

Background: It is not known whether a β-blocker, metoprolol, induces physiological responses through β-arrestins in vivo.
Results: Long-term administration of metoprolol induced cardiac fibrosis in wild type but not β-arrestin2- or GRK5 knock-out mice.
Conclusion: Metoprolol induced cardiac fibrosis in a G protein-independent and GRK5/β-arrestin2-dependent manner.
Significance: Our study provides a physiological significance of β-arrestin-mediated biased signaling pathway by a β-blocker in vivo.
Keywords: Bret, Cardiovascular, Cell Signaling, Fibrosis, Receptors, GRK, β-Arrestin, β-Blocker
Abstract
G-protein coupled receptors (GPCRs) have long been known as receptors that activate G protein-dependent cellular signaling pathways. In addition to the G protein-dependent pathways, recent reports have revealed that several ligands called “biased ligands” elicit G protein-independent and β-arrestin-dependent signaling through GPCRs (biased agonism). Several β-blockers are known as biased ligands. All β-blockers inhibit the binding of agonists to the β-adrenergic receptors. In addition to β-blocking action, some β-blockers are reported to induce cellular responses through G protein-independent and β-arrestin-dependent signaling pathways. However, the physiological significance induced by the β-arrestin-dependent pathway remains much to be clarified in vivo. Here, we demonstrate that metoprolol, a β1-adrenergic receptor-selective blocker, could induce cardiac fibrosis through a G protein-independent and β-arrestin2-dependent pathway. Metoprolol, a β-blocker, increased the expression of fibrotic genes responsible for cardiac fibrosis in cardiomyocytes. Furthermore, metoprolol induced the interaction between β1-adrenergic receptor and β-arrestin2, but not β-arrestin1. The interaction between β1-adrenergic receptor and β-arrestin2 by metoprolol was impaired in the G protein-coupled receptor kinase 5 (GRK5)-knockdown cells. Metoprolol-induced cardiac fibrosis led to cardiac dysfunction. However, the metoprolol-induced fibrosis and cardiac dysfunction were not evoked in β-arrestin2- or GRK5-knock-out mice. Thus, metoprolol is a biased ligand that selectively activates a G protein-independent and GRK5/β-arrestin2-dependent pathway, and induces cardiac fibrosis. This study demonstrates the physiological importance of biased agonism, and suggests that G protein-independent and β-arrestin-dependent signaling is a reason for the diversity of the effectiveness of β-blockers.
Introduction
G protein-coupled receptors (GPCRs)2 mediate physiological responses to a variety of ligands, such as hormones, neurotransmitters, and environmental stimuli, and are tightly regulated by several mechanisms (1). Among the mechanisms, GPCR kinase (GRK) and β-arrestin-mediated events are known as general mechanisms of GPCR functional regulation (2–6). When an agonist binds to GPCR, it activates a cellular signal transduction cascade through G proteins, but also induces GRK/β-arrestin-mediated events to prevent excess stimulation. GRKs phosphorylate the agonist-bound GPCRs, and β-arrestins bind to the phosphorylated receptors, and then inhibit further stimulation of G proteins by the agonist-bound receptors through steric hinderance. As β-arrestins can also bind clathrin and adaptor proteins, the phosphorylated and β-arrestin-bound receptors are internalized into endocytic vesicles (7). The internalized receptors then are recycled back to the plasma membrane or directed to the degradation pathway by undefined processes (8, 9). In addition to the receptor regulation, it has been recently recognized that GRKs and β-arrestins mediate cellular signaling by GPCRs independently of G protein activation (2–4, 10, 11). However, these GRK- and β-arrestin-dependent signaling pathways are largely demonstrated by in vitro cellular systems. To establish the physiological importance of the signaling pathways, it is essential to demonstrate GRK- and β-arrestin-dependent responses in vivo.
β-Adrenergic receptors are members of the G protein-coupled receptor family and are pharmacologically and genetically subdivided into three subtypes: β1, β2, and β3 (12). The β1-adrenergic receptor is involved in the regulation of cardiac functions as well as in the induction and development of various cardiovascular diseases including heart failure (13, 14). During heart failure, a large amount of catecholamine is believed to be released from synaptic ends (15, 16), and exerts harmful effects on the heart. Therefore, administration of β-adrenergic receptor antagonists, β-blockers, is effective for patients in the early stages of heart failure (17). So far, various kinds of β-blockers have been developed, but many basic and clinical studies indicate that the effect of each β-blocker is divergent (18). Several reasons, including receptor selectivity, antioxidant property, and membrane-stabilizing effects have been proposed for the diversity, but there still remains much to be known. Understanding the reasons more properly would help to develop a more efficient β-blocker with fewer side effects.
In addition to the blocking effects, some β-blockers have been shown to evoke signal transduction through the β-adrenergic receptors in G protein-independent and β-arrestin-dependent manner (19–21). Like β-blockers, some agonists for seven-transmembrane receptors have been reported not only to transmit the classic G protein signaling, but also to generate the signaling in a G protein-independent fashion (22–25). These agonists and phenomena are called “biased ligands” and “biased agonism,” respectively (11). To date, the signal transduction pathways by biased ligands have been intensively unveiled by in vitro studies. However, the physiological meaning of the biased ligand-mediated signaling is hardly clarified in vivo.
We found that long-term administration of metoprolol, an inverse agonist for the β1-adrenergic receptor, to mice induced cardiac fibrosis, which is thought to be deleterious for the heart by stiffening it and inhibiting the electrical conductivity between cardiomyocytes. We revealed that this fibrotic pathway was mediated through the β1-adrenergic receptor in a G protein-independent manner. We also discovered that β-arrestin2 and GRK5 are involved in this G protein-independent fibrotic pathway not only in vitro but also in vivo.
