

Abstract
Nuclear, cytoplasmic and mitochondrial proteins are extensively modified by an O-linked β-N-acetylglucosamine (O-GlcNAc) moiety. This sugar modification regulates fundamental cellular processes in response to diverse nutritional and hormonal cues. The enzymes O-GlcNAc transferase (OGT) and O-linked β-N-acetylglucosaminase (O-GlcNAcase) mediate the addition and removal of O-GlcNAc, respectively. Aberrant O-GlcNAcylation has been implicated in a plethora of human diseases, including diabetes, cancer, aging, cardiovascular disease and neurodegenerative disease. As metabolic dysregulation is a vital component of these diseases, unraveling the roles of O-GlcNAc in metabolism is of emerging importance. Here, we review the current understanding of the functions of O-GlcNAc in cell signaling and gene transcription involved in metabolism, and focus on its relevance to diabetes, cancer, circadian rhythm, and mitochondrial function.
Keywords: O-GlcNAc, hexosamine biosynthesis, insulin resistance, cancer metabolism, circadian rhythm, mitochondria
Nutrient-sensing O-GlcNAc modification
Many, if not most, of cytoplasmic and nuclear proteins are modified by a single O-GlcNAc moiety at serine or threonine residues, termed O-GlcNAcylation [1-3]. This dynamic and reversible modification is emerging as a key regulator of diverse cellular processes, such as signal transduction, transcription, translation, and proteasomal degradation [1, 2, 4-10]. Perturbations in protein O-GlcNAcylation are associated with a wide range of human diseases and conditions [1, 11-13].
O-GlcNAcylation often occurs reciprocally or sequentially with serine/threonine phosphorylation. Growing evidence suggests that this sugar modification acts as an “on/off” switch in various cellular pathways that rivals phosphorylation [1, 14]. In sharp contrast to 428 serine/threonine kinases and ∼40 phosphatases encoded in the human genome [15, 16], only two single genes encode the enzymes that are responsible for cyclic O-GlcNAcylation; OGT catalyzes the addition of the GlcNAc moiety from the high-energy donor UDP-GlcNAc to target proteins at the hydroxyl groups of serine or threonine residues, whereas O-GlcNAcase (OGA, NCOAT, or MGEA5) catalyzes the hydrolytic removal of the sugar moiety from proteins (Figure 1).
Figure 1. Schematic diagram of OGT and OGA isoforms.
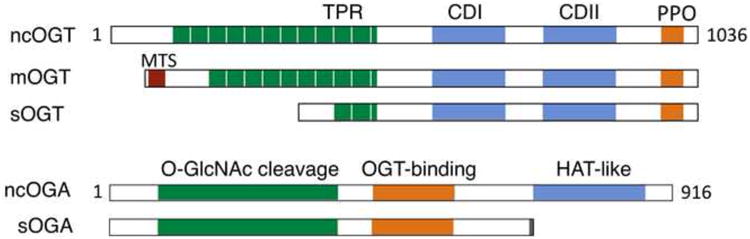
The ogt gene produces multiple OGT isoforms through alternative splicing and multiple start codons [109]. The longest and shortest isoforms (ncOGT and sOGT) are found in both the nucleus and the cytoplasm, whereas an atypical isoform (mOGT) targets the inner mitochondrial membrane [96]. These isoforms are distinct in the number of TPR at the N-terminus. They have a common C-terminal region comprised of the catalytic domains I&II (CDI&II) and the phosphoinositide-binding domain (PPO). The canonical OGA is a nucleocytoplasmic enzyme that contains the N-terminal O-GlcNAc cleavage domain, the OGT-binding region, and the C-terminal histone acetyltransferase (HAT)-like domain. A splicing variant lacking the HAT-like domain appears to reside in the endoplasmic reticulum and lipid droplets [110].
UDP-GlcNAc is derived from extracellular glucose through the hexosamine biosynthesis pathway (HBP). Because UDP-GlcNAc and protein O-GlcNAc levels in the cell fluctuate with the availability of glucose, free fatty acids, uridine and the amino acid glutamine, it has long been speculated that O-GlcNAc serves as a nutrient sensor [17]. The past few years have seen significant advances in our understanding of the functions of O-GlcNAcylation in nutrient sensing and metabolic physiology. In this review, we summarize the current knowledge of transcriptional and post-translational regulation of metabolism by O-GlcNAc signaling, focusing on its relevance to diabetes, cancer, circadian rhythm, and mitochondrial function.
Hexosamine biosynthesis, O-GlcNAc modification and insulin resistance
Nutrient excess is a major culprit of the diabetes epidemic in modern society. There is evidence that high levels of circulating glucose and free fatty acids bring about insulin resistance, a hallmark of type 2 diabetes (T2D), via the HBP [18]. Early studies show that high glucose-induced insulin resistance is associated with elevated UDP-GlcNAc levels in primary adipocytes [19]. In diabetic animals and humans, there are significant increases in global O-GlcNAc levels in tissues and cells such as liver, heart, erythrocytes and leukocytes [20-23]. Glutamine:fructose-6-phosphate amidotransferase (GFAT) is the first and rate-limiting enzyme of the HBP. Transgenic overexpression of GFAT leads to insulin resistance in muscle and adipose tissue [24, 25]. In vivo administration of glucosamine, which enters the hexosamine pathway bypassing GFAT, induces insulin resistance in multiple tissues [26]. Overexpression of OGT in muscle, fat or liver dampens insulin signaling [4, 27]. These studies suggest O-GlcNAc as a nexus between nutrient flux and insulin resistance.
