

Abstract
Muscle weakness and atrophy are important manifestations of multiple sclerosis (MS). To investigate the pathophysiological mechanisms of skeletal muscle change in MS, we induced experimental autoimmune encephalomyelitis (EAE) in Lewis male rats and examined morphological and molecular changes in skeletal muscle. We also treated EAE rats with calpepetin, a calpain inhibitor, to examine its beneficial effects on skeletal muscle damage. Morphological changes in muscle tissue of EAE rats included smaller and irregularly shaped muscle fibers and fibrosis. Western blot analysis demonstrated increased calpain:calpastatin ratio, inflammation-related transcription factors (nuclear factor-κB:inhibitor of κB α ratio), and proinflammatory enzymes (cyclooxygenase-2). TUNEL-positive myonuclei in skeletal muscle cells of EAE rats indicated cell death. In addition, markers of apoptotic cell death (Bax:Bcl-2 ratio and caspase-12 protein levels) were elevated. Expression of muscle-specific ubiquitin ligases (muscle atrophy F-box and muscle ring finger protein 1), was upregulated in muscle tissue of EAE-vehicle animals. Both prophylactic and therapeutic treatment with calpeptin partially attenuated muscle changes noted in EAE animals. These results indicate that morphological and molecular changes including apoptotic cell death and protein breakdown develop in skeletal muscle of EAE animals and that these changes can be reversed by calpain inhibition.
Keywords: muscle atrophy, experimental autoimmune encephalomyelitis, calpain, nuclear factor-κB, apoptosis
Multiple sclerosis (MS) is a T-cell-mediated auto-immune disease of the central nervous system (CNS) with secondary neurodegenerative changes (Guyton et al., 2010). It is characterized by multifocal lesions of demyelination, axonal loss, and astroglial scaring (Vogt et al., 2009). Clinical presentations of MS include impaired vision, paresthesia, and weakness of one or more limbs (Keegan and Noseworthy, 2002; de Haan et al., 2004). The weakness can be associated with impaired muscle function and lead to decreased ambulation (de Haan et al., 2004; Ng et al., 2004). Muscle atrophy in MS is usually considered to be due to disuse, motor control dysfunction in the CNS, or lower motor neuron involvement of spinal cord lesions (Fisher et al., 1983; Vogt et al., 2009). Interestingly, histological studies of skeletal muscle of MS patients showed lymphoplasmocitary vasculitis and inflammatory cells (macrophages) in the intramuscular ends of the motor nerves (Tilbery et al., 1989). In addition, cross-sectional area (CSA) of skeletal muscle fibers of experimental allergic encephalomyelitis (EAE), an animal model of MS, is smaller compared with control (de Haan et al., 2004). These findings suggest the possibility of myogenic changes of skeletal muscle in MS or EAE.
Dysregulation of Th1, Th2, and Th17 cytokines is a key concept for the pathogenesis of MS (McFarland and Martin, 2007). In studies of peripheral blood mononuclear cells (PBMCs) in relapsing-remitting MS (RRMS) patients, mRNA levels of tumor necrosis factor-α (TNF-α), a Th1 cytokine, and lymphotoxin are increased prior to relapse (Rieckmann et al., 1995). We have demonstrated that the proinflammatory cytokines interferon-γ (IFN-γ) and TNF-α cause secondary inflammation, mitochondrial dysfunction, and apoptosis in L6 rat myoblasts in vitro (Nozaki et al., 2010, 2011). These results indicate that circulating cytokines may trigger muscle damage and atrophy in MS and EAE.
Muscle atrophy can be characterized by decreases in muscle fiber size, protein content, and overall number of muscle fibers (Cai et al., 2004; Jackman and Kandarian, 2004). It can result from enhanced protein degradation or reduced protein synthesis or both (Eley and Tisdale, 2007; Li et al., 2008). Activated nuclear factor-κB (NF-κB) may mediate muscle atrophy through induction of protein degradation and ubiquitin-dependent proteolysis (Liu et al., 1996; Han et al., 1999; Oda et al., 2002; Wei et al., 2006). In particular, the ATP-dependent ubiquitin (Ub)-proteasome pathway (UPP) is activated during muscle atrophy and contributes to the intracellular protein degradation (Smith et al., 2008). The UPP is the primary regulator of degradation of both structural and regulatory proteins (Liu et al., 1996; McKinnell and Rudnicki, 2004; Cao et al., 2005). Recently, two inducible components of the UPP, E3 muscle-specific ubiquitin ligases, muscle atrophy F-box (MAFbx, also known as atrogin-1), and muscle ring finger protein 1 (MuRF1), have been identified, which appear to be responsible for the degradation of skeletal muscle protein in various atrophic conditions (Bodine et al., 2001; Cao et al., 2005).
In addition to NF-κB-mediated activation of the UPP, apoptosis may also contribute to atrophy through loss of myofiber segments (hypotrophy) or myofibers (hypoplasia; Dupont-Versteegden, 2005). It has been detected in atrophied muscles as a result of denervation, corticosteroid therapy, limb suspension, and aging (Tews, 2002; Leeuwenburgh, 2003). Myonuclei are removed in an apoptosis-like manner to maintain a constant ratio between myonuclei number and fiber volume in atrophying muscles (Allen et al., 1997; Mitchell and Pavlath, 2004).
In addition to the various signaling pathways described above, calpain has been received recent attention as an important mediator of muscle atrophy. Calpain is an intracellular, nonlysosomal Ca2+-activated cysteine protease. Ubiquitously expressed calpain exists in two forms, μ-calpain and m-calpain, requiring micromolar and millimolar Ca2+ concentrations, respectively. In the setting of muscle atrophy, calpain may be activated because of increased permeability of the muscle cell to Ca2+ and has a preferential site of proteolysis, the Z-disc, which is a dense, multiproteinaceous network located on both sides of the sarcomeres (Bartoli and Richard, 2005). Calpain inhibition has also been shown to stabilize nebulin, an important contributor to sarcomere integrity (Huang and Forsberg, 1998; Smith et al., 2008).
Here we investigated morphological and molecular changes that might occur in skeletal muscle of EAE animals. We specifically focused on the role of calpain and examined whether the inhibition of calpain attenuates changes in muscle of EAE animals.
MATERIALS AND METHODS
Cell Culture and Treatment with IFN-γ and Calpeptin
The L6 rat myoblast cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA). L6 cells were cultured as we described previously (Nozaki et al., 2011). To observe the effect of calpain inhibition, various doses of calpeptin (1 and 5 μM) were added 5 min after IFN-γ (500 U/ml) exposure. Treatment of myoblast cells with IFN-γ and/or calpeptin was carried out for 24 hr at 37°C with 5% CO2 and full humidity. After 24 hr treatment, attached myoblast cells were scraped and mixed with floating cells, centrifuged at 2,000 rpm for 5 min to obtain a pellet, and stored until further use for Western blot analysis.
EAE Induction
Adult male Lewis rats (8 weeks, 180–200 g) were purchased from Charles River Laboratories (Wilmington, MA) and provided with unrestricted access to water and food pellets. To induce EAE, rats were immunized subcutaneously with 0.2 ml complete Freund’s adjuvant (CFA) containing 10 mg/ml Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) and normal saline (NS) containing guinea pig spinal cord homogenate (200 mg/ml) and myelin basic protein (MBP; 200 μg/ml) in a 1:1 emulsion (Guyton et al., 2005, 2010). Control animals received NS/CFA only. Two hours later, all rats received an intraperitoneal (i.p.) injection of 0.125 ml pertussis toxin (1.25 μg/rat). All experiments were performed and approved by the Medical University of South Carolina Institutional Animal Care and Use Committee, in accordance with the Laboratory Welfare Act and the Guide for the care and use of laboratory animals of the National Institutes of Health.
