

Abstract
EML4-ALK gene rearrangements define a unique subset of non-small cell lung cancer (NSCLC) patients and the clinical success of the ALK inhibitor crizotinib in this population has become a paradigm for molecularly-targeted therapy. Here we show that the Hsp90 inhibitor ganetespib induced loss of EML4-ALK expression and depletion of multiple oncogenic signaling proteins in ALK-driven NSCLC cells, resulting in greater in vitro potency, superior antitumor efficacy and prolonged animal survival compared to crizotinib. In addition, combinatorial benefit was seen when ganetespib was used with other targeted ALK agents both in vitro and in vivo. Importantly, ganetespib overcame multiple forms of crizotinib resistance, including secondary ALK mutations, consistent with activity seen in a NSCLC patient with crizotinib-resistant disease. Cancer cells driven by ALK amplification and oncogenic rearrangements of ROS1 and RET kinases were also sensitive to ganetespib exposure. Taken together, these results highlight the therapeutic potential of ganetespib for ALK-driven NSCLC.
Keywords: Hsp90 inhibition, non-small cell lung cancer, anaplastic lymphoma kinase, ganetespib, crizotinib resistance
INTRODUCTION
Non-small cell lung cancer (NSCLC) can be classified into distinct molecular subsets based on specific genomic alterations that drive tumorigenesis (1, 2). Approximately 3-7% of NSCLC tumors are characterized by rearrangement of genes encoding anaplastic lymphoma kinase (ALK), most commonly with echinoderm microtubule-associated protein-like 4 (EML4) (3), resulting in constitutively active kinases with transforming capacity (3, 4). Crizotinib, a dual MET/ALK small molecule tyrosine kinase inhibitor (TKI), was the first ALK-targeted agent evaluated clinically and was recently granted accelerated approval in the United States for the treatment of patients with ALK-positive (ALK+) NSCLC (5, 6). Among these patients, crizotinib therapy has been associated with improved survival compared with that of crizotinib-naïve controls (7), thus providing clinical validation for targeting ALK in ‘oncogene-addicted’ lung tumors with this genotype.
Despite this success, however, durable responses to crizotinib therapy have been hampered by the development of drug resistance, a common feature of many TKI drugs (8). Relapses frequently occur due to a spectrum of newly acquired secondary mutations within the ALK kinase domain (9, 10). Accordingly, considerable effort has been focused on the development of second-generation inhibitors designed to overcome this clinical challenge (11). Importantly, it has now emerged that other ‘ALK-independent’ mechanisms, such as the activation of compensatory signaling pathways, may also confer resistance to targeted ALK agents (6). Strategies to counteract these types of acquired resistance in ALK-driven NSCLC have not yet been established.
Heat shock protein 90 (Hsp90) is a molecular chaperone that plays a central role in regulating the correct folding, stability, and function of numerous ‘client proteins’ (12). Inhibition of Hsp90 activity results in aggregation or proteasomal degradation of these clients, in turn promoting the simultaneous disruption of numerous oncogenic signaling pathways critical for tumor cell proliferation and survival (13, 14). Many of these proteins are kinases that have been shown to be oncogenic drivers in subsets of lung adenocarcinoma, including EGFR, BRAF, HER2, and, notably, the EML4-ALK fusion protein (15-20). Targeting the chaperone function of Hsp90 therefore represents an alternative approach to direct kinase inhibition for therapeutic intervention in ALK-driven cancer. Further, because of the coordinate impact on multiple signal cascades, pharmacological blockade of Hsp90 may also overcome signaling redundancies and drug resistance mechanisms commonly seen in many cancers (21, 22).
Ganetespib is a novel triazolone inhibitor of Hsp90 with superior pharmacologic and biologic properties that distinguish it from other first- and second-generation Hsp90 inhibitors in terms of antitumor activity, potency and safety (23). In human trials, ganetespib has demonstrated promising efficacy in advanced NSCLC patients, including robust single agent activity in individuals with tumors harboring ALK gene rearrangement (24). Given the distinct mechanisms of action of ganetespib and crizotinib for inhibiting ALK, we have undertaken a comprehensive evaluation of the comparative sensitivity of ALK+ NSCLC to each of these treatment modalities. Combined with encouraging early clinical results in NSCLC, our data suggest that ganetespib may offer an alternative, and potentially complementary, strategy to targeted ALK inhibition for inducing substantial antitumor responses and overcoming acquired resistance in ALK+ lung cancer patients.
RESULTS
Loss of viability and client protein expression by ganetespib in EML4-ALK expressing NSCLC cells
The H2228 NSCLC cell line harbors an E6a/b;A20 EML4-ALK fusion protein and, as shown in Fig. 1A, was comparatively more sensitive to the cytotoxic effects of ganetespib than crizotinib (IC50 values 13 vs. 202 nM, respectively). Importantly, these levels are within free (unbound), circulating Cmax levels observed at maximal clinical dosing for both drugs (~200 nM for ganetespib, ~500 nM for crizotinib) (25). Ganetespib (Supplementary Fig. S1) treatment resulted in a robust and dose-dependent destabilization of EML4-ALK, as well as EGFR and MET receptors, all established Hsp90 clients (Fig. 1B). Importantly, targeted degradation of these signaling proteins was accompanied by inactivation of downstream effectors (phosphorylated and total AKT, p-STAT3 and p-ERK) and induction of BIM, an apoptotic marker. A concomitant increase in Hsp70 levels was observed, indicative of Hsp90 inhibition (Fig. 1B).
Figure 1.

Cellular viability, EML4-ALK expression and pathway modulation in ALK-rearranged NSCLC cell lines following ganetespib and crizotinib treatment. A, H2228 cells were treated with increasing concentrations of ganetespib or crizotinib and cell viability assessed at 72 h. B, H2228 cells were exposed to graded concentrations of ganetespib (3.3-100 nM) for 24 h and cell lysates immunoblotted with the indicated antibodies. v represents vehicle control. C, H3122 cells were treated with increasing concentrations of ganetespib or crizotinib and cell viability assessed at 72 h. D, H3122 cells were exposed to graded concentrations of ganetespib (3-500 nM) or crizotinib (10-500 nM) for 24 h and cell lysates immunoblotted with the indicated antibodies.
