

Background: HMGB1 in spontaneously regenerating spinal cord does not trigger the inflammation in contrast to those in injured mammalian cords.
Results: Gecko HMGB1 paralogs failed to interact with TLR2 and TLR4 but do with RAGE receptors to activate the signaling pathway.
Conclusion: HMGB1 is beneficial for spontaneous spinal cord regeneration by eliciting negligible inflammation and promoting oligodendrocyte migration.
Significance: HMGB1 displays distinct functions in regenerative vertebrates.
Keywords: Evolution, Inflammation, Receptor for Advanced Glycation End Products (RAGE), Regeneration, Toll-like receptors (TLR), CNS, HMGB1
Abstract
Uncontrolled, excessive inflammation contributes to the secondary tissue damage of traumatic spinal cord, and HMGB1 is highlighted for initiation of a vicious self-propagating inflammatory circle by release from necrotic cells or immune cells. Several regenerative-competent vertebrates have evolved to circumvent the second damages during the spontaneous spinal cord regeneration with an unknown HMGB1 regulatory mechanism. By genomic surveys, we have revealed that two paralogs of HMGB1 are broadly retained from fish in the phylogeny. However, their spatial-temporal expression and effects, as shown in lowest amniote gecko, were tightly controlled in order that limited inflammation was produced in spontaneous regeneration. Two paralogs from gecko HMGB1 (gHMGB1) yielded distinct injury and infectious responses, with gHMGB1b significantly up-regulated in the injured cord. The intracellular gHMGB1b induced less release of inflammatory cytokines than gHMGB1a in macrophages, and the effects could be shifted by exchanging one amino acid in the inflammatory domain. Both intracellular proteins were able to mediate neuronal programmed apoptosis, which has been indicated to produce negligible inflammatory responses. In vivo studies demonstrated that the extracellular proteins could not trigger a cascade of the inflammatory cytokines in the injured spinal cord. Signal transduction analysis found that gHMGB1 proteins could not bind with cell surface receptors TLR2 and TLR4 to activate inflammatory signaling pathway. However, they were able to interact with the receptor for advanced glycation end products to potentiate oligodendrocyte migration by activation of both NFκB and Rac1/Cdc42 signaling. Our results reveal that HMGB1 does not mediate the inflammatory response in spontaneous spinal cord regeneration, but it promotes CNS regeneration.
Introduction
Inflammatory responses elicited by spinal cord injury (SCI)4 contribute substantially to secondary tissue damage characterized by a series of biochemical events and cellular dysfunction, eventually leading to the formation of an ellipsoidal, loculated cystic cavity (1–3). Controlling the infiltration of immune cells and their excessive releases of the pro-inflammatory cytokines has become the important therapeutic strategies for alleviating functional decline of the SCI (4, 5). The “sterile” inflammatory responses following SCI are significantly associated with the signaling of endogenous damage-associated molecular pattern (DAMP) molecules at sites of tissue damage, which bind with pattern recognition receptors expressed in most cells (6, 7). Ligation of DAMPs with pattern recognition receptors, including most toll-like receptors (TLRs) and advanced glycation end products (RAGE), elicits inflammatory signaling, resulting in the releases of cytokines that augment inflammatory reaction (8, 9). Many DAMPs are nuclear or cytosolic proteins with defined intracellular functions and are released from cells after tissue injury (10). A key DAMP molecule is the nuclear protein high mobility group box 1 (HMGB1, amphoterin) that is passively released from necrotic cells or is actively secreted by neurons and many other cell types upon stimulation (11, 12). HMGB1 promotes immune cell recruitment and activation and as consequences the secretion of a spectrum of inflammatory cytokines, including TNF-α, IL-1, and IL-6 (13–15). In various CNS injury models, inhibition of HMGB1 has been shown to reduce the severity of the lesion and microglial activation (16, 17). As such, HMGB1 has been proposed to be a significant proinflammatory cytokine mediating progressive spinal cord injury (15, 18).