Surprisingly, studies on OGA have produced conflicting results. Pharmacological inhibition of OGA by PUGNAc increases global O-GlcNAc levels and causes insulin resistance in 3T3-L1 adipocytes and skeletal muscle [28, 29]. However, recent studies show that more selective OGA inhibitors such as NButGT and 6-Ac-Cas fail to induce insulin resistance in 3T3-L1 adipocytes and in rodents [30, 31]. To further complicate matters, transient OGA overexpression in the liver improves insulin sensitivity in diabetic db/db mice [32]; however, reduction of O-GlcNAc levels by OGA overexpression or OGT knockdown does not prevent insulin resistance in 3T3-L1 adipocytes [33]. This OGT/OGA paradox in metabolism can also be observed in C. elegans. Forsythe et al. reported that both the ogt- and oga-null worms have elevated stores of glycogen and trehalose, and decreased lipid storage [34, 35]. One compelling explanation for this paradox is that OGT and OGA activity is mutually dependent. Sequential activation of OGT and OGA regulates highly dynamic and cyclic cellular events to maintain metabolic homeostasis. As a result, the effects of manipulating the O-GlcNAc pathway on insulin signaling are dependent on the location, duration, and extent of the intervention.
O-GlcNAc regulation of insulin signaling proteins
Dynamic crosstalk between O-GlcNAcylation and phosphorylation is widely implicated in the regulation of signal transduction and gene transcription [1]. Many insulin signaling proteins, including insulin receptor (IR), Insulin receptor substrate 1 (IRS1) and IRS2, phosphoinositide 3-kinase (PI3K), pyruvate dehydrogenase kinase 1 (PDK1) and the serine/threonine-specific protein kinase AKT, are known to be O-GlcNAcylated, which has been indicated as a negative regulator of insulin signal transduction (Figure 2) [4, 36]. O-GlcNAcylation of IRS1 inhibits phosphorylation at Tyr 608, thereby reducing the interaction with PI3K and the activation of AKT [36, 37]. Negative feedback phosphorylation of IRS1 at multiple serine residues contributes to the termination of insulin signaling. Previous work reveals that O-GlcNAcylation also inhibits IRS1 activity by increasing phosphorylation at Ser 307 and Ser 632/635 [4]. O-GlcNAcylation of AKT at Thr 305 and Thr 312 inhibits Thr 308 phosphorylation and activation by PDK1 [4, 38]. Importantly, the inhibition of IRS1 and AKT by O-GlcNAcylation require the recruitment of OGT to the plasma membrane through the phosphoinositide-binding domain, suggesting that O-GlcNAc serves as a negative feedback mechanism involved in the termination of insulin signaling [4, 39].
Figure 2. O-GlcNAc regulation of insulin signaling.

On binding to insulin, the auto-phosphorylated IR catalyzes tyrosine phosphorylation of IRS proteins, which results in the docking and activation of phosphatidylinositol-3-OH kinase (PI3K). PI3K produces phosphatidylinositol 3,4,5-triphosphate (PIP3), which recruits PDK1 and AKT to the plasma membrane. AKT activated by PDK1 phosphorylates numerous substrates to mediate physiological functions. Subsequently, PIP3-binding OGT attenuates insulin signaling by O-GlcNAcylating IR, IRS, PDK1, and AKT.
AKT phosphorylates AS160, a Rab GAP, to mediate the translocation of the glucose transporter GLUT4 to the membrane. Insulin also activates PKCζ/λ to stimulate the trafficking of GLUT4 vesicles by actin remodeling. O-GlcNAcylation suppresses GLUT4 trafficking by inhibiting Munc18c and possibly regulating PKCζ/λ and actin. O-GlcNAcylation antagonizes insulin's suppression of gluconeogenesis by activating transcription factor and cofactors such as FOXO1, PGC-1α, and CRTC2. SREBP-1c and ChREBP are two key transcription factors that induce expression of lipogenic genes. O-GlcNAcylation regulates lipogenesis by directly stabilizing ChREBP and promoting Srebp-1c transcription through LXR activation. O-GlcNAcylation of GS suppresses glycogen synthesis. The possible role of O-GlcNAcylated GSK3β in glycogen storage has not been explored. In pancreatic β-cells, O-GlcNAcylated Pdx-1 and NeuroD1 promote transcription of the insulin gene.
Red and black hexagons containing letter G indicate positive and negative regulation of the proteins by O-GlcNAcylation, respectively. Grey hexagons note that the role of O-GlcNAcylation is unknown.
Not only does O-GlcNAc modulate the proximal insulin signaling cascade, but also targets specific downstream pathways to elicit diverse physiological responses. As elaborated below, this coordinated action of O-GlcNAc at multiple levels is likely a fine-tuning mechanism governing metabolic homeostasis.
O-GlcNAc regulation of glucose uptake
Translocation of insulin-sensitive glucose transporter GLUT4 to the plasma membrane is required for insulin-induced glucose uptake into adipocytes and muscle cells that normalizes blood glucose levels. There is evidence that the regulation of glucose uptake by O-GlcNAc signaling is independent of early components of insulin signaling [40]. Chen et al. found that glucosamine treatment of 3T3-L1 cells increases O-GlcNAcylation of Munc18c, an essential component of the synaptic vesicle fusion protein complex, and decreases insulin-stimulated trafficking of soluble NSF attachment protein receptor (SNARE) proteins to the plasma membrane [40]. However, it is not known whether O-GlcNAcylation of Munc18c is responsible for the defect in GLUT4 translocation induced by glucosamine.
Atypical protein kinase C (aPKC) isoforms such as PKCζ and PKCλ, are also activated by insulin action, and are proposed to mediate GLUT4 trafficking through actin remodeling and other mechanisms [41, 42]. aPKCs and actin are known as O-GlcNAcylated proteins [43, 44]. Therefore, it is interesting to determine whether O-GlcNAcylation of aPKCs and actin directly regulates GLUT4 translocation.