Calpeptin Treatment and Clinical Evaluation in EAE Rats and Skeletal Muscle Tissue Collection
Animals were treated twice daily with i.p. injections of either vehicle (1.0% DMSO in saline) or calpeptin (250 μg/kg) from day 1 post-EAE induction to the day of sacrifice (prophylactic) or from the day of EAE onset to the day of sacrifice (therapeutic). Control animals were treated with vehicle or calpeptin as in the prophylactic group. No morphological or molecular differences were noted between control animals receiving vehicle and those receiving calpeptin, so we divided the rats into four groups as follows: 1) control rats (control, n = 5), 2) EAE rats treated with vehicle (EAE-vehicle, n = 8), 3) EAE rats treated with calpeptin prophylactically (EAE-calpeptin prophylactic, n = 4), and 4) EAE rats treated with calpeptin therapeutically (EAE-calpeptin therapeutic, n = 4). All rats were monitored daily for weight loss and clinical disability until day of sacrifice. The signs of clinical disability resulting from EAE were rated based on the following grades: 0, no change; 1, limp tail; 2, hind limb weakness with difficulty righting; 3, hind limb partial paralysis; 4, hind limb complete paralysis with front limb weakness; and 5, quadriplegic or moribund. To obtain samples from animals with maximal damage, the rats were sacrificed under anesthesia (95 mg/kg ketamine, 5 mg/kg xylazine) 2–5 days after the onset of EAE. Bilateral quadriceps femoris muscle tissues were removed and separated. A portion of muscle tissue was processed for histological analysis and terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) assay, and the remaining sample was used for Western blot analysis.
Histological Analysis
Muscle samples were cut to an appropriate shape and embedded into 10% tragacanth (Sigma-Aldrich, St. Louis, MO) gum on an embedding block (Thomas Scientific, Swedesboro, NJ). The muscle block was gently immersed in isopentane (Fisher Scientific, Pittsburgh, PA), cooled by dry ice for 20–30 seconds, and stored at −70°C. Prior to cutting, samples were warmed to −22°C, and thin sections (10 μm) were cut using a Reichart-Jung cryostat (Cryocut 1800; Leica, Wetzlar, Germany). Muscle sections were stained for morphological analysis using a standard hematoxylin and eosin (H&E) protocol. Slides were viewed under a fluorescence microscope at ×200 (Carl Zeiss Meditec, Dublin, CA).
Determination of Muscle Fiber Diameter
To analyze muscle atrophy quantitatively, muscle fiber diameter was determined in H&E-stained sections using previously described methods (Ashley et al., 2007; Cornachione et al., 2011). Briefly, images were loaded into NIH Image J (http://rsb.info.nih.gov/ij/), and the outer boundaries of muscle fibers were manually traced. Image J was then used to determine the lesser diameter of each fiber. The lesser diameter was chosen because it corrects for any imperfections in transverse sectioning of muscle samples. Ten fields were counted for each condition, allowing for measurement of at least 130 fibers (n = 130) per treatment group. The lesser diameter is expressed as mean (μm) ± SEM.
TUNEL Assay
To detect cell death in skeletal muscle tissue, we used the TUNEL assay. Skeletal muscle tissues were sliced and postfixed in 4% methanol-free formaldehyde (in PBS) for 25 min at 4°C, followed by rinsing twice in PBS for 5 min. Then, muscle tissue slices (10 μm) were immersed in 0.2% Triton X-100 in PBS for 5 min and washed twice in PBS for 5 min each. As we previously described (Ray et al., 2000), tissue sections on each slide were saturated with 100 μl of an equilibration buffer (EB: 200 mM K-cacodylate, 25 mM Tris-HCl, pH 6.6, 0.2 mM dithiothreitol [DTT], 0.25 μg/μl bovine serum albumin [BSA], 2.5 mM CoCl2) under a plastic coverslip (Promega, Madison, WI) to prevent evaporation for 10 min. This was replaced by 50 μl TUNEL reaction mixture, which was freshly prepared by mixing 45 μl EB, 5 μl 10× DNA labeling solution (2 mM each of dATP, dCTP, dGTP, 1.9 mM dTTP and 0.1 mM alkali-stable DIG-11-dUTP; Roche Molecular Biochemicals, Indianapolis, IN), and 1 μl 25 U/μl of terminal deoxynucleotidyl transferase (TdT; Promega). The TUNEL reaction mixture was evenly spread under a plastic coverslip, and air bubbles were carefully removed. Immediately, all slides were incubated at 37°C for 1 hr in a humidified OmniSlide Thermal Cycler (Hybaid Ltd.). The reaction was terminated by immersing the slides in 2× sodium chloride and sodium citrate (2× SSC: 300 mM NaCl, 30 mM Na-citrate, pH 7.4) for 15 min at room temperature. The slides were washed three times in PBS for 5 min to remove unincorporated DIG-11-dUTP. To prevent rapid photo-bleaching of fluorochromes during microscopic examination, one drop (approximately 25 μl) of Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA) was added and evenly dispersed over the tissue sections with a glass coverslip (22 × 22 mm). All slides were kept in a dark box and observed under a fluorescence microscope at ×200 (Carl Zeiss Meditec).
Protein Extraction and Western Blot Analysis
We detected changes in protein levels by using previously described methods (Nozaki et al., 2010, 2011). Collected L6 myoblast cell pellets or skeletal muscle tissues were homogenized in ice-cold buffer (50 mM Tris-HCl, pH 7.4, 1 mM PMSF, and 5 mM EGTA) to extract protein. Protein content was determined using the standard Lowry protein assay. Then, each sample was diluted with the same volume of sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 5 mM β-mercaptoethanol, and 10% glycerol). Protein samples containing 12 μg were separated on 4–20% linear gradient SDS-PAGE gels according to the standard procedure (Bio-Rad, Temecula, CA). After electrophoresis, proteins were transferred onto PVDF membranes for immunoblot analysis. The blots were probed at 4°C overnight with primary IgG antibodies against calpastatin, NF-κB, I-κB α, STAT3, COX-2, IL-2, Bax, Bcl-2, caspase-12, caspase-3, MAFbx, and MuRF1 (Santa Cruz Biotechnology, Santa Cruz, CA) and calpain. Glyceraldeyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology) was used to standardize the loading of cytosolic protein. Horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary IgG antibody (MP Biomedicals, Solon, OH) was applied to the blots at 37°C for 1 hr. For subsequent detection of specific proteins, the enhanced chemiluminescence (ECL) system was used (GE Health Care, Piscataway, NJ). Blots were immediately processed to digital images using the FluorChem FC2 system (AlphaInnotech, San Leandro, CA). Protein bands were quantified in Image J. Compared with control, the percentage change in amount of protein was calculated.
Statistical Analysis
Results were assessed in StatView software (Abacus Concepts, Berkeley, CA) and compared by one-way ANOVA with Fisher’s protected least significant difference (PLSD) post hoc test at a 95% confidence interval. Data are presented as mean ± SEM of separate experiments (n = 4). Significant differences between control and EAE-vehicle groups were indicated by *P < 0.05 or **P < 0.01. Significant differences between EAE-vehicle and EAE-calpeptin groups were indicated by #P < 0.05 or ##P < 0.01.
RESULTS
Clinical Disability and Morphological Changes in Skeletal Muscle of EAE Rats
Changes in body weight and clinical disability were assessed in controls and EAE animals treated with either vehicle or calpeptin according to the same methods used in our previous study. Doses of calpeptin were chosen based on efficacious effects achieved in that study (Guyton et al., 2010). As expected, control animals steadily gained body weight (74 ± 6.3 g) and did not show clinical signs of EAE during the experimental period. On the other hand, EAE rats receiving vehicle alone demonstrated much smaller gains in body weight (4 ± 1.2 g) after EAE induction. EAE rats treated with calpeptin showed marked restoration of weight gain (40 ± 18.4 g in EAE-CP prophylactic and 35 ± 14.1 g in EAE-CP therapeutic). Furthermore, we identified the typical signs of clinical disability, including weakness of the tail and forelimbs, difficulty righting, and complete paralysis of hind limbs (clinical score 4–5) in EAE-vehicle rats, as described from our previous study (Guyton et al., 2010). Animals receiving prophylactic or therapeutic calpeptin treatment showed only mild weakness of the tail and/or hind limbs and no difficulty in locomotion (clinical score 0–1). One EAE-CP animal briefly showed difficulty in righting (clinical score 3), which dissipated on day 2 after EAE onset.
H&E staining was used to examine the morphological changes in skeletal muscle in EAE rats (Fig. 1A). Various morphological changes were observed, including smaller muscle fiber size, irregular fiber morphology, and fibrosis in EAE rats treated with vehicle, whereas control animals showed typical muscle fiber morphology. EAE-induced morphological changes in skeletal muscle were attenuated in EAE animals with calpeptin treatment prophylactically and therapeutically.
