

Abstract
The cancer drug discovery field has placed much emphasis on the identification of novel and cancer-specific molecular targets. A rich source of such targets for the design of novel anti-tumor agents is the ubiqutin-proteasome system (UP-S), a tightly regulated, highly specific pathway responsible for the vast majority of protein turnover within the cell. Because of its critical role in almost all cell processes that ensure normal cellular function, its inhibition at one point in time was deemed non-specific and therefore not worth further investigation as a molecular drug target. However, today the proteasome is one of the most promising anti-cancer drug targets of the century. The discovery that tumor cells are in fact more sensitive to proteasome inhibitors than normal cells indeed paved the way for the design of its inhibitors. Such efforts have led to bortezomib, the first FDA approved proteasome inhibitor now used as a frontline treatment for newly diagnosed multiple myeloma (MM), relapsed/refractory MM and mantle cell lymphoma. Though successful in improving clinical outcomes for patients with hematological malignancies, relapse often occurs in those who initially responded to bortezomib. Therefore, the acquisition of bortezomib resistance is a major issue with its therapy. Furthermore, some neuro-toxicities have been associated with bortezomib treatment and its efficacy in solid tumors is lacking. These observations have encouraged researchers to pursue the next generation of proteasome inhibitors, which would ideally overcome bortezomib resistance, have reduced toxicities and a broader range of anti-cancer activity. This review summarizes the success and limitations of bortezomib, and describes recent advances in the field, including, and most notably, the most recent FDA approval of carfilzomib in July, 2012, a second generation proteasome inhibitor. Other proteasome inhibitors currently in clinical trials and those that are currently experimental grade will also be discussed.
Keywords: Ubiquitin-proteasome system (UP-S), inhibitors of the 20S proteasome, bortezomib, drug discovery, cancer, natural compounds, clinical studies
THE PROTEASOME
From Therapeutically Untouchable to Prime Molecular Drug Target
Today the proteasome is widely accepted as the primary means by which protein homeostasis is maintained within the cell and that it is essential for the occurrence and progression of almost all normal cell processes. However, this was not always so. With the discovery of the lysosome as a proteolytic organelle, in the early 1950s, it became only logical to suggest that all intracellular proteolysis occurs at the hands of the proteases within this compartment. Fast forward 20–30 years and this simple concept is heavily complicated by much accumulated evidence declaring the presence of an entirely new, very specific, and non-lysosomal protein degradation system dependent on a novel post-translational phenomena termed polyubiquitination. The discovery of the ubiquitin-proteasome system is accredited to Aaron Ciechanover, Avram Hershko and Irwin Rose; their work was acknowledged with the 2004 Nobel Prize in Chemistry. The 26S proteasome (Fig. 1) was first described as a giant protease having multiple subunits, including ring-shaped 19S regulatory and 20S catalytic core components, further composed of numerous polypeptide subunits [1–3]. Seven α subunits make up each of the two identical outer rings of the 20S catalytic core, while seven β subunits form each of the inner two identical rings, together they are arranged in a stacked cylindrical structure with a narrow pore through which a particular protein substrate could pass and reach their ultimate fate: degradation [1]. The high level of specificity of this system is made possible by the series of events that occur prior to the proteasome's activity, the process of polyubiquitination. The ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and the ubiquitin E3 ligases are present in a hierarchal nature and are responsible for the ATP-dependent activation of ubiquitin and ultimate transfer of this moiety to particular protein substrates. The ubiquitin E3 ligases not only dictate which proteins will fall victim to the proteasome by polyubiquitination at the specific lysine 48 residue, but also regulate protein trafficking events through the monoubiquitination of lysine 63 and other non-traditional lysine residues [3].
Fig. (1).

The 26S Proteasome and cross-sectional view of the bortezomib (BTZ) binding site. The 26S proteasome contains ring-shaped 19S regulatory and 20S catalytic core components, which are further composed of 7 α subunits (making up each of the two identical outer rings of the 20S catalytic core) and 7 β subunits (forming each of the inner two identical rings). Together they are arranged in a stacked cylindrical structure with a narrow pore through which a particular protein substrate can pass and reach their ultimate fate: degradation. Also depicted is a magnified cross-sectional view of the β subunits and the particular β1 and β5 bortezomib binding sites.
Because the proteasome is essential for many key cellular regulatory mechanisms, is ubiquitously expressed, and in fact can constitute up to 1% of the cellular protein content in eukaryotes [1, 4], it was a reasonable assumption that modulating its function was an untouchable strategy in the design and development of novel agents for the treatment of various diseases, particularly cancer. In other words, the ubiquitin-proteasome system is absolutely vital for the identification and removal of misfolded, damaged, or toxic proteins; it was therefore thought not to be logical to inhibit its function. However, this again was found not to be so. Proteasome inhibitors have in fact been developed and in the last decade have translated into the clinic, the most notable being bortezomib (Fig. 1 and 2), the first FDA approved proteasome inhibitor as an anticancer agent for the treatment of relapsed multiple myeloma and mantle cell lymphoma in 2003 and most recently carfilzomib (Table I), a second generation product of this class of compounds FDA approved for patients with multiple myeloma progression while on, or after treatment with, bortezomib and an immunomodulatory agent. Contrary to what may have been thought, the use of proteasome inhibitors for the treatment of cancer has proven to induce a pro-apoptotic response that is relatively cancer cell-specific. Furthermore, proteasome inhibition has been shown to be a valuable strategy in the sensitization of cancer cells to the effects of traditional chemotherapies and radiation. Several mechanisms have been proposed to explain these observed effects, such as: down regulation of NFκB and other anti-apoptotic proteins, activation of the tumor suppressor protein p53, and modulation of cell cycle proteins and other pro-apoptotic factors. Interestingly, the effects of proteasome inhibition appear to be selective for tumor cells over normal cells and ongoing research will help to elucidate the mechanism(s) responsible for why this is the case. Herein we take a look at the evolution and future outlook of proteasome inhibitors and the rationale behind their design and development, focusing on a comparison to the field's prototype, bortezomib.
Fig. (2).
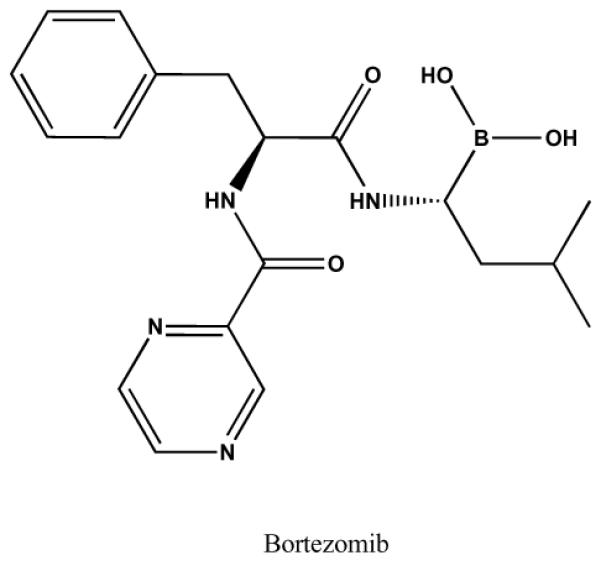
Chemical structure of bortezomib.
Table I.
Bortezomib Clinical Trials
| Trial | Summary | Result | Reference |
|---|---|---|---|
| Phase II CREST |
|
|
[48,49] |
| Phase II SUMMIT |
|
|
[50] |
| Phase III APEX |
|
|
[51,52] |
| Phase III VISTA |
|
|
[53,54] |
| Phase III PINNACLE |
|
|
[60,61] |
| Phase III VERTICAL |
|
|
[64] |
Proteasome Inhibitors Overview: Endogenous, Natural and Synthetic
Prior to the approval of bortezomib by the FDA, many other proteasome inhibitors were developed and used experimentally. Based on earlier studies suggesting the use of small peptide substrates as templates for the design of proteasome inhibitors, many peptide aldehydes were synthesized toward the goal of developing potent and specific inhibitors [5–8]. Among this class of inhibitors are calpain inhibitors I and II, leupeptin, and MG-132 [5, 9–11], all of which have been tested in vitro and in vivo. While these inhibitors are quite potent, their structures also cause them to be multi-specific, with reactivity toward not only the active proteasomal subunits (β1, β2, and β5), but also against serine and cysteine proteases. In an effort to increase selectivity, peptide boron esters were synthesized and demonstrated both greater specificity and higher potency [12–14]. Further derivations of this class, including dipeptide boron esters such as bortezomib, have proven even more successful than their parent structures. While these classes of inhibitors are reversible, some irreversible inhibitors have also been investigated, including several serine protease inhibitors, such as 3,4-dichloroisocoumarin [15–17] as well as chloromethyl and diazomethyl ketones [18]. These classes are limited, however, in their broad reactivity and low potency [18, 19]. Additionally, cysteine protease inhibitors such as peptide vinyl sulfones have also been shown to be potent irreversible inhibitors of the proteasome [20–23]. Presently, the design of new synthetic proteasome inhibitors is based not only on the structure of previously investigated compounds, but also on the structure of the proteasome itself. This rational drug design strategy should yield more potent and specific inhibitors.
The health benefits of several natural compounds have also long been recognized and many investigators have looked to these natural compounds for their potential to specifically inhibit the proteasome. In fact, many of these natural products are much more selective and potent than the synthetically created compounds described previously. One such product is lactacystin (Fig. 3), a Streptomyces metabolite, which was initially investigated for its ability to inhibit cell cycle progression, but was later found to possess the ability to inhibit all proteasomal catalytic activities [22, 24–26]. Another class of natural proteasome inhibitors includes epoxomicin (Fig. 3) and eponemycin, which contain epoxyketone groups at their C-termini. Investigation into the biological targets of these compounds began after the observation that both are able to inhibit tumor growth [27, 28] and that eponemycin is a potent inhibitor of angiogenesis [29]. This class is highly specific and extremely potent towards the proteasome, due to its unusual mode of action [30]. Importantly, chemists have taken advantage of nature's design of these potent inhibitors to synthesize derivatives, which have aided in the identification of the modes of action of these compounds, as well as produced even more potent and specific inhibitors [30–35]. Thus, basing drug design on the structure of the proteasome as well as the structure of potent natural inhibitors may yield the most promising compounds. Additionally, studies have also investigated the green tea polyphenol, epigallocatechin gallate (EGCG) (Fig. 3), as well as its synthetic analogs, grape polyphenols, soy isoflavones, and other microbial products for their proteasome inhibitory abilities, which are described in further detail below.
Fig. (3).

Chemical structures of natural compounds with proteasome-inhibitory activity.