Structurally, HMGB1 proteins are characterized by the presence of two L-shaped DNA-binding elements (HMG box A and B), each made up of 80 amino acids arranged in three positively charged α-helices, and a C-terminal extremely negatively charged domain (19, 20). By binding to the minor groove of double-stranded DNA, the protein participates in nucleosome formation and regulation of gene transcription (21, 22). HMGB1 of immunocompetent cells, including monocytes, macrophages, neutrophils, and dendritic cells, can shuttle from the nucleus to the cytoplasm, where it is further packaged into secretory vesicles and released into extracellular milieu under stimulation of exogenous pathogenic products (23). Alternatively, the protein is released by somatic cells undergoing necrosis and elicits progression of inflammation (11, 24–27). The extracellular HMGB1 mediates inflammatory signaling or other cellular responses, including proliferation, migration, and neurite outgrowth, through its interaction with RAGE, TLR2, and TLR4, etc., resulting in activation of transcription factor NFκB and mitogen-activated protein kinases (MAPK) (28–31). The peptide corresponding to the first 20 amino acids of the B-box domain is able to mimic the inflammatory potential of HMGB1 in vivo (32, 33), and a substitution of one amino acid might affect the interaction of HMGB1 with TLR4, providing a rational design and development of therapeutics for use in sterile and infectious inflammation (34).
Two rounds of genome duplication early in vertebrate evolution, followed by a single round of genome duplication in a common ancestor of sample teleosts, resulted in the occurrence of HMGB1 paralogs in basal vertebrate fishes (35). Recently, HMGB1 paralogs have been identified in amphibian and mammalian species, including the commonly used model organisms Xenopus (GenBankTM accession numbers U21933 and BC054148), mouse (GenBankTM accession numbers XP_889413 and NP_034569), and rat (GenBankTM accession numbers XP_003753270 and NP_037095). During evolution, the duplicated genes have usually undergone degeneration, neofunctionalization, or subfunctionalization, which might be associated with differential or complementary regulation of physiological functions (36, 37). A comparative study of the two paralogs of HMGB1 is therefore indispensable to clarify each physiological role and the associated molecular signaling, especially in their mediation of inflammatory responses spotlighted by clinical intervention. The neglected nuance might be beneficial for refining our knowledge on these proteins, thereby improving therapeutical development. The reptile is the lowest amniote located at a significantly evolutionary position bridging lower vertebrates and mammals. Similar to fishes and amphibians, several species in the taxa are capable of regenerating complex body structures, including significant portions of their central nervous system in tailed adulthood (38–41). However, the mechanisms for these animals to circumvent secondary tissue damage by limiting the vicious self-propagating cycle of inflammation are still poorly understood. Given the evidence that mammalian HMGB1 mediates CNS inflammation (18, 42), we speculate that the regenerative animals might share distinct HMGB1 regulatory mechanisms by limiting excessive inflammatory responses to facilitate spinal cord regeneration, i.e. either by differentially spatial-temporal regulation of two paralogs following injury or by alternative molecular signaling. To address this question, we investigated the distinct roles of two paralogs of Gekko japonicus HMGB1 (gHMGB1) following tail amputation. We have revealed that the two paralogs of HMGB1 are broadly retained from fish onward during evolution. gHMGB1 paralogs displayed differential responses to the challenges of infectious stimulation and spinal cord injury. The expression of gHMGB1 has been selectively switched in the traumatic spinal cord and has not mediated inflammatory responses, but it facilitates the functional recovery by promoting migration of oligodendrocytes through specific interaction with RAGE.
EXPERIMENTAL PROCEDURES
Animals
Adult G. japonicus were used as described by Wang et al. (43). Briefly, adult animals were fed mealworms ad libitum and housed in an air-conditioned room with a controlled temperature (22–25 °C) and saturated humidity. Anesthesia was induced by cooling the animals on ice prior to tail amputation. Amputation was performed at the sixth caudal vertebra, identified based on the special tissue structure present at that position (41), by placing a slipknot of nylon thread and pulling gently until the tail was detached, thus mimicking the autotomy undergone for natural defense.
For lipopolysaccharide (LPS) treatment, LPS (Sigma) was dissolved in normal saline and injected intraperitoneally (7.5 mg/kg body weight) according to the protocol of Hasegawa et al. (44). Control experiments were performed by injection of an equivalent amount of normal saline.
