

Abstract
Two novel bifunctional N-methylhydroxamate-isocyanate linkers 20 and 21 were prepared in good yield and high purity from the corresponding amine salts using a biphasic reaction with phosgene. The facile ring opening reaction of N-Boc lactams using the anion of O-benzylhydroxylamine gave the protected amino hydroxamates 6a and 6c in good yields. The selective methylation of the hydroxamate nitrogen in the presence of the N-Boc group in these intermediates could be readily accomplished. The utility of the linkers was clearly demonstrated by the synthesis of the carbamate-tethered trishydroxamic acid 27 and the urea-tethered 29
Keywords: polyhydroxamate chelators, isocyanate, carbamate, urea
The monobasic hydroxamate ligand is well-known for its ability to complex hard metal ions such as Fe(III) and the actinides(IV). In fact, many natural siderophores used by bacteria and fungi to procure iron essential for their growth are polyhydroxamates.1 In many synthetic hydroxamate siderophores that have been reported, a tripodal platform is employed to attach three hydroxamates and obtain a chelator that can achieve hexadentate coordination of the ferric ion.2 One popular tripodal platform for the tethering of three ligand arms is tris(2-amino)ethylamine, TREN.3
In addition to their role as iron chelators, hydroxamic acids have received much attention due to their potential therapeutic applications.4 The trihydroxamate siderophore Desferrioxamine, is used for the treatment of iron overload in patients with β-thalassemia.5 Many hydroxamic acids have been shown to inhibit enzymes such matrix metalloproteinases and histone deacetylases.6 The recent approval of suberoylanilide hydroxamic acid (SAHA) for the treatment of cutaneous T-cell lymphoma along with the other potential pharmaceutical uses of hydroxamic acids, clearly demonstrates the need to develop new methods to incorporate hydroxamic acids into a variety of structures.
Our group has been interested in the design and synthesis of chelators for the specific binding of ferric ion and actinides, for therapeutic and environmental remediation applications.7 While numerous synthetic siderophores have been studied, to our knowledge, none of them have carbamate and urea linkages generated in the process of attaching the hydroxamic acid ligand moiety to the platform. The most common linkage seen in synthetic and natural hydroxamate siderophores is amides. Our interest in developing methodology for the attachment of hydroxamates on various scaffolds using urea and carbamate linkages was stimulated by two reasons. The first was, of course, to create a new class of compounds for biological and metal complexation studies. But another important reason is that ureas and carbamates have been incorporated into scaffolds to generate ditopic chelators where an anion and cation are bound simultaneously.8 Polyhydroxamates anchored on ureas and carbamates could provide binding sites for various anions in addition to the cation of interest (ditopic chelators) as well as changing H-bonding, geometry, and lipophilicity/hydrophobicity of the chelator.9 Carbamate (polyurethanes) linkages have been shown to be useful to manipulate properties of dendrimers in order to meet specific application needs.10 In this communication, we report the preparation of new bifunctional hydroxamate-isocyanate linkers and their successful coupling chemistry to prepare polyhydroxamates with urea and carbamate tethers.
The key to our approach was the preparation of bifunctional hydroxamate-isocyanate linkers that can be coupled to amines and alcohols (Figure 1). The isocyanate linkers should be available from the corresponding protected amines. The question was the best way to prepare the protected amine hydroxamate intermediates. Two alternate routes were envisioned for this purpose, one that used the corresponding linear amine ester and the other that begins with protected N-Boc lactams.
Figure 1.

Proposed synthetic scheme
Several methods exist for the preparation of hydroxamic acids (both protected and unprotected) but one of the more common routes involves coupling of activated acid derivatives with hydroxylamines or protected hydroxylamines.11,12 The direct reaction of esters with hydroxylamine under basic conditions is a useful reaction but the products are difficult to purify particularly when they are polyhydroxamic acids. A recent publication that described the preparation of O-benzyl hydroxamates by the reaction of methyl esters and lactones with O-benzyl hydroxylamine in the presence of excess lithium hexamethyldisilylamide (LHMDS)13 is an attractive direct route. Although not reported or known, we envisioned that protected amino hydroxamates could be prepared by an analogous reaction of O-benzyl hydroxylamine with N-Boc protected lactams.