Next we examined the comparative effects of ganetespib and crizotinib in the ALK-driven H3122 cell line which is dependent on the E13;A20 EML4-ALK variant for growth and survival. Ganetespib was acutely cytotoxic to these cells, and with 30 times greater potency than crizotinib (10 vs. 300 nM; Fig. 1C). When expression changes in client proteins and signaling pathways were examined in this line (Fig. 1D), we found that ganetespib exposure at concentrations ≥ 30 nM resulted in the complete loss of phosphorylated EML4-ALK protein expression, as well as the active (phosphorylated) forms of STAT3, AKT and ERK. Targeted degradation of EGFR and MET were seen at the same concentrations, as well as negative effects on the mTOR signaling pathway, evidenced by loss of p-p70S6K and p-4E-BP1 expression (Fig. 1D). Consistent with the potent cytotoxic activity of ganetespib, a robust increase in BIM and cleaved PARP expression were observed. In contrast, crizotinib displayed far weaker activity in terms of effector signaling and activation of apoptotic pathways. At least a ten-fold higher concentration of crizotinib (300 nM) was required to significantly reduce phosphorylated EML4-ALK levels, and this was not complete until 500 nM (Fig. 1D). These same maximal concentrations only achieved relatively modest effects compared to ganetespib on the blockade of downstream ERK, AKT and mTOR signaling, as well as apoptotic induction. Taken together, these data show that ganetespib displays greater in vitro potency than crizotinib in ALK+ NSCLC cells.
Ganetespib suppresses tumor growth and extends survival in ALK+ NSCLC xenografts
Ganetespib and crizotinib were highly efficacious in nude mouse models of ALK+ NSCLC, each inducing similar degrees of tumor regression (Supplementary Fig. S2). Crizotinib administered at its maximally tolerated dose (MTD) of 200 mg/kg 5x/week p.o. over a 3 week cycle to mice bearing H3122 xenografts resulted in 24% tumor regression. Ganetespib treatment at its MTD of 150 mg/kg weekly resulted in a similar degree (27%) of tumor shrinkage. In order to more robustly evaluate potential differences in antitumor activity in vivo, we subsequently performed experiments in tumor-bearing SCID mice. Animals bearing H3122 xenografts were dosed intravenously (i.v.) with ganetespib at 50 mg/kg once a week (Fig. 2A). This regimen resulted in significant tumor growth inhibition (T/C value 21%) over a 3 week period. Treating at MTD (data not shown) or splitting the dose into two consecutive day dosing of 25 mg/kg each week resulted in a minor improvement in efficacy (T/C value 10%) and disease stabilization (Fig. 2A). Importantly, all treatment regimens were well tolerated, with no toxicity or changes in body weight seen after 3 weeks of dosing (data not shown). In contrast, crizotinib was less efficacious in the same model. As shown in Figure 2B, 5x/week p.o. dosing of crizotinib at 50 mg/kg resulted in a T/C value of only 55%. When the crizotinib dose was doubled to 100 mg/kg no substantial improvement in efficacy was seen, however significant losses of body weight were more frequently observed; thus this dose was determined to be the MTD (data not shown). Representative images of H3122 xenograft tumors 50 days post-treatment with ganetespib and crizotinib are shown in Figure 2C.
Figure 2.

Ganetespib suppresses tumor growth and extends survival in ALK+ NSCLC xenografts. A, SCID mice bearing H3122 xenografts (n=7/group) were i.v. dosed with 50 mg/kg ganetespib once weekly, or on a weekly 2×25 mg/kg consecutive day dosing regimen, as indicated, for 3 weeks. % T/C values are indicated to the right of each growth curve and the error bars are the SEM. B, SCID mice bearing H3122 xenografts (n=7/group) were p.o. dosed with 50 mg/kg crizotinib 5X/week over a 3 week cycle. C, Representative images of tumors from vehicle, ganetespib (2X/week) or crizotinib (50 mg/kg) treated animals at day 50. D, Pharmacodynamic analysis of client protein modulation in H3122 xenografts. SCID mice bearing established H3122 tumors were treated with vehicle or a single bolus injection of ganetespib at 50 mg/kg at the indicated time points between 24 and 96 h. Mice were also treated a single bolus injection of crizotinib (50 mg/kg) for 24 h. Tumors were resected and the levels of the indicated proteins determined by immunoblotting. E, Kaplan-Meier analysis of overall survival in the H3122 xenograft model. Beginning 12 days after tumor cell implantation SCID mice bearing H3122 xenografts (n=7/group) were dosed with vehicle, 50 mg/kg ganetespib 1x/week i.v, or 50 mg/kg crizotinib 5X/week p.o and animal survival monitored until day 75.
To determine whether these tumor responses correlated with target modulation in vivo, we performed pharmacodynamic analysis in additional mice bearing H3122 xenografts (Fig. 2D). Animals were treated with a single bolus injection of ganetespib at the effective 50 mg/kg dose and tumors harvested at 24, 48, 72 and 96 h after treatment. For comparison, animals were treated with a single injection of vehicle or crizotinib at 50 mg/kg and tumors collected 24 h later. EML4-ALK and downstream ERK signaling were degraded and deactivated, respectively, within 24 h following ganetespib treatment. Importantly, these effects were sustained over time, as recovery did not occur until 72 h. Similar kinetics were observed for the targeted destabilization of the Hsp90 clients EGFR and MET, as well as their effector signaling intermediates p-STAT3 and p-AKT. Loss of these signaling cascades was associated with a corresponding increase in BIM protein expression, indicative of intratumoral apoptotic induction. In stark contrast, single dose crizotinib had negligible effects on ERK activity at 24 h, nor any of the other cascades. Overall, these data show that single dose ganetespib exerts durable suppressive effects on ALK signaling in human tumor xenografts, destabilizing both the fusion kinase and its effectors for up to 72 h.
Having shown that ganetespib displayed greater antitumor activity than crizotinib in SCID mice bearing ALK+ NSCLC xenografts, we next extended the time on study to measure overall survival, with an endpoint defined as animal death, cavitating tumors or tumors > 1.5 cm. Animals were dosed with either once weekly i.v. ganetespib (50 mg/kg), crizotinib 50 mg/kg 5 times per week p.o., or vehicle alone (Fig. 2E). Fifty percent of animals treated with vehicle died by day 40, while crizotinib extended survival of 50% of the animals to 54 days. After 75 days, all animals within the ganetespib group had survived.