All experiments were conducted in accordance with guidelines of the National Institutes of Health (Guide for the Care and Use of Laboratory Animals, 1985), and the Guidelines for the Use of Animals in Neuroscience Research by the Society for Neuroscience. Experiments were approved according to the Animal Care and Use Committee of Nantong University and the Jiangsu Province Animal Care Ethics Committee. All geckos (n = 15) were anesthetized on ice prior to sacrifice.
Surveys of HMGB1 Paralogs in Vertebrates
A cDNA library of the brain and spinal cord from G. japonicus was constructed according to the methods described previously (45). In a large scale sequencing of the cDNA library, more than 5000 clones were analyzed for coding probability with the DNATools program (46). Sequences of HMGB1 paralogs were found based on BLASTP alignment against genome databases deposited at the National Center for Biotechnology Information (47). Phylogenetic tree was constructed using the PHYML implementation of Maximum-Likelihood, with the GTR (CDS sequences) and JTT (protein sequences) substitution model (48).
Preparation of Recombinant Proteins in Eukaryotic Cells and Immunization of Rabbit
Recombinant constructs of gHMGB1 paralogs, gHMGB1a and gHMGB1b, and their mutations, gHMGB1ma and gHMGB1mb, in the pRAG2a expression vector were transfected into 293T cells using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. After a 48-h culture in a 37 °C humidified incubator with 8% CO2, cells were removed by centrifugation at 10,000 rpm for 20 min. Both the cell supernatants and homogenate were filtered through a 0.45-μm filter prior to purification by Ni2+-nitrilotriacetic acid chromatography kit (Qiagen). The process for production of the recombinant proteins referred to the manufacturer's instructions. Purity of the recombinant proteins was subsequently determined by separating each protein in a 12% SDS-PAGE and staining with Coomassie Brilliant Blue R-250.
Rabbits were immunized subcutaneously with 100 μg of recombinant HMGB1 mixed in 0.5 ml of Gerbu adjuvant (Biotech, Poway, CA). Three booster injections were given at 2-week intervals using the same antigen dose. After the final booster dose, rabbits were bled, and sera were separated and stored at −20 °C. Antibody titer was determined using an ELISA, and antibody reactivity was confirmed in a Western blot analysis. Mouse HMGB1 (mHMGB1) recombinant protein and polyclonal antibody were also prepared in parallel as positive controls.
Quantitative Real Time-PCR (Q-PCR)
Total RNA was prepared with TRIzol (Invitrogen) from different tissues, including the brain, spinal cord, kidney, liver, ovary, testis, lung, and spleen of adult geckos. Total RNAs were also extracted from 0.5-cm spinal cord segments of 20 geckos amputated from the sixth caudal vertebra at 1 and 3 days and 1 and 2 weeks, respectively.
For Q-PCR examination of gHMGB1 temporal expression, the first-strand cDNA was synthesized using Omniscript reverse transcription kit (Qiagen) in a 20-μl reaction system containing 2 μg of total RNA, 0.2 units/μl Moloney murine leukemia virus reverse transcriptase, 0.5 mm dNTP mix, 1 μm oligo(dT) primer. The cDNA was diluted 1:5 before use in Q-PCR assays. The sequence-specific primers and TaqMan probe were designed and synthesized by Invitrogen. The primer pair and probe for gHMGB1a are as follows: forward primer 5′-GAG TGA GGA GGC TGC GTA TCG-3′, reverse primer 5′-CCC TTT GCC CAT ACT GAT GAT ATG TC-3′, and TaqMan probe 5′-CCG CTC ACA GCC ATT GCA GTG CAT TGG-3′; gHMGB1b, forward primer 5′-GAG CGG AGA GAG TGA GGA GGC TG-3′, reverse primer 5′-CGG CTT CTT AGG ATC TCC TTT GCC C-3′, and TaqMan probe 5′-GAG GAG GCT GCG TCT GGC TCC CGC TCT C-3′. Q-PCRs were performed in a final volume of 20 μl (1 μl of cDNA template and 19 μl of Q-PCR buffer containing 2.5 mmol/liter MgCl2, 0.2 mmol/liter dNTPs, antisense and sense primers 0.5 μmol/liter, TaqMan probe 0.4 μmol/liter, DNA polymerase 0.2 μl, and 1× DNA polymerase buffer). The Rotor-Gene 5 software (Corbett Research, Rotor-Gene, Australia) was used for real time PCR analysis. Reactions were processed using one initial denaturation cycle at 93 °C for 2 min followed by 40 cycles of 93 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s. Fluorescence was recorded during each annealing step. At the end of each PCR run, data were automatically analyzed by the system and amplification plots obtained. gHMGB1 full-length plasmid was used to prepare standard curves and used as a specificity control for real time PCR. The expression levels of the gHMGB1 cDNA were normalized to an endogenous EF-1α cDNA using forward primer 5′-CCT TCA AAT ATG CCT GGG T-3′, reverse primer 5′-CAG CAC AGT CAG CTT GAG AG-3′, and TaqMan probe 5′-TTG GAC AAG CTG AAG GCA GAA CGT G-3′. In addition, a negative control without the first-strand cDNA was also performed. The statistical analyses were done by STATA 7.0 software.
