

Background: von Willebrand factor (VWF) protects factor VIII (FVIII) from rapid clearance and degradation.
Results: Mass spectrometric footprinting revealed that FVIII protects Lys-773 and the N-terminal Ser-764 of VWF from chemical modification. VWF(S764A) showed increased and VWF(K773A) showed decreased FVIII binding.
Conclusion: The N terminus of VWF is critical for FVIII binding.
Significance: This study sheds new light on the mechanism of FVIII-VWF complex assembly.
Keywords: Blood Coagulation Factors, Factor VIII, Mass Spectrometry (MS), Surface Plasmon Resonance (SPR), von Willebrand Factor, Chemical Footprinting
Abstract
Complex formation between coagulation factor VIII (FVIII) and von Willebrand factor (VWF) is of critical importance to protect FVIII from rapid in vivo clearance and degradation. We have now employed a chemical footprinting approach to identify regions on VWF involved in FVIII binding. To this end, lysine amino acid residues of VWF were chemically modified in the presence of FVIII or activated FVIII, which does not bind VWF. Nano-LC-MS analysis showed that the lysine residues of almost all identified VWF peptides were not differentially modified upon incubation of VWF with FVIII or activated FVIII. However, Lys-773 of peptide Ser-766–Leu-774 was protected from chemical modification in the presence of FVIII. In addition, peptide Ser-764–Arg-782, which comprises the first 19 amino acid residues of mature VWF, showed a differential modification of both Lys-773 and the α-amino group of Ser-764. To verify the role of Lys-773 and the N-terminal Ser-764 in FVIII binding, we employed VWF variants in which either Lys-773 or Ser-764 was replaced with Ala. Surface plasmon resonance analysis and competition studies revealed that VWF(K773A) exhibited reduced binding to FVIII and the FVIII light chain, which harbors the VWF-binding site. In contrast, VWF(S764A) revealed more effective binding to FVIII and the FVIII light chain compared with WT VWF. The results of our study show that the N terminus of VWF is critical for the interaction with FVIII and that Ser-764 and Lys-773 have opposite roles in the binding mechanism.
Introduction
Coagulation factor VIII (FVIII)2 and von Willebrand factor (VWF) play a critical but distinct role in the arrest of bleeding at sites of vessel injury. VWF contributes in this process to the initial platelet plug formation, and FVIII acts as a cofactor for activated factor IX in the membrane-bound activated factor X-generating complex (1, 2). Despite their distinct roles, FVIII circulates in plasma in a tight complex with VWF. In this complex, FVIII is protected from proteolytic degradation, premature binding to its ligands, and rapid clearance from the circulation (3–6). The importance of VWF for FVIII biology is exemplified by the idea that defective FVIII binding of VWF is associated with low FVIII plasma levels, resulting in a hemophilia A-like phenotype. This bleeding disorder is referred to as VWF type 2N disease (5, 7). Despite this well known clinical phenomenon, detailed molecular insight into how VWF binds FVIII is still lacking.
FVIII is synthesized as a single-chain protein encompassing the domains A1-a1-A2-a2-B-a3-A3-C1-C2. a1, a2, and a3 are spacer regions that are rich in acidic amino acid residues. Because of limited proteolysis of the B domain, FVIII circulates in plasma as a heterogeneous heterodimeric protein comprising a heavy chain (domains A1-a1-A2-a2-B) that is noncovalently linked to a light chain (domains a3-A3-C1-C2) (2). It has been shown that VWF binds to the light chain of FVIII (8, 9). For high affinity interaction with FVIII, VWF requires the presence of the a3 region and several sites in the C1 and C2 domain (9–11). Upon activation of FVIII, the acidic a3 region is removed from FVIII, leading to dissociation of the FVIII-VWF complex. Additional cleavages at the C-terminal site of the a1 and a2 regions bisect the A1 and A2 domains and lead to removal of the heterogeneous B domain. The fully activated heterotrimer (FVIIIa) binds via its C domains to procoagulant surfaces, thereby forming a platform for effective generation of FXa by FIXa (2, 12, 13).
The highly glycosylated VWF also comprises multiple domains, which are generally represented in the literature as D1-D2-D′-D3-A1-A2-A3-D4-B-C1-C2-CK (1, 14). Recently, the domain organization within VWF has been further refined in an elegant study by Zhou et al. (15). Besides a number of adjustments in the domain organization between the A3 and CK domains of VWF, these authors showed that the D′ domain can be further subdivided in a trypsin inhibitor-like (TIL) domain and an E repeat.
VWF circulates in plasma as multimers of varying length. The VWF monomers in a multimer are head-to-head and tail-to-tail attached via disulfide linkage between two CK domains and two D3 domains. Disulfide bridge formation between the D3 domains at the N-terminal side of VWF is mediated by the propeptide (domains D1-D2), which is removed from mature VWF through proteolytic cleavage by furin (14).
A major binding site for FVIII has been identified within the D′-D3 region, which includes the first 310 amino acids of mature VWF (16, 17). It has been shown that an isolated dimeric D′-D3 fragment competes with immobilized VWF for FVIII binding. Furthermore, the binding of FVIII to VWF is effectively blocked by antibodies with epitopes that are located within this fragment (16). The importance of the D′-D3 region is further signified by the observation that a spectrum of VWF type 2N mutations has been identified throughout this region (see the ISTH-SSC VWF Database) (5). However, it seems unlikely that all of the identified mutated amino acid residues contribute directly to FVIII binding.