Distinct actions of ganetespib and crizotinib lead to favorable combinatorial activity
Given the distinct mechanisms of action of ganetespib and crizotinib on ALK inhibition, we examined whether combining the two compounds would lead to increased activity. Indeed, as shown in Figure 3A, concurrent administration of low (IC20) doses of ganetespib and crizotinib to H3122 cells substantially increased cell death in vitro. Importantly, similar combinatorial benefit was observed when ganetespib was dosed in combination with the structurally unrelated ALK inhibitors ASP3026 and CH542802.
Figure 3.

Enhanced activity of ganetespib in combination with ALK inhibitors in vitro and in vivo. A, H3122 cells were treated with the indicated concentrations of ganetespib, crizotinib, ASP3026, CH5424802 either as single agents or in combination. Cell viability was determined at 72 h. B, Combination of ganetespib and crizotinib induces enhanced antitumor efficacy in vivo. SCID mice bearing H3122 xenografts (n=7/group) were i.v. dosed with 25 mg/kg ganetespib once weekly, 100 mg/kg crizotinib 5x/week p.o., or the combination, as indicated. % T/C values are indicated to the right of each growth curve and the error bars are the SEM.
To evaluate whether the effects on cell viability seen in vitro translated to improved combinatorial efficacy in vivo, xenograft-bearing mice were treated with ganetespib and crizotinib, both as single agents and in combination. As shown in Figure 3B, once weekly administration of ganetespib at 25 mg/kg was comparable to 5x/week dosing of crizotinib at its MTD, with each compound inducing a similar degree of tumor suppression (T/C values of 41% and 39%, respectively). Consistent with the in vitro findings, concurrent treatment with both drugs resulted in a significant enhancement of antitumor activity, inhibiting tumor growth by 93%. In addition, combination treatment was well tolerated, with no significant changes in body weights seen after 3 weeks of treatment (Supplementary Fig. S3). In fact, combination treatment appeared better tolerated than crizotinib monotherapy, strongly suggesting that there is no additional toxicity conferred by the addition of ganetespib to the regimen. Thus, ganetespib and crizotinib, when combined, displayed superior antitumor efficacy compared to monotherapy in H3122 NSCLC xenografts.
Ganetespib overcomes acquired crizotinib resistance
As has been the clinical experience for other TKIs, prolonged exposure to crizotinib may ultimately give rise to acquired resistance, thereby diminishing the efficacy of long-term treatment. An important consideration, therefore, was whether crizotinib-resistant NSCLC cells remained sensitive to ganetespib. To determine this experimentally, we generated crizotinib-resistant H3122 cells (H3122 CR1) by continuous selective culture in 1 μM crizotinib. Endogenous expression of EML4-ALK was reduced in the resistant line compared to parental H3122 cells, yet the fusion protein remained sensitive to ganetespib-induced destabilization (Fig. 4A).
Figure 4.

Ganetespib retains potency against crizotinib-resistant NSCLC tumor phenotypes. A, Parental H3122 and crizotinib-resistant H3122 CR1 cells were treated with ganetespib at either 25 or 100 nM for 24 h and the levels of EML4-ALK protein determined by immunoblotting. B, H3122 and H3122 CR1 cells were treated with increasing concentrations of ganetespib or crizotinib and cell viability was assessed after 72 h. C, Table of IC50 cytotoxicity values in H3122 and H3122 CR1 cells in response to ganetespib or ALK inhibitor exposure. D, Light micrographs of cellular morphology of H3122 and H3122 CR1 cells. Scale bar, 50 μM.
We next compared the activities of ganetespib and crizotinib using the H3122 and H3122 CR1 lines (Fig. 4B). As expected, crizotinib treatment resulted in dose-dependent cytotoxicity in the parental line, but had no effect on H3122 CR1 cells. In contrast, despite a small shift in IC50 values, ganetespib retained full potency in both cell lines, irrespective of crizotinib resistance status. Indeed, H3122 CR1 cells remained several fold more sensitive to ganetespib compared to the sensitivity of the parental line to crizotinib. Moreover, H3122 CR1 cells were insensitive to other ALK inhibitors (CH5424802, ASP3026 and TAE684) yet succumbed to Hsp90 inhibition by ganetespib (Fig. 4C).
Interestingly, the H3122 CR1 line exhibited a more fibroblastic morphology than the parental, typical of enhanced epithelial plasticity (Fig. 4D). We therefore performed a reverse phase array comparing the expression of proteins between the H3122 and H3122 CR1 cells (Supplementary Fig. S4A). Epithelial markers such as E cadherin and P cadherin were downregulated, as well as other receptor tyrosine kinases including IGF-1R and VEGFR2. Concomitant upregulation of Snail, Notch 1, Caveolin and Src were also seen. These changes, consistent with an epithelial-mesenchymal transition (EMT), were confirmed by Western blot (Supplementary Fig. S4B), which also revealed an increase in vimentin expression in H3122 CR1 cells. Further, H3122 CR1 cells demonstrated increased migratory capacity in a scratch assay (Supplementary Fig. S4C), and this effect could be blocked with low-dose ganetespib treatment (Supplementary Fig. S4D). Taken together, these data strongly suggest that prolonged crizotinib exposure selected for a population of cells with mesenchymal characteristics and a more aggressive phenotype.
Ganetespib retains activity against NPM-ALK-transformed cells bearing secondary ALK mutations that confer crizotinib resistance
One common mechanism leading to acquired resistance to ALK TKIs is the emergence of secondary point mutations within the kinase domain (9). To determine the potential impact of such mutational changes on ganetespib activity, we performed experiments in BaF3 cells oncogenically transformed by engineered expression of the lymphoma-associated NPM-ALK fusion kinase. NPM-ALK-expressing BaF3 cells were exposed in culture to a variety of concentrations of crizotinib until the emergence of viable cell pools, which were then subjected to limiting dilution to isolate crizotinib-resistant clones. As shown in Figure 5A, a spectrum of point mutations located in the ALK kinase domain and involving 15 different substitutions were associated with crizotinib resistance. The crizotinib-sensitive parental NPM-ALK/BaF3 cells used in these experiments demonstrated a crizotinib IC50 value of ~370 nM. By contrast, the clones harboring the various ALK mutations exhibited varying degrees of resistance, with relative IC50 values ranging from approximately 1.6-fold (E1408K, E1132K) to 4-5-fold (G1202R; L1196M) higher (Fig. 5A). We then examined whether crizotinib-resistant NPM-ALK/BaF3 cells demonstrated sensitivity to Hsp90 inhibition. Crizotinib-sensitive NPM-ALK/BaF3 cells were also sensitive to ganetespib (IC50 value 21 nM). Importantly, all of the crizotinib-resistant NPMALK/BaF3 clones retained high sensitivity to ganetespib; indeed, the IC50 values were essentially identical to that of NPM-ALK/BaF3. Consistent with these observations, NPM-ALK protein degradation following ganetespib treatment showed similar dose-dependent responses regardless of the presence or identity of the crizotinib-resistance mutation (Fig. 5B). The kinetics of protein degradation for NPM-ALK/BaF3 and NPM-ALK/BaF3 containing the L1196M gatekeeper mutation following exposure to ganetespib (250 nM) is shown in Figure 5C.
