

Abstract
The Notch signaling pathways are known to play critical roles during pancreatic development, but it remains unclear what functions are important in the adult organ. One area of debate is the role of Notch signaling in the development of pancreatic ductal adenocarcinoma (PDAC) and proposed precursor lesions, pancreatic intraepithelial neoplasia (PanIN). Initial studies revealed that Notch signaling is reactivated during PDAC initiation and development, suggesting Notch promotes PDAC and may therefore represent a target for drug development. However, more recent work reveals a tumor suppressive role for Notch receptors in the context of PanIN development. Here, we summarize the current literature describing Notch signaling in the development of PDAC, and discuss the potential of the Notch pathway as a therapeutic target.
Keywords: pancreatic ductal adenocarcinoma, Notch, PanIN, gamma-secretase inhibitor, Kras
The development of pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal types of cancer – the number of patients diagnosed with the disease annually is nearly equal to the number of mortalities [1]. PDAC is believed to evolve through the progression of non-invasive precursor lesions, including pancreatic intraepithelial neoplasia (PanIN), which are classified into four stages of increasing nuclear and architectural abnormalities: PanIN-1A, -1B, -2, and -3 [2]. Similar to other epithelial cancers, the progression of PanIN lesions coincides with specific genetic events [3]. The most commonly mutated oncogene in PDAC is KRAS; activating mutations are found in greater than 95% of PDAC cases [4]. KRAS mutations are also found in PanIN lesions, with approximately 36% of PanIN-1A lesions and nearly 87% of PanIN-2 and -3 lesions harboring a KRAS mutation [2]. The near universal presence of KRAS mutations in PDAC and their high prevalence in PanIN lesions suggests activation of KRAS is both an initiating event and necessary for the development of PDAC, making this an attractive candidate for a targeted therapy. However, efforts to develop inhibitors directly targeting Kras to date have failed to result in clinically effective treatment options [5]. Thus, focus has shifted from directly targeting KRAS to identifying and targeting pathways that KRAS relies upon to effect transformation.
One such candidate is the Notch signaling pathway. Notch signaling has previously been shown to be required for Ras-induced transformation of fibroblasts [6] and Hras driven tumorigenesis in a mouse mammary tumor model [7]. Furthermore, the Notch signaling pathway appears to be activated in human pancreatic cancer, given that the expression of receptors, ligands, and downstream targets are induced compared to normal epithelial tissue [8]. However, recent studies utilizing mouse models have revealed both oncogenic and tumor suppressive roles for Notch signaling in PDAC development. These conflicting results necessitate further investigation into the role of Notch and strategies for Notch inhibition to treat PDAC.
Mouse models of PDAC
Proof that mutations in KRAS are a critical event in PDAC development came from mouse models in which expression of oncogenic Kras (KrasG12D) at endogenous levels was targeted to pancreatic epithelium using the pancreatic-specific promoters Pdx-1 or p48/Ptf1a [9]. Pdx-1;Cre;LSL-KrasG12D and p48+/Cre;LSL-KrasG12D mice developed the full spectrum of PanIN lesions with complete penetrance. These mouse models demonstrated that physiological levels of KrasG12D are sufficient to drive PanIN development and are excellent model systems for investigating the molecular events that occur in the human disease.
Although termed “ductal adenocarcinoma”, the cell of origin for PDAC remains elusive. The Pdx1-Cre;LSL-KrasG12D model is a valuable tool to study PanIN progression, but it renders the identification of the cell of origin for PDAC challenging because all pancreatic epithelial cells undergo Kras activation, including acinar, ductal, and centroacinar cells, as well as the cells comprising islets. Both PanIN lesions and PDAC express markers of ductal differentiation and morphologically resemble ductal cells, initially leading researches to believe PDAC initiates in ductal cells. However, targeted expression of oncogenic Kras under control of the ductal-specific cytokeratin 19 promoter failed to produce PanIN lesions in a mouse model [10]. More recent studies confirm that ductal cells expressing KrasG12D are largely refractory to PanIN development [11].
In contrast, mouse models have demonstrated that targeting oncogenic Kras expression to acinar and centroacinar cells[LG1][JE2] of the exocrine compartment in the pancreas, as well as endocrine lineages, results in the development of PanIN lesions. The first study to demonstrate that the exocrine lineage gives rise to PanIN lesions utilized Nestin-Cre transgenic mice [12]. Nestin is expressed later than Pdx-1 during pancreatic development, and marks exocrine progenitor cells. Targeting oncogenic Kras to a Nestin-positive cell lineage resulted in the development of PanIN lesions at the same frequency as in Pdx1-Cre;LSL-KrasG12D mice. Additional mouse models have identified the cell types within the exocrine compartment that are susceptible to oncogenic Kras-induced PanIN formation. Embryonic expression of an oncogenic allele of Kras (KrasG12V) in acinar and centroacinar cells leads to the development of PanIN lesions and PDAC [13], although mature acinar and centroacinar cells in adult animals were refractory to Kras-driven PanIN development unless the mice were subjected to chronic chemically-induced pancreatitis. Studies employing a second-generation mouse model found that targeting the expression of a different Kras mutation (KrasG12D) to adult acinar cells did result in the spontaneous induction of PanIN lesions [14]. Overall, both studies conclude that acinar cells expressing oncogenic Kras give rise to PanINs, suggesting that acinar cells are a potential cell of origin for PDAC development.