EXPERIMENTAL PROCEDURES
Animals and Administration of β-Blockers
We purchased ddY, C57BL/6J mice, and Sprague-Dawley rats from KYUDO (Japan). GRK5-, GRK6-, and β-arrestin2-KO mice were obtained by Drs. R. J. Lefkowitz and R. T. Premont (Duke University). Administration of β-blockers was started at 6 weeks of age of male mice. β-Blockers (metoprolol, 30 mg/kg/day; carvedilol, 30 mg/kg/day; propranolol, 10 mg/kg/day) or saline were orally administered twice a day for 3 months. All experiments using mice and rats were approved by the guidelines of Kyushu University.
Isolation of Cardiac Cells and Transfection
Rat neonatal cardiomyocytes and cardiac fibroblasts were isolated as described previously (26, 27). In brief, 1-day-old Sprague-Dawley rats are deeply anesthetized and their hearts were expeditiously extirpated on ice. The atria of the extirpated hearts are quickly removed by using sterilized forceps and scissors. The remaining hearts were cut into small pieces and digested by collagenase. The digested cells were plated on 10-cm plates for 1 h at 37 °C and nonattached cells or attached cells were used as cardiomyocytes or cardiac fibroblasts, respectively. The cardiomyocytes are plated on gelatin-coated 6-well plates (3 × 104 cells/well). When siRNAs were transfected into cardiomyocytes, Lipofectamine 2000 was used. Plasmid DNAs were transfected into the rat neonatal cardiomyocytes or H9c2 cells by electroporation and into HEK293 cells by FuGENE6 (Roche Applied Science).
BRET Assay
To evaluate the interaction between β1-adrenergic receptor and β-arrestin2, or the conformational changes in β-arrestin2, BRET experiments were performed as described previously (28, 29). The plasmids of β1-adrenergic receptor-Rluc, GFP2-β-arrestin1, and β-arrestin2-GFP2 were transfected into HEK293 cells, H9c2 cells, or rat neonatal cardiomyocytes. Rluc-β-arrestin2-GFP2 was expressed in HEK293 or H9c2 cells. These cells stably expressed the β1-adrenergic receptor. Forty-eight hours after the transfection, the cells were collected and washed by DMEM/F12 (Invitrogen). The cells were suspended in the assay buffer (0.1 g/liter of CaCl2, 0.1 g/liter of MgCl2·6H2O, 1 g/liter of d-glucose, and 2 μg/ml of aprotinin in PBS) to adjust 5 × 104 cells/ml. Then, 25 μl of the cell suspension was distributed in a 96-well white microplate and 25 μl of the drug solution was added. After a 5-min incubation, DeepBlueCTM was added at a final concentration of 5 μm and the 400 nm (Rluc) and 515 nm (GFP2) emissions were immediately measured using a Multilabel Reader Mithras LB 940 (Berthold Technologies).
FRET Assay
The FRET probe to monitor the changes of the concentration of cAMP was constructed as reported previously (30). HEK293 cells stably overexpressing the β1-adrenergic receptor were plated on poly-l-lysine-coated 35-mm glass-bottom base dishes and transfected with the FRET probe. Two days after transfection, the culture medium was replaced by phenol red-free medium (DMEM/F-12) containing 0.1% fetal bovine serum, and more than 3 h later, the cells were imaged by the microscopy (Olympus IX-81) in a heated chamber kept at 37 °C. In some experiments of β-blocker stimulation, drugs other than β-blockers were added before the stimulation. ICI118551 (50 μm) or pertussis toxin (100 ng/ml) were added 10 min or 16 h before the stimulation of β-blockers, respectively. 3-Isobutyl-1-methylxanthine (1 mm) was added simultaneously with each β-blocker. The images for CFP, YFP, and DIC were taken every 1 min. Image analysis was performed by MetaMorph software (Universal imaging).
Histological Analysis
Hearts from mice were extirpated and fixed by paraformaldehyde. The fixed hearts were embedded and sectioned (3 μm thickness). The heart sections were stained with hematoxylin and eosin (H&E), Masson's trichrome, or picrosirius red. The digital images for the stained sections were taken by a Biozero microscope (BZ-8000, Keyence). The degree of fibrosis in the images was estimated by BZ-analyzer (BZ-9000, Keyence).
Real Time RT-PCR
Total RNA was extracted from mouse hearts using RNeasy fibrous tissue kit (Qiagen) according to the manufacturer's instructions. Real time RT-PCR was performed as described previously by TaqMan RT-PCR (26). The results were normalized to 18 S RNA or Gapdh. Sequences for PCR primers and TaqMan probes were described in a previous report (26).
Echocardiographic and Hemodynamic Measurement
Left ventricular functions were assessed by transthoracic echocardiography. Recordings were performed using Nemio GX image analyzing system (SSA-580A, Toshiba Medical Systems). Under anesthesia, a micromanometer and conductance 1.4F catheter (SPR-671, Millar Instruments) was inserted into left ventricle. After stabilization, the signals were recorded. The signals were analyzed by Digi-Med Heart Performance Analyzer (Micro-Med) and Digi-Med System Integrator (Micro-Med).
Statistical Analysis
The results are presented as mean ± S.E. from at least three independent experiments. Statistical comparisons were performed with Student's t test (for two groups) or analysis of variance followed by Student-Newman-Keuls procedure (for multiple groups).