We have previously employed a chemical footprinting mass spectrometry approach to identify amino acid residues that contribute to the stability of FVIIIa (19). We have now utilized this same approach to identify amino acid regions of VWF that contribute to FVIII binding. Our findings revealed that Ser-764 and Lys-773 at the N terminus of VWF are protected from chemical modification in the presence of FVIII. Employing site-directed mutagenesis, we subsequently demonstrated that these residues play critical but surprisingly opposite roles in the interaction with FVIII.
EXPERIMENTAL PROCEDURES
Materials and Proteins
HEPES was from Serva (Heidelberg, Germany), NaCl was from Fagron (Rotterdam, The Netherlands), and Tris-HCl was from Invitrogen. All other chemicals were from Merck unless indicated otherwise. The purification of VWF and the construction of VWF variants have been described (20). Plasma-derived FVIII was purified from Aafact® (Sanquin Research) as described by Meems et al. (13). The light chain of FVIII was obtained from plasma-purified FVIII as described (4). Thrombin was purified as described by Mertens et al. (21). Antibody CLB-CAg 12 is described by Stel et al. (22).
Chemical Modification of VWF in the Presence FVIII and FVIIIa
Tandem mass tag (TMT) modification of the lysine residues of VWF in the presence of FVIII or FVIIIa was repeated in three independent experiments and was performed essentially as described by Bloem et al. (19). Briefly, 35 nm VWF was incubated with 70 nm FVIII in buffer containing 50 mm HEPES (pH 7.4), 150 mm NaCl, and 5 mm CaCl2 for 15 min at 37 °C. To activate FVIII, 2 nm thrombin was added for 2 h at 37 °C. Thrombin activation was terminated by the addition of 1.6 units/ml hirudin. The mixture comprising VWF and FVIIIa was subsequently modified with TMT-127 (Thermo Fisher Scientific). For incubation of VWF with non-FVIIIa, 35 nm VWF and 70 nm FVIII were first incubated with hirudin and then with thrombin, followed by chemical modification with TMT-126. The TMT-modified proteins were pooled at a 1:1 molar ratio, and the free cysteines were alkylated (19). To assess the effect of increasing concentrations of VWF and a fixed concentration of FVIII on the modification of VWF with TMT, we incubated 70 nm FVIII or thrombin-FVIIIa with 35, 70, 140, or 280 nm VWF. We subsequently modified the proteins with TMT-126 (FVIII) and TMT-127 (FVIIIa) as described above. The protein mixtures were cleaved overnight at 37 °C with either 0.1 μg of chymotrypsin or 30 ng of trypsin. The peptides obtained were concentrated and washed using a C18 ZipTip (Millipore Corp.) according to the instructions of the manufacturer.
A unique property of the TMTs is that identical VWF peptides carrying TMT-126 or TMT-127 exhibit an identical mass. However, fragmentation of these peptides using higher energy collision dissociation in a mass spectrometer generates a reporter ion from the TMTs as well as a mass spectrum of the peptide. The intensity of the signals of the reporter ions within each mass spectrum allows calculation of the TMT-127:TMT-126 ratio of the lysine-containing peptides. As such, whether a lysine residue of VWF is protected from modification by FVIII can be assessed.
Mass Spectrometry Analysis
VWF peptides were separated by reverse-phase chromatography and directly sprayed in an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) essentially as described (19, 23). During reverse-phase chromatography, we used a 40-min gradient from 2 to 35% (v/v) acetonitrile with 0.5% (v/v) acetic acid. Collision-induced dissociation spectra to identify the peptides and higher energy collision-induced dissociation spectra to identify the reported groups were obtained as described by Bloem et al. (19, 24). The TMT-127:TMT-126 ratios thereof were assessed using Proteome Discoverer 1.2 software. The SEQUEST search algorithm and UniprotKB protein database 25.H_sapiens.fasta were used for peptide identification. We utilized the following selection criteria to filter the peptides. (i) A maximum false discovery rate of 5% was allowed, employing a decoy database that comprised the reversed sequences of 25.H_sapiens.fasta; (ii) lysine residues within the peptide are all modified by a TMT label (+225.1558 Da), and the cysteines are alkylated; and (iii) the N termini of peptides may be modified by TMT.
FVIII-VWF Competition Assay
5 μg/ml VWF was immobilized on a MaxiSorp microtiter plate (Nunc, Roskilde, Denmark). 0.06 nm FVIII heterodimer or 1.5 nm FVIII light chain was incubated with immobilized VWF for 2 h at 37 °C in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm CaCl2, 2% human serum albumin, and 0.1% Tween 20 in the presence of increasing concentrations of WT VWF, VWF(K773A), and VWF(S764A). The plate was subsequently washed three times with 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm CaCl2, 2% human serum albumin, and 0.1% Tween 20. Residual FVIII binding to immobilized VWF was detected using HRP-labeled CLB-CAg 12 antibody as described (25).