The development of lesions from adult acinar cells expressing KrasG12D highlights the plastic nature of mature pancreatic acinar cells and suggest that acinar-to-ductal metaplasia, the replacement of acinar tissue with ductal lesions, may be a precursor to the development of PanIN lesions. This process has been proposed to occur through a variety of mechanisms, including selective proliferation of ductal cells, differentiation of a stem cell population, or transdifferentiation of acinar cells to ductal cells [15]. In vitro, acinar cells can transdifferentiate to a ductal phenotype [16], and recent studies have shown that oncogenic Kras expression is sufficient to induce transdifferentiation in vitro even in the absence of exogenous growth factors [17, 18].
Acinar-to-ductal transdifferentiation has also been documented to occur in vivo using lineage tracing models [19] when metaplasia is induced by chronic pancreatitis. However, acinar-to-ductal transdifferentiation only accounts for a minority of metaplastic lesions, approximately 12% [19], and therefore it is possible that either expansion of ductal cells or transdifferentiation of centroacinar cell populations accounts for increased metaplastic ductal lesions. To this point, a mouse model relying on pancreatic-specific Pten deletion provides evidence that the expansion of centroacinar cells accounts for ductal metaplasia [20]. Observations of human tumor samples suggest that acinar-to-ductal metaplasia does occur [21], but it remains unknown if these lesions progress to PanIN development. Recently, atypical flat lesions (AFLs) arising in areas of ADM in Ptf1a-Cre;KrasG12D mice have been proposed as direct PDAC precursors and as an alternative to the PanIN-PDAC progression model [22]. More work is needed to identify ALFs in human tumors and determine their relationship to PanINs. In addition to acinar and centroacinar cells, endocrine cells are also susceptible to oncogenic Kras-induced transformation in the context of pancreatic injury [23].
These mouse models bring to light two important aspects of PDAC development. First, oncogenic Kras appears to be crucial for the deregulation of pancreatic differentiation. Multiple pancreatic cell lineages, including both exocrine and endocrine cells, can give rise to PanIN lesions when expressing oncogenic Kras. Second, pancreatitis provides a permissive environment for the development of PDAC and may be required in the absence of additional genetic lesions, such as mutations in tumor suppressor genes. This conclusion is especially relevant to human disease because chronic pancreatitis is a strong risk factor for the development of PDAC [24]. Many questions remain concerning how changes induced by pancreatitis affect the different cell lineages and the cellular environment to promote PanIN development, but recent work examining the effects of pancreatitis on acinar cells has revealed potential mechanisms. In the context of caerulein-induced pancreatitis, acinar cells decrease the expression of acinar markers and assume a genetic program resembling embryonic pancreatic precursors [25]. In wild-type mice, the acinar compartment is rapidly repopulated following the cessation of caerulein pancreatitis. However, acinar cells expressing oncogenic Kras fail to regenerate and remain in a persistent state of dedifferentiation, ultimately resulting in PanIN development. The mechanism responsible for blocked regeneration appears to be inhibition of β-catenin signaling [26]. Further work is needed to establish how oncogenic Kras and pancreatitis synergize to promote PanIN development in the endocrine compartment.
Notch signaling in the pancreas
The Notch signaling pathway is a highly evolutionarily conserved pathway that mediates cell-to-cell communication (Figure 1). Among the diverse functions regulated by Notch signaling, some of the most well-documented include the maintenance of stem cell populations, determination of cell fate decisions, and the regulation of proliferation and apoptosis. Transcriptional targets of Notch signaling appear to be context-dependent. Two of the best characterized targets are basic helix-loop-helix transcriptional repressors, the hairy enhancer of split (HES) and hairy-related transcription factor (HEY) families.
Figure 1. Gamma-secretase activity is required during activation of the Notch signaling pathway.

In mammals, the main components of the Notch pathway include the 4 transmembrane Notch receptors (Notch1–4) and their cognate ligands, Delta-like (DLL) 1, -3, and -4, and Jagged1 and -2. Notch receptors expressed on the cell surface bind to the Delta and Jagged family of ligands on an adjacent cell, after which Notch undergoes a series of proteolytic cleavages. The first cleavage is mediated by either tumor-necrosis factor-α-converting enzyme (TACE) or ADAM10, followed by a second cleavage mediated by the gamma-secretase complex. This final cleavage releases the Notch intracellular domain (NICD) from the cell membrane, allowing this domain to translocate to the nucleus. Once in the nucleus, NICD binds to the transcription factor CSL, displacing corepressors and recruiting transcriptional activators, including the coactivator Mastermind-like1 (Maml). Gamma-secretase inhibitors (GSIs) block the cleavage of Notch receptors by the gamma-secretase complex, inhibiting the release of NICD from the cell membrane.