Fig. 1.
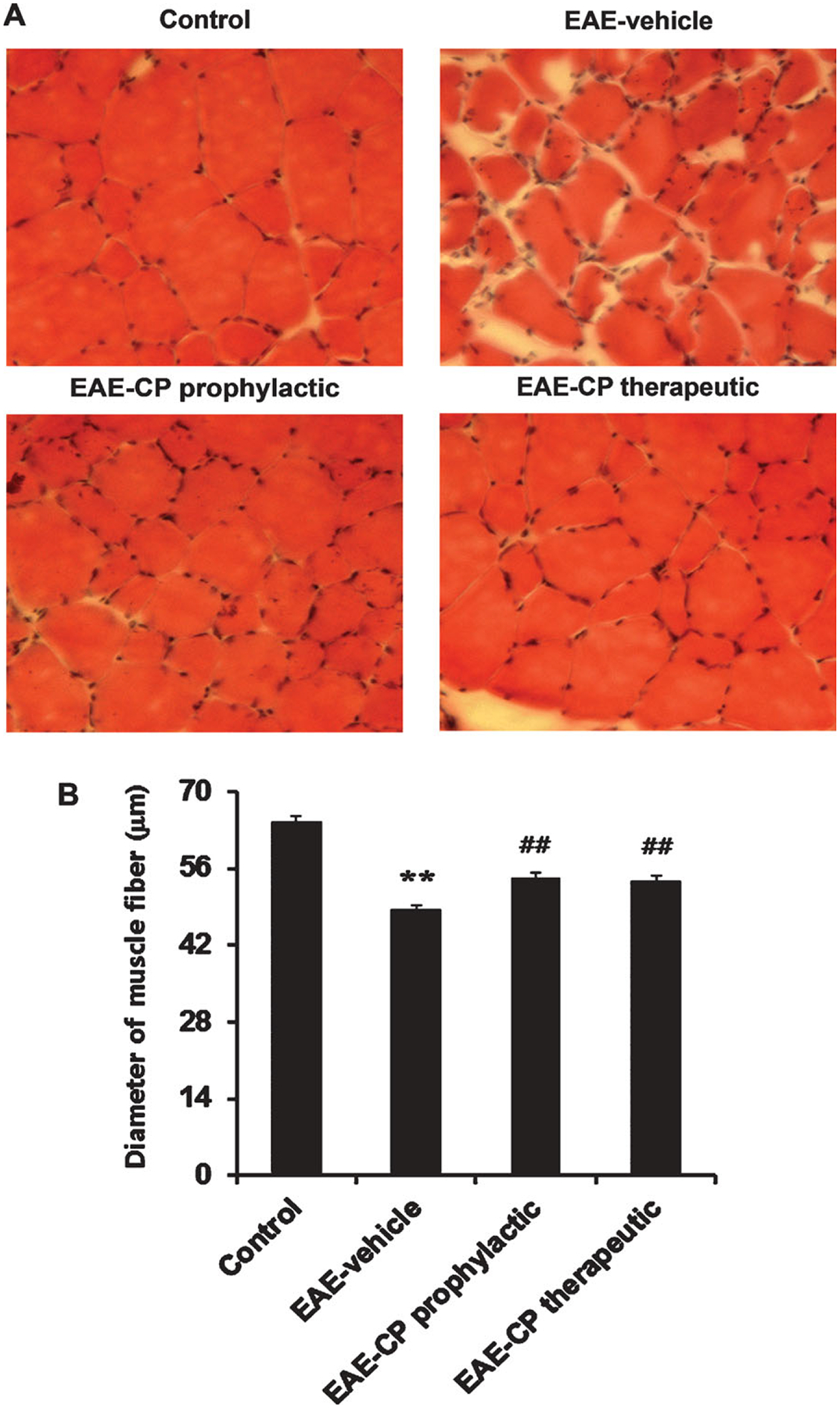
Morphological alterations in skeletal muscle of EAE rats. A: H&E staining demonstrated various changes, including smaller, irregularly shaped muscle fibers and fibrosis in thin section slices (10 μm) obtained from EAE rats treated with vehicle prophylactically or therapeutically compared with control rats. B: Assessment of muscle atrophy in EAE animals by measurement of muscle fiber diameter. A significant reduction in fiber diameter was noted in EAE-vehicle animals. These changes were attenuated in EAE animals treated with calpeptin prophylactically or therapeutically. *P < 0.05 vs. control, **P < 0.01 vs. control; #P < 0.05 vs. EAE-vehicle, ##P < 0.01 vs. EAE-vehicle. CP, calpeptin. ×200.
To provide an objective, quantitative measure of skeletal muscle atrophy in EAE rats, we also measured muscle fiber diameter in H&E-stained muscle sections (Fig. 1B). The average lesser diameter of quadriceps femoris muscle fibers in control animals was measured to be approximately 64.5 ± 1.1 μm. EAE-vehicle demonstrated a significant reduction (to ~78% of the control value) in muscle fiber diameter (48.4 ± 0.8 μm) compared with control animals (P < 0.01). Calpeptin treatment both prophylactically (54.2 ± 0.9 μm) and therapeutically (53.5 ± 1.0 μm) significantly restored fiber diameter (~84% of control in EAE-CP prophylactic, ~83% of control in EAE-CP therapeutic), indicating significant reduction in muscle atrophy following calpeptin administration (P < 0.01).
Calpeptin Attenuated Calpain Expression and Secondary Inflammatory Changes in Skeletal Muscle of EAE Rats
To examine whether EAE induction and calpeptin treatment causes molecular changes in skeletal muscle, Western blot analysis was carried out on skeletal muscle samples obtained from control and EAE animals that had been treated with vehicle or calpeptin (Fig. 2). The calpain:calpastatin ratio was significantly increased in EAE-vehicle rats compared with controls (P < 0.01). In contrast, it was significantly decreased in both EAE-calpeptin prophylactic and therapeutic groups compared with EAE-vehicle group, respectively (P < 0.01; Fig. 2A).
Fig. 2.