Northern Blotting
Northern blotting was performed as described by Wang et al. (39). A total of 4 μg of RNA extracted from brain, spinal cord, kidney, liver, ovary, and testis of adult geckos was electrophoresed and blotted onto a nylon membrane (Osmonics Inc.). The blots were hybridized at high stringency with digoxigenin-labeled gHMGB1a or gHMGB1b riboprobe (1 μg/ml in DIG Easy Hyb) for 15 h at 58 °C and washed twice in 2× SSC with 0.1% SDS at 25 °C for 5 min each and twice in 0.1× SSC with 0.1% SDS at 65 °C for 20 min each. They were then incubated in a blocking solution, pH 7.5, containing 100 mm maleic acid, 150 mm NaCl, and 1% blocking reagent (Roche Applied Science) followed by incubation in the blocking solution plus anti-digoxigenin-alkaline phosphatase (Roche Applied Science) diluted 1:10,000, for 1 and 2 h, respectively, at room temperature. After washing with 100 mm maleic acid buffer, pH 7.5, containing 150 mm NaCl and 0.3% Tween 20, and then 100 mm Tris-HCl buffer, pH 9.5, containing 100 mm NaCl, the hybridized bands were visualized by CDP-Star (Roche Applied Science) and recorded by exposure to x-ray film.
Plasmid Construction, Cell Culture, and Treatment
Open reading frame of gHMGB1a or gHMGB1b was constructed into pEGFP-N1 vector. gHMGB1a mutation (gHMGB1ma, K90E) and gHMGB1b mutation (gHMGB1mb, E90K) were made by PCR-based site-directed mutagenesis (QuikChangeTM, Stratagene) using primers 5′-CAA AAA AGA AGT TCG AGG ACC CAA ATG CAC-3′ and 5′-GTG CAT TTG GGT CCT CGA ACT TCT TTT TTG-3′ for gHMGB1ma and primers 5′-AAG AAG TTC GAG GAT CCC AAT GCA CCC AAG-3′ and 5′-CTT GGG TGC ATT GGG ATC CTC GAA CTT CTT-3′ for gHMGB1mb.
Murine macrophage RAW 264.7 cells, human neuroblastoma cell line SH-SY5Y (Chinese Academy of Sciences, Shanghai Institutes for Biological Sciences Cell Resource Center), or gecko oligodendrocyte cell line Gsn3 (49) were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% (v/v) fetal bovine serum in a 37 or 30 °C humidified incubator with 5% CO2. Cells were treated with 1, 5, and 10 μg/ml recombinant proteins or commercial human HMGB1 (hHMGB1, Sigma), respectively. Alternatively, RAW 264.7 or SH-SY5Y cells were transfected with pEGFP-gHMGB1a, pEGFP-gHMGB1b, pEGFP-gHMGB1ma, or pEGFP-gHMGB1mb plasmids by FuGENE HD transfection reagent (Roche Applied Science) following the manufacturer's instructions. Cells were also counterstained with the Hoechst 33342 (1 μg/ml) for 10 min at 37 °C or stained with annexin V-PE for 10 min at 20–25 °C and mounted on slide glasses with mounting medium. Images were captured on a Nikon Diaphot microscope.