Figure 5.
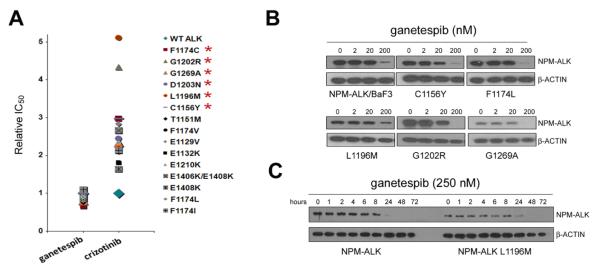
Crizotinib-resistant NPM-ALK mutants retain sensitivity to ganetespib. A, Ganetespib and crizotinib sensitivity was assessed in crizotinib-sensitive NPM-ALK BaF3 cells and crizotinib-resistant NMP-ALK mutants. Relative IC50 values are plotted based on the sensitivity of the parental NPM-ALK BaF3 line to each compound. Clinically relevant mutations in NSCLC are indicated by asterisks. B, Dose-response analysis of ganetespib. Crizotinib-sensitive NPM ALK/BaF3 cells (upper left panel) together with NPM-ALK/BaF3 containing the indicated amino acid substitutions known to confer crizotinib resistance were incubated for 24 h with the range of ganetespib concentrations indicated, and the stability of the NPM-ALK protein expressed in each line assessed by immunoblotting. C, Kinetics of ganetespib-associated NPM-ALK protein degradation. The NPM-ALK protein degradation response of crizotinib-sensitive NPM-ALK/BaF3 and NPM-ALK/BaF3 containing the L1196M gatekeeper mutation to incubation in 250 nM ganetespib for the indicated times is shown.
Clinical activity of ganetespib in crizotinib-resistant NSCLC
In a recent Phase 2 trial of ganetespib monotherapy in patients with advanced NSCLC (24), 8 patients (8%) were identified as harboring ALK gene rearrangements. All were crizotinib-naïve. Of these patients, four achieved an objective partial response (PR), three demonstrated stable disease (SD) and one experienced disease progression at 16 weeks. The median progression-free survival observed for the four patients with PR was 8.1 months, significantly better than for patients without ALK rearrangement. The 50% objective response rate combined with the overall 88% disease control rate within this subset therefore provides clinical validation for the therapeutic potential of ganetespib in ALK+ NSCLC. Further compelling evidence is provided by the computed tomography (CT) scans shown in Figure 6. The images were obtained from a 24 year old male with ALK+ NSCLC who had progressed while on crizotinib after 12 months of therapy. Sequencing performed upon re-biopsy confirmed the presence of a secondary G1269A mutation within the ALK domain sequence. Lung lesions were clearly apparent at baseline (Fig. 6A) and imaging revealed marked tumor shrinkage following one cycle (3 once-weekly doses at 200 mg/m2) of ganetespib monotherapy (Fig. 6B).
Figure 6.
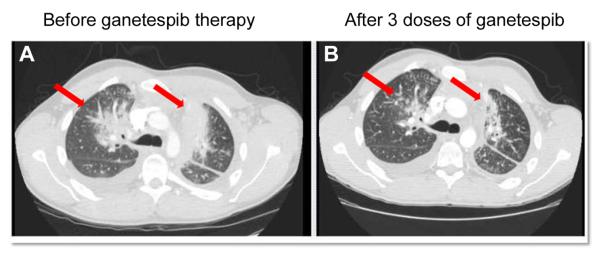
Response of ALK-rearranged, crizotinib-resistant NSCLC after one cycle of ganetespib. CT scans taken at A, baseline and B, after one cycle (200 mg/m2, 1X/week for 3 weeks) of ganetespib. Arrows depict locations of tumor masses.
Ganetespib is active in tumor cells driven by ALK amplification and additional oncogenic kinase fusions
ALK gene amplification represents another event that can contribute to crizotinib resistance in NSCLC and, importantly, can co-exist with ALK mutation (8). To examine targeted inhibitory effects in ALK-amplified tumor cells we treated the NB-39-nu neuroblastoma cell line (which expresses 30-40 copies of the ALK gene per cell) with graded concentrations of ganetespib or crizotinib and found that these cells were acutely sensitive to ganetespib exposure (IC50, 10 nM) and comparatively less so to crizotinib (IC50, 240 nM) (Fig. 7A). Ganetespib treatment resulted in a dose-dependent degradation of oncogenic ALK, as well as loss of downstream effector signaling (activated STAT3, AKT and ERK) and concomitant induction of the apoptotic markers BIM and cleaved PARP (Fig. 7B). Ganetespib also reduced viability in NB69 neuroblastoma cells which are wild-type for ALK (no amplification, no mutation), with an IC50 value of 21 nM (data not shown). Thus, while ALK amplification itself was not the primary determinant of ganetespib sensitivity in neuroblastoma cells, the data clearly show that targeted Hsp90 inhibition results in potent destabilization of amplified ALK in cells dependent on this driver.
Figure 7.