O-GlcNAc regulation of gluconeogenesis
It has long been postulated that O-GlcNAc mediates glucose toxicity in diabetes. Diabetic liver is characterized by uncontrolled gluconeogenesis, which contributes to hyperglycemia. Many transcription factors and cofactors that regulate gluconeogenesis are modified by O-GlcNAc. Housley et al. reported that the peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) facilitates OGT targeting to forkhead box protein O1 (FOXO1), where OGT O-GlcNAcylates and activates FOXO1. Recent work by Ruan et al. reveals that OGT forms a nutrient-sensitive complex with host cell factor C1 (HCF-1) to O-GlcNAcylate PGC-1α. O-GlcNAcylated PGC-1α recruits the deubiquitinase BAP1 to remove poly-ubiquitin from PGC-1α and thereby stabilizes PGC-1α [20]. The effectiveness of glucose to suppress gluconeogenesis is lost in diabetic animals because HCF-1 is highly expressed in the liver of mice fed a high fat diet (HFD) and db/db diabetic mice. Knockdown of OGT and HCF-1 improves glucose homeostasis in db/db mice [20].
It has been well established that cAMP response element-binding protein (CREB) regulated transcription coactivator 2 (CRTC2) co-activates CREB to induce the transcription of gluconeogenic genes [45]. O-GlcNAcylation at Ser 70 and Ser 171 increases CRTC2 activity by disrupting 14-3-3 protein binding, while OGA overexpression in the liver disrupts O-GlcNAcylation of CRTC2 in HFD and db/db mice and lowers the gluconeogenic profile [32]. Collectively, O-GlcNAcylation of gluconeogenic transcription factor and cofactors promotes glucose production in the liver, suggesting that pharmacological inhibition of hepatic O-GlcNAc signaling may be a new strategy for treating T2D.
O-GlcNAc regulation of glycogen synthesis
Glycogen synthase (GS) controls the final step of glycogen synthesis, which is stimulated by insulin through GSK3β inhibition. Activation of GS is reduced in insulin resistance and diabetes. O-GlcNAcylation of GS is associated with decreased enzymatic activity [46]. High glucose increases GS O-GlcNAcylation and suppresses GS activity, which in turn brings about blood glucose retention. This vicious cycle is another example of O-GlcNAc-mediated glucose toxicity. GSK3β is also modified by O-GlcNAc and inhibition of GSK3β alters global O-GlcNAc levels [47, 48]; however, the role of its O-GlcNAcylation has not been explored.
O-GlcNAc regulation of lipogenesis
Sterol responsive element binding protein-1c (SREBP-1c) is a master lipogenic transcription factor regulated by insulin. Fatty acid synthesis is controlled not only by insulin but also by glucose. Carbohydrate-responsive element-binding protein (ChREBP) and liver X receptor (LXR) have been proposed as glucose sensors that regulate lipogenesis. Glucose regulates the levels of O-GlcNAcylation of LXR and its activity on Srebp-1c transcription, an LXR target gene [49]. ChREBP is also subject to O-GlcNAc modification, which stabilizes the ChREBP protein and increases its transcription activity on lipogenic genes [50]. In db/db mice, ChREBP O-GlcNAcylation is elevated in the liver, and OGA overexpression prevents hepatic steatosis in these mice. Although diabetic animals and humans show insulin resistance in the liver, hepatic lipogenic pathways are paradoxically hyperactivated. Glucose toxicity mediated by O-GlcNAcylation of key regulators of lipogenesis might be one of the underlying mechanisms for such paradox.
O-GlcNAc regulation of β-cell function
In addition to its roles in insulin signal transduction, O-GlcNAc signaling also regulates insulin production and islet survival in the pancreas. OGT is highly abundant in β-cells in the islet [51] and O-GlcNAc levels in β-cells are sensitive to glucose [52], implying that O-GlcNAc may function as a glucose sensor to regulate insulin secretion. Indeed, expression of the GFAT transgene in β-cells increases hexosamine flux and results in hyperinsulinemia [53], whereas OGA overexpression in β-cells decreases insulin mRNA levels, islet insulin content, and circulating insulin levels [54]. Neurogenic differentiation 1 (NeuroD1) and pancreatic and duodenal homeobox 1 (Pdx-1) are two β-cells-specific transcription factors that have been demonstrated to play crucial roles in glucose-induced insulin gene transcription [55]. High glucose induces NeuroD1 O-GlcNAcylation and subsequent translocation into the nucleus, while on low glucose OGA interacts with and deglycosylates NeuroD1, which is likely to account for NeuroD1 export into the cytoplasm [56]. Pdx-1 is also O-GlcNAcylated, which correlates with increased DNA binding ability and insulin secretion [57]. These data provide evidence for the functional roles of O-GlcNAc in insulin production and secretion.
O-GlcNAc signaling also mediates glucose toxicity in pancreatic β-cells. Elevating cellular O-GlcNAc levels by the OGA inhibitor PUGNAc or glucosamine infusion decreases glucose-stimulated insulin secretion and β-cell survival [58-60]. However, there is evidence that the adverse effect of adenoviral overexpression of GFAT on β-cell function may not be ascribed to protein O-GlcNAcylation [26, 53]. It is possible that O-GlcNAc stimulates insulin secretion at physiological conditions and the onset of T2D, while O-GlcNAc promotes β-cell dysfunction upon chronic exposure to hyperglycemia at the late stage of diabetes.
O-GlcNAc signaling in cancer metabolism
O-GlcNAc as a biomarker of human cancer
Dysregulation of O-GlcNAc signaling has been implicated in human cancer. Aberrant O-GlcNAcylation is a clinical characteristic of chronic lymphocytic leukemic patients [61]. Global O-GlcNAc levels and expression of OGT are significantly elevated in human lung and colon cancer tissues [62]. O-GlcNAc levels are also increased in hepatocellular carcinoma tissues and, interestingly, low expression of OGA is suggested as a prognostic marker for tumor recurrence [63]. Another study suggests that urinary content of OGT and OGA may be used as a prognostic tool for bladder cancer [64]. The Reginato group described a critical role of OGT in breast and prostate cancer growth and invasion [65, 66]. Therefore, protein O-GlcNAcylation is a promising biomarker and a potential driver for various human cancers.