Western Blot
Protein was extracted from cells with a buffer containing 1% SDS, 100 mm Tris-HCl, 1 mm PMSF, and 0.1 mm β-mercaptoethanol, following treatment with 1 μg/ml recombinant proteins or commercial hHMGB1 for 24 and 48 h. Alternatively, the cells were transfected with pEGFP-gHMGB1a, pEGFP-gHMGB1b, pEGFP-gHMGB1ma, or pEGFP-gHMGB1mb plasmids for 24 and 48 h and then were collected by flow cytometry (BD Biosciences, FACSAria). Protein concentration of each specimen was detected by the Bradford method to maintain the same loads. Protein extracts were heat-denatured at 95 °C for 5 min, electrophoretically separated on 10% SDS-PAGE, and transferred to PVDF membranes. The membranes were subjected to the reaction with a 1:1000 dilution of primary antibodies in TBS buffer at 4 °C overnight, followed by a reaction with secondary antibody conjugated with goat anti-rabbit HRP dilution 1:1000 (Santa Cruz Biotechnology) at room temperature for 2 h. After the membrane was washed, the HRP activity was detected using an ECL kit. The image was scanned with a GS800 Densitometer Scanner (Bio-Rad), and the data were analyzed using PDQuest 7.2.0 software (Bio-Rad). β-Actin (1:5000) was used as an internal control. Antibodies used in Western blot are as follows: gHMGB1a and gHMGB1b; cleaved caspase 3 (Asp-175, Cell Signaling); RAGE, TLR2, TLR4, p-CREB, and CREB (Santa Cruz Biotechnology); JNK, p-JNK, p65NFκB, p-ERK1/2, ERK1/2, p-p38, p38, and β-actin (Abcam).
RhoA, Rac1, or Cdc42 activation was determined using the rhotekin-RBD that specifically binds activated Rho and the PBD-PAK that has a high affinity for both GTP-Rac and GTP-Cdc42 (RhoA/Rac1/Cdc42 Activation Assay Combo Biochem kit, Cytoskeleton, Denver). In brief, the cells were lysed with ice-cold cell lysis buffer containing 50 mm Tris-HCl, pH 7.4, 2 mm MgCl2, 1% Nonidet P-40, 10% glycerol, 100 mm NaCl, and protease inhibitor mixture (Roche Diagnostics) and centrifuged for 5 min at 14,000 × g. The equivalent protein amounts of lysate (500 μg of total cell protein) were performed using pulldown assay with 50 μg of rhotekin-RBD beads and 20 μg of PAK-PBD beads and rotated for 60 min at 4 °C. The beads were washed three times with lysis buffer and heated for 5 min at 100 °C in SDS-PAGE sample buffer, and then analyzed for bound RhoA, Rac1, and Cdc42 molecules by Western blotting using anti-RhoA antibody (1:500, Cytoskeleton), anti-Rac1 antibody (1:500, Cytoskeleton), or anti-Cdc42 antibody (1:500, Cytoskeleton).
Assays for TNF-α and IL-1α
For examination of TNF-α and IL-1α transcriptional expression, murine macrophage RAW 264.7 cells following transfection with plasmids at 1, 2, or 3 days and subsequent selection by flow cytometry (BD FACSAria) were determined by RT-PCR using mouse TNF-α- and IL-1α-specific primers (forward 5′-CCA CCA TCA AGG ACT CAA AT-3′ and reverse 5′-GGT CAC CAA ATC AGC GTT-3′ for TNF-α and forward 5′-GCA CCT TAC ACC TAC CAG-3′ and reverse 5′-TGA ATA GAC TCC CGA AAT-3′ for IL-1α). Mouse GAPDH amplified from the same samples using gene-specific primers (forward 5′-TGA AGT CGC AGG AGA CAA CC-3′ and reverse 5′-GGT GGA GCC AAA AGG GTC A-3′) served as housekeeping controls. PCR parameters were 26 (TNF-α) or 38 (IL-1α) cycles of denaturation at 95 °C for 30 s, primer annealing at 50 °C for 30 s, and primer extension at 72 °C for 30 s. A final extension of 10 min was performed at 72 °C before storing the samples at 4 °C.