RESULTS
Administration of Metoprolol to Wild Type Normal Mice Induces Cardiac Fibrosis
During the analysis of the effects of β-blockers on heart failure, we found that long-term treatment with metoprolol, but not with propranolol and carvedilol, to wild type normal ddY mice induced perivascular fibrosis in their hearts (Fig. 1, A and B). The metoprolol-induced cardiac fibrosis was confirmed by quantitative mRNA analysis, which demonstrates that the expression of fibrotic genes, such as Col1a (encoding Collagen 1α (Col1a)) and Ctgf (encoding connective tissue growth factor (CTGF)) (31, 32), was increased in metoprolol-treated mice (Fig. 1C). Although increased fibrosis with metoprolol was observed with ddY mice, knock-out mice are not available for ddY mice. We then examined the effects of metoprolol on cardiac fibrosis in C57BL/6J mice (Fig. 1D) and found that metoprolol-induced cardiac fibrosis is independent of strain. We next examined whether metoprolol-induced fibrosis affects cardiac morphology and functions. Echocardiography and catheter measurements of metoprolol-treated mice revealed that ventricular wall thickness is decreased (supplemental Fig. S1). Among the parameters of the cardiac functions, one of the best indicators for defects of cardiac functions by fibrosis is dP/dtmin (26, 33). The value of dP/dtmin was significantly decreased by metoprolol treatment (Fig. 1E), suggesting that metoprolol-induced cardiac fibrosis is associated with a decrease in dP/dtmin. However, metoprolol did not induce hypertrophy.
FIGURE 1.

Metoprolol-induced cardiac fibrosis. A, picrosirius red staining of perivascular region of left ventricles of ddY mice after treatment of saline (Control (Con)) or β-blockers (Metoprolol (Met), Propranolol (Pro), Carvedilol (Car)). Paraffin sections of hearts (3 μm thickness) were prepared from mice, to which each β-blocker was administered. The sections were stained with picrosirius red and observed under microscopy. Representative images are shown. Scale bar, 15 μm. B, quantification of collagen volume fraction from A (n = 5). **, p < 0.01. C, up-regulation of mRNA expressions of angiotensin converting enzyme (ACE), Periostin, Collagen 1a (Col1a), α-skeletal actin, and Ctgf (CTGF) in mouse hearts by β-blockers. The expression levels of the fibrotic genes were evaluated by real-time RT-PCR. The fold-increases were calculated by the values of saline-treated mice set as 1 (n = 3–5). **, p < 0.01. D, Masson's trichrome staining of perivascular and interstitial regions of left ventricles from saline- or metoprolol-treated C57BL/6J mice (n = 5–6). Typical images are shown. Arrows indicate the fibrotic region. Scale bar: 15 μm on perivascular images, 30 μm on interstitial images. Collagen volume fraction on each image was quantified and the data were shown below the images. *, p < 0.05. E, hemodynamic parameters (dP/dtmax or dP/dtmin) of the saline- or metoprolol-treated C57BL/6J mice. Saline or metoprolol was administered to mice for 3 months. After the administration, they were subjected to hemodynamic measurements. The parameters dP/dtmax or dP/dtmin represent the maximal rate of pressure development, maximal rate of decay of pressure, respectively. Error bars show S.E. (n = 15). *, p < 0.05; **, p < 0.01.
Metoprolol-induced Fibrotic Response Was Independent of G-protein Signaling
To investigate the molecular mechanism of cardiac fibrosis with metoprolol, we first examined whether metoprolol couples with Gs or Gi. HEK293 cells stably expressing the β1-adrenergic receptor were established, and cAMP accumulation by various β-blockers was then monitored in the presence of ICI118551, a β2-adrenergic receptor-selective blocker. ICI118551 was included to exclude the involvement of the β2-adrenergic receptor. In this study, cAMP accumulation was measured by a fluorescence resonance energy transfer probe, as described elsewhere (30). Fig. 2A shows that several β-blockers increased cAMP accumulation, but metoprolol did not. This result indicates that metoprolol does not activate Gs.
FIGURE 2.

Metoprolol-induced fibrotic responses did not depend on G protein signaling. A, cAMP accumulation in HEK293 cells expressing β1-adrenergic receptors by β-blockers (10 μm) in the presence of ICI118551 (50 μm). HEK293 cells stably expressing β1-adrenergic receptor were transiently transfected with a FRET-based cAMP biosensor. After 15 min of pre-treatment with ICI118551, β-blocker stimulation was performed on the cells and the changes of the fluorescence were followed under microscopy. non, nonstimulated cell; Iso, isopreterenol; Met, metoprolol; CGP, CGP20712A; Bis, bisoprolol; ICI, ICI18,551; Pro, propranolol; Car, carvedilol; Lab, labetalol; Pin, pindolol. ΔFRET ratios were determined at 5 min after the stimulation. The experiments were repeated three times and each result was obtained from 13 to 20 cells. **, p < 0.01; ***, p < 0.001. B, pertussis toxin (PTX) treatment did not influence cAMP accumulation by metoprolol. PTX treatment (100 ng/ml) was performed 16 h before the co-addition of 3-isobutyl-1-methyxanthine (IBMX) (1 mm) and β-blockers (Metoprolol, CGP-20712A) (10 μm) on HEK293 cells stably expressing β1-adrenergic receptor, and cAMP accumulation was determined by FRET using microscopy. The experiments were repeated three times and each result was obtained from 6 cells.
We also examined whether metoprolol activates Gi. Inclusion of a phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine, increased cAMP accumulation. Metoprolol inhibited cAMP accumulation (Fig. 2B), consistent with the report that metoprolol is an inverse agonist (34). The decreased cAMP accumulation by metoprolol was not affected by pertussis toxin treatment, indicating that metoprolol did not activate Gi (Fig. 2B). CGP-20712A is another inverse agonist and inhibited cAMP response similarly to metoprolol (35). These results demonstrate that metoprolol does not require Gs and Gi to induce a fibrotic response.