With the success of synthetic and natural proteasome inhibitors, the existence of an endogenous proteasome inhibitor would clearly validate proteasome inhibition as a viable strategy. Many cellular proteins, such as p53 and NFκB have inhibitory counterparts (MDM2 and IκB-α, respectively), thus it is no surprise that early reports suggested the presence of a potential endogenous inhibitor of the proteasome [36, 37], which was later identified as delta-aminolevulinic acid dehydratase (ALAD) [38], a critical component of the heme biosynthetic pathway. ALAD functions as an octomer consisting of eight identical subunits, with two or three zinc molecules bound to each subunit. In the heme pathway, ALAD catalyzes the condensation of two aminolevulinic acid (ALA) molecules into porphoblinogen (PBG). Importantly, the binding of zinc (Zn) is crucial to the activity of the enzyme, while lead (Pb) binding has been shown to be inhibitory [39–43].
In addition to its role in heme biosynthesis, ALAD has also been shown to be identical to the inhibitory CF-2 (conjugate-degrading factor-2) component of the proteasome [38], as well as an inhibitor of calpain [44]. ALAD and CF-2 have common N-terminal amino acid sequences and isoelectric points, specific antibody cross-reactivity, similar proteasome inhibitory and dehydratase activities, and migrate identically on native and SDS-PAGE gels [38]. The CF-2 inhibitory subunits were shown to exist in an ubiquitinated form within the proteasome complex [45], however, a recent report has shown that ALAD is a proteasome-interacting protein (PIP) rather than a component of the proteasome [46]. Interestingly, ubiquitination of the proteasome inhibitor/ALAD caused a 90% loss of ALAD and its proteasome inhibitory activity [47]. The relationship between ALAD and the proteasome must further be investigated. The activation of ALAD as an endogenous inhibitor of the proteasome may be a promising target in the treatment of human cancers as well as other diseases involving the proteasome.
A Look at the Proteasome Inhibitor Bortezomib: The Good, the Bad, and the Ugly
Bortezomib (Velcade®) (Figs. 1 and 2) is the first proteasome inhibitor approved by the FDA. In 2003, bortezomib received fasttrack approval for the treatment of refractory multiple myeloma (MM) based on the data presented from two independent phase II trials (Table I): the CREST trial [48, 49] and the SUMMIT trial [50]. Bortezomib was first synthesized in 1995 by Myogenics and passed along to several companies until finally landing in the hands of Millennium Pharmaceuticals in 1999 with a low priority tag. That is, until, one of its first patient volunteers in a MM trial achieved complete response (CR) and was still alive four years later. The drug is a C-terminal boronic acid and it is the boron atom that is in fact essential for inhibiting proteasome activity because of its ability to specifically and tightly bind the β5 catalytic subunit. While multiple mechanisms are probably responsible for its anti-tumor effect, it is likely that the inhibition of degradation of proapoptotic proteins initiates the programmed cell death (apoptosis) response specifically in tumor cells which are otherwise dependent on the suppression of such proteins for survival.
Bortezomib was first approved for use in those patients who had received at least two prior lines of therapy, then in 2005 for those who had at least one prior therapy (as a result of the APEX trial) (Table I) [51, 52]) and in 2008, bortezomib successfully became the front-line therapy for newly diagnosed MM patients based on the phase III VISTA trial (Table I) [53, 54]. Naturally, combinations of bortezomib with other commonly used chemotherapeutic agents, such as dexamethasone, have been explored. The combination of bortezomib and high-dose dexamethasone as a first-line option for MM patients was indeed studied in a phase II trial and the results have revealed that bortezomib, with or without dexamethasone, is an effective and well-tolerated first choice regimen for these patients [55, 56]. The addition of a third drug to this mix, like the widely used doxorubicin, has also been examined. This combination was given before stem cell transplantation and the results reported show a 90% response rate in newly diagnosed MM patients with well-tolerated and manageable toxicities [57, 58]. Likewise, the combination of bortezomib, thalidomide and dexamethasone as front-line therapy in newly diagnosed MM patients has also shown a favorable outcome [59]. Moreover, after the 2006 phase II PINNACLE trial (Table I) of 155 relapsed or refractory mantle cell lymphoma (MCL) patients treated with bortezomib monotherapy showed an overall response rate (ORR) of 32% the FDA extended its approval to cover MCL [60, 61]. In another phase II trial, the combination of bortezomib with gemcitabine was investigated and found to be effective in patients with relapsed or refractory MCL with an ORR of 60% [62]. MCL accounts for just 5% of all B-cell lymphomas with diffuse large B-cell lymphoma and follicular lymphoma being the two major types, accounting for 30–40% and 20% respectively [63].
There is great interest in expanding the use of bortezomib for the treatment of other hematological malignancies, such as different types of B-cell lymphomas as well as some advanced stage solid tumors. In the phase II VERTICAL trial, a combination of bortezomib, bendamustine, and rituximab was evaluated and proved to be highly active (ORR of 88% and complete response (CR) of 53%) in patients with relapsed or refractory follicular lymphoma [64]. In a phase I/II B-cell lymphoma study, bortezomib was introduced to the standard CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) plus rituximab regimen (R-CHOP), which produced an evaluable ORR of 100% with 86% CR/unconfirmed CR. The ORRs were 91% and 72% in MCL patients respectively. These results strongly support the push for entry of bortezomib plus the R-CHOP regimen into phase III trials [65].
In solid tumor cases however, the efficacy of bortezomib has been disappointing. Its use has been extensively investigated in several solid tumor types, but its success has fallen short when compared to hematological malignancies. For example, in a study including patients with castration resistant metastatic prostate cancer neither bortezomib alone, nor combined with prednisone, exhibited significant antitumor effects [66]. Similarly, the combination of docetaxel and bortezomib as a potential first-line treatment showed no improved efficacy versus docetaxel alone [67]. In heavily pre-treated metastatic breast cancer patients, bortezomib plus pegylated liposomal doxorubicin was well tolerated but had minimal activity [68]. A trial testing bortezomib monotherapy in chemotherapy-naïve patients with advanced stage non-small cell lung cancer was terminated in the first stage due to lack of response in all patients [69]. Bortezomib monotherapy was also inactive in patients with unresectable or metastatic gastric and gastroesophageal junction adenocarcinoma [70]. Another disappointing example is the failure of a first-line regimen including bortezomib, paclitaxel and carboplatin in the treatment of patients with metastatic esophageal, gastric and gastroesophageal cancer [71]. Therefore it is obvious that more effort is required, both in the lab and clinic, to best determine the place for bortezomib in the treatment of solid tumors. The online United States National Cancer Institute (NCI) clinical trials database has just under 200 currently active trials listed studying bortezomib's potential in the treatment of these various cancer types, although delineating the mechanism by which bortezomib fails in these cases may be the first critical step in improving its efficacy.
After weighing its clinical success and failure, it is important to sort through both bortezomib's advantages and disadvantages in order to better understand what qualities to avoid, which to keep, and which to improve on for the development of the next generation of proteasome inhibitors.
First, as a proteasome inhibitor, bortezomib exhibits favorable selectivity towards tumor cells over normal cells, which is an important criterion for being a good anti-cancer drug. Numerous studies have proved the selectivity of bortezomib in tumor cells versus normal cells. Multiple factors have been implicated in contributing to this tumor cell selectivity. (i) Cancer cells are rapidly dividing cells compared to non-cancerous cells. Therefore, they more heavily rely on proteasomal turnover of cell cycle regulatory proteins to promote cell cycle progression than normal cells. (ii) Cancer cells appear to generate misfolded or damaged proteins much faster than non-cancerous cells due to their uncontrolled cell proliferation. (iii) The amount of proteasomes and/or the proteasomal activity has been shown to be up-regulated in many types of cancers and this up-regulation is important for the maintenance of the malignant phenotype [72, 73].
Secondly, the outstanding efficacy of bortezomib in the treatment of MM and MCL has been observed in clinical trials and more importantly, these clinical trials suggest that bortezomib holds activity in about one third of patients who were heavily pre-treated. This could be attributed to the fact that bortezomib is targeting a unique, previously unaffected target, the proteasome, as the mechanism of action of bortezomib differs from any existing chemotherapeutic agents [74, 75].
Thirdly, bortezomib is capable of enhancing the sensitivity of cancer cells to conventional chemotherapeutic agents or radiation. In several previously discussed clinical trials, bortezomib was introduced into standard therapy and largely improved the outcome in terms of ORR, time to progression, etc. The chemo- and radio-sensitizing effect of bortezomib could once again be attributed to its unique target and mechanism of action. It is conceivable that two drugs with different targets or mechanisms of action may have better efficacy and less toxicity than two drugs with the same target or similar mechanism of action [76, 77].
With the good comes the bad, and some disadvantages and/or limitations of bortezomib have indeed been observed during its preclinical and clinical development.
First, although it is generally well-tolerated, bortezomib still generates some toxicity, and in some cases the regimen must be discontinued. The most frequently occurring side effects are nausea, diarrhea and fatigue. More serious adverse drug reactions include peripheral neuropathy, thrombocytopenia, neutropenia and lymphopenia. It is estimated that more than 40% of patients will present with peripheral neuropathy [78]. Somewhat redeeming in this regard is the newly approved subcutaneous administration of bortezomib, which has been found to significantly alleviate peripheral neuropathy compared to the traditional intravenous injection [79].
Another shortcoming of bortezomib is its narrow therapeutic window. According to a phase I trial, the therapeutic dose of bortezomib is 1.3 mg/m2 and the dose-limiting toxic effects are observed with only a slightly higher dose of 1.5 mg/m2. However, if bortezomib dose reduction is required, the most recent phase II CREST trial, which compared two doses of bortezomib (1.3 mg/m2 vs. 1.0 mg/m2), reports that the 1.0 mg/m2 dose still offers patients a substantial survival benefit [49].
Furthermore, despite the appreciable therapeutic outcome of bortezomib, like almost all anti-cancer drugs, drug resistance becomes a major problem after some time. Bortezomib resistance has been observed in even those newly diagnosed patients who received bortezomib monotherapy for the first time. These clinical observations indicate that bortezomib resistance could be either acquired or inherent. The results from cell-based studies suggest that bortezomib resistance could occur either at the level of the proteasome itself or further downstream. For example, increased mRNA and protein expression of the β5 subunit was observed in bortezomib-resistant leukemic cell lines as well as patient samples [80–82]. Mutations in β5 which impair bortezomib binding have also been reported [80, 83]. In addition, constitutively active NFκB pathway, downstream of the UP-S, was found in some bortezomib-resistant cell lines [84]. Whether or not any of the above is relevant to clinical bortezomib resistance in MM patients remains to be determined.
Moreover, during the clinical application of bortezomib, it was noticed that some natural products, including green tea polyphenols, are able to reduce the efficacy of bortezomib. This was due to the direct interaction between the boronic acid structure of bortezomib and the catechol structure of green tea polyphenols, resulting in the formation of a borate ester and therefore inactivation of bortezomib [85]. Further clinical studies are needed for evidence-based recommendations of green tea consumption in patients receiving bortezomib treatment; fortunately though, this issue only affects boronic acid-based proteasome inhibitors.
Finally, as discussed earlier, the efficacy of bortezomib, either alone or in a multi-drug regimen, is unsatisfactory in the treatment of solid tumors. Addressing this issue will largely extend the application of proteasome inhibitors in cancer treatment.