The preparation of protected aminohydroxamate 2 from ester 1 following a published protocol was investigated (Scheme 1). A solution of LHMDS (4eq) in THF was added to a suspension of O-benzyl hydroxylamine hydrochloride in THF at −78 °C. After stirring for 15 minutes, a solution of the methyl ester 1 in THF was added dropwise to the benzyloxyamine solution in THF at −78 °C. The reaction was stirred at this temperature for two hours, quenched with saturated NH4Cl and the product extracted into ethyl acetate. Using this method, pure O-benzyloxy hydroxamate 2 could be prepared in multigram quantities after recrystallization from ethyl acetate.
Scheme 1.

It was of interest to examine whether we could extend this reaction and its value by preparation of protected N-methyl hydroxamate 3 in one step by using N-methyl O-benzylhydroxylamine in the coupling reaction. Using a similar experimental procedure, 1 was reacted with the anion of N-methyl O-benzyl hydroxylamine (Scheme 1). This led to the formation of the desired product 3 in moderate yields (the best yield was 52%) after careful chromatographic purification. Unfortunately, a significant quantity of the amide by-product 4, was also formed in this reaction.14 A number of experimental conditions (reaction time and temperature, and number of equivalents of LHMDS) were examined to suppress the unwanted amide formation and obtain high yields of the desired product 3. Due to difficulties in the separation of 3 and 4, it was not possible to scale up the reaction to obtain multigram quantities of 3.
Given the fact that N-Boc lactams are readily accessible, a second approach involving ring opening of this class of substrates with the anion of O-benzylhydroxylamine to prepare protected amino hydroxamates, 6, of different chain lengths was attractive. It was thought that these substrates would undergo facile ring opening upon nucleophilic attack on the lactam carbonyl with benzyloxyamine anion, as the N-Boc should be a good leaving group.
It was gratifying to observe that lactam 5a readily reacted with the anion of benzyloxyamine under conditions similar to those described before (Scheme 2). The desired product 6a was obtained in 77% yield after chromatographic purification. The seven membered lactam 5c, also underwent facile ring opening to give 6c in high yield. Surprisingly, the same conversion with δ-lactam 5b proceeded in only modest yields (36%). Many attempts to improve the yield of this reaction were not successful and the reason for this unique behavior is not understood. The ring opening reaction also worked well with Vince lactam, 7, though in this example, it was necessary to conduct the reaction at −42°C in order to make it go to completion. Vince lactam has been shown to be an important synthon for the preparation of a variety of pharmaceutically active molecules including carbocyclic nucleoside analogues.15
Scheme 2.

In order to access N-methyl analogs of the bifunctional hydroxamate isocyanate linkers, the ring opening reaction of 5a using the anion of N-methyl O-benzylhydroxylamine was also studied. Despite examining a variety of reaction conditions, only the starting lactam was recovered from the reaction. This forced us to examine the direct alkylation of the hydroxamate nitrogen as a route to N-methyl hydroxamates. The benzyloxy hydroxamate 2 was treated with excess methyl iodide and K2CO3 in acetonitrile between 40–45 °C. Alkylation of the hydroxamate was found to be much more facile than alkylation of the N-Boc moiety. The crude product was purified using column chromatography to give 3 in 75% yield. The selective methylation of the hydroxamate nitrogen also occurred readily on other substrates (Table 1).
Table 1.
Methylation of representative hydroxamates and their deprotection

| ||||
|---|---|---|---|---|
| Yield | Yield | |||
| 2 | 75% | 3 | quant |
 11 |
| 6a | 80% |
 9 |
94% |
 12 |
| 8 | 84% |
 10 |
||
Having established convenient methods to prepare the protected amino hydroxamates needed for our study, we proceeded forward on the synthesis of the desired bifunctional isocyanate linkers. Deprotection of the Boc group in hydroxamate 2 with TFA in dichloromethane gave the corresponding trifluoroacetate salt 13. The excess TFA was removed in vacuo and trituration of the crude solid with ether gave the desired product in high yield and purity. Using a similar protocol, the N-methyl hydroxamate derivatives 3 and 9 could also be readily deprotected to obtain the desired salts in high purities and yield. The salts were directly used in the next step, the conversion of the amine to the desired isocyanates.
