

SUMMARY
Aims: Lacosamide (LCM; SPM 927, Vimpat®) is an antiepileptic drug (AED) used as adjunctive treatment for adults with partial‐onset seizures. LCM has a different mode of action from traditional sodium channel blocking AEDs in that it selectively enhances slow inactivation of sodium channels without affecting fast inactivation. Initial investigations suggested that LCM might have an additional mode of action by binding to the collapsin response mediator protein 2 (CRMP‐2), which is further investigated here. Methods: LCM binding to native and cloned human CRMP‐2 was determined using radioligand binding experiments and surface plasmon resonance measurements. Results: No specific binding of [3H]LCM (free concentration 100–1450 nM) to isolated or membrane bound human CRMP‐2 expressed in mammalian cell systems and bacteria was observed. Surface plasmon resonance analysis also showed that LCM, over a concentration range of 0.39–100 μM, does not specifically bind to human CRMP‐2. Conclusion: The diverse drug binding methods employed here are well suited to detect specific binding of LCM to CRMP‐2 in the micromolar range, yet the results obtained were all negative. Results of this study suggest that LCM does not specifically bind to CRMP‐2.
Keywords: CRMP‐2, Epilepsy, lacosamide, mechanism of action
Introduction
Lacosamide (LCM, SP927, Vimpat®) is an antiepileptic drug (AED) marketed as adjunctive therapy for patients with partial‐onset seizures who are 17 years or older [1, 2]. The precise mechanism by which LCM exerts its antiepileptic effect in humans remains to be fully elucidated; however, preclinical electrophysiological studies have demonstrated that LCM targets voltage‐gated sodium channels and acts by specifically enhancing slow inactivation without affecting fast inactivation of the channel [3, 4].
It has been proposed that LCM may also bind to collapsin response mediator protein 2 (CRMP‐2), an axonal growth and guidance protein [5, 6, 7]. The collapsin response mediator proteins (CRMP‐1–5) are involved in many cellular and molecular events such as apoptosis, proliferation, cell migration and differentiation [8]. CRMP proteins are intracellular phosphoproteins implicated in the mediation of neurotrophic signaling (e.g., via semaphorin 3A) and neuronal outgrowth [9]. Highest levels of CRMP isoforms are expressed during development and CRMP‐2 remains the most abundantly expressed in adult brain, specifically in regions controlling axonal outgrowth and synaptic rearrangement [10]. Disruptions in normal neurotrophic signaling can lead to axonal defects, including inappropriate neuronal rearrangements and abnormal sprouting and/or demyelination (events also associated with epilepsy), providing a plausible argument for CRMP‐2 as a therapeutic target in epilepsy [5, 11]. Consistent with this, downregulation of CRMP‐2 expression in patients with temporal lobe epilepsy has been observed [12].
A distinct “CRMP‐2 mechanism” for LCM in addition to its “sodium channel mechanism” had originally been proposed based on preliminary proteomic approaches using LCM “fishhook” experiments and [14C]LCM binding to CRMP‐2 expressed in Xenopus laevis oocytes membranes, where an apparent dissociation constant of ∼5 μM was reported [5]. Subsequent studies using LCM affinity baits and in silico docking to CRMP‐2, suggested a direct binding of LCM to CRMP‐2. The investigators proposed, without providing direct experimental evidence, that the binding of LCM to CRMP‐2 may mediate the effects of LCM on voltage‐gated sodium channel slow inactivation [6, 7]. However, in our attempt to further clarify the binding properties of LCM to CRMP‐2 using various biochemical and biophysical approaches, we could not confirm the results of the initial binding studies. Therefore, our findings are inconsistent with a specific binding site for LCM on CRMP‐2.
This report provides the results of multiple binding experiments and also includes a critical evaluation of the previously reported data that suggested a direct interaction between these two molecules.
Methods
Reagents
LCM (formerly harkoseride), the R‐enantiomer of 2‐acetamido‐N‐benzyl‐3‐methoxypropionamide, was synthesized at UCB Pharma (Monheim, Germany). [3H]LCM (31 Ci/mmol) was custom synthesized by RC Tritec AG (Teufen, Switzerland). Stock solution was in ethanol, at a concentration of 1 mCi/mL, and was stored at −20°C.