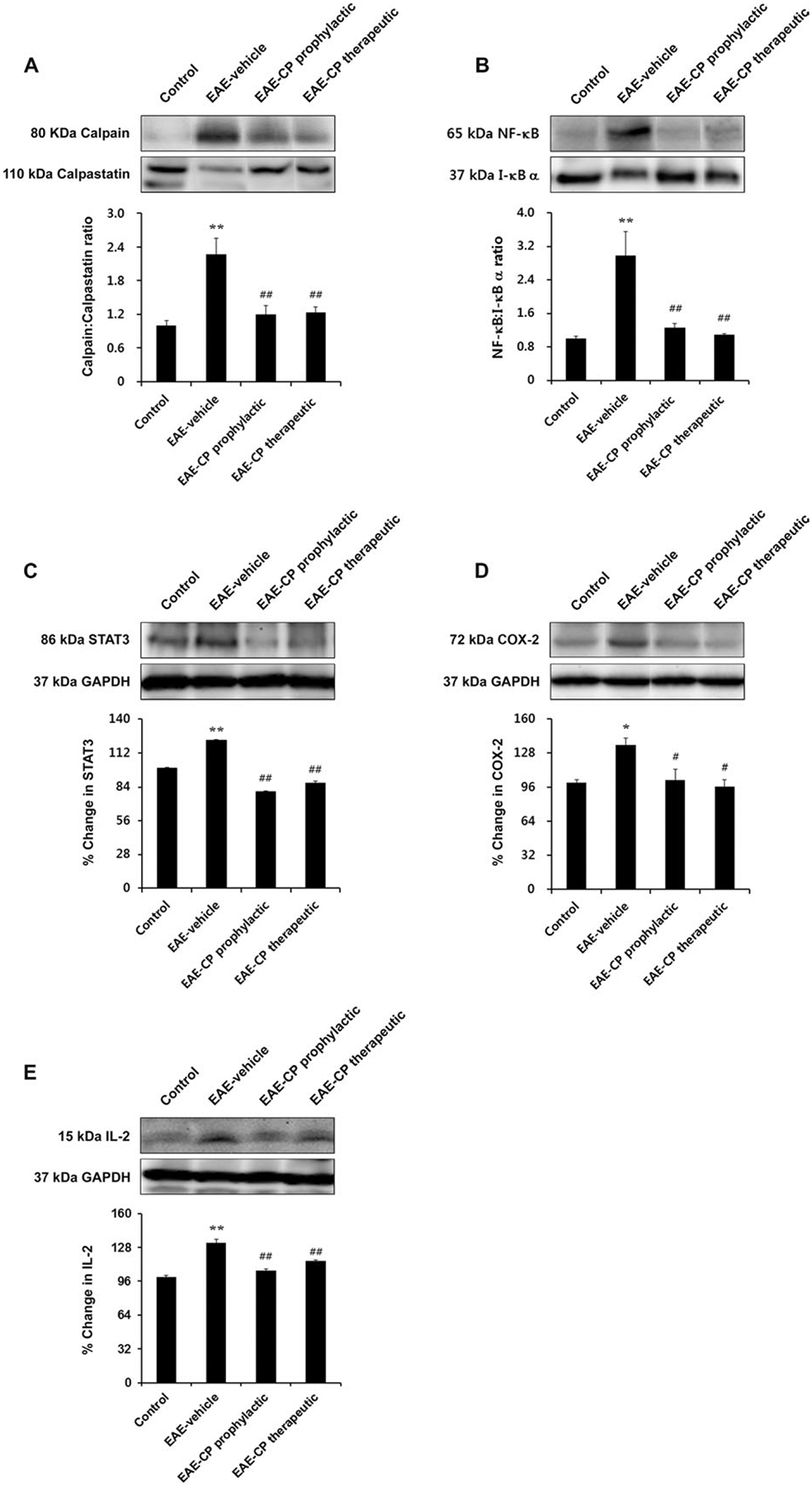
Changes in calpain and inflammation-related protein expression in skeletal muscle of EAE rats. Calpain:calpastatin ratio, NF-κB:I-κB α ratio, STAT3, COX-2, and IL-2 expression were increased in EAE rats, whereas calpeptin treatment reduced these proinflammatory changes. Representative Western blot and determination of 80-kDa calpain:110-kDa calpastatin ratio (A), 65-kDa NF-κB:37-kDa I-κB α ratio (B), percentage change of 86-kDa STAT3 expression (C), 72-kDa COX-2 expression (D), and 15-kDa IL-2 expression (E). *P < 0.05 vs. control, **P < 0.01 vs. control; #P < 0.05 vs. EAE-vehicle, ##P < 0.01 vs. EAE-vehicle. CP, calpeptin.
To examine whether EAE causes secondary inflammatory responses in muscle, we also measured NF-κB, I-κB α, and protein levels. As expected, NF-κB:I-κB α ratios were significantly increased in the EAE-vehicle group compared with control (P < 0.01; Fig. 2B). Changes in this ratio were reversed after calpeptin treatment (both prophylactic and therapeutic) compared with vehicle-treated EAE rats (P < 0.01). To determine whether these changes were specific to NF-κB or might involve other proinflammatory transcription factors, we also measured expression of signal transducer and activator of transcription 3 (STAT3; Fig. 2C). A significant increase in STAT3 protein levels was observed in EAE-vehicle animals compared with control (P < 0.01). However, calpeptin treatment drastically reduced expression of STAT3 in both EAE-CP prophylactic and EAE-CP therapeutic animals (P < 0.01).
We also measured protein levels of other proinflammatory factors, including COX-2 and interleukin-2 (IL-2) in skeletal muscle from EAE animals. COX-2 protein expression was significantly upregulated in EAE-vehicle animals compared with control (P < 0.05). In contrast, expression of COX-2 protein was significantly reduced in after calpeptin treatment both prophylactically and therapeutically (P < 0.05; Fig. 2D). A similar increase in IL-2 levels was noted in EAE-vehicle animals compared with control (P < 0.01) and was significantly attenuated by calpeptin administration (P < 0.01 vs. EAE-vehicle; Fig. 2E).
Calpeptin Reduced Apoptotic Changes in Skeletal Muscle of EAE Rats
To examine whether apoptotic changes occur in skeletal muscle of EAE rats, TUNEL assays were also performed. Little cell death was found in skeletal muscle of control animals. TUNEL-positive myonuclei were found in EAE-vehicle rats, whereas treatment with calpeptin prophylactically and therapeutically attenuated the number of TUNEL-positive myonuclei in EAE animals (Fig. 3A). These results indicate that calpeptin treatment prevents apoptotic changes in skeletal muscle in EAE animals when given prophylatically and, more importantly, after disease onset. To characterize molecular changes associated with apoptosis in skeletal muscle of EAE animals, apoptosis-related protein levels were measured. We demonstrated that EAE-vehicle rats showed upregulation of Bcl-2 and Bax protein levels (Fig. 3B). The highest Bax:Bcl-2 ratio was detected in the EAE-vehicle group (P < 0.01 vs. control), with a significant decrease noted after calpeptin administration prophylactically and therapeutically (P < 0.01). In addition, caspase-12 protein level was significantly elevated in the EAE-vehicle group compared with control (P < 0.01), and prophylactic and therapeutic treatment with calpeptin attenuated the expression of caspase-12 (P < 0.01; Fig. 3C). However, contrary to our expectations, there were no significant changes in 20-kDa active caspase-3 protein levels in EAE-vehicle rats (Fig. 3D).
Fig. 3.
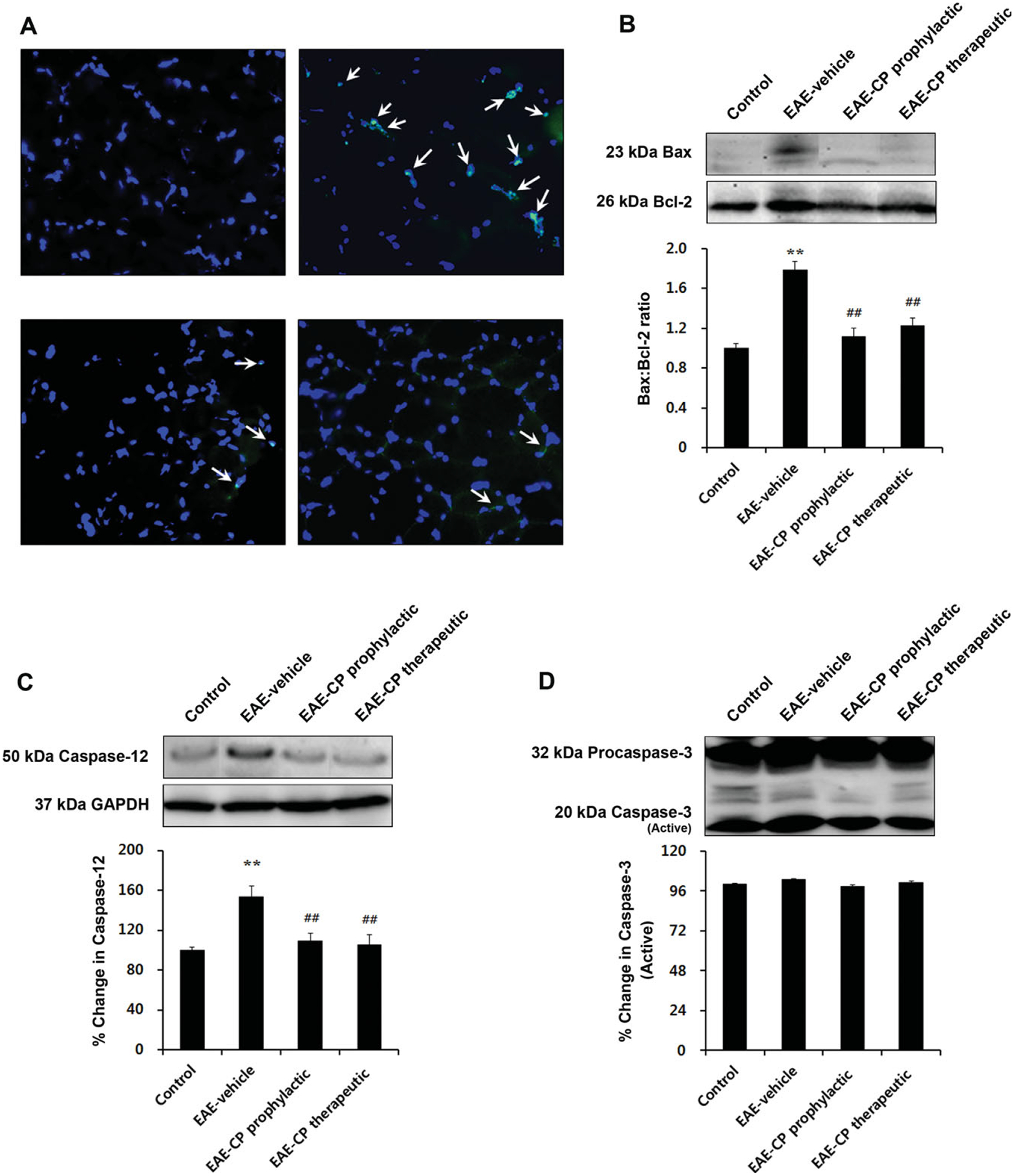
Inhibition of calpain reduced apoptotic changes in skeletal muscle of EAE rats. A: TUNEL-positive myonuclei were detected in muscle of EAE rats treated with vehicle prophylactically or therapeutically. Arrows indicate nicked DNA of apoptotic cells. Posttreatment with calpeptin attenuated these changes. Sections (10 μm) of muscle tissue were costained with TUNEL (green), and DAPI (blue; ×200). Representative Western blot and determination of 23-kDa Bax:26-kDa Bcl-2 ratio (B) and percentage change of 50-kDa caspase-12 (C) and 20-kDa active caspase-3 (D). *P < 0.05 vs. control, **P < 0.01 vs. control; #P < 0.05 vs. EAE-vehicle, ##P < 0.01 vs. EAE-vehicle. CP, calpeptin.