O-GlcNAc in transcriptional regulation of cancer metabolism
Metabolic rewiring is an emerging hallmark of cancer [67]. Cancer cells require increased glucose uptake and aerobic glycolysis (the Warburg effect) to support rapid cell growth and proliferation [68]. This is largely achieved by reprogramming gene expression in the metabolic network. Recent studies reveal the important roles of c-Myc, Sp1, p53 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in this process. All of these transcription factors have been found to be modified by O-GlcNAc, and this modification can regulate their functions either positively or negatively [5, 69-71]. For instance, O-GlcNAcylation of c-Myc and p53 increases the stability and activity of the proteins, whereas O-GlcNAcylation of Sp1 at the activation domain decreases its transcriptional activity [5, 11]. Whether O-GlcNAcylation of these transcription factors contributes to the Warburg effect is an important topic of current investigation.
Recent evidence suggests a role for O-GlcNAc signaling in the regulation of the Warburg effect by NF-κB. The activation of the IKK-NF-κB pathway, by loss of p53, increases the rate of aerobic glycolysis and the expression GLUT3 [72]. High glucose promotes O-GlcNAcylation of IκB kinase κβ (IKKβ) and sustains tumor necrosis factor α (TNFα)-dependent IKKβ activity [73]. Therefore, increased glucose flux via the HBP may contribute to the Warburg effect through O-GlcNAcylation of IKKβ (Figure 3).
Figure 3. Regulation of cancer metabolism by the hexosamine/O-GlcNAc pathway.

In normal cells, p53 suppresses glycolysis by inducing expression of TIGAR and inhibiting expression of glycolytic genes such as hexokinase II, phosphoglycerate mutase, and glucose transporter 3 (GLUT3). p53 also inhibits the diversion of glucose flux to the anabolic pentose phosphate pathway (PPP) by binding to and inhibiting glucose-6-phosphate dehydrogenase (G6PD). Loss-of-function of p53 in tumor cells increases glucose uptake, aerobic glycolysis, and PPP flux. PPP flux produces reducing agents for macromolecule biosynthesis and reactive oxygen species (ROS) neutralization. 2∼5% glucose is shunted to the HBP that produce UDP-GlcNAc, a substrate for OGT. OGT inhibits ubiquitin-mediated degradation of p53 by direct O-GlcNAcylation. OGT also potentiates NF-κB signaling by O-GlcNAcylation and activation of IKKβ, which triggers phosphorylation of IκBα and releasing of NF-κB to the nucleus to promote transcription of glucose transporters and HIF-1. HIF-1 increases expression of glycolytic genes to promote tumor growth. Finally, O-GlcNAcylation of the critical glycolytic enzyme PFK-1 reduces its enzyme activity (red arrow) and redirects glucose flux through the PPP, thereby conferring a selective growth advantage on cancer cells.
In addition to its role in antagonizing NF-κB, p53 also inhibits glycolysis and suppresses tumor growth through the induction of TIGAR, an inhibitor of phosphofructose kinase (PFK) activity [74]. Furthermore, p53 can inhibit the pentose phosphate pathway by directly binding to and inhibiting the activity of glucose-6-phosphate dehydrogenase [75]. Mutations of p53 found in many cancers may orchestrate diverse metabolic pathways to meet the metabolic demand for tumor growth (Figure 3).
O-GlcNAcylation of metabolic enzymes in cancer
Almost all the glycolytic enzymes are O-GlcNacylated. A recent study by Yi et al. reveal that O-GlcNAcylation of PFK-1 decreases its enzymatic activity, thereby decreasing overall glycolytic flux and diverting glucose flux to the pentose phosphate pathway (Figure 3) [8]. This is critical for reducing oxidative stress and supporting rapid cell growth [8]. This study summons more effort to investigate the regulatory roles of direct O-GlcNAcylation of metabolic enzymes in normal physiology and cancer. This may help identify an Achilles' heel of cancer metabolism that could be exploited in future therapeutic interventions.
O-GlcNAc signaling links the circadian clock to metabolism
The circadian clock (Box 1) is emerging as an important mechanism governing metabolism [76]. Many metabolic processes such as hormone secretion, nutrient uptake, gluconeogenesis, and oxidative phosphorylation have a daily cycle [77-79], driven by cell-autonomous clock oscillators in the brain and metabolic tissues [80, 81]. Misalignment of the internal rhythm with the environmental daily cycle contributes to metabolic disorders and cancer [77, 82, 83].
Text Box 1. Anatomical and molecular mechanisms of the circadian clock.
Circadian rhythm is an endogenous oscillation with a period close to 24 hours that is able to be synchronized to environmental cues (e.g. light, temperature, and food), and is temperature-compensated [106]. It promotes an organism to anticipate daily changes in the environment and contributes to the fitness of survival and reproduction [76].
The mammalian circadian clock is built upon molecular oscillators residing in almost all cells [80, 81]. The molecular clockwork, including genetic oscillators and metabolic oscillators, generates circadian rhythm at the cellular level. The genetic oscillator is established by transcriptional-translational negative feedback loops [107]. The BMAL1/CLOCK heterodimer drives expression of Period (Per1, Per2, and Per3) and Cryptchrome (Cry1 and Cry2) genes. PER and CRY feed back to inhibit their own transcription by blocking BMAL1/CLOCK activity. Rhythmic expression of nuclear receptors RAR-related orphan receptor (ROR) and REV-ERB is also controlled by BMAL1/CLOCK. Alternate actions of ROR (activator) and REV-ERB (repressor) drive rhythmic expression of the Bmal1 gene in antiphase to that of Per and Cry genes. The metabolic oscillator is built upon circadian accumulation of cellular metabolites, such as reactive oxygen species that drives the oxidation rhythm of peroxiredoxin proteins [108].
Despite the similar composition of molecular oscillators in different cells and tissues, the mammalian circadian clock is organized hierarchically [80, 81]. Solar light is the dominant synchronizing cue. The central clock residing in the hypothalamic suprachiasmatic nuclei (SCN) senses solar time by retinal innervations via glutamatergic synapses. Peripheral clocks located in virtually all other tissues receive synchronizing cues from the SCN clock and are amenable to the entrainment by food availability and temperature.