Notch signaling regulates multiple cell fate decisions during pancreatic development, but the main function appears to be maintaining a pool of undifferentiated, progenitor-like cells. Deletion of the genes encoding the Notch pathway ligand Dll1 or the DNA-binding protein RBJ-Jκ leads to an accelerated differentiation of pancreatic progenitor cells into endocrine cells [27]. Hes1 knockout mice exhibit a similar phenotype, revealing that the affects of Notch signaling are potentially mediated through this target [28]. As expected, activation of the Notch pathway during development has the converse effect: both exocrine and endocrine differentiation programs are arrested and cells remain in a progenitor-like state [29].
At later stages of pancreatic development, Notch signaling appears to regulate exocrine cell differentiation in a complex manner. Deletion of Rbpj in pancreatic progenitors at embryonic day 10.5 causes a lack of acinar tissue and the appearance of large duct-like structures [30]. Similar results are seen in Jag1-deficient mice, which display malformed pancreatic ducts and acinar cell death [31]. Surprisingly, pancreatic-specific deletion of the Notch1 and Notch2 alleles does not lead to gross abnormalities in development, suggesting a Notch-independent function of Rbpj [30, 32, 33]. Alternatively, activation of Notch3 or Notch4 may compensate for loss of Notch1 and Notch2. Although the previous studies indicate that Notch signaling promotes acinar differentiation, other studies lead to opposing conclusions. Esni et al. have demonstrated that ectopic Notch activation in explant cultures inhibits acinar differentiation [34]. Furthermore, experiments in zebrafish demonstrate that the loss of Notch signaling accelerates acinar differentiation [34]. The discrepancies in these studies may result from loss-of-function versus gain-of-function approaches as well as differences in the model systems. More recently, Notch signaling was shown to initiate ductal cell differentiation over endocrine specification depending on pathway activation thresholds [35]. Clearly, more work is needed to thoroughly understand how Notch signaling regulates exocrine cell differentiation.
Unlike pancreatic development, very little is known about the role of Notch signaling in the adult organ (Figure 2). Most studies suggest that the expression and activation of Notch receptors are downregulated in the adult organ under normal physiological conditions [8, 25]. An exception to this may be terminal duct and centroacinar cells, which express Hes1 [20, 36]. Recent work indicates that Notch signaling in Hes1+ centroacinar cells functions to suppress acinar cell differentiation. Upon deletion of Rbpj in Hes1+ cells, centroacinar cells rapidly transform to acinar cells [36]. However, other studies reveal a distinct function for Notch signaling in the adult pancreas following injury. In response to chemically-induced pancreatitis, the majority of exocrine tissue is lost and the surviving acinar cells induce genes associated with a progenitor-like phenotype, among them Notch1, Notch2, Jagged2, and Hes1 [25]. Deletion of Notch1 in the pancreatic epithelium impairs acinar regeneration following acute pancreatitis, indicating that Notch signaling plays a role in pancreatic homeostasis [32]. Finally, emerging evidence indicates the different Notch receptors may have non-overlapping functions and are expressed in unique cellular compartments of the pancreas [37]. Using transgenic Notch1-GFP and a Notch2lacZ knockin reporter mice it has been shown that Notch1 expression is observed primarily in acinar cells and Notch2 expression is localized to ductal cells. Interestingly, these results contrast with a previous study showing that Notch signaling is restricted to centroacinar cells [38]. The discrepancies described above highlight the need to directly determine where individual Notch receptors are expressed in the adult pancreas. Future studies await the development of antibodies sensitive enough to detect the endogenous proteins in tissue sections.
Figure 2. Notch receptors are expressed in in the mature exocrine pancreas.
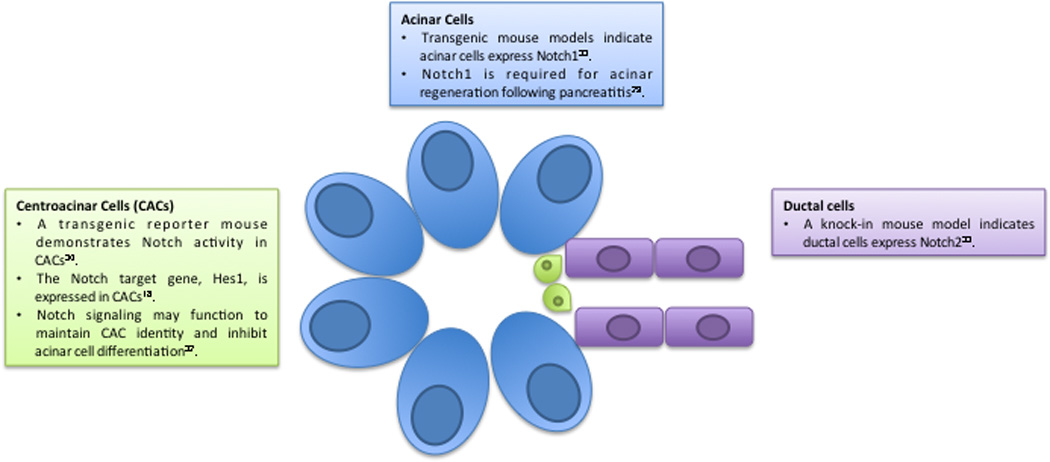
The mature pancreas is composed of exocrine and endocrine compartments, with the exocrine compartment consisting of acinar, ductal, and centroacinar cells. Notch receptors are known to play critical roles during pancreatic development; however their role and expression patterns in the mature pancreas remain to be fully defined. Genetically engineered mouse models as well as immunohistochemical analysis indicate Notch receptors are expressed and activated in mature exocrine cells.
