

Abstract
Diabetes impairs the resolution of periodontal inflammation. We explored pathways altered by inflammation in the diabetic periodontium by using ligatures to induce periodontitis in type-2 diabetic Goto-Kakizaki rats. Ligatures were removed after 7 days, and rats were then treated with TNF inhibitor (pegsunercept) or vehicle alone and euthanized 4 days later. RNA was extracted from periodontal tissue, examined by mRNA profiling, and further analyzed by functional criteria. We found that 1,754 genes were significantly up-regulated and 1,243 were down-regulated by pegsunercept (p < 0.05). Functional analysis revealed up-regulation of neuron-associated and retina-associated gene clusters as well as those related to cell activity and signaling. Others were down-regulated by TNF inhibition and included genes associated with host defense, apoptosis, cell signaling and activity, and coagulation/hemostasis/complement. For selected genes, findings with microarray and rt-PCR agreed. PPAR-α was investigated further by immunohistochemistry due to its anti-inflammatory function and was found to be up-regulated in the gingiva during the resolution of periodontal inflammation and suppressed by diabetes. The results indicate that diabetes-enhanced inflammation both up- and down-regulates genes involved in cellular activity and cell signaling, while it predominantly up-regulates genes involved in the host response, apoptosis, and coagulation/homeostasis/complement and down-regulates mRNA levels of neuron, retina, and energy/metabolism-associated genes.
Keywords: DAVID, diabetes, GSEA, inflammation, microarray, periodontal disease(s)
Introduction
Type 2 diabetes is a metabolic disorder with insulin resistance and decreased β-cell function. It causes a number of complications that involve the retina, kidneys, nervous system, wound healing, and cardiovascular disease. Dysregulation of inflammatory pathways contributes to the development and progression of diabetic complications. Enhanced pro-inflammatory cytokine levels have been found in several diabetic complications, such as nephropathy, which exhibits high levels of TNF-α in the kidneys and the urine of diabetic patients (Wu, Chen et al., 2010).
Dysregulation of TNF is linked to diabetic complications in animal studies. By the use of inhibitors, it has been shown that diabetes-enhanced TNF contributes to impaired wound healing (Goren et al., 2007). Anti-TNF treatment of dermal wounds in type 2 diabetic mice attenuates inflammation, improves re-epithelialization, and restores insulin signaling in wounded tissue (Siqueira et al., 2010). TNF inhibition reduces fibroblast apoptosis and enhances fibroblast proliferation in diabetic wounds to enhance healing (Siqueira et al., 2010). Moreover, it reduces endothelial cell death in the early stages of diabetic retinopathy (Behl et al., 2009) and prevents blood-retinal barrier breakdown (Huang et al., 2011). In diabetic nephropathy, anti-TNF treatment reduces sodium retention, renal hypertrophy, and urinary albumin (DiPetrillo et al., 2003). Other less specific treatments with a component of anti-TNF activity have improved experimental diabetic neuropathy in rats (Chauhan et al., 2012) and limited peripheral neuropathy progression (Sagara et al., 1996). In humans, TNF blockers reduce macular thickness and increase visual acuity in patients with severe diabetic macular edema (Sfikakis et al., 2005).
Diabetes increases the risk and severity of periodontal disease (Mealey et al., 2006). The levels of TNF-α and other inflammatory mediators are elevated in diseased periodontal tissues (Benakanakere and Kinane, 2012). These mediators stimulate the production of enzymes that break down connective tissue and induce bone resorption. Diabetes partly aggravates periodontitis by reducing the capacity to down-regulate inflammation (Graves et al., 2011). Peripheral blood monocytes in individuals with diabetes produce elevated levels of TNF-α and other inflammatory mediators compared with levels found in non-diabetic control individuals (Gacka et al., 2010). In animal studies, diabetes alters the host response to bacterial challenge through prolonged TNF-α expression (Naguib et al., 2004). Diabetes-enhanced inflammation also increases cellular apoptosis and decreases proliferation, reducing the potential repair of damaged periodontal tissue (Liu et al., 2006).