Growing evidence indicates that O-GlcNAcylation mediates the circadian regulation of metabolism. As discussed in previous sections, O-GlcNAcylation fine-tunes nutrient/hormonal signaling pathways. It is possible that circadian clocks drive daily changes in protein O-GlcNAcylation to modulate energy metabolism. Gene expression profiles show that both Ogt and Oga transcripts exhibit circadian rhythm in metabolic tissues (http://bioinf.itmat.upenn.edu/circa). The circadian expression of Gfat-1 in the fat body of fruit flies has been documented [84]. Durgan et al. reported that protein O-GlcNAcylation in the heart changes in a daily manner and is modulated by the heart clock [85]. Recently, Kaasik et al. identified OGT as a direct substrate of GSK3β that exhibits circadian kinase activity in vivo [86], further supporting the idea of circadian changes in protein O-GlcNAcylation. Although the metabolic significance of circadian O-GlcNAcylation is largely unknown, Li et al. demonstrated that genetic manipulation of hepatic OGT expression in mice results in aberrant rhythms of glucose homeostasis [87]. These findings strongly argue for a role of protein O-GlcNAcylation in linking the circadian clock to metabolic outputs.
O-GlcNAcylation also acts as an input pathway to reset the clock. Studies in the past decade have identified many nutrient-responsive proteins as clock regulators [88], which reset the circadian rhythm according to the feeding/fasting cycle in metabolic tissues (Box 1). As an important nutrient-sensing mechanism, O-GlcNAcylation may modify clock proteins to control the internal time. Early studies in Arabidopsis thaliana have shown that the plant OGT (SPINDLY) regulates circadian rhythms [89]. Durgan et al. observed O-GlcNAcylation of two clock proteins and the phase-resetting role of PUGNAc [85], which raises the question as to how O-GlcNAc modulates the function of clock proteins and circadian physiology. To date, circadian changes in O-GlcNAcylation have been found on BMAL1 [85, 87, 90], CLOCK/dClk [86, 87], and PER/dPer [85, 86, 91]. In fruit flies, Kim et al. showed that O-GlcNAcylation of dPer decreases its abundance and delays its nuclear translocation, leading to a long-period behavioral rhythm [91]. In mammals, Li et al. demonstrated that O-GlcNAcylation of BMAL1 and CLOCK is rhythmic and this modification stabilizes BMAL1 and CLOCK by inhibiting their ubiquitination, contributing to increased amplitude of circadian oscillation [87]. Importantly, the actions of O-GlcNAcylation on clock oscillation are responsive to glucose concentration [87]. Kaasik et al. showed that phosphorylation and O-GlcNAcylation compete at serine 662 of PER2 in a glucose-responsive manner [86]. Of note, the serine 662-to-glycine mutation causes Familial Advanced Sleep Phase Disorder [92]. Recent studies show that OGT/HCF-1/BAP1 forms a protein complex to modulate epigenetics and metabolism [20, 93]. The fact that HCF-1 acts as an important modulator of cellular circadian rhythm [94] raises the possibility that the OGT/HCF-1/BAP1 complex interacts with the BMAL1/CLOCK/PER complex to modulate circadian rhythm. The dynamics of O-GlcNAcylation on clock proteins is determined by the metabolic state of the cell, making it amenable to resetting the clock in response to food-derived signals.
In a nutshell, O-GlcNAcylation regulates the temporal organization of metabolism via both the output and input pathways of the circadian clock. A recent chemical screening suggests the potential of circadian therapeutics for metabolic disorders [95]. Further studies on the circadian function of O-GlcNAcylation in metabolism may uncover novel targets for circadian therapies.
Mitochondrial O-GlcNAcylation in metabolic regulation
Mitochondrial OGT (mOGT) arises from a single OGT gene by alternative splicing that generates a unique N-terminus containing mitochondrial targeting sequence and a membrane-spanning helix (Figure 1) [96, 97]. Its nine tetratricopeptide repeat (TPR) motifs through the C-terminus is identical to ncOGT [96]. Overexpression of mOGT is highly cytotoxic and triggers apoptosis [98, 99]. This somehow impedes research on mitochondrial O-GlcNAcylation and its function.
Early studies reported that the levels of mitochondrial O-GlcNAcylation were very low [47, 96]. However, increasing evidence suggests pervasive mitochondrial O-GlcNAcylation [100-103]. This modification may either protect or perturb mitochondrial function. O-GlcNAcylation of voltage-dependent anion channel (VADC) in cardiac myocytes can preserve mitochondrial integrity by interfering with mitochondrial permeability transition pore formation induced by Ca2+ overload and oxidative stress [101, 102]. Increased O-GlcNAcylation of the respiratory chain complex I, III and IV impairs mitochondrial function in cardiac myocytes [103]. Increased O-GlcNAcylation of dynamin-related protein 1 (DRP1) induces translocation of DRP1 from the cytoplasm to mitochondria, which increases mitochondrial fragmentation and decreases mitochondrial membrane potential [104]. Furthermore, low running capacity rats exhibit increased O-GlcNAcylation of cardiac proteins including mitochondrial Complex I and IV, VDAC and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), which may contribute to progressive mitochondrial dysfunction and the pathogenesis of insulin resistance [105]. A large number of mitochondrial proteins associated with OGT was recently identified via a proteomic approach, further suggesting the importance of OGT to mitochondrial function [20].
Although the repertoire of mitochondrial O-GlcNAc targets is rapidly expanding, many critical questions remain to be answered. UDP-GlcNAc is likely limited in mitochondria, and how it transports into mitochondria remains unknown. Although scarce evidence shows direct interaction between mOGT and mitochondrial proteins, it cannot exclude the possibility that those proteins may be O-GlcNAcylated in the cytoplasm prior to entering mitochondria. Despite the fact that OGA activity is detectable in purified mitochondria [103], mitochondrial OGA proteins have not been identified. Finally, whether O-GlcNAcylation of mitochondrial proteins responds to nutritional and hormonal signals and regulates intermediary metabolism is poorly understood.