LCM was dissolved in 100% DMSO to give a 1–10 mM stock solution. Final DMSO concentration in binding assays did not exceed 1%. Two types of scintillation proximity assay (SPA) beads were purchased from GE Healthcare (Diegem, Belgium): glutathione yttrium silicate (YSi) beads (RPNQ0033) and His tag copper polyvinyltoluene (PVT) beads (RPNQ0096). DMEM‐F12 with Glutamax, Dulbecco's phosphate buffered saline (PBS) and pencillin‐streptomycin (pen‐strep) were purchased from Lonza (Verviers, Belgium), geneticin from Invitrogen (Merelbeke, Belgium), FetalClone II from Perbio (Erembodegem, Belgium), and DNAse from Sigma (Bornem, Belgium). Reagents and reference compounds were of analytical grade and obtained from various commercial sources.
CRMP‐2 Expression
Expression and transfection into COS‐7 cells: CRMP‐2 sequences were chemically synthesized by GenScript based on the NCBI number NM001386.4 and cloned in pUC57. All constructs were subcloned in pcDNA3.1 (Invitrogen, Merelbeke, Belgium). COS‐7 cells were cultured in DMEM medium with 10% fetal calf serum, pen‐strep (100 U/mL). All transfections were performed with FUGENE 6 (Roche, Vilvoorde, Belgium) with a DNA ratio 3:1. COS‐7 cells were grown in DMEM F‐12 with Glutamax culture media containing 10% fetal clone II serum, 2% pen‐strep (5000 U/mL) and 0.8% geneticin, at 37°C in a humidified atmosphere of 5% CO2.
Expression of CRMP‐2 in Escherichia Coli: the coding sequence for human CRMP‐2 was subcloned into the expression vectors pEt21b (Novagen, Belgium) and pGEX‐6P1 (Novagen, Belgium), resulting in one construct with a C‐terminal (Ct) hexahistidine His tag and another with an N‐terminal (Nt) glutathione GST tag, respectively. Bacterial pellets were solubilized and CRMP‐2 protein purified by affinity using nickel‐nitrilotriacetic acid resin (Qiagen, Venlo, Netherlands) or Glutathione Sepharose 4B (GE Healthcare, Diegem, Belgium).
Expression of CRMP‐2 in Xenopus oocytes: human mRNA for CRMP2 (20–25 ng; RD‐Biotech, France) was injected into dissected and defolliculated Xenopus oocytes (stage V–VI; purchased from EcoCyte Bioscience, Germany) with an automated micro‐injector (Roboocyte, Multi Channel Systems, Reutlingen, Germany). Protein expression was allowed for 72 hours at 17°C in Barth's solution (pH 7.4) containing (in mM): NaCl (88), KCl (1), NaHCO3 (2.4), Ca (NO3)2 (0.33), CaCl2 (0.41), MgSO4 (0.82), Tris‐HCl (5) and supplemented with pen‐strep (100 U/mL).
Radioligand Binding
Male Oncins France Strain A (OFA) Sprague‐Dawley rats (200–300 g) were obtained from IFFA CREDO (Brussels, Belgium). Brain membrane fractions and homogenates were prepared as previously described [13]. In brief, brain tissues were dissected and homogenized (10% w/v) in 20 mM Tris–HCl buffer (pH 7.4) containing 250 mM sucrose (buffer A). The homogenates were centrifuged at 30,000 g at 4°C for 15 min and the pellets resuspended in the same buffer. After incubation at 37°C for 15 min, the membranes were washed three times using the same centrifugation protocol. The final pellets were resuspended in buffer A at a protein concentration of 10–15 mg/mL and stored in liquid nitrogen. [3H]LCM binding was determined in several brain areas, including the cerebral cortex, striatum, hippocampus, and cerebellum. Membranes and homogenates were incubated for 60 min at 4°C (to reduce the dissociation rate of the ligand) or 25°C in a 50 mM Tris‐HCl buffer (pH 7.4) containing [3H]LCM. At the end of the incubation period, the membrane‐bound radioligand was recovered by rapid filtration through GF/C glass fiber filters presoaked in 0.1% polyethyleneimine. The membranes were washed with 4×2 mL of ice‐cold 50 mM Tris–HCl buffer (pH 7.4). The total filtration procedure did not exceed 10 seconds per sample. Filters were dried and radioactivity determined by liquid scintillation.
SPA (PerkinElmer, Zaventem, Belgium) rely on the use of receptors (membranes or proteins) coupled to PVT or YSi beads that encapsulate a scintillation liquid. The binding of a radioligand at the bead surface can be directly measured by luminescence emitted by the scintillation liquid. Cytosolic or membrane fractions of COS‐7 cells expressing 6×His tagged CRMP‐2 were incubated for 60 min at room temperature under gentle agitation with PVT beads (ratio 1 mg total protein/mg beads), which were then centrifuged at 1000 × g for 10 min, and recovered in binding buffer (50 mM Tris‐HCl pH 7.4, 2 mM MgCl2). Subsequently, 500 μg of beads were incubated at room temperature in a final volume of 25 μL containing 1.2 μM [3H]LCM. After a 60‐min incubation period, radioactivity measurements were performed using a Microbeta Jet (PerkinElmer, Zaventem, Belgium) instrument. The normality of experimental data was verified by a Shapiro–Wilk test, and statistical significance was calculated using a t‐test.