To further investigate the impact of diabetes on the periodontium, we carried out mRNA profiling. We focused on the resolution of periodontitis by treatment of diabetic animals with a TNF inhibitor, pegsunercept, following the cessation of experimental periodontitis. The results indicate that several biological pathways—particularly those involving host defense, cell signaling, and apoptosis—are elevated by prolonged inflammation in diabetic animals, while other pathways—including neuron-associated genes, cell activities such as proliferation and migration, and retina-associated genes—are predominantly down-regulated.
Materials & Methods
Animals
Type-2 diabetic male Goto-Kakizaki (GK) rats were originally obtained by selective breeding of Wistar (WT) rats. Normoglycemic WT rats were used as a control, and both GK and WT rats were purchased from Charles River Laboratories (Wilmington, MA, USA). To induce periodontitis, we placed ligatures around the maxillary 2nd molars after rats were hyperglycemic for 3 to 4 wks (Liu et al., 2006; Pacios et al., 2012). After 7 days, ligatures were removed to allow for resolution of periodontal inflammation. GK rats were treated with the TNF-specific inhibitor pegsunercept, given only once (provided by Amgen, Thousand Oaks, CA, USA) by intraperitoneal injection (4 mg/kg) immediately after ligatures were removed. Control GK rat counterparts were treated with vehicle alone (phosphate-buffered saline, PBS) (Fig. 1). At day 11, 4 GK rats treated with TNF inhibitor and 5 GK rats with vehicle alone were euthanized for microarray analysis. Glycemic levels of GK rats were 436 ± 32 mg/dL, and those of GK rats treated with TNF inhibitor were 452 ± 48 mg/dL at the time of death. All animal procedures were approved by the Institutional Animal Care and Use Committee, and the experiments conformed to ARRIVE guidelines.
Figure 1.

Diagram of the approach used for functional analysis of genes regulated by high levels of TNF in the diabetic periodontium. RNA was isolated from diabetic rats treated with vehicle alone or pegsunercept during the resolution of periodontal inflammation and subjected to mRNA profiling. The distribution of the functional gene clusters was determined by Database for Annotation, Visualization and Integrated Discovery (DAVID) and also by gene set enrichment analysis. The approach identified gene clusters that are up- or down-regulated when TNF is inhibited with pegsunercept in diabetic animals and provides insight as to which gene sets are specifically modulated by diabetes-enhanced inflammation.
mRNA Profiling
We extracted total RNA from the dissected periodontal tissue and determined mRNA levels of 26,214 genes using Affymetrix rat Exon gene 1.0ST array by the Path BioResource, University of Pennsylvania. Partek software was used to standardize and normalize fluorescence intensity. For selected genes, relative mRNA was assessed by real-time PCR (rt-PCR) with primers and probe sets from Applied Biosystems (Foster City, CA, USA) or Roche Applied Science (Indianapolis, IN, USA) (Appendix Table 1). Each value represents the mean of 3 distinct experiments normalized to ribosomal protein L32.
Immunohistochemistry (IHC)
TNF-α contributes to the tissue destruction induced by periodontal bacteria. Periodontal specimens were prepared according to the approach used by Garlet et al. (2007).
In total, 30 GK rats and 22 WT rats were analyzed with IHC. Periodontal tissue was fixed for 2 days with cold 4% paraformaldehyde at 4°C and decalcified in EDTA; 5-µm paraffin sections were prepared. IHC or immunofluorescence with an antibody specific to peroxisome-proliferator-activated receptor-alpha (PPAR-α) (Ab8934, Cambridge, MA, USA) was performed with antigen retrieval (0.01 M citrate) and tyramide signal amplification (Perkin-Elmer, Waltham, MA, USA). Intensity and number of positive cells (Alblowi et al., 2009) of the anti-PPAR-α immunostaining were blindly quantified, confirmed by a second observer, at 600x magnification according to the following scale: 0, no positive staining in the field; 1, 1 to 10% cells immunopositive with light immunostaining; 2, 11 to 25% cells immunopositive with light to moderate immunostaining; 3, 26 to 40% cells immunopositive with moderate immunostaining; 4, 41 to 60% cells positive with heavy immunostaining; and 5, 61% or more cells immunopositive with dark immunostaining.