The conversion of amines to isocyanates is a well-known reaction but can be tricky due to competitive formation of urea formed by reaction of the unreacted amine with the reactive isocyanate product. A procedure for preparing peptidyl isocyanates in high purities and yield by the reaction of amines with phosgene under biphasic reaction conditions was attractive for our system.16
The first reaction to be investigated was the conversion of amine salt 13 to the corresponding isocyanate. Saturated sodium bicarbonate was added to a solution of amine TFA salt 13 in DCM. After stirring for 30 seconds, the layers were allowed to separate and a 20% solution of phosgene (2 eq) in toluene was added to the organic layer at room temperature (Scheme 3). The major product that was isolated was the cyclic benzyloxyhydrouracil 15 and not the desired isocyanate 14. The hydrouracil 15 presumably is formed by the intramolecular cyclization of the initially formed isocyanate 14 with the hydroxamic acid nitrogen.
Scheme 3.

Our results although disappointing are not surprising. It has been reported that the isocyanate 14, prepared from the Curtius reaction of the corresponding acid azide undergoes in situ cyclization albeit at higher temperatures to benzyloxyhydrouracil 15.17 Cleavage of the benzyl protecting group of 15 occurred readily using standard catalytic hydrogenolysis conditions to give the known 3-hydroxydihydrouracil 16.18
We then examined the conversion of the amine TFA salt 17 to its corresponding isocyanate. In this case, it was thought that the isocyanate 17 would be more stable and hence isolable because the undesired intramolecular cyclization to give the corresponding seven membered ring urea 19 would be much less favorable. In fact, treatment of 17 with phosgene under biphasic conditions gave a 2:1 mixture of isocyanate 18 to cyclized product 19, based on proton NMR analysis of the crude product (Scheme 4). The presence of isocyanate, 18, in the product mixture could be easily observed by the characteristic absorption in the IR (2278 cm−1). However, it was not possible to isolate pure isocyanate 18 due to its instability. Our results clearly showed that the desired protected primary hydroxamate-isocyanate linkers were too labile for isolation and hence not useful for our purpose.
Scheme 4.

It was thought that the isocyanates derived from the secondary hydroxamates would be more stable and less prone to the intramolecular cyclization observed with the primary hydroxamates. The amine TFA salt 11 was treated with phosgene using the procedure described (Scheme 5).16 After workup, the isocyanate 20 was isolated in good yields and in sufficient purity, as judged by TLC analysis and 1H NMR spectra, to use in coupling reactions.19 Reaction of amine TFA salt 12 with phosgene under similar conditions gave the corresponding isocyanate 21.20 Both isocyanates were stable when stored in the refrigerator but it was preferable to use them soon after their preparation.
Scheme 5.

With the bifunctional N-methylhydroxamate-isocyanates 20 and 21 in hand, their reactions with simple alcohols and amines were examined, to determine optimum coupling conditions. No reaction was observed when a small excess of octanol was treated with the isocyanate 20 in anhydrous dichloroethane at room temperature. However, after refluxing the reaction mixture for 24 hours, the desired carbamate 22 was isolated in 71% yield after chromatographic purification (Scheme 6). As expected, coupling of isocyanate 20 with octylamine occurred rapidly at 0 °C to give urea 23 in 85% yield. In this example, the solid urea product was purified by trituration with hexane to remove impurities including excess amine. It is not surprising that isocyanate 20 reacted selectively with 3-aminopropanol at 0 °C in dichloromethane to give urea 24 in high yield and purity. Similarly, reaction of isocyanate 21 with 3-aminopropanol gave the corresponding urea 25 in good yield.
Scheme 6.

The focus now was to demonstrate the use of the bifunctional linkers to prepare new classes of trishydroxamic acid chelators with carbamate and urea linkages. To avoid purification problems, it was important to ensure that their reactions with polyols and polyamines go to completion to minimize products of partial coupling. As mentioned earlier, TREN has been a highly used platform for synthetic siderophores. So it was decided to synthesize the tris-hydroxamato derivatives of TREN and triethanolamine using our linkers. These targets would be structurally similar and would facilitate the comparison of urea vs carbamates in metal ion binding and biological properties.
Triethanolamine was refluxed with excess isocyanate 20 in dichloroethane (Scheme 7). Progress of the reaction was monitored by TLC analysis and stepwise formation of mono, di and tri products was observed. The crude product was purified using column chromatography to give carbamate 26 in 72% yield. Deprotection of carbamate 26, which had limited solubility in methanol, was done by catalytic hydrogenolysis (5% Pd on carbon) in 1:1 DCM/MeOH. This reaction was carefully monitored by TLC analysis to ensure completion. The crude product after filtration was triturated with hot ethyl acetate to give pure tris hydroxamic acid 27.21
Scheme 7.