Bortezomib: A Forerunner, Yet Ancestor
At least five second generation proteasome inhibitors are currently under clinical investigation (Table II). Carfilzomib is an irreversible proteasome inhibitor; since new protein synthesis is required for recovery of proteasome activity, carfilzomib has a greater potency than bortezomib. In addition to targeting the β5 subunit in the constitutive proteasome, carfilmozib also targets the correlated β5i subunit of the immunoproteasome, which appears to be preferentially expressed in MM. Moreover, carfilzomib is shown to be more specific than borzetomib, with little or no off-target activity outside of the proteasome [86]. However, like bortezomib, the administration of carfilzomib is intravenous twice weekly, which may be inconvenient for patients. Carfilzomib has been evaluated in two phase II trials, PX-171-003-A1 [87] and PX-171-004 [88, 89], as monotherapy for the treatment of relapsed and refractory MM patients. Both trials observed durable response to carfilzomib with well-tolerated and manageable side effects, regardless of prior exposure to bortezomib. Analysis of 136 patients in the above two trials indicated that peripheral neuropathy occurred in 15% of patients, among which 9% was attributed to carfilzomib. None of the patients required discontinuation or dose adjustments due to neurotoxicity, allowing long-term treatment and prolonged disease control by carfilzomib [90]. These data also indicate that peripheral neuropathy is not a class effect of proteasome inhibitors [90]. A recent study comparing bortezomib and carfilzomib further pointed out that the neurotoxicity associated with bortezomib was related to its off-target inhibition of HtrA2/Omi, a protease known to be involved in neuronal survival [91]. Carfilzomib has also been evaluated in combination with lenalidomide, and low-dose dexamethasone (CRd) in relapsed, refractory and newly diagnosed MM patients in two separate phase II trials, both of which report that this regimen is highly active and well-tolerated [92, 93]. In newly diagnosed patients, the responses are rapid and improve over time reaching 100%, which compare favorably to the best frontline regimens in MM therapy [92]. In relapsed and/or refractory patients, the ORR was 78% [93]. Based on these encour aging results, the phase III ASPIRE trial of this regimen was approved and designed with the intention of full support by the FDA for its approval for use against relapsed multiple myeloma. As of July 20 2012, it has been announced that carfilzomib has received FDA approval for treatment of patients with multiple myeloma progression while on, or after treatment with, bortezomib and an immunomodulatory agent. Its use in the U.S. was launched as of August 1 2012; indeed, another milestone in the field.
Table II.
Second Generation Proteasome Inhibitors
| Inhibitor | Structure | Mechanism | Clinical Status | Reference |
|---|---|---|---|---|
| Carfilzomib (CFZ) |

|
Irreversibly binds and inhibits the β5 subunit | FDA approved for patients with relapsed and/or refractory MM | [86] |
| NPI-0052, Marizomib, Salino-sporide A |

|
Irreversible, inhibits the catalytic activity of all three 20S proteasomal subunits | Phase I for treatment of MM | [94] |
| MLN9708, Ixazomib |

|
Reversibly inhibits proteolytic activity of the β5 subunit | First oral proteasome inhibitor Phase III for relapsed and/or refractory MM |
[95] |
| CEP18770, Delanzomib |

|
Boronic acid, reversibly targets the β5 subunit | Phase I/II | [96] |
| ONX-0912, Oprozomib |

|
Irreversible inhibitor of the β5 subunit | Phase IB/II | [86] |
Marizomib (NPI-0052) (Table II) is another irreversible proteasome inhibitor administrated intravenously twice weekly. Compared to other proteasome inhibitors, marizomib produces rapid, broad and prolonged inhibition of all three 20S proteasome catalytic activities. Data from a phase I trial reports that responses to marizomib were found in patients with bortezomib-refractory MM. The safety profile of marizomib clearly differs from bortezomib, with no significant treatment-emergent peripheral neuropathy, myelo-suppression or thrombocytopenia reported. The dose limiting toxicities are transient and include: hallucinations, cognitive changes and loss of balance. In addition, marizomib exhibits interesting pharmacokinetic and pharmacodynamic properties and tissue distribution, supporting a possible role for marizomib in patients with different disease characteristics such as extramedullary spread [94].
Ixazomib (MLN-9708) (Table II), a boronate-based reversible proteasome inhibitor, has the advantage of being the first oral proteasome inhibitor to enter clinical investigation in MM patients. Oral administration is not only convenient for the patient, but also seems to produce milder side effects. Data from a phase I trial suggest that single-agent ixazomib may have clinical activity in heavily pretreated relapsed and/or refractory MM patients, describing durable disease control and well tolerated results with infrequent peripheral neuropathy [95].
Delanzomib (CEP-18770) (Table II) is another boronate-based reversible proteasome inhibitor taken orally, although current phase I/II clinical investigations are still using intravenous administration. Delanzomib has shown proteasome-inhibitory activity similar to that of bortezomib in hematologic and solid tumor cell lines, as well as in primary cells from MM patients [96]. Along these lines, Oprozomib (ONX-0912) (Table II) is another borteozmib-like proteasome inhibitor proposed to be taken orally; it is epoxyketone-based and irreversible in action. It exhibits similar potency to carfilzomib in cytotoxicity assays. More excitingly, orally administered oprozomib has equivalent antitumor activity to intravenously administered carfilzomib in human tumor xenograft and mouse syngeneic models [97]. It is currently being investigated in early clinical trials via oral administration twice daily.
Natural Compounds with Proteasome-Inhibitory Activity
Natural products have been used as therapeutic agents worldwide for ages. Even today, bioactive compounds from natural sources continue to play an extremely important role in the process of drug design and discovery due to their enormous structural diversity. In fact, almost half of all drugs discovered between 1940 and 2006 were of natural origin or derivatives of other natural bioactive compounds [98, 99]. They not only act as therapeutic agents directly, but also serve as templates which can be structurally modified in order to produce improved, more specific, safe and potent drugs. Various microbial metabolites and phytochemicals with a polyphenol structure are also thought to inhibit the proteasome based on cell culture and animal studies [100]. This section reviews some of these natural compounds and their proteasome inhibitory potential.
Green Tea Polyphenols
Tea, one of the most popular beverages in the world, is said to have cancer preventive and antioxidant effects due to its polyphenol-containing nature, including (−)-epicatechin (EC), (−)-epigallocatechin (EGC), (−)-epicatechin-3-gallate (ECG), and (−)-epigallocatechin-3-gallate (EGCG). EGCG (Fig. 3) has been shown to inhibit the β5 subunit, chymotrypsin-like (CT-like) activity, of the proteasome in vitro and in vivo with an IC50 value of 86 to 194 nM and 1 to 10 μM respectively where the in vivo concentrations are in accordance with the concentrations found in the serum of green tea drinkers [101]. Protein levels of transcription factor HIF-1α, which controls expression of oxygen regulated genes involved in cell proliferation, cell survival and angiogenesis, were decreased by EGCG in HeLa and HepG2 cells. EGCG inhibited the translational machinery of HIF-1α suggesting that its down regulation started even before its own protein synthesis. EGCG also significantly inhibited VEGF expression at the mRNA and protein levels in HeLa cells [102]. However, EGCG, the most abundant and potent constituent of green tea is unstable under neutral and alkaline conditions. Hence, novel EGCG analogs were synthesized with either elimination of the –OH groups from the B- and/or D-rings of its parent compound or its protection by the acetate group [103, 104]. These novel EGCG analogs inhibited cell proliferation and induced apoptosis in leukemic, prostate and breast cancer cells at greater potency when compared to their parent EGCG compound [104]. Furthermore, a pro-drug of EGCG showed more tumor inhibition as compared to EGCG in nude mice injected with breast cancer MDA-MB-231 cells. However, synthetic methylated tea polyphenols do not have strong proteasome inhibitory activities and therefore are less chemopreventative. Hence, in patients with high catechol-O-methyltransferase (COMT) activity, EGCG is methylated, inactivated and therefore not beneficial [105, 106].
Grape Polyphenols
Grapes are a rich source of flavonoids like resveratrol, apigenin and quercetin (Fig. 3) which have shown antiproliferative activities in vitro in different tumor cell lines from melanoma, prostate and pancreatic cancer [107–109]. Currently, no direct evidence of resveratrol's proteasome inhibitory activity exists; however inhibition of NFκB suggests that it may indeed have proteasome inhibitory properties [110]. Apigenin has been shown to strongly inhibit CT-like activity of the proteasome. In silico docking approaches have shown that the carbonyl carbon of apigenin binds to the β5 subunit CT site in an orientation and conformation that makes it susceptible to nucleophilic attack by threonine 1. Use of apigenin in an assay using purified 20S and 26S proteasome showed inhibition of proteasome activity in intact leukemia Jurkat cells at an IC50 of 1.8 to 2.3 μM and 1 to 10 μM respectively. When leukemia Jurkat cells were treated with apigenin, accumulation of proteasome substrates like Bax and IκB-α as well as caspase 3 activation and apoptotic PARP cleavage was observed. Apigenin was not toxic to normal, non-transformed cells, which further validates its potential as a therapeutic proteasome inhibitor. Quercetin inhibits the purified proteasome at an IC50 of 3.5 μM and the 26S proteasome in intact tumor cells at an IC50 of 2 μM. Like apigenin, in silico analysis also suggests that the carbonyl carbon of quercetin binds to the CT site of the proteasome in a similar fashion making it suitable for a nucleophilic attack by threonine 1. When leukemia Jurkat cells were treated with quercetin, accumulation of proteasome substrates like Bax and IκB-α was observed, while caspase 3 activation and apoptotic PARP cleavage also occured in a dose and time dependent manner [111].
Soybean Isoflavones
Epidemiological studies have indicated that a soy bean-rich diet decreases one's likelihood of developing cancer in their lifetime. Genistein (Fig. 3), a major isoflavone found in soybeans has cancer cell growth inhibitory ability in various cancer cell lines [112–114]. In silico computational docking studies suggest that genistein can interact with the β5 subunit of the proteasome in such a manner that the CT-like activity is shut down. Genistein at the dose of 1 μM, which falls within plasma level range of 0.5 to 2.5 μM, can inhibit approximately 30% of 20S proteasome CT-like activity in a purified system with higher concentrations (50 μM) required to completely abolish it in breast and prostate cancer cells [115–117]. Genistein inhibits NFκB activity, suggesting proteasome inhibition and may have multiple cellular targets of which simultaneous inhibition could be responsible for its cancer fighting mechanism [118, 119].