Bioinformatics and Statistical Analysis
Genes with mRNA levels 1.75-fold higher or lower in the experimental (pegsunercept-treated) vs. control (vehicle-treated) group and with adjusted p < 0.05 were identified (Galindo et al., 2009). Database for Annotation, Visualization and Integrated Discovery (DAVID) was used for functional analysis with an enrichment score with a cut-off point of 1.5. Microarray data were also examined by Gene Set Enrichment analysis (GSEA) software and the Molecular Signature Database (MSig DB), with a threshold of false-discovery rate (FDR) < 25% and p < 0.05 (Fig. 1). With the rt-PCR and immunohistochemical analyses, we used an unpaired t test and one-way analysis of variance (ANOVA) to compare 2 unpaired groups or 3 or more independent groups at a particular time-point, respectively. If the result from the ANOVA test was significant, planned multiple comparisons between groups were performed with the contrast method. Statistical significance was set at a level of 5%.
Results
Diabetes causes prolonged and elevated inflammation during the resolution of periodontitis (Liu et al., 2006; Pacios et al., 2012). We examined the differential mRNAs expression levels in diabetic rats treated with a TNF-specific inhibitor compared with levels in rats treated with vehicle alone. Microarray analysis identified 2,997 up-regulated and 1,243 down-regulated genes in the experimental group that exhibited at least a 1.75-fold change in mRNA levels and an adjusted p value < 0.05. The top 500 up-regulated and down-regulated genes are given in Appendix Table 2. These genes were further examined by DAVID (Fig. 2A), which identified 82 up-regulated and 55 down-regulated gene annotation clusters. The most frequently up-regulated functional subgroups were: neuron-associated genes (51%); genes associated with cell activities (cell cycle, cell migration, endocytosis, cytoskeleton organization, cell adhesion, etc.) (27%); cell signaling other than host defense (8%); and retina-associated genes (7%). Down-regulated functional groups involved: host immune defense (53%); cell signaling other than host defense (15%); cell activities (11%); apoptosis or cell death (7%); energy or metabolism (2%); and coagulation, hemostasis, or wound healing (2%).
Figure 2.

Functional distribution of gene clusters up- or down-regulated by TNF inhibition in diabetic specimens. (A) Functional distribution of gene clusters up- or down-regulated as determined by DAVID. RNA was isolated from diabetic rats treated with vehicle alone or pegsunercept during the resolution of periodontal inflammation and subjected to mRNA profiling. Bars represent the distribution of the functional gene clusters determined by Database for Annotation, Visualization and Integrated Discovery (DAVID). Numbers represent the percentage that each category contributed to the total number of up- or down-regulated gene clusters. Gene clusters that are up-regulated when TNF is inhibited with pegsunercept are shown as positive numbers and those that are down-regulated as negative. Only functional annotation clusters with enrichment scores ≥ 1.5 and biological functional ontologies with adjusted p < 0.05 were selected and are displayed in the Fig. (B) Functional distribution of significantly up- or down-regulated gene sets as determined by GSEA. mRNA profiling data described in Fig. 1 were further analyzed by gene set enrichment analysis (GSEA). Only gene sets with FDR < 25% and nominal p value < 0.05 were considered significant and are displayed in the Fig.
Genes from each cluster described above were manually sorted based on their molecular function, as described by the Gene Ontology database terminology (GOTERM_MF_FAT level 5) (Appendix Table 3). Genes up-regulated by TNF inhibition participated in inter- or intracellular signaling, ion binding, and membrane transporter activity. Molecular terms most commonly observed in the down-regulated subgroups included intercellular signaling, particularly inflammation such as immunoglobulin, cytokine, and chemokine receptor activity.
Using the other approach by GSEA to examine functional changes, we found that 86 gene sets were up-regulated in diabetic rats treated with TNF-inhibitor, and 150 sets were down-regulated (Appendix Table 4, Fig. 2B). Up-regulated gene sets included cell signaling other than host defense (33%), neuronal function (26%), energy or metabolism (19%), and cellular activity such as transmembrane transport, cell cycle, and cell migration (19%). Down-regulated gene sets included: inflammatory host defense response (38%); cell cycle, cell migration, and other cell activities (27%); cell signaling other than host defense (11%); extracellular processes such as coagulation or hemostasis (9%); apoptosis (5%); and gene sets related to energy/metabolism (3%).