Alteration of Protein Degradation-Related Proteins in EAE Rat Skeletal Muscles and L6 Myoblast Cells in Response to Inflammatory Stimulation and Its Attenuation by Calpeptin Treatment
To examine the molecular changes associated with skeletal muscle atrophy after EAE induction, protein expression of MAFbx and MuRF1, muscle-specific ubiqutin-proteasome ligases, were analyzed by Western blotting (Fig. 4). Upregulation of these protein levels indicates severe protein degradation in skeletal muscle (de Haan et al., 2004; McKinnell and Rudnicki, 2004; Cao et al., 2005; Li et al., 2008). We found that both MAFbx and MuRF1 protein levels were markedly increased in muscle samples from EAE-vehicle rats compared with control animals (P < 0.01; Fig. 4A,B). Meanwhile, increased protein levels of these ligases were attenuated with both prophylactic and therapeutic calpeptin treatments (P < 0.01). These results are consistent with our earlier findings of muscle atrophy in EAE rats and its attenuation in EAE-calpeptin animals.
Fig. 4.
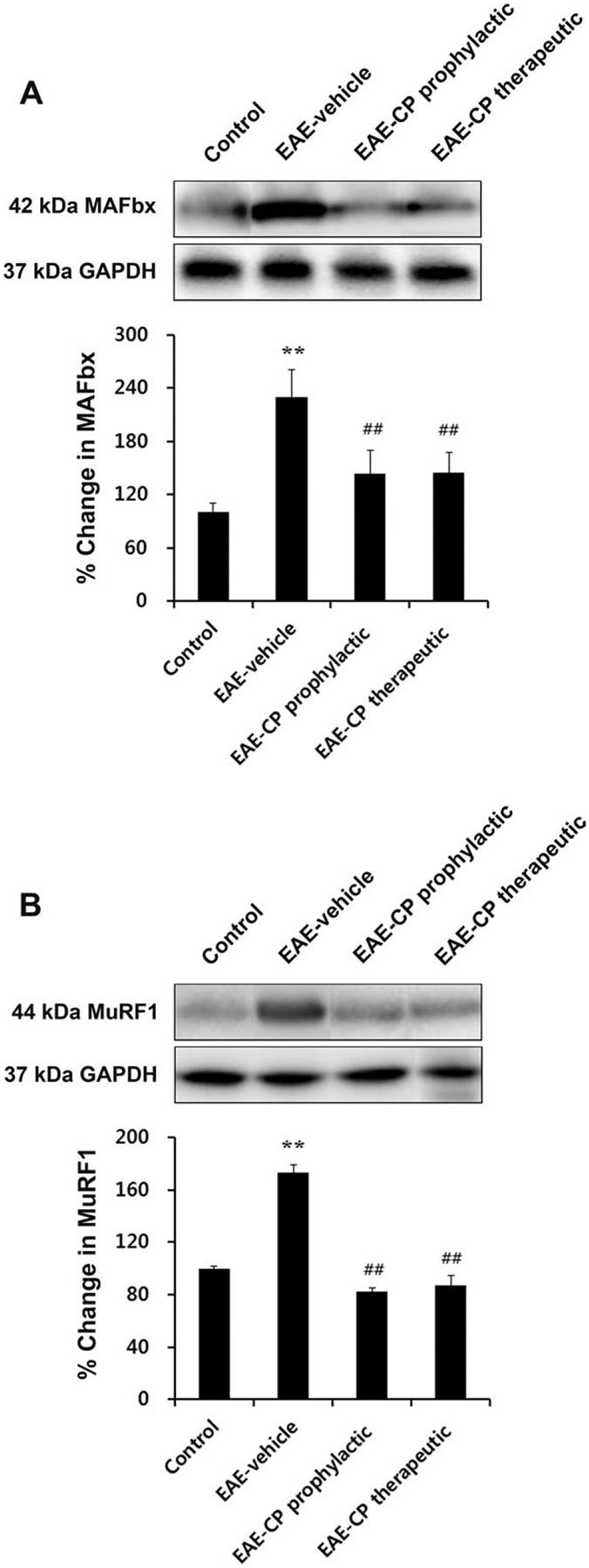
Alteration of protein degradation-related proteins resulting from EAE induction. Posttreatment with calpeptin significantly attenuated muscle-specific E3 ligase expression in EAE rat skeletal muscles. Representative Western blot and determination of percentage changes in 42-kDa MAFbx (A) and 44-kDa MuRF1 (B). *P < 0.05 vs. control, **P < 0.01 vs. control; #P < 0.05 vs. EAE-vehicle, ##P < 0.01 vs. EAE-vehicle. CP, calpeptin.
To identify whether inflammatory stimulation in vitro induces muscle-specific ubiquitin-proteasome ligases, we treated L6 rat myoblast cells with IFN-γ (500 U/ml) and examined the expression of MAFbx and MuRF1 proteins (Fig. 5). Both MAFbx and MuRF1 protein were significantly elevated with IFN-γ treatment alone (P < 0.01). Calpeptin treatment (1 and 5 μM) significantly attenuated the protein levels of MAFbx (P < 0.05) and MuRF1 (P < 0.05 in 1 μM and P < 0.01 in 5 μM; Fig. 5A,B).
Fig. 5.