Western Blot
Cellular fractions were solubilized in LDS sample buffer (Nupage, Invitrogen) and separated (20 μg/lane) on a Novex 4–12% Bis‐Tris gel (Nupage, Invitrogen). The proteins were identified in the presence of a rabbit polyclonal anti‐CRMP2 antibody (AB9218, Chemicon; dil 1:1000) and a secondary anti‐rabbit IgG monoclonal peroxidase antibody (A1949‐1VL, Sigma; dil 1:5000). The protein bands were revealed with an ECL Plus Western Blotting detection system (RPN2132, GE Healthcare) using a digital camera (G:Box, Syngene).
Surface Plasmon Resonance
All experiments were performed on a Biacore® T100 instrument using the CM5 series S‐sensors (GE Healthcare Ltd., Bucks, UK). The assay format consisted of titrating LCM over the immobilized human CRMP‐2 and human serum albumin (HSA) surfaces. His‐tagged (Ct) CRMP‐2 and HSA were immobilized on a CM5 series S‐sensor via amine coupling chemistry in 10 mM sodium acetate pH 4.5. CRMP‐2 was immobilized to a level of ≈18700 binding response units (RU) and HSA immobilized onto a separate flow cell to a level of ≈10800 binding response units. A blank surface, which allows determination of the nonspecific binding (NSB) to the chip surface, was prepared by repeating the procedure but omitting protein. HBS‐P+ buffer (10 mM HEPES, 150 mM NaCl, P20 0.05%, pH 7.4) was used as the running buffer at a flow rate of 10 μL/min. LCM (0.39–100 μM) was titrated over the sensor at a flow rate of 30 μL/min. The surface was re‐equilibrated with buffer for 6 min before the next cycle.
The titration was repeated twice more, reversing the order of titration between runs, to negate any bias in the assay order. Background subtracted binding curves, with double referencing, were analyzed using the evaluation software (Biacore, version 2.0.1, GE Healthcare Ltd, Bucks, UK) following standard procedures.
Results
In this study, [3H]LCM (31 Ci/mmol) was used to characterize the binding of LCM to native or cloned CRMP‐2. [3H]LCM was used at concentrations 300 nM throughout the studies in order to ensure the identification of binding sites in the low micromolar range. Specific binding was defined as the difference between [3H]LCM total binding and non specific binding (NSB) in the presence of unlabelled LCM.
[3H]LCM Binding to Rat Brain Membranes
CRMP‐2 was shown to be expressed in adult rat brain structures, and experiments on rat brain tissues should thus provide information on the binding characteristics of LCM to native CRMP‐2. Figure 1 shows the results of total and NSB of [3H]LCM to rat brain membranes. No significant specific binding was observed. Binding of [3H]LCM (30nM) to crude rat cerebral cortex homogenates (preparation retaining the cytosolic CRMP‐2) was also assessed and again no significant specific binding was observed (Total binding: 680 ± 61dpm/assay, NSB: 642 ± 41dpm/assay, P > 0.05).
Figure 1.
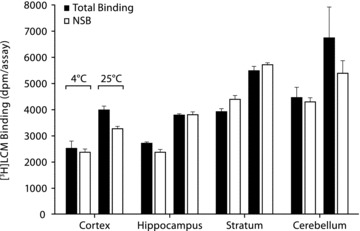
[3H]LCM shows no specific binding to rat brain membranes. Binding of [3H]LCM (300 nM) to rat brain membranes (200 μg/assay) at 4°C and 25°C was determined by manual filtration in a final volume of 50 μL using a 50‐mM Tris‐HCl buffer (pH 7.4). NSB was determined in the presence of 1 mM unlabeled LCM. Data are presented as mean ± SEM (n = 3).