Surface Plasmon Resonance Analysis
A BiacoreTM 3000 system (GE Healthcare) was employed to analyze FVIII association with and dissociation from the VWF variants essentially as described (25, 26). WT VWF, VWF(K773A), and VWF(S764A) were immobilized on a CM5 sensor chip at a density of 2000 response units using the amine coupling kit as described by the manufacturer. Increasing concentrations of FVIII and the FVIII light chain were passed over the immobilized VWF variants at a flow rate of 20 μl/min in buffer containing 20 mm HEPES (pH 7.4), 150 mm NaCl, 5 mm CaCl2, and 0.005% Tween 20. The sensor chip surface was regenerated by three washes with the same buffer containing 1 m NaCl. Association and dissociation curves were corrected for binding in the absence of immobilized VWF, which was <5% of the binding response in the presence of the VWF variants. To obtain the binding response at equilibrium, the association curves were fitted to a one-site association equation employing nonlinear regression as described (25). The response at equilibrium was subsequently plotted as a function of the employed FVIII or FVIII light chain concentration.
Activation of FVIII by FXa in the Presence of the VWF Variants
FVIII was activated by FXa as described by Brinkman et al. (27). Briefly, 0.3 nm FVIII, 200 nm FX, 0.3 nm FIXa, and 25 μm phospholipid vesicles comprising 15% phosphatidylserine, 20% phosphatidylethanolamine, and 65% phosphatidylcholine were incubated in the presence of the VWF variants (15 nm) in buffer containing 25 mm HEPES, 140 mm NaCl, 5 mm CaCl2, 5.4 mm KCl, 0.6 mm MgCl2, 1 mg/ml glucose, and 0.2% (w/v) BSA (Merck). FVIII was activated by 1 nm FXa. Assessment of the amount of FXa generated as a function of time and the preparation of phospholipids are described by Bloem et al. (19).
RESULTS
FVIII Protects the N-terminal VWF Ser-764–Arg-782 Region from Chemical Modification
We chemically modified the lysine residues and the N terminus of VWF with TMT-126 in the presence of FVIII and with TMT-127 in the presence of FVIIIa, which does not bind VWF. The TMT-modified proteins were mixed at a 1:1 molar ratio, processed by chymotrypsin or trypsin, and analyzed by nano-LC-MS. If FVIII protects a lysine of VWF from chemical modification, we would expect identified peptides including this lysine residue to exhibit enhanced incorporation of TMT-127. As a control experiment, we also modified VWF with TMT-126 and TMT-127 in the absence of FVIII and FVIIIa. No difference was expected in the incorporation of TMT-127 and TMT-126 in this experimental setup.
The identified peptides of VWF covered 62% of the amino acid sequence of mature VWF and included 40 of 89 lysine residues. Most of the lysine-containing peptides did not display any change in TMT-127:TMT-126 ratio upon incubation of VWF with FVIII and FVIIIa (Fig. 1 and supplemental Tables S1 and S2). However, D′ domain peptide Ser-766–Leu-774, which included TMT-modified Lys-773, exhibited a TMT-127:TMT-126 ratio of ∼1.5 (Fig. 1A and supplemental Fig. S1). An even more pronounced incorporation of TMT-127 was observed for the peptide that comprised the first 19 amino acid residues of mature VWF (Ser-764–Arg-782) (Fig. 1B). This peptide included TMT-modified N-terminal Ser-764 as well as TMT-modified Lys-773 (supplemental Fig. S2).
FIGURE 1.

The N terminus of VWF is differentially modified by TMT in the presence of FVIII. White bars, 35 nm VWF was incubated with TMT-126 in the presence of 70 nm FVIII and with TMT-127 in the presence of 70 nm FVIIIa as described under “Experimental Procedures.” Gray bars, as a control, 35 nm VWF was modified with TMT-126 and TMT-127 in the absence of FVIII or FVIIIa. The modified proteins were mixed at a 1:1 molar ratio, cleaved by chymotrypsin (A) or trypsin (B), and analyzed by nano-LC-MS. Shown is the mean TMT-127:TMT-126 ratio plus S.D. of lysine-containing peptides for which this ratio was determined three or more times.
In a complementary experiment, we monitored the change in the TMT-127:TMT-126 ratio of the N-terminal peptides Ser-766–Leu-774 and Ser-764–Arg-782 using increasing concentrations of VWF and a fixed concentration of FVIII and FVIIIa (Fig. 2). The data show that the TMT-127:TMT-126 ratio approached a value of 1 for both peptides upon increasing the VWF concentration. This demonstrates that FVIII did not affect the TMT modification of the Ser-764–Arg-782 region under conditions in which there was not enough FVIII available to occupy all of the putative FVIII-binding regions on VWF. These findings show that FVIII protected the VWF Ser-764–Arg-782 region from chemical modification by TMT-126. This implies that this region contributes to FVIII binding.
FIGURE 2.

Changing the molar ratio of FVIII and VWF affects TMT modification of Ser-764 and Lys-773. Increasing concentrations of VWF were incubated with TMT-126 in the presence of 70 nm FVIII and with TMT-127 in the presence of 70 nm FVIIIa as described under “Experimental Procedures.” The indicated molar ratios of FVIII and VWF were employed in the experimental setup. The labeled proteins were mixed, proteolyzed, and analyzed by nano-LC-MS. A, TMT-127:TMT-126 ratio of peptide Ser-764–Arg-782. B, TMT-127:TMT-126 ratio of peptide Ser-766–Leu-774. The modified residues Ser-764 and Lys-773 are indicated by asterisks.