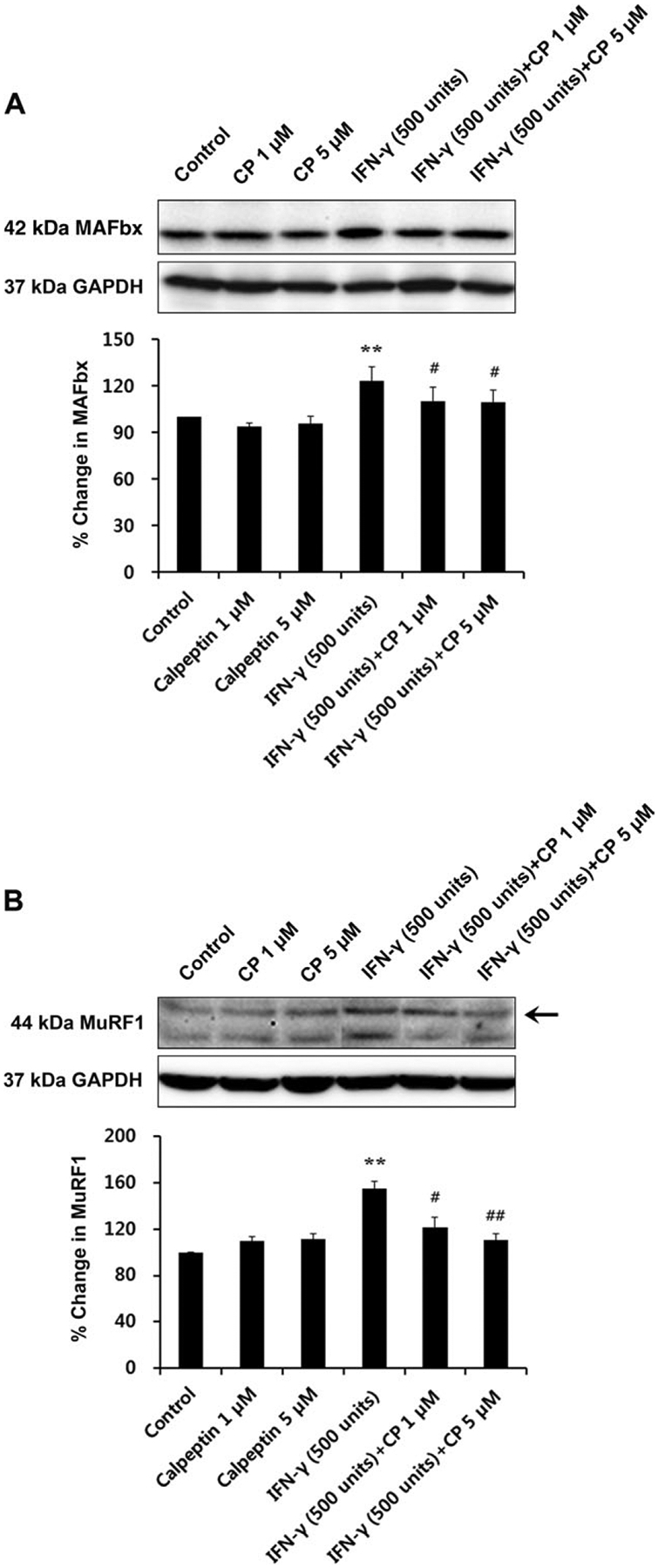
Changes in degradation-related protein expression by inflammatory stimulation (IFN-γ 500 U/ml) and their attenuation by calpeptin treatment (1 and 5 μM) in L6 rat skeletal myoblast cells. Representative Western blot and determination of percentage changes in 42-kDa MAFbx (A) and 44-kDa MuRF1 (B; n = 3 per group). *P < 0.05 vs. control, **P < 0.01 vs. control; #P < 0.05 vs. IFN-γ 500 U/ml, ##P < 0.01 vs. IFN-γ 500 U/ml. CP, calpeptin.
DISCUSSION
We studied skeletal muscle of EAE rats and found various morphological changes, such as smaller, irregularly shaped muscle fibers and fibrosis. In addition, we also noted upregulation of a variety of proinflammatory factors, apoptotic markers, and muscle-specific ubiqutin-proteasome ligases indicating potential mechanisms of muscle atrophy in EAE rats. Muscle atrophy in vehicle-treated EAE rats was associated with complete paralysis of the hind limbs and tail and weakness of the front limbs. Although these clinical manifestations have been attributed primarily to the CNS involvement, we anticipate that part of the weakness seen in EAE rats might be due to skeletal muscle changes and atrophy (Polfliet et al., 2002; Budde et al., 2008; Guyton et al., 2010).
We found that the average lesser diameter of muscle fibers was significantly decreased in EAE-vehicle rats compared with control animals, and calpeptin treatment both prophylactically and therapeutically significantly attenuated these changes, indicating that calpeptin administration attenuates muscle atrophy. Although we attempted to demonstrate inflammatory cell infiltration in muscle tissue from EAE animals, we found no significant differences between groups. Previous studies have demonstrated that inflammatory cell infiltration occurs around motor neurons and in the motor endplate in both EAE and MS (Vogt et al., 2009). However, to our knowledge, there is no evidence to support inflammatory cell infiltration into isolated muscle tissue in EAE animals at the acute phase. Therefore, it is possible that circulating cytokines are responsible for part of the muscle damage noted in EAE animals.
We have demonstrated upregulation of calpain and increased calpain:calpastin ratio in skeletal muscle sample of EAE rats. Calpastatin is an endogenous inhibitor of calpain and also is degraded as a suicide substrate by calpain upon Ca2+ influx and calpain activation (Blomgren et al., 1999). Therefore, increased calpain:calpastatin ratio indicates activation of calpain in EAE skeletal muscle. In addition, various morphological and molecular changes observed in the skeletal muscle of EAE rats were attenuated by calpeptin treatment. These findings suggest that calpain activation is an upstream mediator of the various molecular changes seen in skeletal muscle. Calpain is also a known activator of several proinflammatory transcription factors, including NF-κB through degradation of its inhibitor I-κB α (Han et al., 1999; Schaecher et al., 2004). Calpain inhibition has also been shown to reduce protein degradation (Huang and Forsberg, 1998; Purintrapiban et al., 2003; Xiao et al., 2003). Calpain may regulate the UPP at multiple levels, at least in part through the increased expression of E3 ubiquitin ligases (Bartoli and Richard, 2005; Cao et al., 2005; Smith et al., 2008). Calpain activation may also contribute to muscle atrophy through induction of apoptosis (Dupont-Versteegden, 2005, 2006). Our group has identified a role for calpain in the CNS pathogenesis of MS and EAE, including demyelination (Shields and Banik, 1999), axonal damage, loss of neurons and oligodendrocytes (Guyton et al., 2005), and modulation of proteins involved in apoptotic pathways (Das et al., 2008). In addition, calpain inhibition has been shown to attenuate disease severity in EAE (Guyton et al., 2005, 2010).
We identified increased expression of proinflammatory transcription factors, including NF-κB and STAT3, in EAE animals. NF-κB is involved in the regulation of COX-2, which is responsible for formation of inflammatory mediators, including prostaglandins, prostacyclin, and thromboxane (Nozaki et al., 2010). To this end, we observed increased COX-2 expression following disease onset in EAE animals. In addition, NF-κB/COX-2 signaling induces proinflammatory molecules, including cytokines (TNF-α, IL-1β, IL-2, IL-6, etc.), chemokines, and molecules for cell adhesion, and tissue-degrading enzymes such as matrix metalloproteinases (MMPs; Kumar et al., 2004; Li et al., 2008). Our studies demonstrate increased IL-2 expression in skeletal muscle of EAE rats. In addition, activation of NF-κB is required for inflammatory cytokine-induced loss of skeletal muscle proteins (Ladner et al., 2003; Li et al., 2008) and has been identified in inflammatory myopathies (Monici et al., 2003; Creus et al., 2009). Patients with MS occasionally complain of diffuse pain, which is difficult to explain by its CNS pathology. Thus, activation of NF-κB and resultant inflammatory changes in skeletal muscle may contribute to diffuse muscle pain and muscle atrophy in MS. We found that calpeptin treatment in EAE animals reversed all proinflammatory changes (NF-κB:I-κB α ratio, COX-2, IL-2, STAT3 expression) noted in skeletal muscle. Together, these findings indicate a role for the anti-inflammatory effects of calpain inhibition by calpeptin in preventing muscle atrophy.
In this study, EAE induction caused upregulation of MAFbx and MuRF1 protein levels in skeletal muscle, and these changes were attenuated with calpeptin treatment. Calpain-mediated pathways and the UPP are thought to work synergistically for proteolysis during muscle atrophy (Hasselgren and Fischer, 2001; Jackman and Kandarian, 2004). The proteasome is unable to degrade intact myofibrils, so muscle proteins (actin, myosin, etc.) must be released from the sarcomere prior to ubiquitination and degradation by the 26S proteasome. Ca2+-mediated activation of calpain contributes to the release of these proteins from the sarcomere (Solomon and Goldberg, 1996; Williams et al., 1999; Hasselgren and Fischer, 2001). Thus, it is possible that calpain initiates myofibril degradation and that the proteasome further removes all myofibrillar fragments to recycle amino acids (Solomon and Goldberg, 1996; Huang and Forsberg, 1998; Bartoli and Richard, 2005; Smith et al., 2008). Calpain inhibition can prevent muscle weakness and disruption of sarcomere structure during hind limb suspension (Salazar et al., 2010). In addition, calpastatin overexpression decreases muscle atrophy by 30% during the 10-day unloading period (Tidball and Spencer, 2002).