Notch and cancer
The Notch signaling pathway exerts both oncogenic and tumor suppressive functions, depending on the cellular context. Notch receptors have been identified as oncogenes in multiple tumors, including leukaemia, breast, colorectal, cervical, lung, and oral squamous cell carcinoma (reviewed in [39]). Although few activating mutations in Notch receptors have been identified in solid human tumors [40], high levels of NOTCH1 and JAG1 expression correlate with poor patient prognosis in lung and breast cancer [41, 42]. In human samples, Notch pathway components are highly expressed in pancreatic adenocarcinoma [8], renal cell carcinoma [43] and prostate cancer [44] compared with control tissue. Furthermore, expression of NUMB, a negative regulator of Notch activity, is frequently lost in breast and non-small cell lung cancer [45]. These data imply that Notch pathway activation is induced in a variety of solid human malignancies by mechanisms other than activating mutations.
The finding that Notch signaling is activated in multiple human tumors has led to interest in therapeutically targeting these pathways. The most widely used method to globally inhibit Notch signaling is the use of gamma-secretase inhibitors (GSIs), which block the cleavage of Notch at the cell membrane, inhibiting release of the transcriptionally active NICD subunit. Recently, this approach has proven effective in inhibiting lung tumor progression in a mouse model [46]. Although GSIs are currently being tested in clinical trials for multiple types of cancer, previous clinical studies of treatments for Alzheimer’s disease reveal the compounds have significant toxicity, including gastrointestinal bleeding. Given that Notch signaling is required to maintain undifferentiated cells in intestinal crypts [47], it has been suggested that on-target effects, that is, the global Notch inhibition by GSIs, cause this toxicity. A second caveat to the use of GSIs is that the gamma-secretase complex is responsible for catalyzing the proteolysis of over 100 additional substrates in addition to Notch receptors [48], making it difficult to attribute anti-tumor effects exclusively to a blockade in Notch signaling. Finally, Notch receptors also have known tumor suppressor functions, discussed below, that might be impaired by GSI treatment.
In response to the above issues, monoclonal antibodies capable of selectively inhibiting specific Notch receptors have been developed. Antibodies that specifically antagonize Notch1 or Notch2 inhibit tumor growth in vivo by decreasing cell proliferation and increasing apoptosis [49]. Furthermore, treatment with either the anti-Notch1 or anti-Notch2 antibody alone does not result in severe intestinal toxicity. Based on their efficacy and limited toxicity, Notch antibodies are currently being developed as treatments for multiple cancers, including T-ALL and solid tumors [50].
Although Notch was originally identified as an oncogene, recent studies have also demonstrated tumor suppressive effects for Notch receptors, illustrating the highly context dependent nature of the pathway. The first conclusive evidence showing Notch1 acts as a tumor suppressor came from studies in skin, where the loss of both Notch1 alleles leads to the development of spontaneous basal cell carcinomas in mice [51]. Although initial studies indicated that Notch1 functions as a tumor suppressor in a cell-autonomous manner, more recent work has highlighted a non-cell-autonomous mechanism [52].
Notch1 may function as a tumor suppressor in human skin cancers as well. Multiple components of the Notch signaling pathway, including NOTCH1, NOTCH2, and JAGGED1, show reduced expression in human basal cell carcinoma samples [53]. Further evidence supporting a role for NOTCH in human skin cancers came from the results of clinical trials studying semagacestat, a GSI, as a treatment for Alzheimer’s disease. The trial revealed that patients taking the GSI had an increased risk of developing skin cancer [54].