VWF(S764A) and VWF(K773A) Exhibit an Opposite Effect on FVIII Binding
We next assessed whether the chemically modified Ser-764 and Lys-773 residues contribute directly to FVIII binding. To this end, we constructed and purified two variants of VWF in which these residues were replaced with alanines. The purified proteins revealed a similar multimerization pattern as WT VWF. We first evaluated the efficiency by which these variants competed with immobilized VWF for binding FVIII. As the FVIII light chain comprises the main binding region for VWF (8), we also included the light chain in the assay (Fig. 3A). 6 ± 2 nm WT VWF was required to reduce the binding of the FVIII light chain to immobilized WT VWF by 50%. Intriguingly, the required concentration to reach half-maximum inhibition was markedly lower for VWF(S764A) (3 ± 1 nm) and higher for VWF(K773A) (12 ± 5 nm). Compared with WT VWF, this implies that VWF(S764A) binds more effectively to the FVIII light chain. VWF(K773A) was less effective in FVIII light chain binding.
FIGURE 3.

VWF(S764A) is more effective and VWF(K773A) is less effective than WT VWF in competing with immobilized VWF for binding FVIII. Increasing concentrations of VWF(S764A) (▴), WT VWF (●), and VWF(K773A) (♦) were incubated with the FVIII light chain (A) or the FVIII heterodimer (B) in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm CaCl2, 2% human serum albumin, and 0.1% Tween 20. Residual FVIII or FVIII light chain binding to immobilized VWF was assessed using peroxidase-labeled monoclonal antibody CLB-CAg 12 as described under “Experimental Procedures.” C, multimer pattern of the purified proteins that were employed in the analysis, with WT VWF (I), VWF(K773A) (II), and VWF(S764A) (III).
We subsequently repeated the experiments using the FVIII heterodimer (Fig. 3B). Lower concentrations of WT VWF and its variants were required to reach half-maximum inhibition. This observation confirms the earlier finding that VWF binds with higher affinity to intact FVIII than to its light chain (9). However, the same trend was observed for the FVIII binding efficiencies of the VWF variants. 2 ± 1 nm VWF(S764A) was required to reduce the binding of FVIII to immobilized VWF by 50%. For VWF(K773A) and WT VWF, these values were 8 ± 3 and 3 ± 1 nm, respectively. The observed trend in the data reveals a differential FVIII binding efficiency of the VWF variants in the order VWF(S764A) > WT VWF > VWF(K773A).
The Equilibrium Dissociation Constant of the FVIII-VWF(S764A) Complex Is Reduced Compared with That of the FVIII-VWF(K773A) Complex
We employed surface plasmon resonance analysis to gain additional insight in the FVIII binding efficiency of the VWF variants. To this end, we immobilized the VWF variants on the surface of a CM5 sensor chip and assessed the binding of increasing concentrations of the FVIII light chain (Fig. 4). The data reveal effective association of the FVIII light chain with all of the VWF variants. An accurate assessment of the equilibrium dissociation constant (KD) of the FVIII light chain-VWF complex assembly is, however, not straightforward, as VWF multimers comprise multiple binding sites for FVIII. Incomplete dimerization of the D3 domains within these multimers has been suggested to affect FVIII binding (28). To still gain insight into the KD values of the FVIII light chain-VWF complex assemblies without the risk of over-interpretation of the data, we estimated the binding response at equilibrium for each FVIII light chain concentration. The resulting binding response was subsequently plotted as a function of the employed FVIII light chain concentration (Fig. 4, insets). The concentration required to reach half-maximum binding (which reflects KD) was 3 ± 0.3 nm for the FVIII light chain-WT VWF complex, 1 ± 0.1 nm for the FVIII light chain-VWF(S764A) complex, and 16 ± 3 nm for the FVIII light chain-VWF(K773A) complex. The data reveal that VWF(S764A) bound with the highest affinity to the FVIII light chain, followed by WT VWF and then by VWF(K773A).
FIGURE 4.

VWF(S764A) exhibits a higher affinity and VWF(K773A) exhibits a lower affinity for the FVIII light chain compared with WT VWF. 2000 response units of WT VWF (A), VWF(S764A) (B), and VWF(K773A) (C) were immobilized on the surface of a CM5 sensor chip. Increasing concentrations of the FVIII light chain were subsequently perfused over the immobilized VWF variants in buffer containing 20 mm HEPES (pH 7.4), 150 mm NaCl, 5 mm CaCl2, and 0.005% Tween 20. Binding is expressed in response units. The insets show the estimated equilibrium binding responses as a function of the employed FVIII light chain concentration. Half-maximum binding reflects the equilibrium dissociation constant (KD) of the FVIII light chain-VWF variant complex.
The abovementioned approach was repeated using the intact FVIII heterodimer (Fig. 5). The equilibrium dissociation constants obtained for the complexes between the VWF variants and the FVIII heterodimer were reduced compared with those of the complexes with the FVIII light chain (Fig. 5, insets). VWF(S764A) exhibited again the highest binding affinity (KD = 0.13 ± 0.02 nm) for FVIII, followed by WT VWF (KD = 0.44 ± 0.05 nm) and then by VWF(K773A) (KD = 0.68 ± 0.06 nm). The results together confirm that the FVIII binding efficiency of VWF(S764A) is increased and that of VWF(K773A) is decreased.
FIGURE 5.