The demonstration of increased TUNEL-positive nuclei, Bax:Bcl-2 ratio, and caspase-12 protein expression in skeletal muscle of EAE rats suggests that apoptosis contributes to atrophy in EAE muscle. Bax is a mitochondrial, proapoptotic protein whose expression is increased in apoptotic muscle (Tews et al., 1997a; Alway et al., 2003a,b; Persinger et al., 2003). The increased Bax:Bcl-2 ratio noted herein is indicative of a mitochondria-directed apoptosis. Interestingly, levels of the antiapoptotic protein Bcl-2 were also upregulated in skeletal muscle of EAE rats. Controversy exists about the role of the Bcl-2-family proteins in muscle atrophy. Bcl-2 protein level in atrophied skeletal muscle was decreased in brachial plexus injury (Jin et al., 2001) but elevated in motor neuron disease (Tews et al., 1997a), denervation (Tews et al., 1997b), and myopathies (Olive and Ferrer, 1999) and was unchanged in heart failure (Persinger et al., 2003). Caspase-12 is a main enzyme involved in the initiation of apoptosis, and its activation occurs independently of the mitochondrial pathway (Dupont-Versteegden, 2005). Dysfunction of the sarcoplasmic reticulum leads to activation of caspase-3 following calpain-mediated activation of caspase-12 (Dupont-Versteegden, 2006). In addition to calpain, caspase-3 has also been implicated as a protease responsible for the process of myofibrillar protein breakdown, resulting in further muscle atrophy (Hasselgren and Fischer, 2001; Du et al., 2004). However, apoptosis can result in the absence of activated caspase, and caspase activation does not always trigger cell death (Garrido and Kroemer, 2004). Although caspase-3 is often used as a surrogate for apoptosis, this might not be justified in muscle (Dupont-Versteegden, 2006). We examined the caspase-3 protein levels in skeletal muscle of EAE rats and found that there was no significant difference compared with controls in any experimental group. Caspase-3 activity was increased in denervation-induced muscle atrophy (Siu and Alway, 2005), although it showed no change in muscle atrophy induced by hind limb suspension (Leeuwenburgh et al., 2005; Siu et al., 2005). Thus, these results indicate that the caspase activation may have distinct roles in the various models of muscle atrophy. In addition, caspase-independent apoptotic pathway can be involved in muscle atrophy through the release of apoptosis-inducing factor (AIF) and endonuclease G (EndoG) from mitochondria (Dupont-Versteegden, 2005, 2006).
We have identified histological and molecular changes in skeletal muscle of EAE rats, although their direct causative factors and mechanisms remain unknown. To study our hypothesis that circulating, proinflammatory cytokines may trigger muscle atrophy in MS/EAE by affecting muscle degradation, we examined MAFbx and MuRF1 protein levels in L6 rat myoblast cells treated with IFN-γ and found their expression to be increased. In addition, posttreatment with calpeptin attenuated changes favoring protein degradation. These results suggest that proinflammatory cytokines activate calpain to promote muscle degradation. Similar results were seen in muscle samples of EAE rats, so the results obtained using our in vitro model support our earlier hypothesis. Because EAE produces weight loss, the possibility of starvation-mediated muscular atrophy cannot be ignored. However, we do not think that the morphological and molecular changes seen in our study result from this phenomenon, because we sacrificed animals in the acute phase after disease onset.
In conclusion, we have demonstrated that morphological and molecular changes develop in skeletal muscle of EAE rats. These changes were attenuated by the calpain inhibitor calpeptin. Our results suggest that calpain may play a pivotal role in the pathogenesis of skeletal muscle damage in EAE, so calpain may be a potential target in EAE therapy.
Contract grant sponsor:
National Institutes of Health; Contract grant number: NS-056176; Contract grant number: NS-065456; Contract grant number: NS-041088; Contract grant number: NS048117; Contract grant sponsor: Department of Neurosciences, Medical University of South Carolina.
REFERENCES
- Allen DL, Linderman JK, Roy RR, Grindeland RE, Mukku V, Edgerton VR. 1997. Growth hormone/IGF-I and/or resistive exercise maintains myonuclear number in hindlimb unweighted muscles. J Appl Physiol 83:1857–1861. [DOI] [PubMed] [Google Scholar]
- Alway SE, Degens H, Krishnamurthy G, Chaudhrai A. 2003a. Denervation stimulates apoptosis but not Id2 expression in hindlimb muscles of aged rats. J Gerontol Ser A Biol Sci Med Sci 58:687–697. [DOI] [PubMed] [Google Scholar]
- Alway SE, Martyn JK, Ouyang J, Chaudhrai A, Murlasits ZS. 2003b. Id2 expression during apoptosis and satellite cell activation in unloaded and loaded quail skeletal muscles. AmJ Physiol Regul Integr Comp Physiol 284:R540–R549. [DOI] [PubMed] [Google Scholar]
- Ashley Z, Sutherland H, Lanmuller H, Russold MF, Unger E, Bijak M, Mayr W, Boncompagni S, Protasi F, Salmons S, Jarvis JC. 2007. Atrophy, but not necrosis, in rabbit skeletal muscle denervated for periods up to one year. Am J Physiol Cell Physiol 292:C440–C451. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Richard I. 2005. Calpains in muscle wasting. Int J Biochem Cell Biol 37:2115–2133. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Hallin U, Andersson AL, Puka-Sundvall M, Bahr BA, McRae A, Saido TC, Kawashima S, Hagberg H. 1999. Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. J Biol Chem 274:14046–14052. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. 2001. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294:1704–1708. [DOI] [PubMed] [Google Scholar]
- Budde MD, Kim JH, Liang HF, Russell JH, Cross AH, Song SK. 2008. Axonal injury detected by in vivo diffusion tensor imaging correlates with neurological disability in a mouse model of multiple sclerosis. NMR Biomed 21:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. 2004. IKK-beta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119:285–298. [DOI] [PubMed] [Google Scholar]
- Cao PR, Kim HJ, Lecker SH. 2005. Ubiquitin-protein ligases in muscle wasting. Int J Biochem Cell Biol 37:2088–2097. [DOI] [PubMed] [Google Scholar]
- Cornachione AS, Benedini-Elias PC, Polizello JC, Carvalho LC, Mattiello-Sverzut AC. 2011. Characterization of fiber types in different muscles of the hindlimb in female weanling and adult Wistar rats. Acta Histochem Cytochem 44:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creus KK, De Paepe B, De Bleecker JL. 2009. Idiopathic inflammatory myopathies and the classical NF-kappaB complex: current insights and implications for therapy. Autoimmun Rev 8:627–631. [DOI] [PubMed] [Google Scholar]
- Das A, Guyton MK, Matzelle DD, Ray SK, Banik NL. 2008. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res 86:2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan A, van der Vliet MR, Hendriks JJ, Heijnen DA, Dijkstra CD. 2004. Changes in characteristics of rat skeletal muscle after experimental allergic encephalomyelitis. Muscle Nerve 29:369–375. [DOI] [PubMed] [Google Scholar]
- Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE. 2004. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest 113:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont-Versteegden EE. 2005. Apoptosis in muscle atrophy: relevance to sarcopenia. Exp Gerontol 40:473–481. [DOI] [PubMed] [Google Scholar]
- Dupont-Versteegden EE. 2006. Apoptosis in skeletal muscle and its relevance to atrophy. World J Gastroenterol 12:7463–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley HL, Tisdale MJ. 2007. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J Biol Chem 282:7087–7097. [DOI] [PubMed] [Google Scholar]
- Fisher M, Long RR, Drachman DA. 1983. Hand muscle atrophy in multiple sclerosis. Arch Neurol 40:811–815. [DOI] [PubMed] [Google Scholar]