In addition to non-melanoma skin cancers, Notch has been implicated as a tumor suppressor in prostate cancer, hepatocellular carcinoma, and small cell lung cancer [55], and loss-of-function mutations have been identified in human chronic myelomonocytic leukemia (CMML) and squamous cell carcinoma samples [56–58]. Somatic inactivating mutations in NICASTRIN and APH1, components of the gamma secretase complex, as well as mutations in NOTCH2 and MAML1 were identified in a panel of human CMML samples [56]. Furthermore, mouse models of CMML show that loss of Notch signaling alters hematopoietic stem cell differentiation, resulting in the accumulation of monocyte progenitors and a CMML-like disease. A similar study analyzing human head and neck squamous cell carcinoma (HNSCC) tumors identified inactivating mutations in NOTCH1 in 15% of patients, with the majority of the mutations occurring in the same region of the protein, N-terminal to the transmembrane domain. Importantly, 9 of the 21 samples with NOTCH1 mutations in this study possess inactivating mutations in both alleles, supporting the notion that NOTCH1 acts as a classical tumor suppressor [57]. Finally, loss-of-function mutations in NOTCH1 and NOTCH2 are also seen in human cutaneous squamous cell carcinoma and lung squamous cell carcinoma [58]. Interestingly, the majority of these mutations are heterozygous, implying NOTCH may act as a haploinsufficient tumor suppressor.
Notch signaling and pancreatic cancer
The role of Notch signaling in pancreatic cancer remains unresolved – evidence supporting both oncogenic and tumor suppressive functions exists (Figure 3). In support of an oncogenic role for Notch signaling in PDAC, multiple pathway components are upregulated in pancreatic cancer samples compared to normal pancreatic epithelium, including Notch receptors, ligands, and targets [8]. Furthermore, analysis of the Pdx1-Cre;LSL-KrasG12D mouse model revealed increased expression of the Notch target Hes1 in PanIN lesions compared to normal ducts [9]. Although these results do not establish a causative role for Notch signaling, they do indicate that the pathway is inappropriately activated during PDAC development. However, a more recent global genomic analysis of the core signaling pathways activated in a panel of human pancreatic cancer samples revealed no evidence for overexpression of Notch receptors or classical target genes [59].
Figure 3. Mouse models reveal dual roles for Notch receptors.

Mouse models reveal that Notch receptors either promote or inhibit PanIN development depending on context. Models differ on the Notch receptor being targeted, timing of genetic events, and the cell types targeted. Additional studies employing gamma secretase inhibitors (GSIs) reveal Notch receptors function to inhibit PanIN advancement early during disease progression and appear to play a minimal role in advanced PDAC stages.
Studies relying on mouse models have also revealed both oncogenic and tumor suppressive functions for the Notch receptors. Recently, work from our group demonstrated that Notch1 suppresses PanIN formation in a mouse model of PDAC (Pdx1-Cre;LSL-KrasG12D;Notch1lox/lox) [33], a result supported by findings showing that deletion of both Notch1 alleles in ptf1a+/Cre;LSL-KrasG12D mice caused a slight decrease in median survival [37]. By contrast, other groups have identified an oncogenic role for Notch1:, coactivation of KrasG12D and Notch1-ICD in mature acinar cells led to significantly higher numbers of PanIN lesions compared to activation of KrasG12D alone [60]. However, expression of Notch1-ICD, in the absence of oncogenic Kras, fails to alter acinar cell differentiation or induce PanIN lesions, suggesting that activation of Notch1 alone is not sufficient to drive tumorigenesis. Several differences exist between these models that may account for the conflicting conclusions, including the timing of Cre-mediated recombination and overexpression of the activated form of Notch1 to supraphysiological levels. This possibility was recently illustrated in mammary epithelial cultures, where varying levels of NICD expression caused distinct responses: high NICD levels inhibited proliferation, whereas low Notch activity caused a hyperproliferative response [61], illustrating the crucial nature of Notch levels in regulating downstream effector pathways.
The role of Notch signaling in PanIN development has also been investigated using GSIs [38]. Pdx1-Cre;LSL-KrasG12D;p53lox/+ mice treated with a GSI are refractory to PDAC development, supporting an oncogenic role for Notch signaling. Although this study proposes Notch signaling is required for PanIN progression, more work is needed to identify which Notch receptor is responsible for inhibiting tumor progression and if other GSI targets are involved. Dissecting which Notch receptors are involved in PDAC is especially relevant given recent work revealing that individual receptors have opposing roles in pancreatic cancer development. Whereas deletion of both Notch1 alleles in mice expressing oncogenic Kras accelerates PanIN progression and causes a slight decrease in median survival, deleting Notch2 prolongs survival and delays PDAC development, shifting the spectrum of lesions towards development of mucinous cystic-like neoplasms [33, 37]. The opposing outcomes observed upon Notch1 or Notch2 ablation may be explained by unique downstream targets or differential expression patterns of the receptors. Clearly, the above results indicate the different Notch receptors possess discrete functions in PDAC development, warranting caution in using a global Notch inhibition approach for treatment purposes. An additional explanation for the differences between the studies is that Notch signaling possesses opposing functions during PanIN initiation and progression. In the Pdx1-Cre;LSL-KrasG12D;Notch1lox/lox model, Notch is inactivated concurrently with activation of K-rasG12D at the point of PanIN initiation, whereas in the Pdx1-Cre;LSL-KrasG12D;p53lox/+ mice the GSI treatment is initiated after PanIN lesions have been established. Therefore, the possibility cannot be excluded that Notch signaling may function to inhibit PanIN development early but act to promote PanIN progression at later stages once lesions are established. A more recent study examining the role of Notch signaling in radiologically evident pancreatic tumors demonstrated that GSI treatment failed to extend lifespan [62], implying that Notch signaling may not be involved in the maintenance of advanced pancreatic tumors. Hence, more studies are needed to define the effects of GSI treatment on PanIN initiation and progression before these drugs should be considered for clinical use.