Natural Compounds of Microbial Origin
Some of the natural compounds tested for anti-cancer activity were identified from microorganisms (Fig. 3). For example, lactacystin, isolated from streptomyces, forms a covalent bond with the N-terminal threonine residue of the proteasome thereby inhibiting its activity [31]. Omuralide (Fig. 3), a derivative of lactacystin, is prepared by eliminating the n-acetyl cysteine moiety from the lactone ring and behaves in a similar fashion [120, 121]. Furthermore, PS-519 is prepared based on the structure of omuralide where an n-propyl group is substituted for a methyl group; it has proteasome-inhibitory potential but has recently entered clinical trials for the treatment of acute stroke [122]. Marizomib (Salinosporamide A, Table II), found in marine bacteria, is structurally similar to omularide, but is almost four times more potent than omuralide in inhibiting proteasomal CT-like activity [123, 124]. Marizomib also inhibits the trypsin-like activity of the proteasome and exhibits strong cytotoxic activity against the HCT116 human colon carcinoma cell line [125]. Interestingly, marizomib can overcome bortezomib resistance in relapsed or refractory MM and shows improved proapoptotic activity over bortezomib in an in vitro chronic lymphocytic leukemia cell line system. In addition, its oral administration is clinically favorable [126]. Combinations of marizomib and histone deacetylase inhibitors, MS-275 or valproic acid, have been studied and the observed results report these combinations to have even better apoptotic inducing activity than the bortezomib and MS-275 or valproic acid combinations [127]. Furthermore, in vitro studies using MM cell lines testing marizomib combined with lenalidomide have resulted in a favorable synergistic cytotoxic response [128]. Belactosins A and C (Fig. 3) isolated from the streptomyces species have identical β-lactone rings and so are similar to omuralide in their ability to inhibit proteasomal CT-like activity [129, 130].
Epoxomicin, isolated from actinomycetes, can inhibit the proteasome due to the presence of α-β-epoxyketone moiety, which forms a morpholine adduct with the N-terminal threonine residue of the proteasome core [131]. Carfilzomib (Table II), an irreversible proteasome inhibitor was developed based on the structure of epoxomicin, and is even more selective for the proteasome's CT-like activity than bortezomib [132]. It is active against bortezomib resistant MM cell lines and when combined with dexamethasone, has synergistic and cytotoxic effects. Carfilzomib is clinically well tolerated and as previously mentioned, does not have the undesirable neuropathic side effects commonly seen with bortezomib in MM patients [133]. With its recent FDA approval, more positive clinical data is anticipated in the coming years.
Syringolin A (Fig. 3), isolated from the plant pathogen pseudomonas syringae, irreversibly inhibits all types of proteasomal activity. It also inhibits cell proliferation and induces apoptosis in malignant cells. The x-ray crystallography structure of syringolin A binding to the yeast proteasome shows Michael type 1, 4-addition to the vinyl ketone moiety of the 14 member ring to the hydroxyl group of the threonine residue [134, 135].
TMC-95A (Fig. 3), isolated from apiospora montagnei inhibits all proteasome activities in the nanomolar range [136, 137]. X-ray crystallography studies show that TMC-95A and the core particle of the yeast proteasome bind to each other via hydrogen bonding [138, 139]. Thus, proteasome inhibitors of natural origin by themselves or in combination could be used as cancer therapies. In depth knowledge of the chemical structures of these bioactive molecules and the proteasome, including the study of their interaction, may lead to better proteasome inhibitors with less off target effects and desirable pharmacokinetic properties [140].
OTHER MECHANISMS OF UP-S INHIBITION
Inhibitors of Ubiquitin E3 Ligases as Anti-Cancer Drugs
Ubiquitin E3 ligases (Table III) are a group of enzymes a part of the UP-S, which bind specific protein substrates and promote their degradation by the transfer of ubiquitin from a thioester intermediate to form an amide linkage [141, 142]. E3 ubiquitin ligases can be classified into three major groups: N-end rule ubiquitin li-gases [143], HECT (Homologous to E6AP C-Terminus) type E3 ligases [144] and the RING (Really Interesting New Gene) family of E3 ligases [145, 146]. N-end rule E3 ligases consist of two types: Type I, which target protein substrates specific to the N-terminal destabilizing residues like Arg, Lys and His and type II, which target substrates with Phe, Leu, Trp, Ile and Tyr residues [143, 147]. Proteolytic cleavage exposes the internal destabilizing residues at the N-terminus of the substrate and subsequent E3 ligase binding leads to polyubiquitination and proteasomal degradation.
Table III.
Investigational Ubiquitin E3 Ligases as Specific Targets for Inhibition
| Target E3 Ligase | Inhibitory Agents | Mechanism of Inhibition | Reference |
|---|---|---|---|
| Mdm2/Hdm2 | Nutlins (Nutlin-3) 1–3 compounds Mdm2 antisense oligonucleotides for gene silencing |
Stabilization of p53 | [148] |
| SCF | Protac-1 Methionine Aminopeptidase-2 chimera with ovalicin |
Inhibition of angiogenesis | [165] |
| CHIP | Geldanamycins | Induction of chaperon complex that down-regulates Her2/Neu | [169] |
| E6-AP/E6 | Zinc Ejectors (C16) | Stabilization of p53 | [171] |
| c-Cbl | Trastuzumab (Herceptin) | Degradation of RTKs and termination of signaling | [154] |
| BRCA1/BARD1 | wt-BRCA1 expression using viral vectors | Sensitization of tumor suppressor function and DNA repair. | [157] |
| Efp | Antisense oligonucleotide | Stabilization of 14-3-3 | [160] |
| BCA2 | Disulfiram (Antabuse) and analogues | Affects EGFR receptor mediated endocytosis | [162] |
The HECT type E3 ligases contain a C-terminal region (approx. 350 amino acids) homologues to E6-AP with a conserved active site cysteine near the C-terminus which forms the thioester linkage with ubiquitin.
The third and largest type of E3 ligases are the RING finger domain containing type. RING finger E3 ligases possess autoubiquitination activity in addition to their ability to ubiquitinate other protein substrates. The RING finger is a specialized zinc finger with the consensus sequence: C-X2-C-X [9–39]-C-X[1–3]-H-X[2–3]-CX2-C-X[4–48]-C-X2-, about 40–60 amino acids in linear sequence; binding two Zn atoms per molecule and forming a cross-brace structure.
E3 ubiquitin ligases regulate almost all cellular processes including the cell cycle, transcription, DNA repair, signal transduction, endocytosis, cellular transport and development. Recent discoveries in cancer research have made it very clear that E3 ligases play a vital role in cancer development and progression and that their inhibition results in growth suppression and apoptosis of neoplastic cells. Thus, E3 ligases represent a group of potentially `drugable' target enzymes for mechanism-based drug discovery in the ubiquitin proteasome system (Table III).
An example of a protein that plays a pivotal role in many cancers and is regulated by E3 ligases is the tumor suppressor protein p53, which is primarily regulated by the RING-type E3 ligase Mdm2 and HECT- type E3 ligase E6-AP, both of which influence its degradation by the proteasome [148–151] (Table III). As in the vast majority of cancers, Mdm2 regulates p53 by either inhibiting its transcriptional activity or by targeting it for proteosomal degradation. Since Mdm2 promotes p53 degradation, it has gained much attention as a targeted cancer therapy. The first Mdm2 inhibitors involved antisense oligonucleotide and gene therapy methods, which were unsuccessful due to the lack of effective delivery methods. As a result, three distinct compounds representing a benzsulfonamide, a urea analogue and an imidazole derivative were identified by Lai et al [152] as inhibitors of human Mdm2 (HDM2). Of these, Nutlin-3 has desirable pharmacological properties and has proven to be effective in the inhibition of tumor growth in preclinical studies. Although it is currently in phase I clinical trials for the treatment of retinoblastoma, its effect on normal tissue and cell types needs to be further investigated [153].
C-cbl is another RING finger E3 ligase that contains a SH2 domain which recognizes phosphorylated tyrosine kinase receptors and is in fact involved in EGFR endocytosis [154] (Table III). Overexpression of EGFR, HER2/Neu, and EGF heterodimer signaling is a major contributor to uncontrolled growth in many cancer types. HER2 antibody trastuzumab (Herceptin) has been shown to direct HER2 to the C-cbl regulated pathway leading to its subsequent degradation [155, 156].
The BRCA1 RING E3 ligase plays a critical role in DNA repair and transcriptional control processes. A mutation in its RING domain is associated with familial breast and ovarian carcinomas [157] (Table III). BRCA1 in complex with BARD1 ubiquitinates p53 and therapeutic approaches to express wt-BRCA1 have been evaluated in ovarian cancer. Gene therapy using viral vectors was indeed a promising idea based on data generated from xenograft models and phase I trials. However, phase II trials have failed to produce positive numbers in those patients evaluated with metastatic cancer [158, 159].
Efp is an estrogen-inducible RING E3 ligase that ubiquitinates tumor suppressor 14-3-3σ proteins (Table III). Efp is one of several known estrogen receptor (ER) target genes and plays a critical role in mammary tumors, use of antisense oligonucleotides in MCF7 xenografts has shown a marked reduction in tumor growths [160, 161].
Breast Cancer Associated gene 2 (BCA2) is a zinc-binding E3 ligase found to be overexpressed in breast cancer and involved in the EGFR trafficking pathway [162] (Table III). We have tested disulfiram (DSF, Antabuse) and its analogues as zinc chelating substances for the inhibition of BCA2 by targeting its RING domain [162, 163]. DSF is a FDA approved drug for chronic alcoholism and has been found to be a potent inhibitor of the proteasome when complexed to copper or zinc [164]. DSF is a readily available, inexpensive drug with very few adverse side effects whose anticancer activity has been attributed to its ability to inhibit E3 ligases and the proteasome.
There are at least 38 F-box proteins in the human genome believed to target many functionally and structurally diverse substrates. For example, the SCF (Skp-1-Cullin-F-box protein) contains a variable F-box protein unit that binds to target proteins for ubiquitination (Table III). Sakamato et al designed the Protein Targeting Chimeric Molecule1 (Protac1) to recruit MetAP-2 to SCF. One domain of Protac-1 contains the IκB-α phosphopeptide that is recognized by F-box beta-TRCP, whereas the other domain is composed of the angiogenesis inhibitor ovalicin [165]. Thus, this bispecific compound leads to increased degradation of pro-angiogenic peptide MetAP-2 and also inhibits angiogenesis after ovalicin activation [166]. U-box E3 ligase CHIP together with Hsp90 mediates the ubiquitination of glucocorticoid receptor [167] and the CHIP/Hsp90 complex acts as a quality control ligase that selectively degrades abnormally folded proteins [168] [169]. Thus, modulating the HER2/Neu interaction with CHIP by geldanamycin (GA) and 17-AAG enhances its destabilization and affects HER2 signaling [170]. These compounds are under Phase I/II clinical trials. HECT-type E3 ligase E6-AP was the first one to be described in targeting p53 for rapid degradation and this process is initiated by the E6 gene product of HPV-16 [171]. Therapies targeting E6-AP are therefore limited to cancers caused by HPV infections, such as cervical cancers. As zinc binding is a requirement for E6 interaction with E6-AP, the zinc ejector 4,4'-dithiodimorpholine (C16) was discovered as an agent for selectively inhibiting E6-AP activity in HPV-positive tumors. Thalidomide, which has been in clinical practice for many years, has also been recently identified as an E3 ligase inhibitor.