[3H]LCM Binding to CRMP‐2 Expressed in Oocyte Membranes
Previous investigations indicated that [14C]LCM binds to oocyte membranes expressing CRMP‐2 [5]. The same approach was used to further characterize the binding properties of LCM using [3H]LCM (1000‐fold higher specific activity than [14C]LCM) and mRNA injection to maximize the expression levels of CRMP‐2. In this system, CRMP‐2 is efficiently expressed in the soluble and membrane fractions of oocytes (Figure 2, insert), and binding experiments were subsequently performed on CRMP‐2 associated membrane fractions (Figure 2). No significant difference was observed between total and NSB levels in control and CRMP‐2 injected oocytes. Binding experiments have also been performed in the presence of different buffer compositions (e.g., HEPES 15 mM, KCl 135 mM, CaCl2 1 mM, NaCl 4 mM, MgCl2 2 mM, pH 7.2) to mimic the intracellular ion composition (Total binding: 2484 ± 920 dpm/assay, NSB: 3499 ± 847 dpm/assay, P > 0.05) and also such experimental conditions did not reveal a specific interaction between LCM and CRMP‐2.
Figure 2.
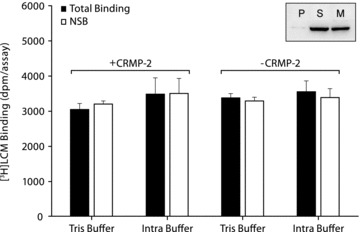
[3H]LCM does not specifically bind to CRMP‐2 expressed in oocyte membrane fractions. Binding of [3H]LCM (969 nM) to oocyte membrane fractions (100 μg/assay) expressing human CRMP‐2. Total binding (gray bars) and NSB (open bars) were determined on membrane fractions from CRMP‐2 and control (–CRMP‐2) injected oocytes. Binding experiments were performed in a final volume of 30μL in either Tris buffer (Tris 10 mM, NaCl 150 mM, pH 7.4) or a buffer composition mimicking the intracellular environment (HEPES 15 mM, KCl 135 mM, CaCl2 1 mM, NaCl 4 mM, MgCl2 2 mM: pH 7.2). NSB was determined in the presence of 1mM unlabeled LCM. Data are presented as mean ± SEM (n = 3–5). Insert: Western blot analysis of CRMP‐2 expression in oocyte fractions: pellet (P), supernatant (S) and membranes (M) (20 μg/sample).
[3H]LCM Binding to CRMP‐2 Expressed in Mammalian Cell Systems
CRMP‐2 is a cytosolic phosphoprotein, thus raising the possibility that soluble CRMP‐2 could be a preferred binding partner for LCM. Therefore, a binding assay was designed where tagged CRMP‐2 was expressed in COS‐7 cells and subsequently coupled to SPA beads for convenient use in a homogeneous binding assay. The SPA presents the advantage that binding of a radiolabeled ligand can be detected without using a separation step of the unbound ligand. The choice of the COS‐7 cell system was based on previous studies showing that CRMP‐2 expression in these cells is able to mediate semaphorin 3A‐induced cell contraction, thus demonstrating that the protein can be efficiently expressed and processed in such a cellular system [14]. Both the N‐ and C‐terminal tagged proteins (either His or GST) were used in case one of the two positions would be deleterious to the affinity of [3H]LCM. The tagged isoforms of CRMP‐2 were expressed for 48 h in COS‐7 cells and the presence of the protein was detected by Western Blot in the cytosolic and the membrane fractions (Figure 3A). CRMP‐2 was subsequently coupled to SPA beads and incubated in the presence of [3H]LCM. Both total binding and NSB of [3H]LCM, measured for each CRMP‐2 construct isolated from COS‐7 cytosolic or membrane fractions, are shown in Figure 3B. No differences between total and NSB in any of the tested samples were observed, indicating that [3H]LCM does not specifically bind to CRMP‐2 expressed in COS‐7 cells. Additional binding experiments performed on COS‐7 cell membrane fractions, using the manual filtration method, also showed no binding of [3H]LCM (600 nM) to CRMP‐2 (Total binding: 3453 ± 195 dpm/assay, NSB: 3583 ± 381 dpm/assay, P > 0.05).
Figure 3.