A final area of debate is the role of Notch signaling in acinar-to-ductal metaplasia (ADM). Notch signaling has been implicated in ADM given that ectopic expression of NICD promotes transdifferentiation in explant culture models [8, 63]. Notch is activated by epidermal growth factor receptor (EGFR) activation, and is required for growth factor induced ADM [8]. However, this does not appear to be the case in vivo because expression of Notch1-ICD alone has no affect on acinar cell differentiation. Further, the Notch pathway is consistently upregulated and activated in the absence of EGFR. [60, 64]. This result is support by work showing that deletion of Notch1 has no effect on ADM in vitro [18]. One difference between these studies is the status of Kras: the Miyamoto model induces ADM using ectopic EGFR activation in the context of wildytpe Kras, while subsequent studies utilize KrasG12D expression. These results suggest that activation of Kras and downstream signaling pathways are capable of overriding a requirement for Notch signaling. Furthermore, in Pdx1-Cre;LSL-KrasG12D;p53lox/+ mice, treatment with a GSI does not suppress the abundance of metaplastic ducts, despite decreasing the prevalence of PanINs. Finally, in a slightly different in vitro model of ADM using isolated rat acinar cells, inhibition of Notch signaling by a GSI increased the proliferation of metaplastic exocrine cells in a Hes1-independent manner [65]. Finally, Recent studies have demonstrated that the ductal fate determinant Sox9 is a key regulator of oncogenic Kras induced acinar reprogramming [11]. As Notch signaling controls Sox9 expression, this may represent a critical effector of Notch-mediated ADM.
Overall, additional studies are needed to clarify the role of the various Notch receptors in the development of PDAC. As is the case in the pancreas, there is evidence from other organs indicating that Notch receptors have distinct functions that are cell-type and context dependent. In the skin, chimeric deletion of Notch1 leads to the spontaneous development of epidermal tumors, whereas deletion of either Notch2 or Notch3 has no phenotypic effect [52]. Recent studies analyzing Notch transcriptional complexes have revealed differential responses for Notch1 and Notch2 depending on promoter architecture, further supporting distinct roles for each receptor [66].
Finally, there is a need to carefully evaluate the assays currently used to assess Notch activity and develop readouts specific to the different receptors. For example, Hes1 is commonly used as a surrogate for activation of the signaling pathway, but Hes1 expression does not necessarily correlate with Notch activation and does not distinguish between Notch receptors. Indeed, the inhibition of Notch signaling in the pancreas does not lead to decreased Hes1 expression [33, 37, 65], and Hes1 expression fails to recapitulate the effects of activated Notch expression [8, 38]. Finally, Hes1 can be activated in a Notch-independent manner [67]. Hence, it will be beneficial to identify all downstream effectors of Notch signaling specific to the pancreas in order to accurately assess pathway activation.
Several lines of evidence indicate a functional interaction between Notch and Ras in development and cancer. Notch and Ras have been shown to cooperate or antagonize one another depending on cellular context [68]. Notch signaling is required for Ras-induced transformation of fibroblasts and for tumor formation in a mouse mammary tumor model [6, 7]. In contrast, Notch1 deletion is required for primary keratinocytes expressing oncogenic Hras to form tumors when injected into nude mice [51]. Given that Kras and Notch signaling are deregulated in PDAC, it is imperative to determine if functional interactions exist between these two critical signaling pathways.
Concluding remarks
There is now considerable interest in the use of GSIs to treat pancreatic cancer and other malignancies. However, conflicting results on the function of Notch signaling in PDAC warrant further investigation into the long-term effects of this class of compounds. Preclinical animal models testing GSI treatment for pancreatic cancer have demonstrated mixed results, showing that early treatment with GSIs inhibits PanIN development [38], whereas treatment of advanced tumors fails to prolong survival [62]. Given that most patients are initially diagnosed at an advanced stage of the disease, it is possible that treatment with GSIs alone will fail to significantly improve patient outcomes.
A more significant concern is that the use of GSIs may increase the risk of developing PanIN lesions and, ultimately, PDAC. This concern is strengthened by recent phase III clinical trials testing GSI treatment for Alzheimer’s, the results of which showed that patients receiving the drug had a higher incidence of developing skin carcinomas, presumably due to inhibition of Notch signaling [54]. Thus, it is conceivable that GSI treatment may increase the risk for developing PanIN lesions. One population specifically at risk would be patients suffering from pancreatitis, a common risk factor for the development of PDAC. Therefore, it is imperative to fully understand the function of Notch signaling pathways during PanIN development to better inform treatment decisions with GSIs.
