Abstracts) 22, 14S‐5609. [Google Scholar]
- 33. O'Neill AJ, Prencipe M, Dowling C, Fan Y, Mulrane L, Gallagher WM et al (2011) Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer. 10, 126. doi: 10.1186/1476-4598-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clarke SJ, Rivory LP (1999) Clinical pharmacokinetics of docetaxel. Clin. Pharmacokinet. 36, 99–114. [DOI] [PubMed] [Google Scholar]
- 35. Gridelli C, Rossi A, Venturino P, De Marinis F (2011) Treatment, Rationale, and Study Design of TALISMAN Study: a randomized phase II open‐label study of second‐line erlotinib versus intermittent erlotinib dosing with docetaxel in the treatment of former‐smoker men affected by recurrent squamous non–small‐cell lung cancer. Clin. Lung Cancer 12, 70–73. [DOI] [PubMed] [Google Scholar]
- 36. Langlois NEI, Eremin O, Heys SD (2000) Apoptosis and prognosis in cancer: rationale and relevance. J. R. Coll. Surg. Edinb. 45, 211–219. [PubMed] [Google Scholar]
- 37. Biscotti CV, Hart WR (1998) Apoptotic bodies: a consistent morphologic feature of endocervical adenocarcinoma in situ. Am. J. Surg. Pathol. 22, 434–439. [DOI] [PubMed] [Google Scholar]
- 38. Grant DS, Williams TL, Zahaczewsky M, Dicker AP (2003) Comparison of antiangiogenic activities using Paclitaxel (TAXOL) and Docetaxel (TAXOTERE). Int. J. Cancer 104, 121–129. [DOI] [PubMed] [Google Scholar]
- 39. Fabbri F, Carloni S, Brigliadori G, Zoli W, Lapalombella R, Marini M (2006) Sequential events of apoptosis involving docetaxel, a microtubule‐interfering agent: a cytometric study. BMC Cell Biol. doi: 10.1186/1471-2121/7/6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsushima T, Nakashima M, Oshima K, Abe Y, Nishimura J, Nawata H et al (2001) Receptor binding cancer antigen expressed on SiSo cells, a novel regulator of apoptosis of erythroid progenitor cells. Blood 98, 313–321. [DOI] [PubMed] [Google Scholar]
- 41. Ofir R, Sedman R, Rabinski T, Krup M, Yavelsky V, Weinstein Y et al (2002) Taxol‐induced apoptosis in human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase‐3 and caspase‐9 independent. Cell Death Differ. 9, 636–642. [DOI] [PubMed] [Google Scholar]
- 42. Blanc C, Deveraux QL, Krajewski S, Jänicke RU, Porter AG, Reed JC et al (2000) Caspase 3 is essential for procaspase 9 processing and cisplatin induced apoptosis of MCF‐7 breast cancer cells. Cancer Res. 60, 4386–4390. [PubMed] [Google Scholar]
- 43. Kagawa S, Gu J, Honda T, McDonnell TJ, Swisher SG, Roth JA et al (2001) Deficiency of caspase‐3 in MCF7 cells blocks bax‐mediated nuclear fragmentation but not cell death. Clin. Cancer Res. 7, 1474–1480. [PubMed] [Google Scholar]
- 44. Muhamad S, Pihie AHL, Latif J, Rha C‐K, Sambandan TG (2011) Induction of apoptosis in MCF‐7 via the caspase pathway by longilactone from Eurycoma longifolia Jack. Res. Pharm. Biotechnol. 3, 1–10. [Google Scholar]
- 45. Mooney LM, Al‐Sakkaf KA, Brown BL, Dobson PR (2002) Apoptotic mechanisms in T47D and MCF‐7 human breast cancer cells. Br. J. Cancer 87, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gooch JL, Yee D (1999) Strain specific differences in formation of apoptotic DNA ladders in MCF‐7 breast cancer cells. Cancer Lett. 144, 31–37. [DOI] [PubMed] [Google Scholar]
- 47. Özlem DI, Meltem DK, Ufuk G (2011) Two different docetaxel resistant MCF‐7 sublines exhibited different gene expression pattern. Mol. Biol. Rep. doi: 10.1007/s11033-011-1123-5. [DOI] [PubMed] [Google Scholar]
- 48. Fauzee NJS, Pan J, Wang YL (2010) PARP and PARG inhibitors‐new therapeutic targets in cancer treatment. Pathol. Oncol. Res. 16, 469–478. [DOI] [PubMed] [Google Scholar]
- 49. Perry DK, Smyth MJ, Wang HG (2002) Bcl2 acts upstream of the PARP protease and prevents its activation. Cell Death Differ. 4, 29–33. [DOI] [PubMed] [Google Scholar]
- 50. Fukazawa T, Maeda Y, Matsuoka J, Tanaka N, Tanaka H, Durbin ML et al (2009) Drug‐regulatable cancer cell death induced by BID under control of the tissue‐specific, lung cancer‐targeted TTS promoter system. Int. J. Cancer 125, 1975–1984. [DOI] [PubMed] [Google Scholar]
- 51. Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501. [DOI] [PubMed] [Google Scholar]
- 52. Fauzee NJS, Dong Z, Wang YL (2011) Taxanes: promising anti‐cancer drugs. Asian Pasific J. Cancer Prev. 12, 837–851. [PubMed] [Google Scholar]