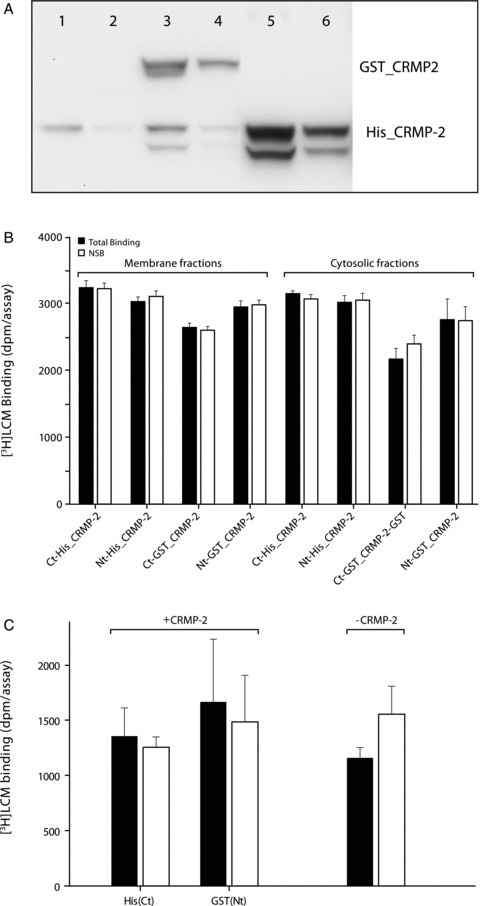
(A) Western blot analysis of CRMP‐2 expression in COS cells: Analysis of soluble and membrane fractions, respectively (20 μg/lane), of untransfected cells (1,2), GST‐tagged CRMP‐2 (3,4) and His‐tagged CRMP‐2 (5,6). (B) Binding of [3H]LCM to His‐ or GST‐tagged CRMP‐2 proteins using a scintillation proximity assay: CRMP‐2 proteins expressed in cytosolic or membrane fractions of COS‐7 cells were coupled to Copper‐PVT beads and YSi beads, respectively and incubated for 60 min (25°C) in the presence of 1200 nM [3H]LCM. Radioactivity coupled to the beads was measured by luminescence. Data represent total binding (gray bars) and NSB (open bars) in the presence of 1 mM unlabeled LCM (mean ± SD; n = 4). Ct‐His_CRMP‐2 and Nt‐GST_CRMP‐2 are the C‐ and N‐terminal tagged versions, respectively. (C) [3H]LCM does not bind to purified CRMP‐2: His(Ct) and GST(Nt) CRMP‐2 were expressed in E. Coli and coupled to SPA beads. Binding experiments were performed at 25°C during 1 h in the presence of [3H]LCM (969 nM) and in a final volume of 30 μL in Tris buffer (Tris 10 mM, NaCl 150 mM: pH 7.4). NSB was determined in the presence of 1 mM unlabeled LCM. Data are presented as mean ± SEM (n = 10). Total binding (gray bars), NSB (open bars).
[3H]LCM Binding to Purified CRMP‐2
Because the determination of specific binding of [3H]LCM to CRMP‐2 may be limited by the amount of protein present in the previous assays, purified CRMP‐2 was isolated to maximize the number of potential binding sites and to reduce the signal‐to‐noise ratio in the next experiments. Human CRMP‐2 was expressed and purified with a Ct‐His and an Nt‐GST tag in E. Coli as described in the Methods Section. The same approach as in the COS‐7 cell experiments was adapted by coupling the purified tagged protein to affinity beads and performing a manual filtration binding assay in the presence of [3H]LCM. The amount of protein coupled to the beads was estimated to be at least 100 ng/250 μg of beads (based on protein band intensity on Western Blot compared with known quantities of CRMP‐2, data not shown). No difference in specific binding levels of [3H]LCM to the CRMP‐2‐coupled and control beads was observed (Figure 3C), suggesting that LCM does not bind to a specific site on CRMP‐2. The same assay was repeated using different buffer conditions (Total binding: 6020 ± 9878 dpm/assay, NSB: 786 ± 305 dpm/assay, P > 0.05) and incubation at 37°C (Total binding: 398 ± 59 dpm/assay, NSB: 441 ± 381 dpm/assay, P > 0.05), but results also suggested the absence of a specific binding site.
LCM Binding to CRMP‐2 by Surface Plasmon Resonance
Surface plasmon resonance (Biacore technology) monitors the binding between biomolecules by refractive index change, which is proportional to mass change, in real time and without the requirement for labeling [15]. CRMP‐2 and HSA, as a nonspecific binding control, were immobilized on separate surfaces of a sensor. LCM in solution was passed over the proteins, and any concentration‐dependent complex formation was monitored as the association phase. Potential dissociation of LCM from the protein was measured by replacing the drug solution with buffer under flow conditions.
The binding of unlabeled LCM in a concentration range of 0.39–100 μM to CRMP‐2 and HSA was evaluated. Based on the protein immobilization levels, and taking into account the difference in the molecular sizes of the interacting partners (1:1 stoichiometry), the theoretical maximal response (Rmax) of LCM binding to CRMP‐2 or HSA may be calculated as 75 and 40.5 response units (RU), respectively. Figure 4 shows the binding levels, expressed in RU, of LCM to CRMP‐2 or HSA recorded from report points measured 5 seconds before the end of LCM injection. The binding levels of LCM to CRMP‐2 or HSA, up to concentrations of 100μM, remain very small and represent only ∼6% of the predicted Rmax value indicating that LCM shows minimal NSB to CRMP‐2 and HSA proteins.