Metoprolol Induced the Expression of Fibrotic Genes in Cardiomyocytes
We examined whether metoprolol up-regulates the expression of fibrotic genes in cardiomyocytes and cardiac fibroblasts. Real time PCR experiments revealed that treatment of rat neonatal cardiomyocytes with metoprolol increased the expression of fibrotic genes, Tgfβ (encoding TGF-β), Ctgf, and Col1a (Fig. 3A). On the other hand, carvedilol stimulation did not induce expression of Tgfβ in rat neonatal cardiomyocytes (supplemental Fig. S2). In contrast to the results in cardiomyocytes, metoprolol did not increase the expression of fibrotic genes in cardiac fibroblasts, which mainly contribute to cardiac fibrosis (Fig. 3B). These results suggest that the induction of fibrotic genes by metoprolol is evoked through the β-adrenergic receptor in cardiomyocytes.
FIGURE 3.

Metoprolol up-regulated expressions of fibrotic genes in rat neonatal cardiomyocytes but not in cardiac fibroblasts. Metoprolol-induced induction of fibrotic genes in rat neonatal cardiomyocytes (A) or cardiac fibroblasts (B) were determined (n = 3 for cardiomyocytes, and n = 8 for cardiac fibroblast). Rat neonatal cardiomyocytes or cardiac fibroblasts were stimulated with 10 μm metoprolol. The expression levels of fibrotic genes (TGF-β, Ctgf (CTGF), and Col1a (Col1a)) after the stimulation were determined by real time RT-PCR. *, p < 0.05.
Metoprolol-induced Response through β1-Adrenergic Receptor Depends on β-Arrestin2
Recent reports demonstrated that β-blockers activate β-adrenergic receptor-mediated and G protein-independent signaling (19–21). In several studies, G protein-independent signaling requires β-arrestins. Thus, we examined whether the induction of cardiac fibrosis by metoprolol treatment required β-arrestins. At first, we examined whether β-arrestin1/2 interacts with the β1-adrenergic receptor using by BRET assay. The BRET assay is a powerful technique to detect the weak interaction between an activated receptor and its downstream molecules (24). The β1-adrenergic receptor tagged with Renilla luciferase (Rluc) and β-arrestin1 fused with GFP2 or β-arrestin2 fused with GFP2 (supplemental Fig. S3A) were transiently co-expressed in HEK293 cells. The metoprolol-stimulated BRET signal was then measured. Stimulation of the cells with isoproterenol, a full agonist for β-adrenergic receptors, strongly increased the BRET ratio in both cells expressing β1-adrenergic receptor-Rluc and GFP2-β-arrestin1 or expressing β1-adrenergic receptor-Rluc and β-arrestin2-GFP2. When metoprolol were added to the cells, the BRET ratio was increased only in cells expressing β1-adrenergic receptor-Rluc and β-arrestin2, although the interaction is fairly weak compared with that of isoproterenol (Fig. 4A). This result indicates that the β1-adrenergic receptor interacts weakly with β-arrestin2 but not β-arrestin1 by the binding of metoprolol to the receptor. As HEK293 cells are different from cardiomyocytes in some aspects, we further performed similar experiments with cardiomyocytes and a rat heart myoblast cell line, H9c2. Isoproterenol increased the BRET ratios in H9c2 cells (Fig. 4B) and cardiomyocytes (Fig. 4C). Metoprolol also increased the BRET ratios in both cells, although the extents of metoprolol-induced BRET ratios in H9c2 cells were small as compared with those in HEK293 cells. The reason of small changes in metoprolol-induced BRET ratios in H9c2 cells is unknown. As isoproterenol and metoprolol activate β-arrestin2 through the β1-adrenergic receptor, we next examined the conformational changes of β-arrestin2 by metoprolol and isoproterenol stimulation by a BRET assay using a β-arrestin2-based biosensor, Rluc-β-arrestin2-GFP2 (28) (supplemental Fig. S3B). The biosensor was reported to detect the conformational changes of β-arrestin2 caused by both G protein-dependent and -independent pathways through GPCRs such as the angiotensin II receptor (28, 36). As shown in Fig. 4, D and E, the BRET ratios were increased by isoproterenol stimulation of HEK293 cells expressing the β1-adrenergic receptor and H9c2 cells expressing the β1-adrenergic receptor. In contrast, metoprolol stimulation decreased it, indicating that conformation of β-arrestin2 by metoprolol stimulation was different from that by isoproterenol stimulation (Fig. 4, D and E).
FIGURE 4.