Deubiquitinating Enzyme (DUB) Inhibitors as Anti-Cancer Agents
Recently, DUBs have been studied as potential molecular targets for UP-S inhibitor development. USP7 (Herpes virus associated USP, HAUSP) is critical in cancer progression as it deubiquitinates HDM2 and HDMX resulting in destabilization of p53 and repression of its transactivation activity [172]. High throughput screens have identified HBX 41,108 as an inhibitor of USP7 that stabilizes p53 and induces apoptosis. Another screen has recently identified USP8 [173], which has demonstrated stabilization of p53 and induction of p21 [173]. USP8 plays a key role in receptor endocytosis and its knockdown results in ubiquitinated EGFR present in the endosome. HBX 90, 397 is also an identified inhibitor of USP8 [174]. WP1130 (degrasyn) is a small molecule compound that inhibits Janus activated kinase 2 (Jak2) activity. WP1130 is found to inhibit several DUBs like USP9x, USP5, and USP14 and can be of therapeutic value because of its pro-apoptotic properties [175]. Another DUB inhibitor with broad specificity is PR-619, having a reversible mechanism of inhibition and was discovered using ubiquitin CHOP reporter technology [176, 177].
The E3 ubiquitin ligases and their corresponding DUBs regulate many biological processes due to their timely and ultra-specific ubiquitination and subsequent degradation of many key cellular proteins. Although a better understanding of E3 ligase mechanisms of action is still required, it is now clear that because of their subsrate specificity, E3 ligase inhibitors can be used as promising anti-cancer drugs with high levels of specificity and lower toxicity. Many E3 ligase inhibitors have been successful in a preclinical setting and are being evaluated for cancer treatment. Also, recent research in DUB mechanisms of action has become a promising avenue for the development of new drug targets.
THE FUTURE OF PROTEASOME INHIBITION AS AN ANTI-CANCER STRATEGY
Importance and Perspective
Since its discovery, the proteasome has gained considerable currency and has indeed most recently been validated as a novel, valuable molecular target in the treatment of various cancers. A large amount of data exists which supports the idea that suppression of proteasome activity, by synthetic or natural compounds, is effective in inducing cancer cell death in vitro and suppressing tumor growth in vivo, with minimally toxic effects to nearby healthy cells. Bortezomib, the first FDA-approved proteasome inhibitor, has become a staple in the field and deservingly so; it has achieved great success in the treatment of MM and MCL. However, resistance, dose limiting toxicities, and interaction with some natural compounds limit its use and therefore compromise its clinical benefits. Furthermore, bortezomib's potency against solid tumors has been rather disappointing. The involved molecular mechanisms are currently unclear. However, one contributing factor may be induction of stress granule formation by bortezomib in solid tumors, involving phosphorylation of translation initiation factor eIF2α by heme-regulated inhibitor kinase (HRI) [178]. Other possibilities may include mutations or over-expression of the proteasome β5 subunit, increased levels of proteasome activity, overexpression of a proteasome downstream effector (e.g., Bcl-2 or NF-κB), or altered regulation of some chaperone proteins, when compared to hematologic malignancies. While, bortezomib's shortcomings in this regard have herein been outlined, the mechanism responsible for the little effect observed in solid tumors remains to be determined. There is therefore more work to be done; the development of a new generation of proteasome inhibitors is absolutely necessary.
The successes and limitations of bortezomib as the first proteasome inhibitor anticancer drug have taught us useful lessons and encouraged researchers to search for the next generation proteasome inhibitors that could have improved properties, reduced toxicities and broader anticancer activities. The structure-activity relationships need to be established for newly developed proteasome inhibitors in various types of cancers in hopes of uncovering what is responsible for the differential response seen in blood and solid tumors to proteasome inhibitor therapy. Also, continuous exploration of effective combinations including proteasome inhibitors and current chemotherapeutic agents is essential to optimize clinical outcome. The underlying culprit for success in all of these areas is a better understanding of the proteasome itself and the ubiquitination system, fundamental to its operation. Only when we have achieved full understanding of the UP-S can we propose mechanism(s) accountable for proteasome inhibition-induced cell death, or resistance, observed in vitro and in vivo. Until then, much work remains to be done.
ACKNOWLEDGEMENTS
This work was partially supported by grants from the National Cancer Institute (1R01CA120009, 3R01CA120009-04S1 and 5R01CA127258-05, to QPD).
Footnotes
CONFLICT OF INTEREST The authors confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Gerards WL, de Jong WW, Boelens W, Bloemendal H. Structure and assembly of the 20S proteasome. Cell Mol Life Sci. 1998;54(3):253–62. doi: 10.1007/s000180050147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arrigo AP, Tanaka K, Goldberg AL, Welch WJ. Identity of the 19S `prosome' particle with the large multifunctional protease complex of mammalian cells (the proteasome) Nature. 1988;331(6152):192–4. doi: 10.1038/331192a0. [DOI] [PubMed] [Google Scholar]
- [3].Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438–44. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- [4].Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition. J Pept Sci. 2009;15(2):58–66. doi: 10.1002/psc.1107. [DOI] [PubMed] [Google Scholar]
- [5].Harding CV, France J, Song R, et al. Novel dipeptide aldehydes are proteasome inhibitors and block the MHC-I antigen-processing pathway. J Immunol. 1995;155(4):1767–75. [PubMed] [Google Scholar]
- [6].Iqbal M, Chatterjee S, Kauer JC, et al. Potent inhibitors of proteasome. J Med Chem. 1995;38(13):2276–7. doi: 10.1021/jm00013a002. [DOI] [PubMed] [Google Scholar]
- [7].Vinitsky A, Cardozo C, Sepp-Lorenzino L, Michaud C, Orlowski M. Inhibition of the proteolytic activity of the multicatalytic proteinase complex (proteasome) by substrate-related peptidyl aldehydes. J Biol Chem. 1994;269(47):29860–6. [PubMed] [Google Scholar]
- [8].Wilk S, Figueiredo-Pereira ME. Synthetic inhibitors of the multicatalytic proteinase complex (proteasome) Enzyme Protein. 1993;47(4–6):306–13. doi: 10.1159/000468688. [DOI] [PubMed] [Google Scholar]
- [9].Lee DH, Goldberg AL. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem. 1996;271(44):27280–4. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- [10].Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78(5):773–85. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- [11].Rock KL, Gramm C, Rothstein L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78(5):761–71. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- [12].Adams J, Stein R. Novel inhibitors of the proteasome and their therapeutic use in inflammation. Annu. Rep. Med. Chem. 1996;(31):279–88. [Google Scholar]
- [13].Iqbal M, Chatterjee S, Kauer JC, et al. Potent a-ketocarbonyl and boronic ester derived inhibitors of proteasome. Bioorg Med Chem Lett. 1996;6(287–290) [Google Scholar]
- [14].McCormack T, Baumeister W, Grenier L, et al. Active site-directed inhibitors of Rhodococcus 20 S proteasome. Kinetics and mechanism. J Biol Chem. 1997;272(42):26103–9. doi: 10.1074/jbc.272.42.26103. [DOI] [PubMed] [Google Scholar]
- [15].Harper JW, Powers JC. Reaction of serine proteases with substituted 3-alkoxy-4-chloroisocoumarins and 3-alkoxy-7-amino-4-chloroisocoumarins: new reactive mechanism-based inhibitors. Biochemistry. 1985;24(25):7200–13. doi: 10.1021/bi00346a028. [DOI] [PubMed] [Google Scholar]
- [16].Kam CM, Fujikawa K, Powers JC. Mechanism-based isocoumarin inhibitors for trypsin and blood coagulation serine proteases: new anticoagulants. Biochemistry. 1988;27(7):2547–57. doi: 10.1021/bi00407a042. [DOI] [PubMed] [Google Scholar]
- [17].Orlowski M, Michaud C. Pituitary multicatalytic proteinase complex. Specificity of components and aspects of proteolytic activity. Biochemistry. 1989;28(24):9270–8. doi: 10.1021/bi00450a006. [DOI] [PubMed] [Google Scholar]
- [18].Savory PJ, Djaballah H, Angliker H, Shaw E, Rivett AJ. Reaction of proteasomes with peptidylchloromethanes and peptidyldiazomethanes. Biochem J. 1993;296(Pt 3):601–5. doi: 10.1042/bj2960601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Powers JC, Kam CM. Isocoumarin inhibitors of serine peptidases. Methods Enzymol. 1994;244:442–57. doi: 10.1016/0076-6879(94)44033-6. [DOI] [PubMed] [Google Scholar]
- [20].Bromme D, Klaus JL, Okamoto K, Rasnick D, Palmer JT. Peptidyl vinyl sulphones: a new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem J. 1996;315(Pt 1):85–9. doi: 10.1042/bj3150085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Palmer JT, Rasnick D, Klaus JL, Bromme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J Med Chem. 1995;38(17):3193–6. doi: 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- [22].Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H. Covalent modification of the active site threonine of proteasomal beta subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc Natl Acad Sci USA. 1997;94(13):6629–34. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bogyo M, Shin S, McMaster JS, Ploegh HL. Substrate binding and sequence preference of the proteasome revealed by active-site-directed affinity probes. Chem Biol. 1998;5(6):307–20. doi: 10.1016/s1074-5521(98)90169-7. [DOI] [PubMed] [Google Scholar]
- [24].Omura S, Fujimoto T, Otoguro K, et al. Lactacystin, a novel microbial metabolite, induces neuritogenesis of neuroblastoma cells. J Antibiot (Tokyo) 1991;44(1):113–6. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]
- [25].Omura S, Matsuzaki K, Fujimoto T, et al. Structure of lactacystin, a new microbial metabolite which induces differentiation of neuroblastoma cells. J Antibiot (Tokyo) 1991;44(1):117–8. doi: 10.7164/antibiotics.44.117. [DOI] [PubMed] [Google Scholar]
- [26].Craiu A, Gaczynska M, Akopian T, et al. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem. 1997;272(20):13437–45. doi: 10.1074/jbc.272.20.13437. [DOI] [PubMed] [Google Scholar]
- [27].Hanada M, Sugawara K, Kaneta K, et al. Epoxomicin, a new antitumor agent of microbial origin. J Antibiot (Tokyo) 1992;45(11):1746–52. doi: 10.7164/antibiotics.45.1746. [DOI] [PubMed] [Google Scholar]