Figure 4.
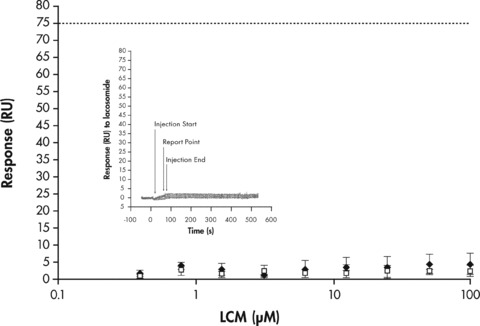
Analysis of LCM binding to immobilized CRMP‐2 or HSA using Biacore. Surface plasmon resonance report point data (n = 3, ±SD) for the binding of LCM (0.39–100μM) to immobilized CRMP‐2 (black diamonds) or HSA (open squares). CRMP‐2 and HSA were immobilized at binding response levels of ∼18700 RU and ∼10800 RU, respectively. LCM was titrated over the immobilized protein surfaces. Report points (n = 3, ± SD) for the binding of LCM or HSA to CRMP‐2 were recorded 5 seconds before the end of the injection (see insert). The predicted Rmax binding levels of LCM to CRMP‐2 and HSA based on 1:1 stoichiometry (horizontal dotted lines) are 75 RU and 40.5 RU, respectively. Insert: SPR overlaid sensorgrams depicting the interaction of LCM (0.39–100 μM) to the immobilized human CRMP‐2.
Discussion
The use of radioligand binding experiments is a very useful approach to characterize the binding site and pharmacological interactions of drugs with their target receptors [16]. In this study, a number of different biochemical and biophysical approaches were used to characterize the binding of LCM to native and cloned CRMP‐2. Results from all the different experimental approaches taken (Table 1), suggest that LCM does not bind specifically to CRMP‐2.
Table 1.
Summary of assay conditions used to investigate LCM binding to CRMP‐2
| Ligand | Tissues /protein | Binding material | Method |
|---|---|---|---|
| [3H]LCM (3–600 nM) | Native CRMP‐2 from rat brain | Membranes and Homogenates | Manual filtration |
| [3H]LCM (600–1200 nM) | Untagged CRMP‐2, GST‐ and His‐tagged (Nt and Ct) CRMP‐2 expressed in mammalian cells (COS) and Xenopus oocytes | Intact cells | Manual filtration |
| Membrane fractions | Centrifugation | ||
| Cytosolic fractions | Scintillation Proximity Assay | ||
| CRMP‐2 coupled to beads | |||
| GST‐ and His‐tagged (Nt and Ct) CRMP‐2 expressed in E.Coli | CRMP‐2 coupled to beads | Manual filtration | |
| Scintillation Proximity Assay | |||
| LCM (0.39–100 μM) | Histidine (Ct) CRMP‐2 expressed in E. Coli | Purified protein | Surface Plasmon Resonance |
| LCM (5 mM) | Tagged CRMP‐2 | CRMP‐2 co‐crystal | X‐ray structure* |
LCM=lacosamide; CRMP‐2 = collapsing response mediator protein 2; Nt = N‐terminus; Ct = C‐terminus; GST = Glutathione S‐Transferase; His = Histidine.
*Personal communication (Dr. Petri Kursula, Oulu University, Finland)
CRMP‐2 is the most highly expressed CRMP isoform in the adult brain [8], and recent reports indicate that LCM “affinity bait” analogues are able to interact with CRMP‐2 in brain extracts [6, 7]. However, experiments designed and conducted to determine binding of [3H]LCM to rat brain structures (Figure 1) did not reveal any specific binding. These negative findings may potentially be explained by the relative high NSB levels of [3H]LCM in rat brain membranes (∼2000–6000 dpm/assay, Figure 1) or attributed to the low levels of CRMP‐2 expression in native tissues. To address this latter issue, further studies were performed on the binding of LCM to cloned and expressed CRMP‐2, which offered increased protein expression levels and access to a more diverse set of experimental protocols (e.g., use of tagged forms of CRMP‐2) for the detection of specific LCM‐binding.
A previous study investigating LCM binding to CRMP‐2 expressed in oocyte membranes, suggested that LCM binds to CRMP‐2 with an affinity of ∼5 μM [5]. Because further characterization of a potential LCM binding site was hampered by the low specific activity of [14C]LCM, we further evaluated the binding of LCM to CRMP‐2 in oocyte membranes by using [3H]LCM (having a 1000‐fold higher specific activity than [14C]LCM) and mRNA injection to maximize protein expression levels (Figure 2, insert). Although free [3H]LCM concentrations (∼1 μM) were high enough to allow detection of a specific binding site in the low‐micromolar range, the previous findings, using [14C]LCM binding to CRMP‐2 in oocyte membranes, could not be confirmed.