Since neuronal genes as a group were particularly affected by TNF inhibition, we examined them in more depth using rt-PCR. Among the up-regulated neuronal genes, microtubule-associated protein 2 (MAP2), synaptophysin (SYP), synuclein-alpha (SNCA), syntaxin-binding protein 1 (STXBP1), neurofilament light polypeptide (NEFL), neuropeptide Y (NPY), and tachykinin, precursor 1 (TAC1), were further validated (Table). The overall pattern between the groups was similar, although the magnitude of the difference was typically underestimated in the microarray data. The mRNA levels of GK rats treated with vehicle relative to that of L32 are given in the Table as well. Generally, the expression levels of these genes were 0 to 91% of the level of L32, which represents low to moderate mRNA levels.
Table.
Fold Increase in mRNA Levels of Selected Genes Determined by Microarray and rt-PCR
| Gene | Fold Increase TNF Inhibitor/ Veh (Microarray) | Fold Increase TNF Inhibitor/ Veh (rt-PCR) | Relative mRNA Level |
|---|---|---|---|
| MAP2 | 3.57 | 9.38 | 0.0009 |
| SYP | 24.66 | 416.05 | 0.0003 |
| SNCA | 5.46 | 27.54 | 0.14 |
| STXBP1 | 6.97 | 31.55 | 0.26 |
| NEFL | 16.26 | 160.05 | 0.004 |
| NPY | 1.94 | 5.53 | 0.91 |
| TAC1 | 3.04 | 255.11 | 0.34 |
| PPAR-α | 7.3 | 19.94 | 0.000011 |
RNA was isolated from rats treated with TNF inhibitor compared with vehicle alone, and the relative mRNA level of each gene was assessed by real-time PCR or by microarray. The relative mRNA level of each gene was based upon real-time PCR results from GK rats treated with vehicle relative to the mRNA level of a housekeeping gene, ribosomal protein L32.
Because of the potential importance of PPAR-α in the dysregulation of inflammation, PPAR-α protein level was examined by IHC (Fig. 3; Appendix Fig.). In the gingival connective tissue, the number of cells with PPAR-α protein was low at baseline (day 0) and when periodontitis was induced (day 7) in normoglycemic and diabetic rats. When periodontitis was resolved by the removal of ligatures, there was a 3-fold increase in the relative expression of PPAR-α in normoglycemic rats on day 11 (p < 0.05). In contrast, no change was observed in the diabetic animals (p > 0.05). However, when diabetic rats were treated with TNF-inhibitor, there was a significant increase (p < 0.05), so that the diabetic rats with TNF-inhibitor behaved similarly to the normal animals. Similar trends were observed in the periodontal ligament (PDL).
Figure 3.
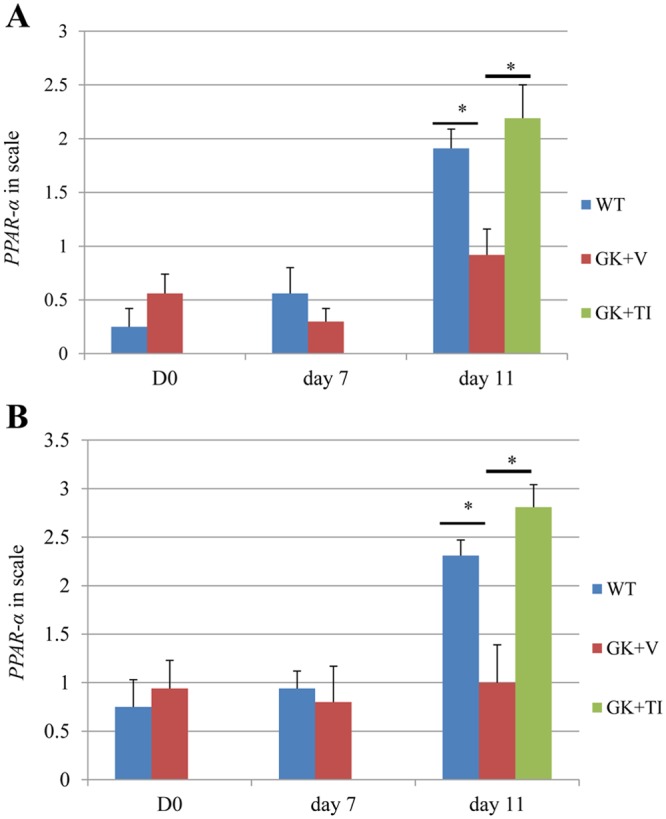
Up-regulation of PPAR-α protein levels in periodontal tissues is reduced by diabetes during the resolution phase of inflammation. Periodontal tissue samples at days 0, 7, and 11 were obtained from normal wild-type rats (blue bars), diabetic rats treated with vehicle (red bars), and diabetic rats treated with TNF-inhibitor (green bars). Immunohistochemistry with antibody specific for PPAR-α was carried out and analyzed according to the following scale: 0, no positive staining in the field; 1, 1 to 10% immunopositive cells with light immunostaining; 2, 11 to 25% immunopositive with moderate immunostaining; 3, 26 to 40% immunopositive with moderate immunostaining; 4, 41 to 60% immunopositive with dark immunostaining; and 5, 61% or higher immunopositive with dark immunostaining. (A) Immunostaining in gingival connective tissue. (B) Immunostaining in periodontal ligament space. Data are mean ± SEM. *Significant difference (p < 0.05). Each group at each time-point included from 6 to 8 animals. WT, Wistar rats; GK + V, GK rats treated with vehicle only; GK+TI, GK rats treated with TNF-specific inhibitor.