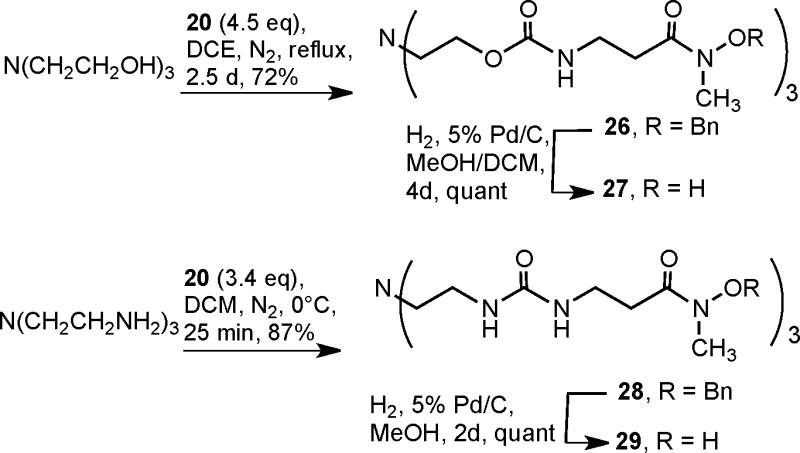
Reaction of TREN with a slight excess of isocyanate 20 at 0°C in dichloromethane gave the protected trishydroxamic acid with urea linkages 28 in 87% yield after chromatographic purification (Scheme 7). Debenzylation of 28 in neat methanol gave the water soluble tris urea hydroxamate chelator 29 in quantitative yield after trituration of the crude product with ethyl acetate.22
In conclusion, bifunctional N-methylhydroxamate-isocyanate linkers 20 and 21 were prepared in good yields and high purity from the corresponding amine salts using biphasic reaction conditions with phosgene and sodium bicarbonate. Our studies showed that the corresponding primary hydroxamate-isocyanate analogs can not be isolated as they underwent intramolecular cyclization to give cyclic N-benzyloxyheterocycles. A key finding is that the facile ring opening reaction of N-Boc lactams using the anion of O-benzylhydroxylamine can be exploited to prepare protected amino hydroxamates. These intermediates can be selectively methylated on the hydroxamate nitrogen in good yields and then readily deprotected. The utility of the bifunctional linkers was clearly demonstrated by the synthesis of the carbamate tethered trishydroxamic acid 27 and the urea tethered 29. The methodology described in this paper will provide access to new classes of carbamate and urea tethered hydroxamate siderophores.
Acknowledgments
This research was supported by grants from the National Institutes of Health under Grant no. 1SC3GM084809 and GM07667-35 (fellowship to ET).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.a) Hider RC, Kong X. Nat Prod Rep. 2010;27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]; b) Sandy M, Butler A. Chem Rev. 2009;109:4580. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miethke M, Marahiel MA. Microbiol Molecular Biol Rev. 2007:413. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) d’Hardemare AM, Torelli S, Serratrice G, Pierre J-L. BioMetals. 2006;19:349. doi: 10.1007/s10534-005-2997-2. [DOI] [PubMed] [Google Scholar]; b) Meijler MM, Arad-Yellin R, Cabantchik I, Shanzer A. J Am Chem Soc. 2002;124:12666. doi: 10.1021/ja027013s. [DOI] [PubMed] [Google Scholar]
- 3.a) Xu J, O’Sullivan B, Raymond KN. Inorg Chem. 2002;41:6731. doi: 10.1021/ic025610+. [DOI] [PubMed] [Google Scholar]; b) Karunaratne V, Hoveyda HR, Orvig C. Tetrahedron Lett. 1992;33:1827. [Google Scholar]; c) Streater M, Taylor PD, Hider RC, Porter J. J Med Chem. 1990;33:1749. doi: 10.1021/jm00168a033. [DOI] [PubMed] [Google Scholar]
- 4.a) Ji C, Juarez-Hernandez RE, Miller MJ. Future Med Chem. 2012;4:297. doi: 10.4155/fmc.11.191. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kovacic P, Edwards CL. Journal of Receptors and Signal Transduction. 2011;31(1):10. doi: 10.3109/10799893.2010.497152. [DOI] [PubMed] [Google Scholar]; c) Frederick RE, Mayfield JA, DuBois JL. Biometals. 2009;22:583. doi: 10.1007/s10534-009-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wencewicz TA, Möllmann U, Long TE, Miller MJ. Biometals. 2009;22:633. doi: 10.1007/s10534-009-9218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Crisponi G, Remelli M. Coord Chem Rev. 2008;252:1225. [Google Scholar]; f) Muri EMF, Nieto MJ, Williamson JS. Medicinal Chemistry Reviews-Online. 2004;1:385. [Google Scholar]