VWF(S764A) exhibits a higher affinity and VWF(K773A) exhibits a lower affinity for the FVIII heterodimer compared with WT VWF. 2000 response units of WT VWF (A), VWF(S764A) (B), and VWF(K773A) (C) were immobilized on the surface of a CM5 sensor chip. Increasing concentrations of the FVIII heterodimer were subsequently passed over the immobilized VWF variants in buffer containing 20 mm HEPES (pH 7.4), 150 mm NaCl, 5 mm CaCl2, and 0.005% Tween 20. Binding is expressed in response units. The insets show the estimated equilibrium binding responses as a function of the employed FVIII heterodimer concentration. Half-maximum binding reflects the equilibrium dissociation constant (KD) of the FVIII-VWF variant complex.
VWF(S764A) and VWF(K773A) Make Opposite Contributions to Protection of FVIII against Activation by FXa
Although FVIII light chain cleavage by thrombin is accelerated by VWF, the time to reach full cofactor activity is not changed in the presence of VWF. Cleavage of the heavy chain therefore seems to be the rate-limiting step during FVIII activation by thrombin (29). However, VWF has been shown to protect FVIII against premature activation by FXa (27, 30). We therefore assessed the effect of the VWF variants on the activation of FVIII by FXa. To this end, 0.3 nm FVIII was activated by FXa in the presence of the VWF variants (Fig. 6). The data show that the amount of FXa generated as a function of time was reduced in the presence of VWF(S764A) and increased in the presence of VWF(K773A). This finding implies that the differential affinity of the VWF variants for FVIII translates into a differential protection of FVIII against premature proteolysis by FXa.
FIGURE 6.
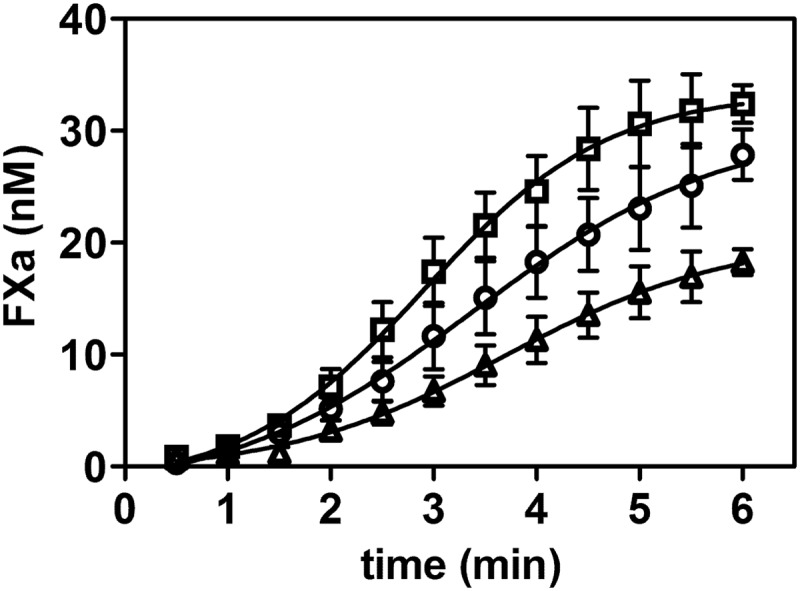
VWF variants offer a differential protection of FVIII against proteolytic activation by FXa. 0.3 nm FVIII was incubated with 200 nm FX, 0.3 nm FIXa, and 25 μm phospholipid vesicles comprising 15% phosphatidylserine, 20% phosphatidylethanolamine, and 65% phosphatidylcholine in the presence of 15 nm VWF(K773A) (□), WT VWF (○), or VWF(S764A) (▵). FVIII was activated by 1 nm FXa, and the newly formed FXa was assessed as a function of time.
DISCUSSION
We previously used TMTs in a chemical footprinting approach to identify lysine residues of FVIII that contribute to the stability of FVIIIa (19). We have now employed this approach to identify critical lysine residues of VWF that contribute to the interaction with FVIII. The NH2 groups of these residues are expected to be susceptible to chemical modification only in the absence of FVIII. However, the dynamic equilibrium between the FVIII-VWF complex and the unbound proteins will also allow for a partial labeling of these critical residues. In addition, VWF is a highly complex glycoprotein that comprises multimers that vary in size. It has been shown that these multimers adopt a globular configuration in the absence of blood (shear) flow (31). Part of the FVIII-binding regions may therefore be buried within the globular VWF protein. Whereas FVIII cannot interact with these buried regions, the small TMT labels may readily penetrate the protein core, thereby modifying residues that contribute to FVIII binding despite the presence of FVIII. The fact that we identified a differential modification of the VWF Ser-764–Arg-782 region in the presence of FVIII therefore emphasizes the critical importance of this region for FVIII binding.
The direct contribution of the side chain of Lys-773 to FVIII binding is demonstrated by the observation that introduction of alanine at position 773 led to a reduced ability to compete with immobilized VWF for binding FVIII and the FVIII light chain (Fig. 3). In addition, surface plasmon resonance analysis showed a decreased FVIII binding affinity of VWF(K773A) (Figs. 4 and 5). However, changing the N-terminal Ser-764 to Ala did not result in a deletion of the N-terminal NH2 group, which was targeted by the chemical labels. Intriguingly, our data reveal that alanine at position 764 led to an increased FVIII binding efficiency (Figs. 3–5). This finding suggests that the small hydrophobic side chain of an alanine may contribute to the direct interaction with FVIII or that the alanine places the N terminus in a more favorable conformation for enhanced interaction with FVIII.