Discussion
The resolution of inflammation following periodontitis is an active process that involves lipoxins and resolvins and a reduction in pro-inflammatory cytokine expression (Kayal et al., 2010; Hasturk et al., 2012). Over-expression of TNF-α prevents down-regulation of inflammation in the periodontium and prolonged high levels of apoptosis (Graves et al., 2006; Pacios et al., 2012). Our previous findings showed that the largest effect of the TNF-inhibitor in the diabetic group was during the resolution of inflammation (Pacios et al., 2012). Therefore, the microarray analysis was focused on day 11. Our findings suggest the presence of biologically important pathways significantly affected by inflammation in diabetic animals during the resolution of periodontitis. Diabetes-enhanced inflammation decreases mRNA levels of genes associated with neurons, cell signaling, and cell activities such as migration. In contrast, diabetes-enhanced inflammation stimulates the host response, inter- or intracellular cell signaling, extracellular processes including coagulation, or hemostasis, apoptosis, and energy/metabolism-related pathways.
Several neuron-associated genes were up-regulated when TNF was inhibited in the diabetic group. Many of these genes are expressed by cells other than neurons, including fibroblasts (SNCA, NEFL), epithelial cells (STXBP1, TAC1), keratinocytes (TAC1), monocytes (SNCA), neutrophils (TAC1), endothelial cells (TAC1), osteoclasts (TAC1), or osteoblasts (TAC1, NPY) (Wu, Orozco et al., 2009). Moreover, inflammation modulates the expression levels of several of them: NEFL, TAC1, NPY, and SNCA. Many of these genes affect functions that are important in the periodontium. For example, NPY and substance P, a TAC1 gene product, can enhance osteoblast differentiation (Goto et al., 2007; Franquinho et al., 2010). Thus, mRNA levels of neuron-associated genes are affected by diabetes-enhanced inflammation, which may have important ramifications in the periodontium.
We found that an approximately 1.75 greater number of genes were up-regulated by the TNF inhibitor than were down-regulated. Similar differences have been noted by other investigators (Paiva et al., 2012). Findings from DAVID and GSEA showed consistencies in identifying up-regulated gene clusters when inflammation is inhibited, including clusters associated with neuron, cell cycle, cell migration, and other cellular activities. They also identified down-regulated clusters related to host inflammatory responses, apoptosis, and extracellular processes such as coagulation and hemostasis. In contrast, the 2 methods showed some dissimilarities: DAVID showed clusters associated with cell signaling other than host defense and with energy/metabolism to be predominantly or slightly down-regulated by TNF inhibition, while GSEA showed the corresponding clusters to be predominantly or slightly up-regulated. These differences may be related to the slight difference in database sources for gene set/cluster annotations.