Table 1.
Mouse models reveal Notch receptors are implicated in PanIN and PDAC development.
| Receptor | Mouse model | Role in PanIN/PDAC development |
Reference |
|---|---|---|---|
| Notch1 | Pdx1CreERT2;KrasG12D;Rosa26NIC | Notch1-ICD expression accelerates PanIN development | 57 |
| Pdx1-Cre;KrasG12D;Notch1lox/lox | Notch1 deletion accelerates PanIN development | 30 | |
| Ptf1a-Cre;KrasG12D;Notch1lox/lox | Notch1 deletion decreases median survival | 33 | |
| Notch2 | Ptf1a-Cre;KrasG12D;Notch2lox/lox | Notch2 deletion inhibits PanIN development and increases median survival | 33 |
Outstanding Questions Box.
What is the cell of origin for PanIN lesions and ultimately PDAC?
During the development and progression of PanIN the pancreas undergoes a shift from a predominantly acinar epithelium to one composed mainly of ductal structures, but despite the term “ductal adenocarcinoma”, the cell of origin for PDAC remains elusive. Mouse models have demonstrated that targeting oncogenic Kras to both acinar and centroacinar cells, as well as endocrine lineages, can lead to development of PanIN lesions, but more work is needed to identify the cell of origin in the human disease.
Are individual Notch receptors expressed in specific compartments in the adult pancreas?
The location of Notch receptor expression in the adult quiescent pancreas remains undefined. Most studies agree Notch receptor expression in the adult pancreas is reduced to nearly undetectable levels, but recent studies propose that individual Notch receptors are expressed in specific compartments of the adult pancreas. Using transgenic reporter mice, Notch1 appears to be localized mainly to acinar tissue, whereas Notch2 is present in ductal and centroacinar cells [37]. However, these results conflict with a previous study demonstrating Notch signaling is restricted to centroacinar cells [38]. These discrepancies highlight the need to further elucidate where individual Notch receptors are expressed in the adult pancreas.
What are the downstream mediators of Notch signaling in the pancreas?
Notch signaling exerts specific functions depending on the tissue type and developmental stage. The classical mediators of Notch signaling, the Hes and Hey family of transcription factors, are commonly used as surrogates for pathway activation, yet multiple other targets exist. To accurately assess Notch pathway activation during PanIN development and progression, it will be crucial to identify all downstream effectors, as well as receptor-specific targets.
What effect, if any, will GSI treatment have on PDAC patients?
Recent studies show that individual Notch receptors have opposing roles in the context of pancreatic cancer and normal development. Therefore, global inhibition of the signaling pathway by GSIs may have either beneficial or detrimental effects on PDAC progression. Clearly, more studies are needed to clarify the roles of individual Notch receptors in PDAC progression prior to employing GSIs as a therapeutic approach.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ottenhof NA, et al. Molecular characteristics of pancreatic ductal adenocarcinoma. Patholog Res Int. 2011;2011:620601. doi: 10.4061/2011/620601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 4.Almoguera C, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 5.Vakiani E, Solit DB. KRAS and BRAF: drug targets and predictive biomarkers. J Pathol. 2011;223:219–229. doi: 10.1002/path.2796. [DOI] [PubMed] [Google Scholar]
- 6.Weijzen S, et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 2002;8:979–986. doi: 10.1038/nm754. [DOI] [PubMed] [Google Scholar]
- 7.Kiaris H, et al. Modulation of notch signaling elicits signature tumors and inhibits hras1-induced oncogenesis in the mouse mammary epithelium. Am J Pathol. 2004;165:695–705. doi: 10.1016/S0002-9440(10)63333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyamoto Y, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 9.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 10.Brembeck FH, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 11.Kopp JL, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carriere C, et al. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2007;104:4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerra C, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Habbe N, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tosh D, Slack JM. How cells change their phenotype. Nat Rev Mol Cell Biol. 2002;3:187–194. doi: 10.1038/nrm761. [DOI] [PubMed] [Google Scholar]
- 16.Means AL, et al. Pancreatic epithelial plasticity mediated by acinar cell trans-differentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 17.Scotti ML, et al. Protein kinase C iota regulates pancreatic acinar-to-ductal metaplasia. PLoS One. 2012;7:e30509. doi: 10.1371/journal.pone.0030509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avila JL, et al. Notch1 is not required for acinar-to-ductal metaplasia in a model of Kras-induced pancreatic ductal adenocarcinoma. PLoS One. 2012;7:e52133. doi: 10.1371/journal.pone.0052133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strobel O, et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133:1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stanger BZ, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 21.Parsa I, et al. Ductal metaplasia of human exocrine pancreas and its association with carcinoma. Cancer Res. 1985;45:1285–1290. [PubMed] [Google Scholar]
- 22.Aichler M, et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: a comparative study in transgenic mice and human tissues. J Pathol. 2012;226:723–734. doi: 10.1002/path.3017. [DOI] [PubMed] [Google Scholar]