The identity of the amino acid residues of FVIII that interact with the positively charged Lys-773 and N terminus of VWF remains to be determined. It is tempting to suggest that the negatively charged residues within the acidic a3 region of FVIII bind directly to the N terminus of VWF. A chemical footprinting approach that relies on modification of the negatively charged residues of FVIII is required to address this issue. In this study, we did not find any differential modification of the positively charged lysine residues of FVIII that could be attributed to the binding of VWF. The changes that were identified involved the same lysine residues that contribute to the stability of FVIIIa (19). This is not surprising, as we employed thrombin activation of FVIII to separate the constituents of the FVIII-VWF complex in the experimental approach. The unstable FVIIIa readily dissociates itself. This will therefore allow for modification of the lysine residues that contribute to the stability of FVIIIa.
Most of the previously identified VWF type 2N mutations are upstream of the N terminus of VWF (see the ISTH-SSC VWF Database) (5, 7). At first sight, one would assume that these residues contribute to FVIII binding as well. To date, there is no crystal structure of the D′ domain available that can elucidate the molecular mechanisms that underlie these FVIII binding defects. However, Zhou et al. (15) showed that the N-terminal part of the D′ domain (i.e. the TIL domain) shares primary sequence homology with that of Apis mellifera chymotrypsin/cathepsin G inhibitor-1 (AMCI-1), for which an NMR structure is available (32). We therefore constructed a homology model of the TIL domain of VWF by comparative homology modeling using AMCI-1 as a template (Fig. 7) (33). The model reveals that the TIL domain is a flat ellipsoid structure comprising a network of critical cysteine disulfide bonds. The model agrees with the previous experimental studies, which showed that cysteines 767 and 808, 776 and 804, and 810 and 812 are connected (15, 34). Interestingly, the sequence alignment and model also reveal that most of the VWF type 2N mutations within this region are at or close to the critical cysteine residues or involve replacements of residues that are shared between the TIL domain and AMCI-1. It therefore seems feasible that the type 2N mutations may lead to an altered conformation of the TIL domain, thereby affecting FVIII binding, instead of these residues contributing directly to the interaction with FVIII.
FIGURE 7.

Molecular model of the N-terminal part of the D′ domain. A model of the first 64 residues of the D′ domain, i.e. the TIL domain, was constructed by comparative homology modeling with Modeller 9v3. The NMR structure of AMCI-1 (Protein Data Bank code 1CCV) was used as a template (32, 33). Shown are side and front views of the model. S–S bridges between the cysteine residues (yellow) are shown in red. Lys-773 is indicated in blue, and Ser-764 in green. The primary sequence alignment of the TIL domain of VWF and AMCI-1 is shown below the model. Replacements of amino acids that are associated with VWF type 2N disease are indicated above the sequence (see the ISTH-SSC VWF Database).
The model further shows that Ser-764 and Lys-773 may be in close proximity at the edge of the structure (Fig. 7). Interestingly, it suggests that the OH group of Ser-764 may even form an intramolecular hydrogen bond with the backbone carbonyl group of Lys-773. Although it is speculation, direct binding of FVIII to Lys-773 may interfere with this intramolecular interaction, leading to a repositioning of the N terminus. This molecular mechanism may be essential for effective FVIII binding. Replacement of Ser-764 with Ala prevents the intramolecular binding of the OH group and may position the N terminus already in a conformation that favors FVIII binding. If so, this could then explain the enhanced binding affinity of VWF(S764A) for FVIII and the FVIII light chain (Figs. 3–5).
Our finding also reconciles a previous discrepancy in the literature. It has been suggested that a VWF variant that lacks the propeptide does not bind to FVIII at all. As the propeptide mediates dimerization of the N-terminal side of VWF, it was proposed that proper dimerization is a prerequisite for effective FVIII binding (35). In a later study, Bendetowicz et al. (28) showed that the VWF variant that lacks the propeptide does bind FVIII, albeit with a reduced affinity. The apparent discrepancy between these studies has been attributed to the fact that the VWF variant that does not bind FVIII comprises an additional alanine before Ser-764 at its N terminus (28, 35). Our study has shown that a native N terminus, which starts with Ser-764, is critical for FVIII binding. Addition of the extra alanine will most likely prevent interaction of the N terminus with FVIII. Alternatively, this alanine places the N terminus in an unfavorable position for effective FVIII interaction.
It is also remarkable that there is a difference in the binding affinity of VWF for the FVIII light chain and the FVIII heterodimer (Figs. 4 and 5) (9). So far, VWF has been shown to require amino acid regions in the C2 domain, the C1 domain, and the acidic a3 region of the FVIII light chain for high affinity binding (8–11). The increased affinity for intact FVIII could be explained if VWF binds regions outside the FVIII light chain as well. Alternatively, analysis of the available crystal structures of FVIII reveals that the C2 domain is loosely tethered in the structure (18, 36). It cannot be excluded that the dynamics of the C2 domain is more restricted in the intact heterodimer than in the isolated FVIII light chain. The reduced whole-domain dynamics in the heterodimer may facilitate the increased binding affinity of the FVIII-VWF complex. Further studies are required to discriminate between the two possibilities. Irrespective of the outcome of these studies, our present findings suggest that the N terminus of VWF contributes to the direct interaction with the FVIII light chain and the intact FVIII heterodimer.