- 5.a) Tam TF, Leung-Toung R, Li W, Wang Y, Kariman K, Spino M. Curr Med Chem. 2003;10:938. doi: 10.2174/0929867033457593. [DOI] [PubMed] [Google Scholar]; b) Kalinowski DS, Richardson DR. Pharmacological Reviews. 2005;57:547. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 6.a) Tommasi RA, Weiler S, McQuire LW, Rogel O, Chambers M, Clark K, Doughty J, Fang J, Ganu V, Grob J, Goldberg R, Goldstein R, LaVoie S, Kulathila R, Macchia W, Melton R, Springer C, Walker M, Zhang J, Zhu L, Shultz M. Bioorg Med Chem Lett. 2011;21:6440. doi: 10.1016/j.bmcl.2011.08.087. [DOI] [PubMed] [Google Scholar]; b) Pabba C, Gregg BT, Kitchen DB, Chen ZJ, Judkins A. Bioorg Med Chem Lett. 2011;21:324. doi: 10.1016/j.bmcl.2010.11.006. [DOI] [PubMed] [Google Scholar]; c) Rossi C, Porcelloni M, D’Andrea P, Fincham CI, Ettorre A, Mauro S, Squarcia A, Bigioni M, Parlani M, Nardelli F, Binaschi M, Maggi CA, Fattori D. Bioorg Med Chem Lett. 2011;21:2305. doi: 10.1016/j.bmcl.2011.02.085. [DOI] [PubMed] [Google Scholar]; d) Lu Z, Ott GR, Anand R, Liu RQ, Covington MB, Vaddi K, Qian M, Newton RC, Christ DD, Trzaskos J, Duan JJW. Bioorg Med Chem Lett. 2008;18:1958. doi: 10.1016/j.bmcl.2008.01.120. [DOI] [PubMed] [Google Scholar]; e) Ott GR, Azakawa N, Lu Z, Liu RQ, Covington MB, Vaddi K, Qian M, Newton RC, Christ DD, Trzaskos JM, Decicco CP, Duan JJW. Bioorg Med Chem Lett. 2008;18:694. doi: 10.1016/j.bmcl.2008.01.075. [DOI] [PubMed] [Google Scholar]; f) Jones P, Steinkühler C. Curr Pharm Design. 2008;14:545. doi: 10.2174/138161208783885317. [DOI] [PubMed] [Google Scholar]
- 7.a) Liu Y, Jacobs HK, Gopalan AS. Tetrahedron. 2011;67:2206. doi: 10.1016/j.tet.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arumugam J, Brown HA, Jacobs HK, Gopalan AS. Synthesis. 2011:57. doi: 10.1055/s-0030-1258337. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Harrington JM, Chittamuru S, Dhungana S, Jacobs HK, Gopalan AS, Crumbliss AL. Inorg Chem. 2010;49:8208. doi: 10.1021/ic902595c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu Y, Jacobs HK, Gopalan AS. J Org Chem. 2009;74:782. doi: 10.1021/jo802410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Oton F, Tárraga A, Espinosa A, María D, Molina P. Dalton Trans. 2006;30:3685. doi: 10.1039/b603544b. [DOI] [PubMed] [Google Scholar]; b) Tongraung P, Chantarasiri N, Tuntulani T. Tetrahedron Lett. 2003;44:29. [Google Scholar]; c) Beer PC, Webber PRA. Dalton Trans. 2003;11:2249. [Google Scholar]; d) Kirkovits GJ, Shriver JA, Gale PA, Sessler JL. J Inclusion Phenom Macrocyclic Chem. 2001;41:69. [Google Scholar]; e) Scheerder J, van Duynhoven JPM, Engbersen JFJ, Reinhoudt DN. Angew Chem Int Ed. 1996;35:1090. [Google Scholar]