- Garrido C, Kroemer G. 2004. Life’s smile, death’s grin: vital functions of apoptosis-executing proteins. Curr Opin Cell Biol 16:639–646. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. 2005. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res 81:53–61. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Das A, Samantaray S, Wallace GCt, Butler JT, Ray SK, Banik NL. 2010. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res 88:2398–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR. 1999. Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J Biol Chem 274:787–794. [DOI] [PubMed] [Google Scholar]
- Hasselgren PO, Fischer JE. 2001. Muscle cachexia: current concepts of intracellular mechanisms and molecular regulation. Ann Surg 233:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Forsberg NE. 1998. Role of calpain in skeletal-muscle protein degradation. Proc Natl Acad Sci U S A 95:12100–12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman RW, Kandarian SC. 2004. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 287:C834–C843. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu Z, Tian T, Gu Y. 2001. Apoptosis in atrophic skeletal muscle induced by brachial plexus injury in rats. J Trauma 50:31–35. [DOI] [PubMed] [Google Scholar]
- Keegan BM, Noseworthy JH. 2002. Multiple sclerosis. Annu Rev Med 53:285–302. [DOI] [PubMed] [Google Scholar]
- Kumar A, Takada Y, Boriek AM, Aggarwal BB. 2004. Nuclear factor-kappaB: its role in health and disease. J Mol Med 82:434–448. [DOI] [PubMed] [Google Scholar]
- Ladner KJ, Caligiuri MA, Guttridge DC. 2003. Tumor necrosis factor-regulated biphasic activation of NF-kappa B is required for cytokine-induced loss of skeletal muscle gene products. J Biol Chem 278:2294–2303. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh C 2003. Role of apoptosis in sarcopenia. J Gerontol Ser A Biol Sci Med Sci 58:999–1001. [DOI] [PubMed] [Google Scholar]
- Leeuwenburgh C, Gurley CM, Strotman BA, Dupont-Versteegden EE. 2005. Age-related differences in apoptosis with disuse atrophy in soleus muscle. Am J Physiol Regul Integr Comp Physiol 288:R1288–R1296. [DOI] [PubMed] [Google Scholar]
- Li H, Malhotra S, Kumar A. 2008. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med 86:1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZQ, Kunimatsu M, Yang JP, Ozaki Y, Sasaki M, Okamoto T. 1996. Proteolytic processing of nuclear factor kappa B by calpain in vitro. FEBS Lett 385:109–113. [DOI] [PubMed] [Google Scholar]
- McFarland HF, Martin R. 2007. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 8:913–919. [DOI] [PubMed] [Google Scholar]
- McKinnell IW, Rudnicki MA. 2004. Molecular mechanisms of muscle atrophy. Cell 119:907–910. [DOI] [PubMed] [Google Scholar]
- Mitchell PO, Pavlath GK. 2004. Skeletal muscle atrophy leads to loss and dysfunction of muscle precursor cells. Am J Physiol Cell Physiol 287:C1753–C1762. [DOI] [PubMed] [Google Scholar]
- Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. 2003. Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology 60:993–997. [DOI] [PubMed] [Google Scholar]
- Ng AV, Miller RG, Gelinas D, Kent-Braun JA. 2004. Functional relationships of central and peripheral muscle alterations in multiple sclerosis. Muscle Nerve 29:843–852. [DOI] [PubMed] [Google Scholar]
- Nozaki K, Das A, Ray SK, Banik NL. 2010. Calpain inhibition attenuates intracellular changes in muscle cells in response to extracellular inflammatory stimulation. Exp Neurol 225:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Das A, Ray SK, Banik NL. 2011. Calpeptin attenuated apoptosis and intracellular inflammatory changes in muscle cells. J Neurosci Res 89:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda A, Wakao H, Fujita H. 2002. Calpain is a signal transducer and activator of transcription (STAT) 3 and STAT5 protease. Blood 99:1850–1852. [DOI] [PubMed] [Google Scholar]
- Olive M, Ferrer I. 1999. Bcl-2 and Bax protein expression in human myopathies. J Neurol Sci 164:76–81. [DOI] [PubMed] [Google Scholar]
- Persinger R, Janssen-Heininger Y, Wing SS, Matthews DE, LeWinter MM, Toth MJ. 2003. Effect of heart failure on the regulation of skeletal muscle protein synthesis, breakdown, and apoptosis. Am J Physiol Endocrinol Metab 284:E1001–E1008. [DOI] [PubMed] [Google Scholar]
- Polfliet MM, van de Veerdonk F, Dopp EA, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. 2002. The role of perivascular and meningeal macrophages in experimental allergic encephalomyelitis. J Neuroimmunol 122:1–8. [DOI] [PubMed] [Google Scholar]
- Purintrapiban J, Wang MC, Forsberg NE. 2003. Degradation of sarcomeric and cytoskeletal proteins in cultured skeletal muscle cells. Comp Biochem Physiol B Biochem Mol Biol 136:393–401. [DOI] [PubMed] [Google Scholar]
- Ray SK, Schaecher KE, Shields DC, Hogan EL, Banik NL. 2000. Combined TUNEL and double immunofluorescent labeling for detection of apoptotic mononuclear phagocytes in autoimmune demyelinating disease. Brain Res Brain Res Protoc 5:305–311. [DOI] [PubMed] [Google Scholar]
- Rieckmann P, Albrecht M, Kitze B, Weber T, Tumani H, Broocks A, Luer W, Helwig A, Poser S. 1995. Tumor necrosis factor-alpha messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann Neurol 37:82–88. [DOI] [PubMed] [Google Scholar]
- Salazar JJ, Michele DE, Brooks SV. 2010. Inhibition of calpain prevents muscle weakness and disruption of sarcomere structure during hindlimb suspension. J Appl Physiol 108:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaecher K, Goust JM, Banik NL. 2004. The effects of calpain inhibition on IκB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res 29:1443–1451. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. 1999. Pathophysiological role of calpain in experimental demyelination. J Neurosci Res 55:533–541. [DOI] [PubMed] [Google Scholar]
- Siu PM, Alway SE. 2005. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J Physiol 565:309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM, Pistilli EE, Alway SE. 2005. Apoptotic responses to hindlimb suspension in gastrocnemius muscles from young adult and aged rats. Am J Physiol Regul Integr Comp Physiol 289:R1015–R1026. [DOI] [PubMed] [Google Scholar]
- Smith IJ, Lecker SH, Hasselgren PO. 2008. Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab 295:E762–E771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon V, Goldberg AL. 1996. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem 271:26690–26697. [DOI] [PubMed] [Google Scholar]
- Tews DS. 2002. Apoptosis and muscle fibre loss in neuromuscular disorders. Neuromusc Disord 12:613–622. [DOI] [PubMed] [Google Scholar]
- Tews DS, Goebel HH, Meinck HM. 1997a. DNA-fragmentation and apoptosis-related proteins of muscle cells in motor neuron disorders. Acta Neurol Scand 96:380–386. [DOI] [PubMed] [Google Scholar]
- Tews DS, Goebel HH, Schneider I, Gunkel A, Stennert E, Neiss WF. 1997b. DNA-fragmentation and expression of apoptosis-related proteins in experimentally denervated and reinnervated rat facial muscle. Neuropathol Appl Neurobiol 23:141–149. [PubMed] [Google Scholar]
- Tidball JG, Spencer MJ. 2002. Expression of a calpastatin transgene slows muscle wasting and obviates changes in myosin isoform expression during murine muscle disuse. J Physiol 545:819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilbery CP, Atra M, Oliveira AS, Calia LA, Schmidt B. 1989. [Histochemical study of the skeletal muscle in multiple sclerosis]. Arquivos Neuropsiquiatria 47:337–345. [DOI] [PubMed] [Google Scholar]
- Vogt J, Paul F, Aktas O, Muller-Wielsch K, Dorr J, Dorr S, Bharathi BS, Glumm R, Schmitz C, Steinbusch H, Raine CS, Tsokos M, Nitsch R, Zipp F. 2009. Lower motor neuron loss in multiple sclerosis and experimental autoimmune encephalomyelitis. Ann Neurol 66:310–322. [DOI] [PubMed] [Google Scholar]
- Wei W, Yang H, Cao P, Menconi M, Chamberlain C, Petkova V, Hasselgren PO. 2006. Degradation of C/EBPbeta in cultured myotubes is calpain-dependent. J Cell Physiol 208:386–398. [DOI] [PubMed] [Google Scholar]
- Williams AB, Decourten-Myers GM, Fischer JE, Luo G, Sun X, Hasselgren PO. 1999. Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-dependent mechanism. FASEB J 13:1435–1443. [DOI] [PubMed] [Google Scholar]
- Xiao YY, Wang MC, Purintrapiban J, Forsberg NE. 2003. Roles of mucalpain in cultured L8 muscle cells: application of a skeletal muscle-specific gene expression system. Comp Biochem Physiol Toxicol Pharmacol 134:439–450. [DOI] [PubMed] [Google Scholar]