Ganetespib displays potent cellular activity in models of ALK amplification, ROS1 kinase fusion and RET kinase fusion proteins. A, NB-39-nu neuroblastoma cells were treated with increasing concentrations of ganetespib or crizotinib and cell viability assessed at 72 h. B, NB-39-nu cells were exposed to graded concentrations of ganetespib (1-500 nM) for 24 h and cell lysates immunoblotted with the indicated antibodies. v represents vehicle control. C, HCC78 NSCLC and U118-MG glioma cells were treated with increasing concentrations of ganetespib and cell viability assessed at 72 h. D, HCC78 cells were treated with ganetespib at 5, 10, 50 or 100 nM for 24 h and the levels of phosphorylated and total SLC34A2-ROS1 fusion protein determined by immunoblotting. E, TPC1 thyroid carcinoma cells were treated with increasing concentrations of ganetespib and cell viability assessed at 72 h. F, TPC1 cells were treated with increasing concentrations of ganetespib as indicated for 24 h and the levels of phosphorylated and total CCDC6-RET fusion protein, phosphorylated ERK and cleaved PARP expression determined by immunoblotting.
Chromosomal rearrangements involving the ROS1 receptor tyrosine kinase have recently been described in a subset of NSCLCs (26). Here we examined the effect of ganetespib treatment on HCC78 NSCLC cells and U118-MG glioma cells that bear SLC34A2-ROS1 and FIG-ROS1 fusion proteins, respectively (Fig. 7C). Both cells lines were highly sensitive to ganetespib exposure (IC50 values of 17nM and 20 nM), whereas crizotinib displayed significantly weaker activity (data not shown). Dose-dependent degradation of total and phosphorylated forms of the SLC34A2-ROS1 fusion protein in HCC78 cells was confirmed by immunoblotting (Fig. 7D).
Finally, RET kinase fusions, frequently associated with papillary thyroid carcinoma, are emerging as relevant oncogenic drivers in some lung and colorectal cancers (27). The TPC1 thyroid carcinoma cell line expresses a CCDC6-RET fusion protein that has also been detected in NSCLC tumors (28). As shown in Figure 7E, ganetespib potently induces cell death in TPC1 cells (IC50 value 14 nM). Moreover, potent destabilization of the fusion kinase, loss of downstream ERK signaling and induction of apoptosis (shown by elevations in cleaved PARP expression) occurred in these cells in a dose-dependent manner (Fig. 7F). In two thyroid lines lacking RET fusions, HTC/C3 and BHT-101, ganetespib treatment resulted in cytotoxicity IC50 values of 22 and 24 nM, respectively (data not shown). Mutated BRAF, another Hsp90 client, serves as an alternative driver in both these lines thereby accounting for their sensitivity. Overall, however, in tumor cells driven by ALK amplification or chromosomal rearrangements of ROS1 or RET, ganetespib exposure leads to robust loss of the relevant driver and subsequent cell death.
DISCUSSION
Oncogenic gene rearrangements of ALK define a clinically relevant subset of human NSCLCs and the success of crizotinib serves as a paradigm for molecularly-targeted therapy in this malignancy. However, as is the case for many small molecule TKIs, crizotinib responses do not last, which highlights the ongoing challenge of discovering superior treatment options, particularly those that can overcome the invariable development of acquired resistance. EML4-ALK is a highly sensitive client protein of the molecular chaperone Hsp90 (19, 20) and preclinical evidence suggests that disrupting the chaperone function of this molecule can effectively overcome oncogenic ALK activity, including in lines that harbor ALK inhibitor-resistant mutations (8, 29). In addition to direct kinase inhibition, we and others have recently shown that crizotinib-naïve ALK+ lung cancer patients can derive therapeutic benefit from targeted degradation of ALK via Hsp90 blockade, thereby confirming preclinical predictions (24, 30). Here we have provided a compelling rationale for the use of ganetespib as an alternative and potentially complementary strategy for NSCLC patients with ALK-driven disease.
A significant finding of this study was the capacity of ganetespib to overcome crizotinib resistance in ALK+ cancer, as was shown in multiple experimental models and also in the clinical setting. Unlike the case of EGFR, in which a secondary point mutation at the gatekeeper residue (T790M) represents the predominant mechanism of acquired resistance to erlotinib and gefitinib in NSCLC tumors (31, 32), crizotinib resistance arising from ALK mutation appears more analogous to that of imatinib resistance seen in chronic myeloid leukemia patients. There, multiple mutations within the kinase domain of BCR-ABL have been reported that are associated with the development of drug resistance and mutational frequencies may even increase with disease progression (33). To date, a variety of ALK kinase domain mutations at different amino acid sites have been reported in NSCLC patients who exhibited resistance to crizotinib including L1152R, C1156Y, F1174L, L1196M (the ALK gatekeeper), G1202R, D1203N and G1269A, and a number of others that can mediate ALK TKI resistance have been identified through in vitro mutagenesis screens (reviewed in (6, 11, 34)). Importantly, our studies using NPM-ALK-expressing BaF3 cells rendered crizotinib resistant due to these mutations revealed that ganetespib possessed robust cytotoxic activity irrespective of the mutational site or specific amino acid substitution present. This result was strikingly validated by the clinical observations seen in the relapsed NSCLC patient after one year of crizotinib therapy. Despite the presence of a G1269A kinase domain mutation, a single cycle of ganetespib treatment resulted in a marked tumor response and discernible shrinkage of lung lesions, highlighting the therapeutic potential of the drug within this refractory population.
Further, systemic resistance to ALK inhibitors can arise in the absence of secondary ALK kinase domain mutations (10). While the mechanisms remain to be fully elucidated, it appears that ligand-mediated activation of secondary and/or separate oncogenic signaling pathways, in particular EGFR and HER2 (35-37), is one process that may bypass the dependency of tumor cells on ALK signaling and contribute to a resistant phenotype. Moreover the use of ALK-selective inhibitors with increased potency is unlikely to provide any clinical impact for these forms of resistance. In this regard, the broader spectrum of biological activity afforded by Hsp90 inhibition represents a promising strategy to counteract such compensatory mechanisms. These driver kinases are established Hsp90 clients, and our data show that ganetespib exposure results in the simultaneous destabilization of EML4-ALK as well as receptor kinases such as EGFR and MET in ALK+ NSCLC lines both in vitro and in vivo, with concomitant loss of multiple downstream effector signaling pathways. These effects were distinct to those of crizotinib, and this multimodal activity of ganetespib accounts for its superior potency and antitumor efficacy. Although the underlying basis of resistance in the H3122 CR1 line was not identified, sequencing analysis of 12 clones failed to identify any acquired ALK mutations (D. Proia, unpublished results), and amplification of the ALK gene was not present. It is interesting to note that these cells manifested a morphology and molecular profile consistent with having undergone EMT, and this transition has been associated with activation of the EGFR pathway (38, 39). The mechanistic nature of the resistance exhibited by these cells, one that retains sensitivity to ganetespib, is currently under investigation.