To test whether the binding properties of LCM are affected by the nature of the soluble versus the membrane‐associated form of CRMP‐2, we also determined LCM binding to soluble CRMP‐2. Tagged CRMP‐2 was expressed in mammalian cells or bacteria and binding experiments of [3H]LCM to isolated CRMP‐2 were performed by manual filtration and SPA (Figure 3). This latter approach overcomes the need to remove the unbound ligand by filtration and thus represents an alternative method for evaluating receptor‐ligand interactions that display fast dissociation kinetics and which could potentially be missed by manual filtration. Results from the two different techniques evaluating [3H]LCM binding to isolated CRMP‐2 also suggest the absence of a specific binding site (Figure 3).
LCM binding to CRMP‐2 was further evaluated by surface plasmon resonance (Biacore), a technology platform that quantifies the association and dissociation rates of unlabeled LCM binding to captured CRMP‐2 in real‐time. The Biacore data indicated similar binding levels of LCM to CRMP‐2 and HSA and represented only ∼6% of the predicted binding levels based on a 1:1 stoichiometry (Figure 4). We conclude that the low level of binding observed is due to a non‐specific interaction with CRMP‐2 and HSA. Our data are consistent with previous Biacore experiments (Dr. Petri Kursula; personal communication) showing that binding of LCM to CRMP‐2 at high concentrations (>1 mM) is not saturable and hence mediated by nonspecific interactions.
In contrast to the data presented here, previous studies had proposed that LCM may directly bind to CRMP‐2 and modulate the function of voltage‐gated Na+ channels [6, 7]. The findings of these studies were based on affinity‐labeling techniques, using modified LCM analogues or “affinity baits,” which showed that such LCM analogues were able to bind to CRMP‐2 in rat brain extracts. Yet, the interaction of such LCM affinity baits with CRMP‐2 remained very weak since it could only be partially competed at LCM concentrations of 25 mM [7], far higher than the therapeutic dose range of LCM. This is consistent with a nonspecific, or low affinity (millimolar range) interaction. Furthermore, the results reported by Wang et al. [7] were obtained using LCM analogues and thus we cannot exclude that modification of the LCM structure may generate ligands with an altered receptor interaction profile and hence low affinity CRMP‐2 binding properties.
Wang et al. [7] also evaluated LCM binding to CRMP‐2 using an in silico docking approach, based on the CRMP‐2 X‐ray structure [17, 18] and proposed that CRMP‐2 had five binding pockets for LCM. Although molecular docking is a general approach to identify potential binding sites on a target protein, it does not provide experimental evidence for the existence of such sites. Indeed, co‐crystallization of CRMP‐2 in the presence of LCM did not reveal a specific binding pocket for LCM (Dr. Petri Kursula; personal communication) thus further supporting the negative binding data obtained in the present study.
Data obtained by Wang et al. [7] using CRMP‐2 mutants, suggest that this protein may play a role in voltage‐gated sodium channel function. The authors demonstrated that CRMP‐2 expression levels modulate the effects of LCM on sodium channel slow inactivation. The precise mechanistic link that may underlie these observations is not clear, and there is no specific evidence supporting a direct binding of LCM to CRMP‐2. LCM is able to enhance slow inactivation of cloned Nav1.3 and 1.7 channels in human embryonic kidney (HEK) cells [4], a cell line that does not express endogenous CRMP‐2 [19], confirming that LCM is a sodium channel ligand and that it acts independently of CRMP‐2. CRMP‐2 was recently reported to affect the cycling of voltage‐gated calcium channels in neurons [9, 20], an observation that highlights the potential key role of CRMP‐2 in modulating the function and pharmacology of voltage‐gated ion channels. Based on the absence of an LCM binding site on CRMP‐2, we speculate that interaction of CRMP‐2 with voltage‐gated sodium channels may modify the effects of LCM on slow inactivation indirectly; for example, through an allosteric effect on the channel, thereby affecting the affinity of LCM on channel binding.