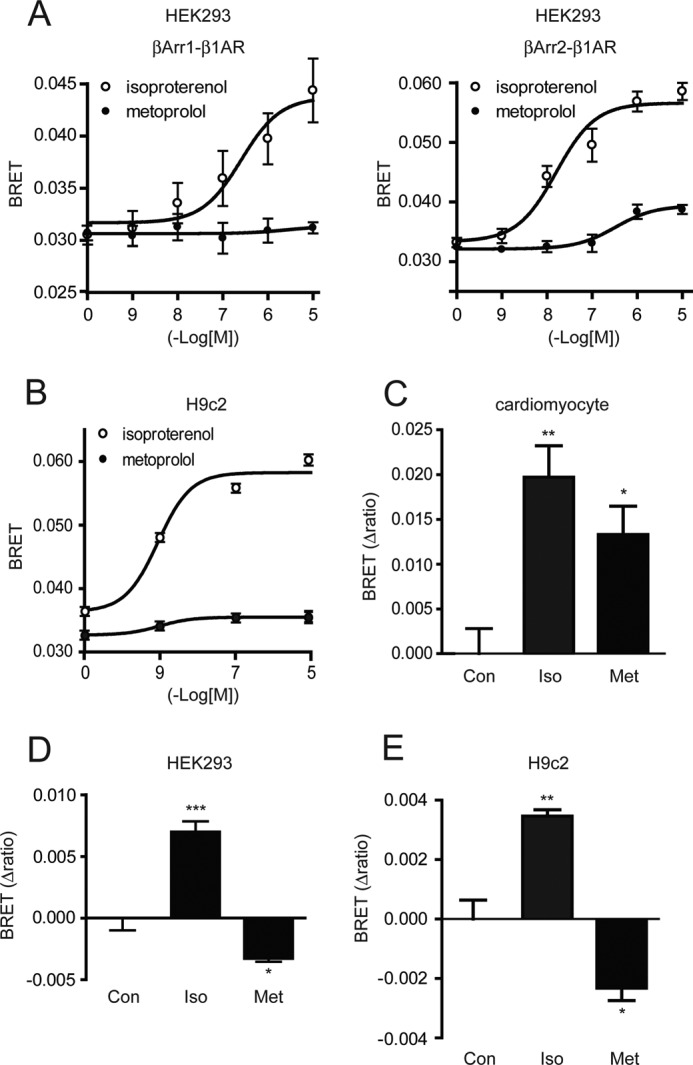
Requirement of β-arrestin2 for metoprolol-induced responses. A–C, the changes in BRET ratios by metoprolol or isoproterenol stimulation for 5 min in HEK293 cells (A), H9c2 cells (B), or cardiomyocytes (C). A, HEK293 cells were transfected with β1-adrenergic receptor-Rluc and GFP2-β-arrestin1 (left panel, βArr1-β1AR) or β1-adrenergic receptor-Rluc and β-arrestin2-GFP2 (right panel, βArr2-β1AR) (n = 4). B and C, H9c2 cells (B) or rat neonatal cardiomyocytes (C) were transfected with β1-adrenergic receptor-Rluc and β-arrestin2-GFP2 (n = 3). They were harvested 48 h after the transfection, washed with PBS, and split into 96-well plates. Then, they were incubated with isoproterenol or metoprolol at the indicated concentrations for 5 min. After the incubation, Rluc substrate, DeepBlueCTM was added to a final concentration of 5 μm. BRET signal was determined by calculating the ratio of the light emitted by the GFP2 and the light emitted by RLuc. D and E, the cells were transfected with Rluc-β-arrestin2-GFP2 (n = 4 for HEK293 cells and n = 3 for H9c2 cells). Then, the changes in BRET ratios of Rluc-β-arrestin2-GFP2 were determined after metoprolol (10 μm) or isoproterenol stimulation (10 μm) for 5 min in HEK293 cells expressing β1-adrenergic receptor (D) or H9c2 cells expressing β1-adrenergic receptor (E). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Metoprolol-induced Fibrosis in Mice Hearts Depends on β-Arrestin2
Next, we examined whether β-arrestin2 is required for metoprolol-induced cardiac fibrosis in vivo. Metoprolol was orally administered to β-arrestin2-KO mice twice a day for 3 months. Cardiac functions and fibrosis were then examined. Metoprolol treatment induced fibrosis in the perivascular and interstitial regions of wild type but not β-arrestin2-KO hearts (Fig. 5, A and B). Up-regulation of a fibrotic gene, Ctgf, with metoprolol was inhibited in β-arrestin2-KO mice (Fig. 5C). Consistent with these results, metoprolol-induced impairment of cardiac function was not observed in β-arrestin2-KO mice (Fig. 5D). These results demonstrate that β-arrestin2 is essential for metoprolol-induced cardiac fibrosis.
FIGURE 5.

Requirement of β-arrestin2 for metoprolol-induced cardiac fibrosis. A and B, cardiac fibrosis in perivascular (A) or interstitial (B) regions of left ventricles of wild type or β-arrestin2-KO mice by metoprolol. Metoprolol was orally administered to wild type or β-arrestin2-KO mice for 3 months. The degrees of their cardiac fibrosis were evaluated by picrosirius staining using paraffin sections of their hearts. *, p < 0.05; ***, p < 0.001. C, CTGF mRNA expression by metoprolol in wild type or β-arrestin2-KO mice. *, p < 0.05. Mouse heart total RNA was prepared from wild-type or β-arrestin2 knock-out mice (n = 3), which underwent metoprolol administration for 3 months. CTGF expression levels were evaluated by real time RT-PCR. D, determination of dP/dtmin of wild type or β-arrestin2-KO mice. The metoprolol-administered wild type or β-arrestin2-KO mice were subjected to hemodynamic measurements (n = 7–12). ***, p < 0.001.
Metoprolol-induced Fibrosis in Mice Hearts Depends on GRK5
As GRK5 and GRK6 are often involved in a G protein-independent and β-arrestin2-dependent pathway (20, 22, 23, 36, 37), we examined their potential involvement in metoprolol-induced cardiac fibrosis. At first, we knocked down GRK5 or GRK6 in HEK293 cells (supplemental Fig. S4, A and B) and measured the interaction between the β1-adrenergic receptor and β-arrestin2 with the BRET assay. Knockdown of GRK5 but not GRK6 inhibited the metoprolol-stimulated interaction of the β1-adrenergic receptor with β-arrestin2 (Fig. 6A). Metoprolol was next administered to GRK5- or GRK6-KO mice. Cardiac dysfunction reflected by a decrease of dP/dtmin was observed in wild type and GRK6-KO mice, but not in GRK5-KO mice (Fig. 6B). Metoprolol-induced cardiac fibrosis (Fig. 6, C and D) was not evoked in β-arrestin2-KO and GRK5-KO mice. These results demonstrate that GRK5 but not GRK6 is required for metoprolol-induced cardiac fibrosis.
FIGURE 6.