Combination treatment of crizotinib with other antineoplastic agents represents a potential approach for inducing durable remissions in ALK+ NSCLC patients, and number of clinical trials are currently ongoing (40), including a phase I study of crizotinib with the dual EGFR/HER inhibitor PF299804 (NCT01121575). This consideration therefore prompted an investigation of combining the two modalities of Hsp90 inhibition and selective ALK targeting. In vitro, combinatorial benefit was seen when ganetespib was used as a co-treatment with crizotinib as well as the structurally unrelated inhibitors ASP3026 and CH542802, both of which have been reported to display superior potency to crizotinib (41, 42). Importantly, this benefit was recapitulated in vivo, where the complementary actions of ganetespib and crizotinib resulted in significantly improved efficacy than either agent alone in ALK-driven H3122 xenografts.
Further, we showed that oncogenic gene arrangements of two additional tyrosine kinases, ROS1 and RET, were also sensitive to Hsp90 inhibition by ganetespib. Similar to ALK, ROS1 has recently been shown to define a genomic subset of NSCLC with distinct clinical characteristics (26). The incidence of ROS1 fusions in lung cancer is 1.6% (43, 44) and, interestingly, cell lines driven by these activating rearrangements are also sensitive to crizotinib. Our results for two different ROS1 fusions, derived from different tumor types, revealed that ganetespib exposure induced degradation of the aberrant kinases with low nanomolar potency, and was again superior to crizotinib. RET kinase fusions have also recently emerged as promising molecular targets in NSCLC (28), where they have been reported to segregate from genetic modifications in EGFR, KRAS, HER2 and ALK (45). As the clinical significance of these oncogenic drivers in NSCLC becomes realized, the data we are presenting here suggest that pharmacological blockade of Hsp90 function warrants investigation as therapeutic approach.
In summary, our data suggest that targeting the Hsp90 chaperone pathway with ganetespib represents a potentially effective strategy for therapeutic intervention in multiple ALK-driven malignancies, in particular NSCLC. The pleiotropic effects of Hsp90 inhibition on both ALK itself as well as other client proteins provides more complete and durable responses compared to direct kinase inhibition. In light of its select advantages over ALK-specific TKIs and maturing clinical profile, these findings are likely to provide a framework for the optimal design of ganetespib-based therapies in the future management of advanced NSCLC.
METHODS
Cell lines, antibodies and reagents
The H2228 and U118-MG cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and the HCC78 and BaF3 cells purchased from the DSMZ (Braunschweig, Germany). All were maintained according to the suppliers’ instructions, authenticated by the routine company DNA typing and were used within 6 months of receipt for this study. H3122 cells were a kind gift from Dr. John Minna from the University of Texas Southwestern Medical Center (Dallas, TX). The NB-39-nu line was obtained from the National Cancer Center Research Institute (Tokyo, Japan) and TPC1 cells were a kind gift from Drs. Hiroshi Sato (Kanazawa University Cancer Research Institute, Japan) and Rebecca Schweppe (University of Colorado, Aurora, CO). No authentication of the lines obtained as gifts were carried out by the authors. All primary antibodies were purchased from Cell Signaling Technology (CST, Beverly, MA) with the exception of the Hsp70 (Enzo Life Sciences, Farmingdale, NY), Claudin (Invitrogen, Grand Island, NY) and GAPDH and Cyclin E antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Ganetespib [3-(2,4-dihydroxy-5-isopropylphenyl)-4-(1-methyl-1H-1,2,4-triazol-5(4H)-one] was synthesized by Synta Pharmaceuticals Corp. Crizotinib, ASP3026, CH5424802 and TAE684 were all purchased from Active Biochem (Maplewood, NJ).
Cell viability assays
Cellular viability was assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Tumor cell lines were seeded into 96-well plates based on optimal growth rates determined empirically for each line. Twenty-four hours after plating, cells were dosed with graded concentrations of ganetespib or crizotinib for 72 h. CellTiter-Glo was added (50% v/v) to the cells, and the plates incubated for 10 min prior to luminescent detection in a SpectraMax Plus 384 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Data were normalized to percent of control and IC50 values were determined using XLFit software.
Western blotting
Following in vitro assays, tumor cells were disrupted in lysis buffer (CST) on ice for 10 min. For the pharmacodynamic analysis, xenograft tumors (average volume of 100-200 mm3) were excised, cut in half, and flash frozen in liquid nitrogen. Each tumor fragment was lysed in 0.5 mL of lysis buffer using a FastPrep-24 homogenizer and Lysing Matrix A (MP Biomedicals, Solon, OH). Lysates were clarified by centrifugation and equal amounts of proteins resolved by SDS-PAGE before transfer to nitrocellulose membranes (Invitrogen, Carlsbad CA). Membranes were blocked with StartingBlock T20 blocking buffer (Thermo Scientific, Cambridge MA) and immunoblotted with the indicated antibodies. Antibody-antigen complexes were visualized using an Odyssey system (LI-COR, Lincoln, NE).
Combinatorial drug effect analysis
For combinatorial analysis, H3122 cells (7.5 × 103) were seeded in 96-well plates and incubated at 37oC, 5% CO2 for 24 h. Drug combinations were applied at a non-constant ratio, using three 1.5 fold serial dilutions above and below the IC50 values for each compound. Cell viability was assessed 72 h after drug addition by Cell Titer-Glo and normalized to vehicle controls. For each combination study, the level of growth inhibition (fraction affected) is plotted relative to vehicle control. Data are presented as one relevant combination point and the corresponding single agent data for each cell line tested.