PPAR-α is a target of fibrates for hypolipidemic drugs. PPAR-α was selected for further study because of its potential function in promoting down-regulation of inflammation (van Bilsen and van Nieuwenhoven, 2010). It is expressed in certain tissues, including the periodontium (Offenbacher et al., 2009; Belfort et al., 2010). We found that PPAR-α protein was expressed in the gingiva and PDL of normal rats during the resolution, but not in diabetic rats. However, when diabetic rats were treated with a TNF-inhibitor, the expression of PPAR-α, at both the protein and mRNA levels, was significantly enhanced. Interestingly, mRNA PPAR-α levels are up-regulated during the resolution of experimental gingivitis in healthy individuals (Offenbacher et al., 2009). PPAR-α inhibits TNF activity and suppresses activation of the pro-inflammatory transcription factors nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) and activator protein 1 (AP-1) activity (Delerive et al., 1999). Thus, PPAR-α may participate in the resolution of periodontitis through its anti-inflammatory properties, but under diabetic conditions its up-regulation is suppressed.
Other studies have examined mRNA profiling of experimental gingivitis in healthy humans (Offenbacher et al., 2009). They noted the down-regulation of immune response genes during gingivitis resolution, similar to the down-regulation in the diabetic group treated with TNF-inhibitor. Proteomic analysis of the gingival fluid samples has been examined in human specimens of acute gingivitis (Grant et al., 2010). A high level of proteins associated with inflammatory response was reported during the induction, which decreased during resolution. Two other studies found an increase in neuron-associated genes when inflammation was induced (Offenbacher et al., 2009; Grant et al., 2010), in contrast to our results, in which the mRNA levels of neuron-associated genes increased when inflammation was inhibited. This difference may be due to the fact that we examined a diabetic animal model while the others examined healthy humans. Alternatively, the specific neuron-associated genes in the 3 studies are different. Moreover, many of the neuron-associated genes in one of the reports exhibited only a slight decrease in mRNA levels during the resolution of gingivitis (Offenbacher et al., 2009).
Prolonged inflammation in diabetes has a broad effect in up-regulating the expression of host response genes and apoptotic genes in the periodontium. This is consistent with findings that diabetes leads to dysregulation of cytokine networks: There is an up-regulation of pro-inflammatory and pro-apoptotic pathways in vivo in diabetic fracture-healing (Kayal et al., 2010). The results presented here indicate that diabetes-enhanced inflammation both up- and down-regulates genes involved in cellular activity and cell signaling, while it predominantly up-regulates genes involved in the host response, apoptosis, and coagulation/homeostasis/complement and down-regulates mRNA levels of neuron, retina, and energy/metabolism-associated genes.
Acknowledgments
We thank Kang I. Ko for help with the manuscript.
Footnotes
This study was supported by NIDCR grant DE017732 and a Research Supplement to Promote Diversity.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Alblowi J, Kayal RA, Siqueira M, McKenzie E, Krothapalli N, McLean J, et al. (2009). High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing. Am J Pathol 175:1574-1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl Y, Krothapalli P, Desta T, Roy S, Graves DT. (2009). FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 58:917-925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfort R, Berria R, Cornell J, Cusi K. (2010). Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab 95:829-836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakanakere M, Kinane DF. (2012). Innate cellular responses to the periodontal biofilm. Front Oral Biol 15:41-55 [DOI] [PubMed] [Google Scholar]
- BioGps (2009). Gene Portal System. [Google Scholar]
- Chauhan N, Taliyan R, Sharma PL. (2012). Effect of dipyrone and thalidomide alone and in combination on STZ-induced diabetic neuropathic pain. Naunyn Schmiedebergs Arch Pharmacol 385:527-538 [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. (1999). Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem 274:32048-32054 [DOI] [PubMed] [Google Scholar]
- DiPetrillo K, Coutermarsh B, Gesek FA. (2003). Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol 284:F113-F121 [DOI] [PubMed] [Google Scholar]
- Franquinho F, Liz MA, Nunes AF, Neto E, Lamghari M, Sousa MM. (2010). Neuropeptide Y and osteoblast differentiation—the balance between the neuro-osteogenic network and local control. FEBS J 277:3664-3674 [DOI] [PubMed] [Google Scholar]