- 9.a) Amendola V, Fabbrizzi L, Mosca L. Chem Soc Rev. 2010;39:3889. doi: 10.1039/b822552b. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Duran E, Nacionales D, Valencia A, Wostenberg C, Marinez ER. Tetrahedron Lett. 2008;49:6410. [Google Scholar]; c) Sun XH, Li W, Xia PF, Luo H, Wei Y, Wong MS, Cheng Y, Shuang S. J Org Chem. 2007;72:2419. doi: 10.1021/jo062258z. [DOI] [PubMed] [Google Scholar]
- 10.Bruchmann B. Macromol Mater Eng. 2007;292:981. [Google Scholar]
- 11.Sandler SR, Karo W. Organic Functional Group Preparations. 2. Vol. 3. Academic Press; San Diego: 1989. pp. 482–522. [Google Scholar]
- 12.a) Massaro A, Mordini A, Reginato G, Russo F, Taddei M. Synthesis. 2007:3201. [Google Scholar]; b) Sibi MP, Hasegawa H, Ghorpade SR. Org Lett. 2002;4:3343. doi: 10.1021/ol0263301. [DOI] [PubMed] [Google Scholar]; c) Sibi MP, Hasegawa H. Org Lett. 2002;4:3347. doi: 10.1021/ol026331t. [DOI] [PubMed] [Google Scholar]; d) Takahashi H, Hitomi Y, Iwai Y, Ikegami S. J Am Chem Soc. 2000;122:2995. [Google Scholar]; e) Pirrung MC, Chau JH-L. J Org Chem. 1995;60:8084. [Google Scholar]
- 13.Gissot A, Volonterio A, Zanda M. J Org Chem. 2005;70:6925. doi: 10.1021/jo0509713. [DOI] [PubMed] [Google Scholar]
- 14.Graham SL, Scholz TH. Tetrahedron Lett. 1990;31:6269. [Google Scholar]
- 15.a) Giorgi G, Guideri L, Ponticelli F. Tetrahedron. 2011;67:1463. [Google Scholar]; b) Wehn PM, DuBois J. Angew Chem Int Ed. 2009;4:3802. [Google Scholar]; c) Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, III, Bennett EM, Aldrich CC. J Med Chem. 2008;51:5349. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Silverman RB, Lu H. J Med Chem. 2006;49:7404. doi: 10.1021/jm0608715. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kudoh T, Fukuoka M, Ichikawa S, Murayama T, Ogawa Y, Hashii M, Higashida H, Kunerth S, Weber K, Guse AH, Potter BVL, Matsuda A, Shuto S. J Am Chem Soc. 2005;127:8846. doi: 10.1021/ja050732x. [DOI] [PubMed] [Google Scholar]
- 16.Nowick JS, Holmes DL, Noronha G, Smith EM, Nguyen TM, Huang SL. J Org Chem. 1996;61:3929. doi: 10.1021/jo960038n. [DOI] [PubMed] [Google Scholar]
- 17.Klötzer W. Monatshefte Fuer Chemie. 1964;95(6):1729. [Google Scholar]
- 18.a) Buess CM, Bauer L. J Org Chem. 1955;20:33. [Google Scholar]; b) Hurd CD, Buess CM, Bauer L. J Org Chem. 1954;19:1140. [Google Scholar]
- 19.Synthesis of isocyanate 20. Saturated NaHCO3 (5 mL) was added to a solution of TFA salt, 11, (0.160g, 0.476 mmol) in DCM (5 mL) and the mixture stirred for 30 seconds. The layers were allowed to separate and a solution of phosgene (1.25 mL, 2.5 mmol, 20% in toluene) was added via syringe to the DCM layer. The reaction was then stirred for 20 minutes until the evolution of gas had ceased. The organic layer was separated and the aqueous layer again extracted with DCM (5 mL). The combined organic layers were washed with saturated NaCl (5 mL) and dried (MgSO4). The isocyanate 20 (0.095 g, 80.5%) was obtained as an oil and was used without purification. IR (Neat) 3032, 2941, 2879, 2274, 1659 cm-1; 1H NMR (300 MHz, CDCl3) δ 2.6 (t, J= 6.6 Hz, 2H), 3.22 (s, 3H), 3.52 (t, J= 6.6 Hz, 2H), 4.84 (s, 2H), 7.37–7.41 (m, 5H); 13C NMR (75 MHz, CDCl3) 32.1, 33.5, 38.4, 76.3, 128.8, 129.2, 129.4, 134.2, 153.7, 171.8.