In vivo xenograft tumor models
Female CD-1 nude and CB.17 (SCID) mice (Charles River Laboratories, Wilmington, MA) at 7-12 weeks of age were maintained in a pathogen-free environment and all in vivo procedures were approved by the Synta Pharmaceuticals Corp. Institutional Animal Care and Use Committee. H3122 NSCLC cells (7.5 × 106) were subcutaneously implanted into the animals. Mice bearing established tumors (~200 mm3) were randomized into treatment groups of 7 and dosed with vehicle, ganetespib (i.v.) or crizotinib (p.o.), both formulated in DRD (10% DMSO, 18% Cremophor RH 40, 3.6% dextrose), using the schedules indicated. Tumor volumes (V) were calculated by caliper measurements of the width (W), length (L) and thickness (T) of each tumor using the formula: V=0.5236(LWT). Tumor growth inhibition was determined as described previously (46). Statistical analyses were performed using a Kruskal-Wallace one-way ANOVA on ranks followed by the Tukey test. For the survival analysis, H3122-implanted SCID mice were randomized into groups of 7 after 12 days, and then dosed with vehicle, 50 mg/kg ganetespib 1X/week, or 50 mg/kg crizotinib 5X/week. Animal survival assessed for 75 days with the endpoints defined as animal death, cavitating tumors or tumors > 1.5 cm. Overall survival is presented using the Kaplan-Meier method.
Selection of NPM-ALK/BaF3 cells for crizotinib resistance and characterization of ganetespib responsiveness of crizotinib-resistant NPM-ALK/BaF3
BaF3 cells engineered to stably express NPM-ALK were used to select for crizotinib-resistance and screening for inhibitor-resistant colonies was performed as previously described (47). No chemical mutagenesis (e.g. with ENU) was used to accelerate the emergence of resistant clones. Clonally-derived, crizotinib-resistant NPM-ALK/BaF3 lines were isolated by limiting dilution, and sequence analysis of the ALK kinase domain was performed to identify the resistance mutations. Cytotoxic IC50 determinations were performed using an XTT Cell Viability Assay Kit (CST) after a 72 h incubation of crizotinib with NPM-ALK/BaF3 clones bearing each of the identified inhibitor-resistant mutations. Further, each putative mutation was confirmed to confer crizotinib resistance by engineering into NPM-ALK cDNA, generating clonal NPM-ALK/BaF3 cell lines to express the mutation, and determining IC50 cytotoxicity values for crizotinib in the clonal lines. Ganetespib cytotoxicity IC50 values for crizotinib-resistant NPM-ALK/BaF3 cells were determined following incubation with graded concentrations of the compound for 72 h. Immunoblotting to assess the degradation of NPM-ALK in response to ganetespib exposure was performed using an ALK 11 rabbit polyclonal anti-serum (48) at a dilution of 1:2000.
Ganetespib trial
This study includes preliminary data of clinical response in one 24 year old male patient enrolled into a Phase II multicenter trial of ganetespib monotherapy in advanced NSCLC (NCT01031225). The patient harbored an EML4 exon 6-ALK translocation and had failed previous crizotinib therapy – subsequent direct ALK exon sequencing identified the presence of a secondary G1269A mutation. The trial was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee at each participating institution; patients were required to provide written informed consent prior to enrollment. The study was sponsored by Synta Pharmaceuticals Corp.
Supplementary Material
Figure S1. Chemical structures of ganetespib and crizotinib.
Figure S2. Ganetespib and crizotinib are similarly efficacious in H3122 xenograft tumors implanted in nude mice. A, Nude mice bearing H3122 xenografts (n=7/group) were i.v. dosed with 150 mg/kg ganetespib once weekly, or p.o. dosed with 200 mg/kg crizotinib 5X/week over a 5 week cycle as indicated. % T/C values are indicated to the right of each growth curve and the error bars are the SEM.
Figure S3. The addition of ganetespib to crizotinib does not result in added toxicity in H3122 xenografts. Mice bearing established H3122 xenografts (n=7/group) were i.v. dosed with 25 mg/kg ganetespib once weekly, 100 mg/kg crizotinib 5x/week p.o., or the combination, as indicated. Body weights were measured 5 times per week. Mean values are plotted against vehicle controls.
Figure S4. Crizotinib-resistant H3122 CR1 cells express an EMT phenotype. A, Fold-changes in protein expression following ganetespib treatment in H3122 and H3122 CR1 cells using reverse phase protein array analysis. B, Whole cell lysates from H3122 and H3122 CR1 cells were immunoblotted with the indicated antibodies. Lysates derived from the NIH-3T3 mouse fibroblast cell line were included as a control for mesenchymal marker expression. C, Confluent cultures of H3122 and H3122 CR1 cells were scraped to generate linear wounds. After wounding, cells were maintained in culture medium and images collected at 0, 8, 24, 48 and 72 h to determine the comparative degrees of cellular migration. D, Effect of ganetespib on migratory capacity of H3122 CR1 cells. H3122 and H3122 CR1 cells were seeded in the wound healing assay. H3122 CR1 cells were also cultured in the presence of increasing concentrations (12.5 and 25 nM) of ganetespib and images collected at 0, 24 and 48 h.
SIGNIFICANCE.
In addition to direct kinase inhibition, pharmacological blockade of the molecular chaperone Hsp90 is emerging as a promising approach for treating tumors driven by oncogenic gene rearrangements of ALK. The bioactivity profile of ganetespib presented here underscores a new therapeutic opportunity to target ALK and overcome multiple mechanisms of resistance in ALK-positive NSCLC patients.
Acknowledgments
Financial Support: All work was funded by Synta Pharmaceuticals Corp.
Abbreviations
- ALK
anaplastic lymphoma kinase
- EGFR
epidermal growth factor receptor
- EML4
echinoderm microtubule-associated protein-like 4
- EMT
epithelial mesenchymal transition
- Hsp90
heat shock protein 90
- MTD
maximally tolerated dose
- NSCLC
non-small cell lung cancer
- TKI
tyrosine kinase inhibitor
Footnotes
Disclosure of Potential Conflicts of Interest: All authors with the exception of Q. Jiang, L. Xue, C. M. Lovly, A. T. Shaw, R. Doebele, D. R. Camidge and S. W. Morris are employees of Synta Pharmaceuticals Corp. There are no other competing interests to declare.