Requirement of GRK5 for metoprolol-induced cardiac fibrosis. A, interaction between β1-adrenergic receptor and β-arrestin2 by metoprolol (10 μm) or isoproterenol (1 nm, 100 nm) stimulation in GRK5- or GRK6-knockdown cells (n = 4). HEK293 cells were transiently co-transfected with siRNA (control siRNA, GRK5 siRNA, or GRK6 siRNA), β1-adrenergic receptor-Rluc, and β-arrestin2-GFP2. Two days after transfection, BRET measurement was performed. *, p < 0.05. B, the changes of dP/dtmin in GRK5- or GRK6-KO mice (n = 7–14). Metoprolol was administered to wild type, GRK5-KO, or GRK6-KO mice for 3 months. After the administration, they were subjected to hemodynamic measurements. *, p < 0.05; **, p < 0.01. C and D, cardiac fibrosis in perivascular (C) or interstitial (D) regions of left ventricles of metoprolol-treated wild type or GRK5-KO mice (n = 5). The degrees of cardiac fibrosis in metoprolol-administered wild type or GRK5-KO mice were evaluated by picrosirius staining using paraffin sections of their hearts. *, p < 0.05; ***, p < 0.001.
DISCUSSION
In this study, we demonstrated that metoprolol is a biased ligand and could induce cardiac fibrosis through a G protein-independent and GRK5/β-arrestin2-dependent pathway (Fig. 7). Accumulating evidence has implicated that β-arrestin-mediated biased signaling is elicited by stimulation of various seven-transmembrane receptors with their corresponding agonists (11, 22–25). However, there are only a few reports that the physiological meaning of this biased signaling are clearly demonstrated in vivo (36, 38). In addition to those agonists, it has been shown that some β-blockers also trigger the β-arrestin-mediated biased signaling. Among β-blockers, carvedilol (21, 39) and alprenolol (20) have been reported to activate intracellular signaling in HEK293 cells through the β2- and β1-adrenergic receptors in a β-arrestin-dependent manner. The β-arrestin-mediated signaling by alprenolol and carvedilol trans-activates the EGF receptor, resulting in ERK activation. However, how such a pathway by two β-blockers is related with physiological or pathological responses still remains to be determined. In this study, we revealed that the G protein-independent and β-arrestin2-dependent pathway evoked by a β-blocker could influence physiological responses, cardiac fibrosis, and impairment of diastolic function. The β-arrestin-mediated biased signaling by metoprolol did not induce EGF receptor transactivation (supplemental Movies S1 and S2). Thus, the β-arrestin-mediated biased pathway seemed to be different from that by carvedilol and alprenolol.
FIGURE 7.

Schematic diagram depicting the model of metoprolol-mediated cardiac fibrosis. Metoprolol induces cardiac fibrosis through β1-adrenergic receptor depending on GRK5 and β-arrestin2.
Metoprolol increased the expression of fibrotic genes in cardiomyocytes but not cardiac fibroblasts in vitro (Fig. 3). This result is consistent with previous reports that metoprolol is a β1-adrenergic receptor-selective antagonist (40) and cardiac fibroblasts express only β2-adrenergic receptor subtype (41). Metoprolol also induced the interaction of β1-adrenergic receptor with β-arrestin2 in different cell types, such as HEK293 cells, H9c2 cells, and cardiomyocytes. From these results, we propose that metoprolol directly acts on β1-adrenergic receptors expressed on cardiomyocytes in vivo. This would induce the release of the fibrotic factors from cardiomyocytes, resulting in the activation of cardiac fibroblasts to induce fibrosis.
GRK5 but not GRK6 is required for the G protein-independent and β-arrestin2-dependent cardiac fibrosis by metoprolol. Among GRK family proteins, GRK5 and GRK6 were reported to be involved in seven-transmembrane receptor-mediated biased signaling that is independent of G protein signaling and dependent on β-arrestins (22, 23, 36, 37). In these reports, the roles of GRK5 and GRK6 seem to be the same and functional differences between two GRKs in the signaling have not been detected. Accordingly, the specific contribution of GRK5 to cardiac fibrosis induced by metoprolol is unique in G protein-independent and β-arrestin-dependent signaling. For agonist-promoted desensitization, the different sets of Ser or Thr on a given GPCR undergo cell- and tissue-specific phosphorylation by different kinases in a barcode-like fashion (42–44). It was recently reported that CCL21 is a natural biased agonsit for CCR7, a seven-transmembrane chemokine receptor, and the ligand recruits β-arrestin2 only by GRK6-promoted phosphorylation of the receptor (45). Our results, together with their report, suggest that different patterns of GPCR phosphorylation by GRK5 and GRK6 determine the downstream signaling through β-arrestins. Specific interaction of β1-adrenergic receptor with β-arrestin2, but not β-arrestin1 by metoprolol stimulation also supports this hypothesis.
Our BRET assay using Rluc-β-arrestin2-GFP2 demonstrated that conformation of β-arrestin2 by metoprolol stimulation was different from that by isoproterenol stimulation. Recent progress with the structural analysis of GPCRs has revealed that GPCRs can form several conformations by ligand binding. The neutral antagonist, the agonist, and the inverse agonist for the β2-adrenergic receptor induced its different conformational states (46). In addition, two distinct conformations were observed in different antagonist-bound β1-adrenergic receptors (47). Thus, metoprolol may induce a specific conformation of the β1-adrenergic receptor to activate the GRK5/β-arrestin2-dependent signaling pathway.
Metoprolol is used as one of the β-blockers for the treatment of various heart diseases and its effectiveness has been verified by several clinical tests (48, 49). In this study, we demonstrated that long-term administration of metoprolol induced cardiac fibrosis and impaired diastolic function. Although metoprolol induced cardiac fibrosis, we think that metoprolol still has a beneficial effect on the treatment of heart failure patients. The hearts of heart failure patients are overstimulated by catecholamine, and metoprolol blocks the action of catecholamine. The degree of metoprolol-induced fibrosis is fairly small when compared with the fibrosis induced by heart failure. Accordingly, a small increase in fibrosis by metoprolol will be overcome by the beneficial effects of the β-blocking action of metoprolol. However, it is interesting to compare the effects of metoprolol to those of carvedilol and other β-blockers on the treatment of the patients who have not suffered from fibrosis to examine the role of β-blocker-induced fibrosis.