- 20.Isocyanate 21. IR (Neat) 3032, 2941, 2879, 2274, 1659 cm-1; 1H-NMR (300 MHz, CDCl3) δ 1.85 (p, J= 6.6 Hz, 2H), 2.44 (t, J= 7.2 Hz, 2H), 3.22 (s, 3H), 3.34 (t, J= 6.6 Hz, 2H), 4.84 (s, 2H), 7.40 (br s, 5H); 13C-NMR (75 MHz, CDCl3) 25.9, 28.8, 33.5, 42.5, 76.2, 122.0, 128.8, 129.1, 129.4, 134.4, 174.0.
- 21.Carbamate 26. IR (Neat) 3333, 2939, 1715, 1651 cm-1; 1H-NMR (300 MHz, CDCl3) δ 2.6 (unres t, 6H), 2.79 (t, J= 5.8 Hz, 6H), 3.20 (s, 9H), 3.39–3.44 (m, 6H), 4.07 (t, J= 5.7 Hz, 6H), 4.81 (s, 6H), 5.52 (br s, 3H), 7.38 (s, 15H); 13C-NMR (75 MHz, CDCl3) δ 32.8, 33.7, 36.6, 54.1, 63.3, 76.6, 129.0, 129.4, 129.6, 134.6, 156.8, 174.1; HRMS(m/z) [MH]+ calcd for C42H60N10O9 849.4618, found 849.4631.Trishydroxamic acid 27. IR (Neat) 3317, 2936, 1704, 1633 cm-1; 1H-NMR (300 MHz, CD3OD) δ 2.65–2.71 (m, 6H), 2.81–2.85 (m, 6H), 3.19 (s, 9H), 3.36 (t, J= 6 Hz, 6H), 4.08 (q, J= 5.6 Hz, 6H); 13C-NMR (75 MHz, CD3OD) δ 34.3, 37.1, 38.5, 55.8, 65.1, 159.6, 174.7; HRMS(m/z) [MH]+ calcd for C21H40N7 O12 582.2729, found 582.2737.
- 22.Synthesis of urea 28. A solution of isocyanate 20 (0.10 g, 0.41 mmol) in DCM (3 mL) was added to tris(2-aminoethylamine) (0.02 g, 0.12 mmol) at 0 °C and the reaction stirred for 20 min. The solvents were removed in vacuo and the resultant residue was washed with ether (3×10 mL). The crude product was purified by silica gel chromatography (Ethyl acetate: MeOH, 7:3) to give the tris urea 28 as a viscous oil (0.088 g, 87%): IR (KBr) 3380, 2927, 1651, 1558 cm-1; 1H NMR (300 MHz, CDCl3) δ 2.50 (unres t, 6H), 2.65 (unres t, 6H), 3.10–3.20 (m, 6H), 3.17 (s, 9H), 3.45 (q, J= 5.6 Hz, 6H), 4.82 (s, 6H), 5.66 (br s, 3H), 5.83 (br s, 6H), 7.37 (s, 15H); 13C NMR (75 MHz, CDCl3) δ 33.2, 33.4, 35.7, 38.5, 55.1, 76.3, 128.7, 128.9, 129.2, 134.4, 159.1, 174.3. HRMS(m/z) [MH]+ calcd for C42H61N10O9 849.4618, found 849.4631.Synthesis of trishydroxamic acid 29. A suspension of urea 28 (0.09 g, 0.10 mmol) and 5% Pd/C (0.01 g) in MeOH (3 mL) was stirred under H2 (balloon) for 2 days. The catalyst was removed through a 0.45 μm nylon filter rinsing with MeOH. The solvent was removed in vacuo. The resultant crude was washed with ether and EtOAc to give trishydroxamic acid 29 as a yellow oil (0.061 g, quant): IR (Neat) 3322, 2923, 1634, 1568 cm-1; 1H-NMR (300 MHz, D2O) δ 2.55–2.63 (m, 3H), 2.71 (t, J= 6.5 Hz, 6H), 3.13 (s, 9H), 3.31–3.26 (m, 9H), 3.41-3.37 (t, J= 5.6 Hz, 6H); 13C NMR (75 MHz, D2O) δ 33.0, 35.9, 36.5, 49.5, 55.4, 161.0, 174.1; HRMS(m/z) [MH]+ calcd for C21H43N10O9 579.3209, found 579.3226.