Concluding remarks
The past decade has witnessed significant advances in our understanding of the versatile roles of posttranslational modifications, such as phosphorylation, acetylation, methylation and ubiquitination, in fundamental cellular processes. Despite the discovery of O-GlcNAc modification three decades ago, we are just beginning to unravel its biological functions at levels ranging from protein structure and interaction to cell signaling and gene regulation to physiology and metabolism. Accumulating evidence suggests that O-GlcNAc acts as a nutrient sensor that couples systemic metabolic status to cellular regulation of signal transduction, transcription and protein degradation. The best established is O-GlcNAc regulation of insulin signaling proteins and transcriptional regulators involved in glucose and lipid metabolism. Only recently, O-GlcNAcylation of transcription factors involved in cancer metabolism and circadian rhythm is being appreciated. As metabolic enzymes are the prevalent targets of O-GlcNAcylation in the cytoplasm, nucleus, and mitochondria, elucidating the roles of O-GlcNAcylation of metabolic enzymes in regulating metabolic flux and epigenetics will be an exciting area of research. In light of the emerging links between protein O-GlcNAcylation and diabetes, cancer, aging, cardiovascular and neurodegenerative diseases, uncovering functions and mechanisms of O-GlcNAc signaling would be crucially important for exploring O-GlcNAc as a therapeutic target for these diseases (Box 2).
Text Box 2. Outstanding Questions.
What are the principle and mechanism by which OGT and OGA recognize numerous proteins that share no apparent motifs?
How does O-GlcNAc integrate nutritional and circadian signals?
How does O-GlcNAc signaling impinge on other nutrient-sensing pathways, such as the mTOR, cAMP, AMPK, and MAPK signaling pathways?
Are OGT and OGA mutually regulated, and if so, how?
Are balanced O-GlcNAc levels critical for cellular processes and metabolic homeostasis?
Is O-GlcNAc “druggable” for human diseases?
Highlights.
O-GlcNAc is a nutrient sensor and regulatory molecular switch.
O-GlcNAc is a negative regulator of insulin signaling.
O-GlcNAc is crucially involved in transcriptional regulation of metabolism and circadian clock.
Aberrant O-GlcNAcylation has implications in a wide range of metabolic diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hart GW, et al. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanover JA, et al. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 3.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- 4.Yang X, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451:964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- 5.Yang X, et al. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci U S A. 2001;98:6611–6616. doi: 10.1073/pnas.111099998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, et al. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell. 2002;110:69–80. doi: 10.1016/s0092-8674(02)00810-3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang F, et al. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell. 2003;115:715–725. doi: 10.1016/s0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- 8.Yi W, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujiki R, et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capotosti F, et al. O-GlcNAc Transferase Catalyzes Site-Specific Proteolysis of HCF-1. Cell. 2011;144:376–388. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 11.Slawson C, et al. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci. 2010;35:547–555. doi: 10.1016/j.tibs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darley-Usmar VM, et al. Protein O-linked beta-N-acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. J Mol Cell Cardiol. 2012;52:538–549. doi: 10.1016/j.yjmcc.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuzwa SA, Vocadlo DJ. O-GlcNAc modification and the tauopathies: insights from chemical biology. Curr Alzheimer Res. 2009;6:451–454. doi: 10.2174/156720509789207967. [DOI] [PubMed] [Google Scholar]
- 14.Copeland RJ, et al. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab. 2008;295:E17–28. doi: 10.1152/ajpendo.90281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alonso A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Moorhead GB, et al. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol. 2007;8:234–244. doi: 10.1038/nrm2126. [DOI] [PubMed] [Google Scholar]
- 17.Zachara NE, Hart GW. O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim Biophys Acta. 2004;1673:13–28. doi: 10.1016/j.bbagen.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 18.Teo CF, et al. Hexosamine flux, the O-GlcNAc modification, and the development of insulin resistance in adipocytes. Mol Cell Endocrinol. 2010;318:44–53. doi: 10.1016/j.mce.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traxinger RR, Marshall S. Coordinated regulation of glutamine:fructose-6-phosphate amidotransferase activity by insulin, glucose, and glutamine. Role of hexosamine biosynthesis in enzyme regulation. J Biol Chem. 1991;266:10148–10154. [PubMed] [Google Scholar]
- 20.Ruan HB, et al. O-GlcNAc Transferase/Host Cell Factor C1 Complex Regulates Gluconeogenesis by Modulating PGC-1alpha Stability. Cell Metab. 2012;16:226–237. doi: 10.1016/j.cmet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fricovsky ES, et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. 2012;303:R689–699. doi: 10.1152/ajpregu.00548.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Springhorn C, et al. Exploring Leukocyte O-GlcNAcylation as a Novel Diagnostic Tool for the Earlier Detection of Type 2 Diabetes Mellitus. J Clin Endocrinol Metab. 2012;97:4640–4649. doi: 10.1210/jc.2012-2229. [DOI] [PubMed] [Google Scholar]
- 23.Park K, et al. Increased expression of beta-N-acetylglucosaminidase in erythrocytes from individuals with pre-diabetes and diabetes. Diabetes. 2010;59:1845–1850. doi: 10.2337/db09-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hebert LF, Jr, et al. Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin Invest. 1996;98:930–936. doi: 10.1172/JCI118876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hazel M, et al. Activation of the hexosamine signaling pathway in adipose tissue results in decreased serum adiponectin and skeletal muscle insulin resistance. Endocrinology. 2004;145:2118–2128. doi: 10.1210/en.2003-0812. [DOI] [PubMed] [Google Scholar]
- 26.Virkamaki A, et al. Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance in multiple insulin sensitive tissues. Endocrinology. 1997;138:2501–2507. doi: 10.1210/endo.138.6.5172. [DOI] [PubMed] [Google Scholar]