REFERENCES
- 1.Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27:4232–5. doi: 10.1200/JCO.2009.23.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramalingam SS, Owonikoko TK, Khuri FR. Lung cancer: New biological insights and recent therapeutic advances. CA Cancer J Clin. 2011;61:91–112. doi: 10.3322/caac.20102. [DOI] [PubMed] [Google Scholar]
- 3.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 4.Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971–6. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 5.Sasaki T, Janne PA. New strategies for treatment of ALK-rearranged non-small cell lung cancers. Clin Cancer Res. 2011;17:7213–8. doi: 10.1158/1078-0432.CCR-11-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camidge DR, Doebele RC. Treating ALK-positive lung cancer-early successes and future challenges. Nat Rev Clin Oncol. 2012;9:268–77. doi: 10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–12. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ardini E, Galvani A. ALK Inhibitors, a Pharmaceutical Perspective. Front Oncol. 2012;2:17. doi: 10.3389/fonc.2012.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–28. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 13.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 14.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65:6401–8. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- 16.Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270:24585–8. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- 17.da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R. Activated BRAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686–91. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- 18.Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, et al. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–8. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Sasaki T, Tan X, Carretero J, Shimamura T, Li D, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res. 2010;70:9827–36. doi: 10.1158/0008-5472.CAN-10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Normant E, Paez G, West KA, Lim AR, Slocum KL, Tunkey C, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30:2581–6. doi: 10.1038/onc.2010.625. [DOI] [PubMed] [Google Scholar]
- 21.Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–9. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 22.Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009;15:9–14. doi: 10.1158/1078-0432.CCR-08-0132. [DOI] [PubMed] [Google Scholar]
- 23.Ying W, Du Z, Sun L, Foley KP, Proia DA, Blackman RK, et al. Ganetespib, a unique triazolone-containing hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther. 2012;11:475–84. doi: 10.1158/1535-7163.MCT-11-0755. [DOI] [PubMed] [Google Scholar]
- 24.Wong K, Koczywas M, Goldman JW, Paschold EH, Horn L, Blackman RK, et al. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) as monotherapy in patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2011;29(Suppl) Abstr 7500. [Google Scholar]
- 25.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71:4920–31. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–43. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28:4953–60. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 33.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–9. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 34.Lovly CM, Pao W. Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4:120ps2. doi: 10.1126/scitranslmed.3003728. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. 2012;18:6219–26. doi: 10.1158/1078-0432.CCR-12-0392. [DOI] [PubMed] [Google Scholar]
- 37.Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, et al. Paracrine Receptor Activation by Microenvironment Triggers Bypass Survival Signals and ALK Inhibitor Resistance in EML4-ALK Lung Cancer Cells. Clin Cancer Res. 2012;18:3592–602. doi: 10.1158/1078-0432.CCR-11-2972. [DOI] [PubMed] [Google Scholar]
- 38.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 39.Andl CD, Rustgi AK. No one-way street: cross-talk between e-cadherin and receptor tyrosine kinase (RTK) signaling: a mechanism to regulate RTK activity. Cancer Biol Ther. 2005;4:28–31. doi: 10.4161/cbt.4.1.1431. [DOI] [PubMed] [Google Scholar]
- 40.Ou SH. Crizotinib: a novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des Devel Ther. 2011;5:471–85. doi: 10.2147/DDDT.S19045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiromitsu S, Mori M, Shimada I, Kondoh Y, Nobuaki S, Soga T, et al. Antitumor activities of ASP3026 against EML4-ALK-dependent tumor models. Mol Cancer Ther. 2011;10(11, Suppl) abstr A227. [Google Scholar]
- 42.Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–90. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 43.Rimkunas VM, Crosby KE, Li D, Hu Y, Kelly ME, Gu TL, et al. Analysis of Receptor Tyrosine Kinase ROS1-Positive Tumors in Non-Small Cell Lung Cancer: Identification of a FIGROS1 Fusion. Clin Cancer Res. 2012;18:4449–57. doi: 10.1158/1078-0432.CCR-11-3351. [DOI] [PubMed] [Google Scholar]
- 44.Stumpfova M, Janne PA. Zeroing in on ROS1 Rearrangements in Non-Small Cell Lung Cancer. Clin Cancer Res. 2012;18:4222–4. doi: 10.1158/1078-0432.CCR-12-1812. [DOI] [PubMed] [Google Scholar]
- 45.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–7. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Proia DA, Foley KP, Korbut T, Sang J, Smith D, Bates RC, et al. Multifaceted intervention by the Hsp90 inhibitor ganetespib (STA-9090) in cancer cells with activated JAK/STAT signaling. PLoS One. 2011;6:e18552. doi: 10.1371/journal.pone.0018552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.von Bubnoff N, Barwisch S, Speicher MR, Peschel C, Duyster J. A cell-based screening strategy that predicts mutations in oncogenic tyrosine kinases: implications for clinical resistance in targeted cancer treatment. Cell Cycle. 2005;4:400–6. doi: 10.4161/cc.4.3.1560. [DOI] [PubMed] [Google Scholar]
- 48.Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–88. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chemical structures of ganetespib and crizotinib.
Figure S2. Ganetespib and crizotinib are similarly efficacious in H3122 xenograft tumors implanted in nude mice. A, Nude mice bearing H3122 xenografts (n=7/group) were i.v. dosed with 150 mg/kg ganetespib once weekly, or p.o. dosed with 200 mg/kg crizotinib 5X/week over a 5 week cycle as indicated. % T/C values are indicated to the right of each growth curve and the error bars are the SEM.
Figure S3. The addition of ganetespib to crizotinib does not result in added toxicity in H3122 xenografts. Mice bearing established H3122 xenografts (n=7/group) were i.v. dosed with 25 mg/kg ganetespib once weekly, 100 mg/kg crizotinib 5x/week p.o., or the combination, as indicated. Body weights were measured 5 times per week. Mean values are plotted against vehicle controls.
Figure S4. Crizotinib-resistant H3122 CR1 cells express an EMT phenotype. A, Fold-changes in protein expression following ganetespib treatment in H3122 and H3122 CR1 cells using reverse phase protein array analysis. B, Whole cell lysates from H3122 and H3122 CR1 cells were immunoblotted with the indicated antibodies. Lysates derived from the NIH-3T3 mouse fibroblast cell line were included as a control for mesenchymal marker expression. C, Confluent cultures of H3122 and H3122 CR1 cells were scraped to generate linear wounds. After wounding, cells were maintained in culture medium and images collected at 0, 8, 24, 48 and 72 h to determine the comparative degrees of cellular migration. D, Effect of ganetespib on migratory capacity of H3122 CR1 cells. H3122 and H3122 CR1 cells were seeded in the wound healing assay. H3122 CR1 cells were also cultured in the presence of increasing concentrations (12.5 and 25 nM) of ganetespib and images collected at 0, 24 and 48 h.