VWF is of principal importance for protection of FVIII in the circulation (3–6). Our results show that it may be feasible to further increase the stability of the FVIII-VWF complex in vivo. Gaining further insight into how VWF binds FVIII may pave the way for the design of variants of both proteins that bind with a markedly enhanced affinity compared with their natural counterparts. We have shown here that the start of the N terminus of VWF constitutes an attractive target to further explore the FVIII-VWF binding mechanism.
This work was supported by Landsteiner Stichting voor Bloedtransfusieresearch Grant LSBR-0730 (to A. B. M.).

This article contains supplemental Figs. S1 and S2 and Tables S1 and S2.
- FVIII
- factor VIII
- FVIIIa
- activated FVIII
- VWF
- von Willebrand factor
- TIL
- trypsin inhibitor-like
- TMT
- tandem mass tag
- AMCI-1
- A. mellifera chymotrypsin/cathepsin G inhibitor-1.
REFERENCES
- 1. Schneppenheim R., Budde U. (2011) von Willebrand factor: the complex molecular genetics of a multidomain and multifunctional protein. J. Thromb. Haemost. 9, 209–215 [DOI] [PubMed] [Google Scholar]
- 2. Fay P. J. (2006) Factor VIII structure and function. Int. J. Hematol. 83, 103–108 [DOI] [PubMed] [Google Scholar]
- 3. Morfini M., Mannucci P. M., Tenconi P. M., Longo G., Mazzucconi M. G., Rodeghiero F., Ciavarella N., De Rosa V., Arter A. (1993) Pharmacokinetics of monoclonally-purified and recombinant factor VIII in patients with severe von Willebrand disease. Thromb. Haemost. 70, 270–272 [PubMed] [Google Scholar]
- 4. Lenting P. J., Donath M. J., van Mourik J. A., Mertens K. (1994) Identification of a binding site for blood coagulation factor IXa on the light chain of human factor VIII. J. Biol. Chem. 269, 7150–7155 [PubMed] [Google Scholar]
- 5. Jacquemin M. (2009) Factor VIII-von Willebrand factor binding defects in autosomal recessive von Willebrand disease type Normandy and in mild hemophilia A. New insights into factor VIII-von Willebrand factor interactions. Acta Haematol. 121, 102–105 [DOI] [PubMed] [Google Scholar]
- 6. Brinkhous K. M., Sandberg H., Garris J. B., Mattsson C., Palm M., Griggs T., Read M. S. (1985) Purified human factor VIII procoagulant protein: comparative hemostatic response after infusions into hemophilic and von Willebrand disease dogs. Proc. Natl. Acad. Sci. U.S.A. 82, 8752–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sadler J. E., Budde U., Eikenboom J. C., Favaloro E. J., Hill F. G., Holmberg L., Ingerslev J., Lee C. A., Lillicrap D., Mannucci P. M., Mazurier C., Meyer D., Nichols W. L., Nishino M., Peake I. R., Rodeghiero F., Schneppenheim R., Ruggeri Z. M., Srivastava A., Montgomery R. R., Federici A. B. (2006) Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J. Thromb. Haemost. 4, 2103–2114 [DOI] [PubMed] [Google Scholar]
- 8. Lollar P., Hill-Eubanks D. C., Parker C. G. (1988) Association of the factor VIII light chain with von Willebrand factor. J. Biol. Chem. 263, 10451–10455 [PubMed] [Google Scholar]
- 9. Saenko E. L., Scandella D. (1997) The acidic region of the factor VIII light chain and the C2 domain together form the high affinity binding site for von Willebrand factor. J. Biol. Chem. 272, 18007–18014 [DOI] [PubMed] [Google Scholar]
- 10. Foster P. A., Fulcher C. A., Houghten R. A., Zimmerman T. S. (1988) An immunogenic region within residues Val1670–Glu1684 of the factor VIII light chain induces antibodies which inhibit binding of factor VIII to von Willebrand factor. J. Biol. Chem. 263, 5230–5234 [PubMed] [Google Scholar]
- 11. Leyte A., van Schijndel H. B., Niehrs C., Huttner W. B., Verbeet M. P., Mertens K., van Mourik J. A. (1991) Sulfation of Tyr1680 of human blood coagulation factor VIII is essential for the interaction of factor VIII with von Willebrand factor. J. Biol. Chem. 266, 740–746 [PubMed] [Google Scholar]
- 12. Lü J., Pipe S. W., Miao H., Jacquemin M., Gilbert G. E. (2011) A membrane-interactive surface on the factor VIII C1 domain cooperates with the C2 domain for cofactor function. Blood 117, 3181–3189 [DOI] [PubMed] [Google Scholar]
- 13. Meems H., Meijer A. B., Cullinan D. B., Mertens K., Gilbert G. E. (2009) Factor VIII C1 domain residues Lys2092 and Phe2093 contribute to membrane binding and cofactor activity. Blood 114, 3938–3946 [DOI] [PubMed] [Google Scholar]
- 14. Sadler J. E. (2009) von Willebrand factor assembly and secretion. J. Thromb. Haemost. 7, 24–27 [DOI] [PubMed] [Google Scholar]
- 15. Zhou Y. F., Eng E. T., Zhu J., Lu C., Walz T., Springer T. A. (2012) Sequence and structure relationships within von Willebrand factor. Blood 120, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foster P. A., Fulcher C. A., Marti T., Titani K., Zimmerman T. S. (1987) A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J. Biol. Chem. 262, 8443–8446 [PubMed] [Google Scholar]