- 27.McClain DA, et al. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U S A. 2002;99:10695–10699. doi: 10.1073/pnas.152346899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vosseller K, et al. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2002;99:5313–5318. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arias EB, et al. Prolonged incubation in PUGNAc results in increased protein O-Linked glycosylation and insulin resistance in rat skeletal muscle. Diabetes. 2004;53:921–930. doi: 10.2337/diabetes.53.4.921. [DOI] [PubMed] [Google Scholar]
- 30.Macauley MS, et al. Inhibition of O-GlcNAcase using a potent and cell-permeable inhibitor does not induce insulin resistance in 3T3-L1 adipocytes. Chem Biol. 2010;17:937–948. doi: 10.1016/j.chembiol.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macauley MS, et al. Elevation of global O-GlcNAc levels in 3T3-L1 adipocytes by selective inhibition of O-GlcNAcase does not induce insulin resistance. J Biol Chem. 2008;283:34687–34695. doi: 10.1074/jbc.M804525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dentin R, et al. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–1405. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- 33.Robinson KA, et al. Reduction of O-GlcNAc protein modification does not prevent insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2007;292:E884–890. doi: 10.1152/ajpendo.00569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forsythe ME, et al. Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc Natl Acad Sci U S A. 2006;103:11952–11957. doi: 10.1073/pnas.0601931103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanover JA, et al. A Caenorhabditis elegans model of insulin resistance: altered macronutrient storage and dauer formation in an OGT-1 knockout. Proc Natl Acad Sci U S A. 2005;102:11266–11271. doi: 10.1073/pnas.0408771102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whelan SA, et al. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked beta-N-acetylglucosamine in 3T3-L1 adipocytes. J Biol Chem. 2010;285:5204–5211. doi: 10.1074/jbc.M109.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein AL, et al. O-linked N-acetylglucosamine modification of insulin receptor substrate-1 occurs in close proximity to multiple SH2 domain binding motifs. Mol Cell Proteomics. 2009;8:2733–2745. doi: 10.1074/mcp.M900207-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PloS one. 2012;7:e37427. doi: 10.1371/journal.pone.0037427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whelan SA, et al. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J Biol Chem. 2008;283:21411–21417. doi: 10.1074/jbc.M800677200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G, et al. Glucosamine-induced insulin resistance is coupled to O-linked glycosylation of Munc18c. FEBS Lett. 2003;534:54–60. doi: 10.1016/s0014-5793(02)03774-2. [DOI] [PubMed] [Google Scholar]
- 41.Liu LZ, et al. Protein kinase Czeta mediates insulin-induced glucose transportthrough actin remodeling in L6 muscle cells. Mol Biol Cell. 2006;17:2322–2330. doi: 10.1091/mbc.E05-10-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Standaert ML, et al. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem. 1999;274:25308–25316. doi: 10.1074/jbc.274.36.25308. [DOI] [PubMed] [Google Scholar]
- 43.Robles-Flores M, et al. Posttranslational modifications on protein kinase c isozymes. Effects of epinephrine and phorbol esters. Biochim Biophys Acta. 2008;1783:695–712. doi: 10.1016/j.bbamcr.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 44.Ramirez-Correa GA, et al. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–1358. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dentin R, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 46.Parker GJ, et al. Insulin resistance of glycogen synthase mediated by o-linked N-acetylglucosamine. J Biol Chem. 2003;278:10022–10027. doi: 10.1074/jbc.M207787200. [DOI] [PubMed] [Google Scholar]
- 47.Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem. 2000;275:10983–10988. doi: 10.1074/jbc.275.15.10983. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, et al. Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol Cell Proteomics. 2007;6:1365–1379. doi: 10.1074/mcp.M600453-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Anthonisen EH, et al. Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose. J Biol Chem. 2010;285:1607–1615. doi: 10.1074/jbc.M109.082685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guinez C, et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes. 2011;60:1399–1413. doi: 10.2337/db10-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanover JA, et al. Elevated O-linked N-acetylglucosamine metabolism in pancreatic beta-cells. Arch Biochem Biophys. 1999;362:38–45. doi: 10.1006/abbi.1998.1016. [DOI] [PubMed] [Google Scholar]
- 52.Liu K, et al. Glucose stimulates protein modification by O-linked GlcNAc in pancreatic beta cells: linkage of O-linked GlcNAc to beta cell death. Proc Natl Acad Sci U S A. 2000;97:2820–2825. doi: 10.1073/pnas.97.6.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang J, et al. Transgenic mice with increased hexosamine flux specifically targeted to beta-cells exhibit hyperinsulinemia and peripheral insulin resistance. Diabetes. 2000;49:1492–1499. doi: 10.2337/diabetes.49.9.1492. [DOI] [PubMed] [Google Scholar]
- 54.Soesanto Y, et al. Pleiotropic and age-dependent effects of decreased protein modification by O-linked N-acetylglucosamine on pancreatic beta-cell function and vascularization. J Biol Chem. 2011;286:26118–26126. doi: 10.1074/jbc.M111.249508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrali SS, et al. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem J. 2008;415:1–10. doi: 10.1042/BJ20081029. [DOI] [PubMed] [Google Scholar]
- 56.Andrali SS, et al. Glucose mediates the translocation of NeuroD1 by O-linked glycosylation. J Biol Chem. 2007;282:15589–15596. doi: 10.1074/jbc.M701762200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao Y, et al. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch Biochem Biophys. 2003;415:155–163. doi: 10.1016/s0003-9861(03)00234-0. [DOI] [PubMed] [Google Scholar]
- 58.Akimoto Y, et al. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology. 2007;17:127–140. doi: 10.1093/glycob/cwl067. [DOI] [PubMed] [Google Scholar]
- 59.Monauni T, et al. Effects of glucosamine infusion on insulin secretion and insulin action in humans. Diabetes. 2000;49:926–935. doi: 10.2337/diabetes.49.6.926. [DOI] [PubMed] [Google Scholar]
- 60.Kang ES, et al. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp Cell Res. 2008;314:2238–2248. doi: 10.1016/j.yexcr.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia. 2010;24:1588–1598. doi: 10.1038/leu.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mi W, et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta. 2011;1812:514–519. doi: 10.1016/j.bbadis.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 63.Zhu Q, et al. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol. 2012;29:985–993. doi: 10.1007/s12032-011-9912-1. [DOI] [PubMed] [Google Scholar]