- Gacka M, Dobosz T, Szymaniec S, Bednarska-Chabowska D, Adamiec R, Sadakierska-Chudy A. (2010). Proinflammatory and atherogenic activity of monocytes in type 2 diabetes. J Diabetes Complications 24:1-8 [DOI] [PubMed] [Google Scholar]
- Galindo RC, Munoz PM, de Miguel MJ, Marin CM, Blasco JM, Gortazar C, et al. (2009). Differential expression of inflammatory and immune response genes in rams experimentally infected with a rough virulent strain of Brucella ovis. Vet Immunol Immunopathol 127:295-303 [DOI] [PubMed] [Google Scholar]
- Garlet GP, Cardoso CR, Campanelli AP, Ferreira BR, Avila-Campos MJ, Cunha FQ, et al. (2007). The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: host protection and tissue destruction. Clin Exp Immunol 147:128-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren I, Muller E, Schiefelbein D, Christen U, Pfeilschifter J, Muhl H, et al. (2007). Systemic anti-TNFalpha treatment restores diabetes-impaired skin repair in ob/ob mice by inactivation of macrophages. J Invest Dermatol 127:2259-2267. 10.1038/sj.jid.5700842 [DOI] [PubMed] [Google Scholar]
- Goto T, Nakao K, Gunjigake KK, Kido MA, Kobayashi S, Tanaka T. (2007). Substance P stimulates late-stage rat osteoblastic bone formation through neurokinin-1 receptors. Neuropeptides 41:25-31 [DOI] [PubMed] [Google Scholar]
- Grant MM, Creese AJ, Barr G, Ling MR, Scott AE, Matthews JB, et al. (2010). Proteomic analysis of a noninvasive human model of acute inflammation and its resolution: the twenty-one day gingivitis model. J Proteome Res 9:4732-4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Liu R, Alikhani M, Al-Mashat H, Trackman PC. (2006). Diabetes-enhanced inflammation and apoptosis—impact on periodontal pathology. J Dent Res 85:15-21 [DOI] [PubMed] [Google Scholar]
- Graves DT, Li J, Cochran DL. (2011). Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res 90:143-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Van Dyke TE. (2012). Paradigm shift in the pharmacological management of periodontal diseases. Front Oral Biol 15:160-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Gandhi JK, Zhong X, Wei Y, Gong J, Duh EJ, et al. (2011). TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophthalmol Vis Sci 52:1336-1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal RA, Siqueira M, Alblowi J, McLean J, Krothapalli N, Faibish D, et al. (2010). TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J Bone Miner Res 25:1604-1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, et al. (2006). Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res 85:510-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealey BL, Oates TW, American Academy of Periodontology (2006). Diabetes mellitus and periodontal diseases. J Periodontol 77:1289-1303 [DOI] [PubMed] [Google Scholar]
- Naguib G, Al-Mashat H, Desta T, Graves DT. (2004). Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol 123:87-92 [DOI] [PubMed] [Google Scholar]
- Offenbacher S, Barros SP, Paquette DW, Winston JL, Biesbrock AR, Thomason RG, et al. (2009). Gingival transcriptome patterns during induction and resolution of experimental gingivitis in humans. J Periodontol 80:1963-1982 [DOI] [PubMed] [Google Scholar]
- Pacios S, Kang J, Galicia J, Gluck K, Patel H, Ovaydi-Mandel A, et al. (2012). Diabetes aggravates periodontitis by limiting repair through enhanced inflammation. FASEB J 26:1423-1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva RT, Saliba AM, Fulco TO, de Souza Sales J, Serra de Carvalho D, Sampaio EP, et al. (2012). A framework to identify gene expression profiles in a model of inflammation induced by lipopolysaccharide after treatment with thalidomide. BMC Res Notes 5:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara M, Satoh J, Wada R, Yagihashi S, Takahashi K, Fukuzawa M, et al. (1996). Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia 39:263-269 [DOI] [PubMed] [Google Scholar]
- Sfikakis PP, Markomichelakis N, Theodossiadis GP, Grigoropoulos V, Katsilambros N, Theodossiadis PG. (2005). Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care 28:445-447 [DOI] [PubMed] [Google Scholar]
- Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N, et al. (2010). Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 53:378-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bilsen M, van Nieuwenhoven FA. (2010). PPARs as therapeutic targets in cardiovascular disease. Expert Opin Ther Targets 14:1029-1045 [DOI] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. (2009). BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10:R130. 10.1186/gb-2009-10-11-r130 Found at http://biogps.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Chen JS, Lu KC, Chen CC, Lin SH, Chu P, et al. (2010). Aberrant cytokines/chemokines production correlate with proteinuria in patients with overt diabetic nephropathy. Clin Chim Acta 411:700-704 [DOI] [PubMed] [Google Scholar]