- 23.Gidekel Friedlander SY, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris JPt, et al. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen JN, et al. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Morris JPt, et al. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Apelqvist A, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 28.Jensen J, et al. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 29.Murtaugh LC, et al. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakhai H, et al. Conditional ablation of Notch signaling in pancreatic development. Development. 2008;135:2757–2765. doi: 10.1242/dev.013722. [DOI] [PubMed] [Google Scholar]
- 31.Golson ML, et al. Ductal malformation and pancreatitis in mice caused by conditional Jag1 deletion. Gastroenterology. 2009;136:1761–1771. doi: 10.1053/j.gastro.2009.01.040. e1761. [DOI] [PubMed] [Google Scholar]
- 32.Siveke JT, et al. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology. 2008;134:544–555. doi: 10.1053/j.gastro.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Hanlon L, et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010;70:4280–4286. doi: 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esni F, et al. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131:4213–4224. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- 35.Shih HP, et al. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopinke D, et al. Ongoing Notch signaling maintains phenotypic fidelity in the adult exocrine pancreas. Dev Biol. 2012;362:57–64. doi: 10.1016/j.ydbio.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazur PK, et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2010;107:13438–13443. doi: 10.1073/pnas.1002423107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plentz R, et al. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2009;136:1741–1749. doi: 10.1053/j.gastro.2009.01.008. e1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranganathan P, et al. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 40.Lobry C, et al. Oncogenic and tumor suppressor functions of Notch in cancer: it's NOTCH what you think. J Exp Med. 2011;208:1931–1935. doi: 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donnem T, et al. Prognostic impact of Notch ligands and receptors in nonsmall cell lung cancer: coexpression of Notch-1 and vascular endothelial growth factor-A predicts poor survival. Cancer. 2010;116:5676–5685. doi: 10.1002/cncr.25551. [DOI] [PubMed] [Google Scholar]
- 42.Reedijk M, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 43.Sjolund J, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest. 2008;118:217–228. doi: 10.1172/JCI32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santagata S, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004;64:6854–6857. doi: 10.1158/0008-5472.CAN-04-2500. [DOI] [PubMed] [Google Scholar]
- 45.Pece S, et al. NUMB-ing down cancer by more than just a NOTCH. Biochim Biophys Acta. 2011;1815:26–43. doi: 10.1016/j.bbcan.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Maraver A, et al. Therapeutic effect of gamma-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell. 2012;22:222–234. doi: 10.1016/j.ccr.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 48.Groth C, et al. Pharmacological analysis of Drosophila melanogaster gamma-secretase with respect to differential proteolysis of Notch and APP. Mol Pharmacol. 2010;77:567–574. doi: 10.1124/mol.109.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 50.Groth C, Fortini ME. Therapeutic approaches to modulating Notch signaling: current challenges and future prospects. Semin Cell Dev Biol. 2012;23:465–472. doi: 10.1016/j.semcdb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicolas M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 52.Demehri S, et al. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thelu J, et al. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002;2:7. doi: 10.1186/1471-5945-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Extance A. Alzheimer's failure raises questions about disease-modifying strategies. Nat Rev Drug Discov. 2010;9:749–751. doi: 10.1038/nrd3288. [DOI] [PubMed] [Google Scholar]
- 55.Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci. 2007;64:2746–2762. doi: 10.1007/s00018-007-7164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klinakis A, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473:230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agrawal N, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang NJ, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108:17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De La OJ, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mazzone M, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A. 2010;107:5012–5017. doi: 10.1073/pnas.1000896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cook N, et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209:437–444. doi: 10.1084/jem.20111923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sawey ET, et al. Matrix metalloproteinase 7 controls pancreatic acinar cell trans-differentiation by activating the Notch signaling pathway. Proc Natl Acad Sci U S A. 2007;104:19327–19332. doi: 10.1073/pnas.0705953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ardito CM, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rooman I, et al. Expression of the Notch signaling pathway and effect on exocrine cell proliferation in adult rat pancreas. Am J Pathol. 2006;169:1206–1214. doi: 10.2353/ajpath.2006.050926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan Z, et al. Characterization of CSL (CBF-1, Su(H), Lag-1) Mutants Reveals Differences in Signaling Mediated by Notch1 and Notch2. J Biol Chem. 2012;287:34904–34916. doi: 10.1074/jbc.M112.403287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hashimoto T, et al. VEGF activates divergent intracellular signaling components to regulate retinal progenitor cell proliferation and neuronal differentiation. Development. 2006;133:2201–2210. doi: 10.1242/dev.02385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sundaram MV. The love-hate relationship between Ras and Notch. Genes Dev. 2005;19:1825–1839. doi: 10.1101/gad.1330605. [DOI] [PubMed] [Google Scholar]