It was recently reported that β-blockers affect calcium signaling through β-arrestin2-biased signaling in central nervous system neurons (39). It was suggested that this β-arrestin2-biased signaling explains some of the adverse effects of β-blockers on central nervous system functions. So far, β-blockers are classified by affinity, subtype selectivity, duration of action and so on (50). In this study, we focused on metoprolol in the activation of β-arrestin2-mediated biased signaling and the induction of cardiac fibrosis. This study, and studies from other groups, will help to establish that β-arrestin2-dependent signaling is another index for classifying β-blockers (20, 21, 39).
Supplementary Material
Acknowledgments
We thank Dr. R. J. Lefkowitz (Duke University) for β-arrestin2-KO mice and Dr. R. T. Premont (Duke University) for GRK5- and GRK6-KO mice. We appreciate the technical support from the Research Support Center, Graduate School of Medical Sciences, Kyushu University.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to M. Na, M. Ni, and H. K.), a Grant-in-aid for Scientific Research on Innovative Areas (to M. Ni), a Grant-in-aid for Scientific Research on Priority Areas (to H. K.), the Takeda Science Foundation and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to M. Na).

This article contains supplemental Figs. S1–S4 and Movies S1 and S2.
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- BRET
- bioluminescence resonance energy transfer
- FRET
- fluorescence resonance energy transfer
- CTGF
- connective tissue growth factor
- Col1a
- collagen 1α
- Rluc
- Renilla luciferase
- KO
- knockout.
REFERENCES
- 1. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 2. Reiter E., Lefkowitz R. J. (2006) GRKs and β-arrestins. Roles in receptor silencing, trafficking, and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 3. DeFea K. A. (2011) β-Arrestins as regulators of signal termination and transduction. How do they determine what to scaffold? Cell. Signal. 23, 621–629 [DOI] [PubMed] [Google Scholar]
- 4. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 5. Premont R. T., Gainetdinov R. R. (2007) Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534 [DOI] [PubMed] [Google Scholar]
- 6. Moore C. A., Milano S. K., Benovic J. L. (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482 [DOI] [PubMed] [Google Scholar]
- 7. Hanyaloglu A. C., von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 8. Shenoy S. K., McDonald P. H., Kohout T. A., Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 9. Cao T. T., Deacon H. W., Reczek D., Bretscher A., von Zastrow M. (1999) A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature 401, 286–290 [DOI] [PubMed] [Google Scholar]
- 10. Ribas C., Penela P., Murga C., Salcedo A., Garcíia-Hoz C., Jurado-Pueyo M., Aymerich I., Mayor F., Jr. (2007) The G protein-coupled receptor kinase (GRK) interactome. Role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta 1768, 913–922 [DOI] [PubMed] [Google Scholar]
- 11. Rajagopal S., Rajagopal K., Lefkowitz R. J. (2010) Teaching old receptors new tricks. Biasing seven-transmembrane receptors. Nat. Rev. Drug. Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rockman H. A., Koch W. J., Lefkowitz R. J. (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212 [DOI] [PubMed] [Google Scholar]
- 13. Brodde O. E. (1993) β-Adrenoceptors in cardiac disease. Pharmacol. Ther. 60, 405–430 [DOI] [PubMed] [Google Scholar]
- 14. Metra M., Cas L. D., di Lenarda A., Poole-Wilson P. (2004) β-Blockers in heart failure. Are pharmacological differences clinically important? Heart Fail. Rev. 9, 123–130 [DOI] [PubMed] [Google Scholar]
- 15. Leimbach W. N., Jr., Wallin B. G., Victor R. G., Aylward P. E., Sundlöf G., Mark A. L. (1986) Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73, 913–919 [DOI] [PubMed] [Google Scholar]
- 16. Thomas J. A., Marks B. H. (1978) Plasma norepinephrine in congestive heart failure. Am. J. Cardiol. 41, 233–243 [DOI] [PubMed] [Google Scholar]
- 17. Bristow M. R. (2000) β-Adrenergic receptor blockade in chronic heart failure. Circulation 101, 558–569 [DOI] [PubMed] [Google Scholar]
- 18. Cruickshank J. M. (2007) Are we misunderstanding β-blockers. Int. J. Cardiol. 120, 10–27 [DOI] [PubMed] [Google Scholar]
- 19. Azzi M., Charest P. G., Angers S., Rousseau G., Kohout T., Bouvier M., Piñeyro G. (2003) β-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 11406–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim I. M., Tilley D. G., Chen J., Salazar N. C., Whalen E. J., Violin J. D., Rockman H. A. (2008) β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc. Natl. Acad. Sci. U.S.A. 105, 14555–14560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wisler J. W., DeWire S. M., Whalen E. J., Violin J. D., Drake M. T., Ahn S., Shenoy S. K., Lefkowitz R. J. (2007) A unique mechanism of β-blocker action. Carvedilol stimulates β-arrestin signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim J., Ahn S., Ren X. R., Whalen E. J., Reiter E., Wei H., Lefkowitz R. J. (2005) Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ren X. R., Reiter E., Ahn S., Kim J., Chen W., Lefkowitz R. J. (2005) Different G protein-coupled receptor kinases govern G protein and β-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. U.S.A. 102, 1448–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shukla A. K., Violin J. D., Whalen E. J., Gesty-Palmer D., Shenoy S. K., Lefkowitz R. J. (2008) Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 9988–9993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishida M., Sato Y., Uemura A., Narita Y., Tozaki-Saitoh H., Nakaya M., Ide T., Suzuki K., Inoue K., Nagao T., Kurose H. (2008) P2Y6 receptor-Gα12/13 signalling in cardiomyocytes triggers pressure overload-induced cardiac fibrosis. EMBO J. 27, 3104–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujii T., Onohara N., Maruyama Y., Tanabe S., Kobayashi H., Fukutomi M., Nagamatsu Y., Nishihara N., Inoue R., Sumimoto H., Shibasaki F., Nagao T., Nishida M., Kurose H. (2005) Gα12/13-mediated production of reactive oxygen species is critical for angiotensin receptor-induced NFAT activation in cardiac fibroblasts. J. Biol. Chem. 280, 23041–23047 [DOI] [PubMed] [Google Scholar]