Given our findings, together with the limitations of the previously reported data [5, 6, 7], we conclude that there is currently no experimental evidence to support a direct binding relationship between LCM and CRMP‐2. The possibility of an indirect functional interaction between these molecules needs to be further explored. As clinicians continue to search for differential qualities in the many available AEDs, including using mechanism‐based rational polytherapy [21], we need to keep in mind that the precise relationship between LCM and CRMP‐2—whether it is ultimately proven to be direct, indirect, or non‐existent— is not yet fully understood.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank Azita Tofighy, PhD of UCB Pharma, for her assistance in the preparation of this manuscript. Jennifer Hepker, PhD of Prescott Medical Communications Group (Chicago, IL), provided editorial assistance during the preparation and submission of the manuscript.
References
- 1. 2008 VIMPAT® (lacosamide) Prescribing Information. UCB, Inc., Smyrna , GA , US ). [Google Scholar]
- 2. 2008 VIMPAT® Summary of Product Characteristics for Lacosamide. UCB Pharma, S.A., Belgium ). [Google Scholar]
- 3. Errington AC, Stohr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage‐gated sodium channels. Mol Pharmacol 2008;73:157–169. [DOI] [PubMed] [Google Scholar]
- 4. Sheets PL, Heers C, Stoehr T, Cummins TR. Differential block of sensory neuronal voltage‐gated sodium channels by lacosamide [(2R)‐2‐(acetylamino)‐N‐benzyl‐3‐methoxypropanamide], lidocaine, and carbamazepine. J Pharmacol Exp Ther 2008;326:89–99. [DOI] [PubMed] [Google Scholar]
- 5. Beyreuther BK, Freitag J, Heers C, Krebsfanger N, Scharfenecker U, Stohr T. Lacosamide: A review of preclinical properties. CNS Drug Rev 2007;13:21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park KD, Morieux P, Salome C, et al Lacosamide isothiocyanate‐based agents: Novel agents to target and identify lacosamide receptors. J Med Chem 2009;52:6897–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Brittain JM, Jarecki BW, et al In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein‐2 identifies a pocket important in modulating sodium channel slow inactivation. J Biol Chem 2010;285:25296–25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J. Collapsin response mediator proteins (CRMPs): Involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol 2003;28:51–64. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Brittain JM, Wilson SM, Khanna R. Emerging roles of collapsin response mediator proteins (CRMPs) as regulators of voltage‐gated calcium channels and synaptic transmission. Commun Integr Biol 2010;3:172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmidt EF, Strittmatter SM. The CRMP family of proteins and their role in Sema3A signaling. Adv Exp Med Biol 2007;600:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin Response mediator Protein‐2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 2011;43:180–191. [DOI] [PubMed] [Google Scholar]
- 12. Czech T, Yang J, Csaszar E, Kappler J, Baumgartner C, Lubec G. Reduction of hippocampal collapsin response mediated protein‐2 in patients with mesial temporal lobe epilepsy. Neurochem Res 2004;29:2189–2196. [DOI] [PubMed] [Google Scholar]
- 13. Wolff C, Gillard M, Fuks B, Chatelain P. [3H]linopirdine binding to rat brain membranes is not relevant for M‐channel interaction. Eur J Pharmacol 2005;518:10–17. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi T, Fournier A, Nakamura F, et al Plexin‐neuropilin‐1 complexes form functional semaphorin‐3A receptors. Cell 1999;99:59–69. [DOI] [PubMed] [Google Scholar]
- 15. Karlsson R, Stahlberg R. Surface plasmon resonance detection and multispot sensing for direct monitoring of interactions involving low‐molecular‐weight analytes and for determination of low affinities. Anal Biochem 1995;228:274–280. [DOI] [PubMed] [Google Scholar]
- 16. Lynch BA, Lambeng N, Nocka K, Kensel‐Hammes P, Bajjalieh S M, Matagne A, Fuks B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA 2004;101:9861–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deo RC, Schmidt EF, Elhabazi A, Togashi H, Burley SK, Strittmatter SM. Structural bases for CRMP function in plexin‐dependent semaphorin3A signaling. EMBO J 2004;23:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stenmark P, Ogg D, Flodin S, et al The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J Neurochem 2007;101:906–917. [DOI] [PubMed] [Google Scholar]
- 19. Kawano Y, Yoshimura T, Tsuboi D, et al CRMP‐2 is involved in kinesin‐1‐dependent transport of the Sra‐1/WAVE1 complex and axon formation. Mol Cell Biol 2005;25:9920–9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD, Khanna R. Regulation of N‐type voltage‐gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein‐2 (CRMP‐2) in sensory neurons. J Cell Sci 2009;122:4351–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sake JK, Hebert D, Isojarvi J, et al A pooled analysis of lacosamide clinical trial data grouped by mechanism of action of concomitant antiepileptic drugs. CNS Drugs 2010;24:1055–1068. [DOI] [PubMed] [Google Scholar]