- 64.Rozanski W, et al. Prediction of bladder cancer based on urinary content of MGEA5and OGT mRNA level. Clin Lab. 2012;58:579–583. [PubMed] [Google Scholar]
- 65.Lynch TP, et al. Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem. 2012;287:11070–11081. doi: 10.1074/jbc.M111.302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caldwell SA, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29:2831–2842. doi: 10.1038/onc.2010.41. [DOI] [PubMed] [Google Scholar]
- 67.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 68.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 69.Chou TY, et al. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem. 1995;270:18961–18965. doi: 10.1074/jbc.270.32.18961. [DOI] [PubMed] [Google Scholar]
- 70.Yang WH, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8:1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 71.Yang WH, et al. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U S A. 2008;105:17345–17350. doi: 10.1073/pnas.0806198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kawauchi K, et al. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611–618. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- 73.Kawauchi K, et al. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A. 2009;106:3431–3436. doi: 10.1073/pnas.0813210106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bensaad K, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 75.Jiang P, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bass J. Circadian topology of metabolism. Nature. 2012;491:348–356. doi: 10.1038/nature11704. [DOI] [PubMed] [Google Scholar]
- 77.Green CB, et al. The meter of metabolism. Cell. 2008;134:728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang X. A wheel of time: the circadian clock, nuclear receptors, and physiology. Genes Dev. 2010;24:741–747. doi: 10.1101/gad.1920710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang X, et al. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126:801–810. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 80.Mohawk JA, et al. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dibner C, et al. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–549. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- 82.Roenneberg T, et al. Social jetlag and obesity. Curr Biol. 2012;22:939–943. doi: 10.1016/j.cub.2012.03.038. [DOI] [PubMed] [Google Scholar]
- 83.Buxton OM, et al. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Science Trans Medicine. 2012;4:129–143. doi: 10.1126/scitranslmed.3003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu K, et al. The circadian clock interacts with metabolic physiology to influence reproductive fitness. Cell Metab. 2011;13:639–654. doi: 10.1016/j.cmet.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Durgan DJ, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–44619. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaasik K, et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li MD, et al. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. 2013;17:303–310. doi: 10.1016/j.cmet.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 89.Olszewski NE, et al. O-GlcNAc protein modification in plants: Evolution and function. Biochim Biophys Acta. 2010;1800:49–56. doi: 10.1016/j.bbagen.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma YT, et al. O-GlcNAcylation of BMAL1 regulates circadian rhythms in NIH3T3 fibroblasts. Biochem Biophys Res Commun. 2013 doi: 10.1016/j.bbrc.2013.01.043. 10.1016/j.bbrc.2013.01.043. [DOI] [PubMed] [Google Scholar]
- 91.Kim EY, et al. A role for O-GlcNAcylation in setting circadian clock speed. Genes Dev. 2012;26:490–502. doi: 10.1101/gad.182378.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu Y, et al. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell. 2007;128:59–70. doi: 10.1016/j.cell.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dey A, et al. Loss of the Tumor Suppressor BAP1 Causes Myeloid Transformation. Science. 2012;337:1541–1546. doi: 10.1126/science.1221711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang EE, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hirota T, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Love DC, et al. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci. 2003;116:647–654. doi: 10.1242/jcs.00246. [DOI] [PubMed] [Google Scholar]
- 97.Hanover JA, et al. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch Biochem Biophys. 2003;409:287–297. doi: 10.1016/s0003-9861(02)00578-7. [DOI] [PubMed] [Google Scholar]
- 98.Lubas WA, et al. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272:9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 99.Shin SH, et al. Elevated O-GlcNAc-dependent signaling through inducible mOGT expression selectively triggers apoptosis. Amino acids. 2011;40:885–893. doi: 10.1007/s00726-010-0719-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gu Y, et al. Altered O-GlcNAc modification and phosphorylation of mitochondrial proteins in myoblast cells exposed to high glucose. Arch Biochem Biophys. 2011;505:98–104. doi: 10.1016/j.abb.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 101.Jones SP, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–1182. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- 102.Ngoh GA, et al. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45:313–325. doi: 10.1016/j.yjmcc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu Y, et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. 2009;284:547–555. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gawlowski T, et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. 2012;287:30024–30034. doi: 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnsen VL, et al. Enhanced cardiac protein glycosylation (O-GlcNAc) of selected mitochondrial proteins in rats artificially selected for low running capacity. Physiol Genomics. 2012 doi: 10.1152/physiolgenomics.00111.2012. 10.1152/physiolgenomics.00111.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Menaker M. Circadian clocks: 50 years on. Cold Spring Harb Symp Quant Biol. 2007;72:655–659. doi: 10.1101/sqb.2007.72.057. [DOI] [PubMed] [Google Scholar]
- 107.Lowrey PL, Takahashi JS. Genetics of Circadian Rhythms in Mammalian Model Organisms. Genetics of Circadian Rhythms. 2011:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Ooijen G, Millar AJ. Non-transcriptional oscillators in circadian timekeeping. Trends Biochem Sci. 2012;37:484–492. doi: 10.1016/j.tibs.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 109.Nolte D, Muller U. Human O-GlcNAc transferase (OGT): genomic structure, analysis of splice variants, fine mapping in Xq13.1. Mamm Genome. 2002;13:62–64. doi: 10.1007/s00335-001-2108-9. [DOI] [PubMed] [Google Scholar]
- 110.Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE 2005. 2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]