- 28. Charest P. G., Terrillon S., Bouvier M. (2005) Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep. 6, 334–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mangmool S., Haga T., Kobayashi H., Kim K. M., Nakata H., Nishida M., Kurose H. (2006) Clathrin required for phosphorylation and internalization of β2-adrenergic receptor by G protein-coupled receptor kinase 2 (GRK2). J. Biol. Chem. 281, 31940–31949 [DOI] [PubMed] [Google Scholar]
- 30. Nikolaev V. O., Bünemann M., Schmitteckert E., Lohse M. J., Engelhardt S. (2006) Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ. Res. 99, 1084–1091 [DOI] [PubMed] [Google Scholar]
- 31. Daniels A., van Bilsen M., Goldschmeding R., van der Vusse G. J., van Nieuwenhoven F. A. (2009) Connective tissue growth factor and cardiac fibrosis. Acta Physiol. 195, 321–338 [DOI] [PubMed] [Google Scholar]
- 32. Ruiz-Ortega M., Rodríguez-Vita J., Sanchez-Lopez E., Carvajal G., Egido J. (2007) TGF-β signaling in vascular fibrosis. Cardiovasc. Res. 74, 196–206 [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto K., Masuyama T., Sakata Y., Nishikawa N., Mano T., Yoshida J., Miwa T., Sugawara M., Yamaguchi Y., Ookawara T., Suzuki K., Hori M. (2002) Myocardial stiffness is determined by ventricular fibrosis, but not by compensatory or excessive hypertrophy in hypertensive heart. Cardiovasc. Res. 55, 76–82 [DOI] [PubMed] [Google Scholar]
- 34. Varma D. R., Shen H., Deng X. F., Peri K. G., Chemtob S., Mulay S. (1999) Inverse agonist activities of β-adrenoceptor antagonists in rat myocardium. Br. J. Pharmacol. 127, 895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Engelhardt S., Grimmer Y., Fan G. H., Lohse M. J. (2001) Constitutive activity of the human β1-adrenergic receptor in β1-receptor transgenic mice. Mol. Pharmacol. 60, 712–717 [PubMed] [Google Scholar]
- 36. Rakesh K., Yoo B., Kim I. M., Salazar N., Kim K. S., Rockman H. A. (2010) β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci. Signal. 3, ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., Rockman H. A. (2007) β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gesty-Palmer D., Flannery P., Yuan L., Corsino L., Spurney R., Lefkowitz R. J., Luttrell L. M. (2009) A β-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci. Transl. Med. 1, 1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tzingounis A. V., von Zastrow M., Yudowski G. A. (2010) β-Blocker drugs mediate calcium signaling in native central nervous system neurons by β-arrestin-biased agonism. Proc. Natl. Acad. Sci. U.S.A. 107, 21028–21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bristow M. R. (2011) Treatment of chronic heart failure with β-adrenergic receptor antagonists. A convergence of receptor pharmacology and clinical cardiology. Circ. Res. 109, 1176–1194 [DOI] [PubMed] [Google Scholar]
- 41. Gustafsson A. B., Brunton L. L. (2000) β-Adrenergic stimulation of rat cardiac fibroblasts enhances induction of nitric-oxide synthase by interleukin-1β via message stabilization. Mol. Pharmacol. 58, 1470–1478 [DOI] [PubMed] [Google Scholar]
- 42. Butcher A. J., Prihandoko R., Kong K. C., McWilliams P., Edwards J. M., Bottrill A., Mistry S., Tobin A. B. (2011) Differential G protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 286, 11506–11518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Torrecilla I., Spragg E. J., Poulin B., McWilliams P. J., Mistry S. C., Blaukat A., Tobin A. B. (2007) Phosphorylation and regulation of a G protein-coupled receptor by protein kinase CK2. J. Cell Biol. 177, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zidar D. A., Violin J. D., Whalen E. J., Lefkowitz R. J. (2009) Selective engagement of G protein-coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. U.S.A. 106, 9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bokoch M. P., Zou Y., Rasmussen S. G., Liu C. W., Nygaard R., Rosenbaum D. M., Fung J. J., Choi H. J., Thian F. S., Kobilka T. S., Puglisi J. D., Weis W. I., Pardo L., Prosser R. S., Mueller L., Kobilka B. K. (2010) Ligand-specific regulation of the extracellular surface of a G protein-coupled receptor. Nature 463, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moukhametzianov R., Warne T., Edwards P. C., Serrano-Vega M. J., Leslie A. G., Tate C. G., Schertler G. F. (2011) Two distinct conformations of helix 6 observed in antagonist-bound structures of a β1-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 8228–8232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Group M. H. S. (1999) Effect of metoprolol CR/XL in chronic heart failure. Metoprolol CR/XL randomized intervention trial in congestive heart failure (MERIT-HF). Lancet 353, 2001–2007 [PubMed] [Google Scholar]
- 49. Waagstein F., Caidahl K., Wallentin I., Bergh C. H., Hjalmarson A. (1989) Long-term β-blockade in dilated cardiomyopathy. Effects of short- and long-term metoprolol treatment followed by withdrawal and readministration of metoprolol. Circulation 80, 551–563 [DOI] [PubMed] [Google Scholar]
- 50. Reiter M. J. (2004) Cardiovascular drug class specificity, β-blockers. Prog. Cardiovasc. Dis. 47, 11–33 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.