- 17. Takahashi Y., Kalafatis M., Girma J. P., Sewerin K., Andersson L. O., Meyer D. (1987) Localization of a factor VIII binding domain on a 34 kilodalton fragment of the N-terminal portion of von Willebrand factor. Blood 70, 1679–1682 [PubMed] [Google Scholar]
- 18. Shen B. W., Spiegel P. C., Chang C. H., Huh J. W., Lee J. S., Kim J., Kim Y. H., Stoddard B. L. (2008) The tertiary structure and domain organization of coagulation factor VIII. Blood 111, 1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bloem E., Meems H., van den Biggelaar M., van der Zwaan C., Mertens K., Meijer A. B. (2012) Mass spectrometry-assisted study reveals that lysine residues 1967 and 1968 have opposite contribution to stability of activated factor VIII. J. Biol. Chem. 287, 5775–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van den Biggelaar M., Bouwens E. A., Voorberg J., Mertens K. (2011) Storage of factor VIII variants with impaired von Willebrand factor binding in Weibel-Palade bodies in endothelial cells. PLoS ONE 6, e24163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mertens K., van Wijngaarden A., Bertina R. M. (1985) The role of factor VIII in the activation of human blood coagulation factor X by activated factor IX. Thromb. Haemost. 54, 654–660 [PubMed] [Google Scholar]
- 22. Stel H. V., Sakariassen K. S., Scholte B. J., Veerman E. C., van der Kwast T. H., de Groot P. G., Sixma J. J., van Mourik J. A. (1984) Characterization of 25 monoclonal antibodies to factor VIII-von Willebrand factor: relationship between ristocetin-induced platelet aggregation and platelet adherence to subendothelium. Blood 63, 1408–1415 [PubMed] [Google Scholar]
- 23. van Haren S. D., Herczenik E., ten Brinke A., Mertens K., Voorberg J., Meijer A. B. (2011) HLA-DR-presented peptide repertoires derived from human monocyte-derived dendritic cells pulsed with blood coagulation factor VIII. Mol. Cell Proteomics 10, M110.002246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dayon L., Pasquarello C., Hoogland C., Sanchez J. C., Scherl A. (2010) Combining low- and high-energy tandem mass spectra for optimized peptide quantification with isobaric tags. J. Proteomics 73, 769–777 [DOI] [PubMed] [Google Scholar]
- 25. Meems H., van den Biggelaar M., Rondaij M., van der Zwaan C., Mertens K., Meijer A. B. (2011) C1 domain residues Lys2092 and Phe2093 are of major importance for the endocytic uptake of coagulation factor VIII. Int. J. Biochem. Cell Biol. 43, 1114–1121 [DOI] [PubMed] [Google Scholar]
- 26. van den Biggelaar M., Meijer A. B., Voorberg J., Mertens K. (2009) Intracellular cotrafficking of factor VIII and von Willebrand factor type 2N variants to storage organelles. Blood 113, 3102–3109 [DOI] [PubMed] [Google Scholar]
- 27. Brinkman H. J., Mertens K., Holthuis J., Zwart-Huinink L. A., Grijm K., van Mourik J. A. (1994) The activation of human blood coagulation factor X on the surface of endothelial cells: a comparison with various vascular cells, platelets and monocytes. Br. J. Haematol. 87, 332–342 [DOI] [PubMed] [Google Scholar]
- 28. Bendetowicz A. V., Morris J. A., Wise R. J., Gilbert G. E., Kaufman R. J. (1998) Binding of factor VIII to von Willebrand factor is enabled by cleavage of the von Willebrand factor propeptide and enhanced by formation of disulfide-linked multimers. Blood 92, 529–538 [PubMed] [Google Scholar]
- 29. Hill-Eubanks D. C., Lollar P. (1990) von Willebrand factor is a cofactor for thrombin-catalyzed cleavage of the factor VIII light chain. J. Biol. Chem. 265, 17854–17858 [PubMed] [Google Scholar]
- 30. Koedam J. A., Hamer R. J., Beeser-Visser N. H., Bouma B. N., Sixma J. J. (1990) The effect of von Willebrand factor on activation of factor VIII by factor Xa. Eur. J. Biochem. 189, 229–234 [DOI] [PubMed] [Google Scholar]
- 31. Schneider S. W., Nuschele S., Wixforth A., Gorzelanny C., Alexander-Katz A., Netz R. R., Schneider M. F. (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. U.S.A. 104, 7899–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cierpicki T., Bania J., Otlewski J. (2000) NMR solution structure of Apis mellifera chymotrypsin/cathepsin G inhibitor-1 (AMCI-1): structural similarity with Ascaris protease inhibitors. Protein Sci. 9, 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eswar N., Eramian D., Webb B., Shen M. Y., Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 34. Dong Z., Thoma R. S., Crimmins D. L., McCourt D. W., Tuley E. A., Sadler J. E. (1994) Disulfide bonds required to assemble functional von Willebrand factor multimers. J. Biol. Chem. 269, 6753–6758 [PubMed] [Google Scholar]
- 35. Leyte A., Voorberg J., Van Schijndel H. B., Duim B., Pannekoek H., Van Mourik J. A. (1991) The pro-polypeptide of von Willebrand factor is required for the formation of a functional factor VIII-binding site on mature von Willebrand factor. Biochem. J. 274, 257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ngo J. C., Huang M., Roth D. A., Furie B. C., Furie B. (2008) Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure 16, 597–606 [DOI] [PubMed] [Google Scholar]