- [28].Sugawara K, Hatori M, Nishiyama Y, et al. Eponemycin, a new antibiotic active against B16 melanoma. I. Production, isolation, structure and biological activity. J Antibiot (Tokyo) 1990;43(1):8–18. doi: 10.7164/antibiotics.43.8. [DOI] [PubMed] [Google Scholar]
- [29].Oikawa T, Hasegawa M, Shimamura M, Ashino H, Murota S, Morita I. Eponemycin, a novel antibiotic, is a highly powerful angiogenesis inhibitor. Biochem Biophys Res Commun. 1991;181(3):1070–6. doi: 10.1016/0006-291x(91)92046-m. [DOI] [PubMed] [Google Scholar]
- [30].Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin: 20S proteasome reveals a molecular basis for selectivity of a ,b-epoxyketone proteasome inhibitors. J Am Chem Soc. 2000;122:1237–8. [Google Scholar]
- [31].Fenteany G, Standaert RF, Reichard GA, Corey EJ, Schreiber SL. A beta-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proc Natl Acad Sci USA. 1994;91(8):3358–62. doi: 10.1073/pnas.91.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Meng L, Kwok BH, Sin N, Crews CM. Eponemycin exerts its antitumor effect through the inhibition of proteasome function. Cancer Res. 1999;59(12):2798–801. [PubMed] [Google Scholar]
- [33].Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci USA. 1999;96(18):10403–8. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sin N, Meng L, Auth H, Crews CM. Eponemycin analogues: syntheses and use as probes of angiogenesis. Bioorg Med Chem. 1998;6(8):1209–17. doi: 10.1016/s0968-0896(98)00089-3. [DOI] [PubMed] [Google Scholar]
- [35].Sin N, Kim KB, Elofsson M, et al. Total synthesis of the potent proteasome inhibitor epoxomicin: a useful tool for understanding proteasome biology. Bioorg Med Chem Lett. 1999;9(15):2283–8. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- [36].Speiser S, Etlinger JD. ATP stimulates proteolysis in reticulocyte extracts by repressing an endogenous protease inhibitor. Proc Natl Acad Sci USA. 1983;80(12):3577–80. doi: 10.1073/pnas.80.12.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li XC, Gu MZ, Etlinger JD. Isolation and characterization of a novel endogenous inhibitor of the proteasome. Biochemistry. 1991;30(40):9709–15. doi: 10.1021/bi00104a020. [DOI] [PubMed] [Google Scholar]
- [38].Guo GG, Gu M, Etlinger JD. 240-kDa proteasome inhibitor (CF-2) is identical to delta-aminolevulinic acid dehydratase. J Biol Chem. 1994;269(17):12399–402. [PubMed] [Google Scholar]
- [39].Tsukamoto I, Yoshinaga T, Sano S. The role of zinc with special reference to the essential thiol groups in delta-aminolevulinic acid dehydratase of bovine liver. Biochim Biophys Acta. 1979;570(1):167–78. doi: 10.1016/0005-2744(79)90211-0. [DOI] [PubMed] [Google Scholar]
- [40].Hernberg S, Nikkanen J, Mellin G, Lilius H. Delta-aminolevulinic acid dehydrase as a measure of lead exposure. Arch Environ Health. 1970;21(2):140–5. doi: 10.1080/00039896.1970.10667211. [DOI] [PubMed] [Google Scholar]
- [41].Thompson J, Jones DD, Beasley WH. The effect of metal ions on the activity of delta-aminolevulinic acid dehydratase. Br J Ind Med. 1977;34(1):32–6. doi: 10.1136/oem.34.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Meredith PA, Moore MR, Goldberg A. The effects of aluminium, lead and zinc on 3-aminolaevulinic acid dehydratase. Biochem. Soc. Trans. 1974;2:1243–5. [Google Scholar]
- [43].Jaffe EK, Martins J, Li J, Kervinen J, Dunbrack RL., Jr. The molecular mechanism of lead inhibition of human porphobilinogen synthase. J Biol Chem. 2001;276(2):1531–7. doi: 10.1074/jbc.M007663200. [DOI] [PubMed] [Google Scholar]
- [44].Murakami K, Etlinger JD. Endogenous inhibitor of nonlysosomal high molecular weight protease and calcium-dependent protease. Proc Natl Acad Sci USA. 1986;83(20):7588–92. doi: 10.1073/pnas.83.20.7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li XS, Etlinger JD. Ubiquitinated proteasome inhibitor is a component of the 26 S proteasome complex. Biochemistry. 1992;31(48):11964–7. doi: 10.1021/bi00163a001. [DOI] [PubMed] [Google Scholar]
- [46].Bardag-Gorce F, French SW. Delta-aminolevulinic dehydratase is a proteasome interacting protein. Exp Mol Pathol. 2011;91(2):485–9. doi: 10.1016/j.yexmp.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Etlinger JD, Li SX, Guo GG, Li N. Phosphorylation and ubiquitination of the 26S proteasome complex. Enzyme Protein. 1993;47(4–6):325–9. doi: 10.1159/000468690. [DOI] [PubMed] [Google Scholar]
- [48].Jagannath S, Barlogie B, Berenson J, et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004;127(2):165–72. doi: 10.1111/j.1365-2141.2004.05188.x. [DOI] [PubMed] [Google Scholar]
- [49].Jagannath S, Barlogie B, Berenson JR, et al. Updated survival analyses after prolonged follow-up of the phase 2, multicenter CREST study of bortezomib in relapsed or refractory multiple myeloma. Br J Haematol. 2008;143(4):537–40. doi: 10.1111/j.1365-2141.2008.07359.x. [DOI] [PubMed] [Google Scholar]
- [50].Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- [51].Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- [52].Richardson PG, Sonneveld P, Schuster M, et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood. 2007;110(10):3557–60. doi: 10.1182/blood-2006-08-036947. [DOI] [PubMed] [Google Scholar]
- [53].San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–17. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- [54].Mateos MV, Richardson PG, Schlag R, et al. Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol. 2010;28(13):2259–66. doi: 10.1200/JCO.2009.26.0638. [DOI] [PubMed] [Google Scholar]
- [55].Jagannath S, Durie BG, Wolf J, et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129(6):776–83. doi: 10.1111/j.1365-2141.2005.05540.x. [DOI] [PubMed] [Google Scholar]
- [56].Jagannath S, Durie BG, Wolf JL, et al. Extended follow-up of a phase 2 trial of bortezomib alone and in combination with dexamethasone for the frontline treatment of multiple myeloma. Br J Haematol. 2009;146(6):619–26. doi: 10.1111/j.1365-2141.2009.07803.x. [DOI] [PubMed] [Google Scholar]
- [57].Oakervee HE, Popat R, Curry N, et al. PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma. Br J Haematol. 2005;129(6):755–62. doi: 10.1111/j.1365-2141.2005.05519.x. [DOI] [PubMed] [Google Scholar]
- [58].Popat R, Oakervee HE, Hallam S, et al. Bortezomib, doxorubicin and dexamethasone (PAD) front-line treatment of multiple myeloma: updated results after long-term follow-up. Br J Haematol. 2008;141(4):512–6. doi: 10.1111/j.1365-2141.2008.06997.x. [DOI] [PubMed] [Google Scholar]
- [59].Cavo M, Tacchetti P, Patriarca F, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–85. doi: 10.1016/S0140-6736(10)61424-9. [DOI] [PubMed] [Google Scholar]
- [60].Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–74. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- [61].Goy A, Bernstein SH, Kahl BS, et al. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Annals of Oncology. 2009;20(3):520–5. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kouroukis CT, Fernandez LA, Crump M, et al. A phase II study of bortezomib and gemcitabine in relapsed mantle cell lymphoma from the National Cancer Institute of Canada Clinical Trials Group (IND 172) Leuk Lymphoma. 2011;52(3):394–9. doi: 10.3109/10428194.2010.546015. [DOI] [PubMed] [Google Scholar]
- [63].Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nature Reviews Cancer. 2005;5(4):251–62. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- [64].Fowler N, Kahl BS, Lee P, et al. Bortezomib, bendamustine, and rituximab in patients with relapsed or refractory follicular lymphoma: the phase II VERTICAL study. J Clin Oncol. 2011;29(25):3389–95. doi: 10.1200/JCO.2010.32.1844. [DOI] [PubMed] [Google Scholar]
- [65].Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(6):690–7. doi: 10.1200/JCO.2010.31.1142. [DOI] [PubMed] [Google Scholar]
- [66].Morris MJ, Kelly WK, Slovin S, et al. A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. J Urol. 2007;178(6):2378–83. doi: 10.1016/j.juro.2007.08.015. discussion 83–4. [DOI] [PubMed] [Google Scholar]
- [67].Hainsworth JD, Meluch AA, Spigel DR, et al. Weekly docetaxel and bortezomib as first-line treatment for patients with hormone-refractory prostate cancer: A minnie pearl cancer research network phase II trial. Clinical Genitourinary Cancer. 2007;5(4):278–83. doi: 10.3816/CGC.2007.n.004. [DOI] [PubMed] [Google Scholar]
- [68].Irvin WJ, Orlowski RZ, Chiu WK, et al. Phase II Study of Bortezomib and Pegylated Liposomal Doxorubicin in the Treatment of Metastatic Breast Cancer. Clinical Breast Cancer. 2010;10(6):465–70. doi: 10.3816/CBC.2010.n.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li TH, Ho L, Piperdi B, et al. Phase II study of the proteasome inhibitor bortezomib (PS-341, Velcade) in chemotherapy-naive patients with advanced stage non-small cell lung cancer (NSCLC) Lung Cancer. 2010;68(1):89–93. doi: 10.1016/j.lungcan.2009.05.009. [DOI] [PubMed] [Google Scholar]
- [70].Shah MA, Power DG, Kindler HL, et al. A multicenter, phase II study of Bortezomib (PS-341) in patients with unresectable or metastatic gastric and gastroesophageal junction adenocarcinoma. Investigational New Drugs. 2011;29(6):1475–81. doi: 10.1007/s10637-010-9474-7. [DOI] [PubMed] [Google Scholar]
- [71].Jatoi A, Dakhil SR, Foster NR, et al. Bortezomib, paclitaxel, and carboplatin as a first-line regimen for patients with metastatic esophageal, gastric, and gastroesophageal cancer - Phase II Results from the North Central Cancer Treatment Group (N044B) J Thoracic Oncol. 2008;3(5):516–20. doi: 10.1097/JTO.0b013e31816de276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pleban E, Bury M, Mlynarczuk I, Wojcik C. Effects of proteasome inhibitor PSI on neoplastic and non-transformed cell lines. Folia Histochemica Et Cytobiologica. 2001;39(2):133–4. [PubMed] [Google Scholar]
- [73].Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov Today. 2003;8(7):307–15. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- [74].Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr Cancer Drug Targets. 2011;11(3):239–53. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23(11):1964–79. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Orlowski RZ. Bortezomib in combination with other therapies for the treatment of multiple myeloma. J Natl Compr Canc Netw. 2004;2(Suppl 4):S16–20. [PubMed] [Google Scholar]
- [77].Reddy N, Czuczman MS. Enhancing activity and overcoming chemoresistance in hematologic malignancies with bortezomib: preclinical mechanistic studies. Annals of Oncology. 2010;21(9):1756–64. doi: 10.1093/annonc/mdq009. [DOI] [PubMed] [Google Scholar]
- [78].Appel A. Drugs: More shots on target. Nature. 2011;480(7377):S40–2. doi: 10.1038/480S40a. [DOI] [PubMed] [Google Scholar]
- [79].Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–40. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- [80].Oerlemans R, Franke NE, Assaraf YG, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112(6):2489–99. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- [81].Lu SQ, Yang JM, Huang CM, Cheng H, Wang JM. Upregulated expression of the PSMB5 gene may contribute to drug resistance in patient with multiple myeloma when treated with bortezomib-based regimen. Experimental Hematology. 2011;39(12):1117–8. doi: 10.1016/j.exphem.2011.09.003. [DOI] [PubMed] [Google Scholar]
- [82].Lu SQ, Chen ZL, Yang JM, et al. Overexpression of the PSMB5 gene contributes to bortezomib resistance in T-lymphoblastic lymphoma/leukemia cells derived from Jurkat line. Experimental Hematology. 2008;36(10):1278–84. doi: 10.1016/j.exphem.2008.04.013. [DOI] [PubMed] [Google Scholar]
- [83].Franke NE, Niewerth D, Assaraf YG, et al. Impaired bortezomib binding to mutant beta 5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26(4):757–68. doi: 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- [84].Markovina S, Callander NS, O'Connor SL, et al. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res. 2008;6(8):1356–64. doi: 10.1158/1541-7786.MCR-08-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Golden EB, Lam PY, Kardosh A, et al. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113(23):5927–37. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- [86].Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103(13):1007–17. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- [87].Samuel D, Martin T, Wang M, et al. Results of PX-171-003-A1, An Open-Label, Single-Arm, Phase 2 (Ph 2) Study of Carfilzomib (CFZ) In Patients (pts) with Relapsed and Refractory Multiple Myeloma (MM) Blood. 2010;116(21):433. [Google Scholar]
- [88].Vii R, Kaufman JL, Jakubowiak AJ, et al. Final Results From the Bortezomib-naive Group of PX-171-004, a Phase 2 Study of Single-Agent Carfilzomib in Patients with Relapsed and/or Refractory MM. Blood. 2011;118(21):369–70. [Google Scholar]
- [89].Stewart K, Siegel D, Wang M, et al. Results of Px-171-004, an Ongoing Open-Label, Phase Ii Study of Carfilzomib in Patients with Relapsed and/or Refractory Multiple Myeloma (R/R Mm) with or without Prior Bortezomib Exposure. Haematologica-the Hematology Journal. 2010;95:452. [Google Scholar]
- [90].Vij R, Wang LH, Orlowski RZ, et al. Carfilzomib (CFZ), a Novel Proteasome Inhibitor for Relapsed or Refractory Multiple Myeloma, Is Associated with Minimal Peripheral Neuropathic Effects. Blood. 2009;114(22):178–9. [Google Scholar]
- [91].Arastu-Kapur S, Anderl JL, Kraus M, et al. Nonproteasomal Targets of the Proteasome Inhibitors Bortezomib and Carfilzomib: a Link to Clinical Adverse Events. Clinical Cancer Research. 2011;17(9):2734–43. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- [92].Jakubowiak AJ, Dytfeld D, Jagannath S, et al. Final Results of a Frontline Phase 1/2 Study of Carfilzomib, Lenalidomide, and Low-Dose Dexamethasone (CRd) in Multiple Myeloma (MM) Blood. 2011;118(21):288–9. [Google Scholar]
- [93].Wang M, Bensinger W, Martin T, Alsina M. Interim results from PX-171-006, a phase (Ph) II multicenter dose-expansion study of carfilzomib (CFZ), lenalidomide (LEN), and low-dose dexamethasone (loDex) in relapsed and/or refractory multiple myeloma (R/R MM) J Clin Oncol. 2011;(suppl):8025. abstr. [Google Scholar]
- [94].Richardson PG, Spencer A, Cannel P, et al. Phase 1 Clinical Evaluation of Twice-Weekly Marizomib (NPI-0052), a Novel Proteasome Inhibitor, in Patients with Relapsed/Refractory Multiple Myeloma (MM) Blood. 2011;118(21):140–1. [Google Scholar]
- [95].Richardson PG, Baz R, Wang LH, et al. Investigational Agent MLN9708, An Oral Proteasome Inhibitor, in Patients (Pts) with Relapsed and/or Refractory Multiple Myeloma (MM): Results From the Expansion Cohorts of a Phase 1 Dose-Escalation Study. Blood. 2011;118(21):140. [Google Scholar]
- [96].Molineaux SM. Molecular Pathways: Targeting Proteasomal Protein Degradation in Cancer. Clinical Cancer Research. 2012;18(1):15–20. doi: 10.1158/1078-0432.CCR-11-0853. [DOI] [PubMed] [Google Scholar]
- [97].Chauhan D, Singh AV, Aujay M, et al. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood. 2010;116(23):4906–15. doi: 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75(3):311–35. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Newman DJ, Cragg GM. Natural product scaffolds as leads to drugs. Future Med Chem. 2009;1(8):1415–27. doi: 10.4155/fmc.09.113. [DOI] [PubMed] [Google Scholar]
- [100].Landis-Piwowar KR, Milacic V, Chen D, et al. The proteasome as a potential target for novel anticancer drugs and chemosensitizers. Drug Resist Updat. 2006;9(6):263–73. doi: 10.1016/j.drup.2006.11.001. [DOI] [PubMed] [Google Scholar]
- [101].Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem. 2001;276(16):13322–30. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- [102].Zhang Q, Tang X, Lu Q, Zhang Z, Rao J, Le AD. Green tea extract and (−)-epigallocatechin-3-gallate inhibit hypoxia- and serum-induced HIF-1alpha protein accumulation and VEGF expression in human cervical carcinoma and hepatoma cells. Mol Cancer Ther. 2006;5(5):1227–38. doi: 10.1158/1535-7163.MCT-05-0490. [DOI] [PubMed] [Google Scholar]
- [103].Landis-Piwowar KR, Kuhn DJ, Wan SB, Chen D, Chan TH, Dou QP. Evaluation of proteasome-inhibitory and apoptosis-inducing potencies of novel (−)-EGCG analogs and their prodrugs. Int J Mol Med. 2005;15(4):735–42. [PubMed] [Google Scholar]
- [104].Kuhn D, Lam WH, Kazi A, et al. Synthetic peracetate tea polyphenols as potent proteasome inhibitors and apoptosis inducers in human cancer cells. Front Biosci. 2005;10:1010–23. doi: 10.2741/1595. [DOI] [PubMed] [Google Scholar]
- [105].Wu AH, Tseng CC, Van Den Berg D, Yu MC. Tea intake, COMT genotype, and breast cancer in Asian-American women. Cancer Res. 2003;63(21):7526–9. [PubMed] [Google Scholar]
- [106].Landis-Piwowar KR, Wan SB, Wiegand RA, Kuhn DJ, Chan TH, Dou QP. Methylation suppresses the proteasome-inhibitory function of green tea polyphenols. J Cell Physiol. 2007;213(1):252–60. doi: 10.1002/jcp.21124. [DOI] [PubMed] [Google Scholar]
- [107].Caltagirone S, Rossi C, Poggi A, et al. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int J Cancer. 2000;87(4):595–600. doi: 10.1002/1097-0215(20000815)87:4<595::aid-ijc21>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- [108].Kampa M, Hatzoglou A, Notas G, et al. Wine antioxidant polyphenols inhibit the proliferation of human prostate cancer cell lines. Nutr Cancer. 2000;37(2):223–33. doi: 10.1207/S15327914NC372_16. [DOI] [PubMed] [Google Scholar]
- [109].Mouria M, Gukovskaya AS, Jung Y, et al. Food-derived polyphenols inhibit pancreatic cancer growth through mitochondrial cytochrome C release and apoptosis. Int J Cancer. 2002;98(5):761–9. doi: 10.1002/ijc.10202. [DOI] [PubMed] [Google Scholar]
- [110].Liao HF, Kuo CD, Yang YC, et al. Resveratrol enhances radiosensitivity of human non-small cell lung cancer NCI-H838 cells accompanied by inhibition of nuclear factor-kappa B activation. J Radiat Res. 2005;46(4):387–93. doi: 10.1269/jrr.46.387. [DOI] [PubMed] [Google Scholar]
- [111].Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol. 2005;69(10):1421–32. doi: 10.1016/j.bcp.2005.02.022. [DOI] [PubMed] [Google Scholar]
- [112].Singletary K, Ellington A. Genistein suppresses proliferation and MET oncogene expression and induces EGR-1 tumor suppressor expression in immortalized human breast epithelial cells. Anticancer Res. 2006;26(2A):1039–48. [PubMed] [Google Scholar]
- [113].Russo A, Cardile V, Lombardo L, Vanella L, Acquaviva R. Genistin inhibits UV light-induced plasmid DNA damage and cell growth in human melanoma cells. J Nutr Biochem. 2006;17(2):103–8. doi: 10.1016/j.jnutbio.2005.05.011. [DOI] [PubMed] [Google Scholar]
- [114].Yu Z, Li W, Liu F. Inhibition of proliferation and induction of apoptosis by genistein in colon cancer HT-29 cells. Cancer Lett. 2004;215(2):159–66. doi: 10.1016/j.canlet.2004.06.010. [DOI] [PubMed] [Google Scholar]
- [115].Kazi A, Daniel KG, Smith DM, Kumar NB, Dou QP. Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem Pharmacol. 2003;66(6):965–76. doi: 10.1016/s0006-2952(03)00414-3. [DOI] [PubMed] [Google Scholar]
- [116].Watanabe S, Yamaguchi M, Sobue T, et al. Pharmacokinetics of soybean isoflavones in plasma, urine and feces of men after ingestion of 60 g baked soybean powder (kinako) J Nutr. 1998;128(10):1710–5. doi: 10.1093/jn/128.10.1710. [DOI] [PubMed] [Google Scholar]
- [117].Uehar M, Arai Y, Watanabe S, Adlercreutz H. Comparison of plasma and urinary phytoestrogens in Japanese and Finnish women by time-resolved fluoroimmunoassay. Biofactors. 2000;12(1–4):217–25. doi: 10.1002/biof.5520120134. [DOI] [PubMed] [Google Scholar]
- [118].Gu Y, Zhu CF, Iwamoto H, Chen JS. Genistein inhibits invasive potential of human hepatocellular carcinoma by altering cell cycle, apoptosis, and angiogenesis. World J Gastroenterol. 2005;11(41):6512–7. doi: 10.3748/wjg.v11.i41.6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li Y, Sarkar FH. Inhibition of nuclear factor kappaB activation in PC3 cells by genistein is mediated via Akt signaling pathway. Clin Cancer Res. 2002;8(7):2369–77. [PubMed] [Google Scholar]
- [120].Dick LR, Cruikshank AA, Grenier L, Melandri FD, Nunes SL, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin: a central role for clasto-lactacystin beta-lactone. J Biol Chem. 1996;271(13):7273–6. doi: 10.1074/jbc.271.13.7273. [DOI] [PubMed] [Google Scholar]
- [121].Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268(5211):726–31. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- [122].Elliott PJ, Zollner TM, Boehncke WH. Proteasome inhibition: a new anti-inflammatory strategy. J Mol Med (Berl) 2003;81(4):235–45. doi: 10.1007/s00109-003-0422-2. [DOI] [PubMed] [Google Scholar]
- [123].Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl. 2003;42(3):355–7. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- [124].Macherla VR, Mitchell SS, Manam RR, et al. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J Med Chem. 2005;48(11):3684–7. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- [125].Chauhan D, Catley L, Li G, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- [126].Ruiz S, Krupnik Y, Keating M, Chandra J, Palladino M, McConkey D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol Cancer Ther. 2006;5(7):1836–43. doi: 10.1158/1535-7163.MCT-06-0066. [DOI] [PubMed] [Google Scholar]
- [127].Miller CP, Ban K, Dujka ME, et al. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood. 2007;110(1):267–77. doi: 10.1182/blood-2006-03-013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2010;115(4):834–45. doi: 10.1182/blood-2009-03-213009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Asai A, Hasegawa A, Ochiai K, Yamashita Y, Mizukami T. Belactosin A, a novel antitumor antibiotic acting on cyclin/CDK mediated cell cycle regulation, produced by Streptomyces sp. J Antibiot (Tokyo) 2000;53(1):81–3. doi: 10.7164/antibiotics.53.81. [DOI] [PubMed] [Google Scholar]
- [130].Asai A, Tsujita T, Sharma SV, et al. A new structural class of proteasome inhibitors identified by microbial screening using yeast-based assay. Biochem Pharmacol. 2004;67(2):227–34. doi: 10.1016/j.bcp.2003.08.035. [DOI] [PubMed] [Google Scholar]
- [131].Wu Q, Huang Z. Anatomic correction of Ebstein anomaly. J Thorac Cardiovasc Surg. 2001;122(6):1237–8. doi: 10.1067/mtc.2001.116463. [DOI] [PubMed] [Google Scholar]
- [132].Kuhn DJ, Chen Q, Voorhees PM, et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitinproteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110(9):3281–90. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].O'Connor OA, Stewart AK, Vallone M, et al. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin Cancer Res. 2009;15(22):7085–91. doi: 10.1158/1078-0432.CCR-09-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Groll M, Schellenberg B, Bachmann AS, et al. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature. 2008;452(7188):755–8. doi: 10.1038/nature06782. [DOI] [PubMed] [Google Scholar]
- [135].Clerc J, Groll M, Illich DJ, et al. Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc Natl Acad Sci USA. 2009;106(16):6507–12. doi: 10.1073/pnas.0901982106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Koguchi Y, Kohno J, Nishio M, et al. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, production, isolation, and biological activities. J Antibiot (Tokyo) 2000;53(2):105–9. doi: 10.7164/antibiotics.53.105. [DOI] [PubMed] [Google Scholar]
- [137].Kohno J, Koguchi Y, Nishio M, et al. Structures of TMC-95A-D: novel proteasome inhibitors from Apiospora montagnei sacc. TC 1093. J Org Chem. 2000;65(4):990–5. doi: 10.1021/jo991375+. [DOI] [PubMed] [Google Scholar]
- [138].Groll M, Koguchi Y, Huber R, Kohno J. Crystal structure of the 20 S proteasome: TMC-95A complex: a non-covalent proteasome inhibitor. J Mol Biol. 2001;311(3):543–8. doi: 10.1006/jmbi.2001.4869. [DOI] [PubMed] [Google Scholar]
- [139].Groll M, Gotz M, Kaiser M, Weyher E, Moroder L. TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasome. Chem Biol. 2006;13(6):607–14. doi: 10.1016/j.chembiol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- [140].Tsukamoto S, Yokosawa H. Inhibition of the ubiquitin-proteasome system by natural products for cancer therapy. Planta Med. 2010;76(11):1064–74. doi: 10.1055/s-0029-1240901. [DOI] [PubMed] [Google Scholar]
- [141].Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17(24):7151–60. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Ciechanover A, Schwartz AL. The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc Natl Acad Sci USA. 1998;95(6):2727–30. doi: 10.1073/pnas.95.6.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Varshavsky A. The N-end rule and regulation of apoptosis. Nat Cell Biol. 2003;5(5):373–6. doi: 10.1038/ncb0503-373. [DOI] [PubMed] [Google Scholar]
- [144].Schwarz SE, Rosa JL, Scheffner M. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J Biol Chem. 1998;273(20):12148–54. doi: 10.1074/jbc.273.20.12148. [DOI] [PubMed] [Google Scholar]
- [145].Saurin AJ, Borden KL, Boddy MN, Freemont PS. Does this have a familiar RING? Trends Biochem Sci. 1996;21(6):208–14. [PubMed] [Google Scholar]
- [146].Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA. 1999;96(20):11364–9. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Varshavsky A. The N-end rule pathway of protein degradation. Genes Cells. 1997;2(1):13–28. doi: 10.1046/j.1365-2443.1997.1020301.x. [DOI] [PubMed] [Google Scholar]
- [148].Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275(12):8945–51. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- [149].Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420(1):25–7. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- [150].Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- [151].Talis AL, Huibregtse JM, Howley PM. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. J Biol Chem. 1998;273(11):6439–45. doi: 10.1074/jbc.273.11.6439. [DOI] [PubMed] [Google Scholar]
- [152].Lai Z, Yang T, Kim YB, et al. Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc Natl Acad Sci USA. 2002;99(23):14734–9. doi: 10.1073/pnas.212428599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Secchiero P, Bosco R, Celeghini C, Zauli G. Recent advances in the therapeutic perspectives of Nutlin-3. Curr Pharm Des. 2011;17(6):569–77. doi: 10.2174/138161211795222586. [DOI] [PubMed] [Google Scholar]
- [154].Miura-Shimura Y, Duan L, Rao NL, et al. Cbl-mediated ubiquitinylation and negative regulation of Vav. J Biol Chem. 2003;278(40):38495–504. doi: 10.1074/jbc.M305656200. [DOI] [PubMed] [Google Scholar]
- [155].Klapper LN, Waterman H, Sela M, Yarden Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000;60(13):3384–8. [PubMed] [Google Scholar]
- [156].Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61(Suppl 2):1–13. doi: 10.1159/000055396. [DOI] [PubMed] [Google Scholar]
- [157].Dong Y, Hakimi MA, Chen X, et al. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol Cell. 2003;12(5):1087–99. doi: 10.1016/s1097-2765(03)00424-6. [DOI] [PubMed] [Google Scholar]
- [158].Tait DL, Obermiller PS, Hatmaker AR, Redlin-Frazier S, Holt JT. Ovarian cancer BRCA1 gene therapy: Phase I and II trial differences in immune response and vector stability. Clin Cancer Res. 1999;5(7):1708–14. [PubMed] [Google Scholar]
- [159].Tait DL, Jensen RA, Holt JT, Johnson DH, Gralow J, King MC. Gene therapy for breast and ovarian cancer with BRCA1. Breast Dis. 1998;10(1–2):89–98. doi: 10.3233/bd-1998-101-211. [DOI] [PubMed] [Google Scholar]
- [160].Horie K, Urano T, Ikeda K, Inoue S. Estrogen-responsive RING finger protein controls breast cancer growth. J Steroid Biochem Mol Biol. 2003;85(2–5):101–4. doi: 10.1016/s0960-0760(03)00209-7. [DOI] [PubMed] [Google Scholar]
- [161].Li X, Cao X, Zhang W, Feng Y. Expression level of insulin-like growth factor binding protein 5 mRNA is a prognostic factor for breast cancer. Cancer Sci. 2007;98(10):1592–6. doi: 10.1111/j.1349-7006.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Burger AM, Gao Y, Amemiya Y, et al. A novel RING-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005;65(22):10401–12. doi: 10.1158/0008-5472.CAN-05-2103. [DOI] [PubMed] [Google Scholar]
- [163].F RK, Buac D, A MB. Disulfiram, and disulfiram derivatives as novel potential anticancer drugs targeting the ubiquitin-proteasome system in both preclinical and clinical studies. Curr Cancer Drug Targets. 2011;11(3):338–46. doi: 10.2174/156800911794519798. [DOI] [PubMed] [Google Scholar]
- [164].Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66(21):10425–33. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- [165].Turk BE, Griffith EC, Wolf S, Biemann K, Chang YH, Liu JO. Selective inhibition of amino-terminal methionine processing by TNP-470 and ovalicin in endothelial cells. Chem Biol. 1999;6(11):823–33. doi: 10.1016/s1074-5521(99)80129-x. [DOI] [PubMed] [Google Scholar]
- [166].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98(15):8554–9. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Hatakeyama S, Nakayama KI. U-box proteins as a new family of ubiquitin ligases. Biochem Biophys Res Commun. 2003;302(4):635–45. doi: 10.1016/s0006-291x(03)00245-6. [DOI] [PubMed] [Google Scholar]
- [168].Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI. U box proteins as a new family of ubiquitin-protein ligases. J Biol Chem. 2001;276(35):33111–20. doi: 10.1074/jbc.M102755200. [DOI] [PubMed] [Google Scholar]
- [169].Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA. 2002;99(20):12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Yang W, Klos KS, Zhou X, et al. ErbB2 overexpression in human breast carcinoma is correlated with p21Cip1 up-regulation and tyrosine-15 hyperphosphorylation of p34Cdc2: poor responsiveness to chemotherapy with cyclophoshamide methotrexate, and 5-fluorouracil is associated with Erb2 overexpression and with p21Cip1 overexpression. Cancer. 2003;98(6):1123–30. doi: 10.1002/cncr.11625. [DOI] [PubMed] [Google Scholar]
- [171].Traidej M, Chen L, Yu D, Agrawal S, Chen J. The roles of E6-AP and MDM2 in p53 regulation in human papillomavirus-positive cervical cancer cells. Antisense Nucleic Acid Drug Dev. 2000;10(1):17–27. doi: 10.1089/oli.1.2000.10.17. [DOI] [PubMed] [Google Scholar]
- [172].Sheng Y, Saridakis V, Sarkari F, et al. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat Struct Mol Biol. 2006;13(3):285–91. doi: 10.1038/nsmb1067. [DOI] [PubMed] [Google Scholar]
- [173].Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60(1–2):61–8. doi: 10.1007/s12013-011-9185-5. [DOI] [PubMed] [Google Scholar]
- [174].Colombo M, Vallese S, Peretto I, et al. Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem. 2010;5(4):552–8. doi: 10.1002/cmdc.200900409. [DOI] [PubMed] [Google Scholar]
- [175].Pham LV, Tamayo AT, Li C, Bornmann W, Priebe W, Ford RJ. Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications. Mol Cancer Ther. 2010;9(7):2026–36. doi: 10.1158/1535-7163.MCT-10-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Udeshi ND, Mani DR, Eisenhaure T, et al. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol Cell Proteomics. 2012;11(5):148–59. doi: 10.1074/mcp.M111.016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Seiberlich V, Goldbaum O, Zhukareva V, Richter-Landsberg C. The small molecule inhibitor PR-619 of deubiquitinating enzymes affects the microtubule network and causes protein aggregate formation in neural cells: Implications for neurodegenerative diseases. Biochim Biophys Acta. 2012;1823(11):2057–68. doi: 10.1016/j.bbamcr.2012.04.011. [DOI] [PubMed] [Google Scholar]
- [178].Fournier MJ, Gareau C, Mazroui R. The chemotherapeutic agent bortezomib induces the formation of stress granules. Cancer Cell Int. 2010;10:12. doi: 10.1186/1475-2867-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]