Two-site sandwich ELISA was used to quantify TNF-α and IL-1α in the extracts of murine macrophage RAW 264.7 cells cultured with 1 μg/ml recombinant proteins for 24 h or segments of 0.5 cm of proximal spinal cord injected with 5 μl of 300 μg/ml recombinant proteins at 24, 48, or 72 h, respectively, according to the manufacturer's instructions (Duo-Set; R&D Systems, Minneapolis, MN).
Tissue Immunohistochemistry
The vertebral segments were harvested, post-fixed, and sectioned. Sections were allowed to incubate with polyclonal rabbit anti-gecko HMGB1a, rabbit anti-gecko HMGB1b antibody (1:100 dilution), polyclonal rat anti-bovine cd11b antibody (1:200 dilution, Millipore), or polyclonal mouse anti-human NF200 antibody (1:200 dilution, Abcam) at 4 °C for 36 h. The sections were further reacted with the FITC-labeled secondary antibody goat anti-mouse IgG (1:400 dilution, Invitrogen) or the TRITC-labeled secondary antibody donkey anti-rabbit IgG (1:400 dilution, Invitrogen) at 4 °C overnight, followed by observation under a confocal laser scanning microscope (Leica, Heidelberg, Germany).
Immunoprecipitation
RAW 264.7 or SH-SY5Y cells were washed twice with cold phosphate-buffered saline and then extracted with lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerolphosphate, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, and complete protease inhibitors from Roche Applied Science). Whole cell extracts were centrifuged at 14,000 rpm for 20 min to remove the debris. The proteins in the supernatant were measured using a Protein Assay Kit II (Bio-Rad). For immunoprecipitation analysis, 500 μg of total cell lysates was precleared with protein A plus G-Sepharose before incubation with specific antibodies, followed by addition of protein A plus G-Sepharose. The precipitated proteins were resolved in 2× SDS-PAGE sample buffer and separated by electrophoresis on 10–12% SDS-PAGE. After transferring onto a polyvinylidene difluoride membrane (Millipore Corp.), they were incubated with anti-His-tag antibody and then with horseradish peroxidase-conjugated secondary antibody (Cell Biolabs, Inc.).
Cell Migration Assay
Migration of oligodendrocyte Gsn3 was studied using 6.5-mm Transwell chambers with 8-μm pores (Corning Costar) as described previously (50). 100 μl of oligodendrocytes (2 × 105 cells/ml) resuspended in DMEM/F-12 were transferred to the top chambers of each transwell and allowed to migrate at 30 °C in 5% CO2 for 36 h, and 600 μl of DMEM/F-12 including 1 μg/ml recombinant proteins or antibodies (1:50) was injected into the lower chambers. The upper surface of each membrane was cleaned with a cotton swab at the indicated time point. Cells adhering to the bottom surface of each membrane were stained with 0.1% crystal violet, imaged, and counted using a DMR inverted microscope (Leica Microsystems). Assays were done three times using triplicate wells.
RESULTS
HMGB1 Paralogs Are Broadly Retained from Fish Onward in the Phylogeny
To identify whether HMGB1 paralogs exist in the reptiles, in which some species are capable of spontaneously regenerating spinal cord, we constructed brain and spinal cord cDNA library from G. japonicus. Two cDNA clones (GenBankTM accession numbers HM239648 and HM239649) were thus obtained by 5′-rapid amplification of cDNA ends, and each encoded protein consisted of two highly conserved HMG boxes and a longest acidic tail, characteristic of the HMGB1 family (51, 52). Therefore, the two clones were designated as gHMGB1a and gHMGB1b, respectively. Our results indicated that reptiles, together with teleosts and amphibians with the ability to regenerate their adult spinal cord, retained HMGB1 paralogs.
A question then arises as to whether chicken and mammals, which have been shown to be a failure in regenerating their adult spinal cord, still keep two paralogs of HMGB1. It is interesting to note that mammalian HMGB1 has two seemingly diametrically opposed functions in promoting regeneration of many tissues and progression of inflammation (53). To address the issues, candidates of the HMGB1 paralogs in human, mouse, rat, and chicken genomic databases were searched using BLASTP alignment. Results have demonstrated that mouse and rat evidently preserve HMGB1 two paralogs with GenBankTM accession numbers as follows: mouse HMGB1-like XP_889413 and mouse HMGB1 NP_034569; rat HMGB1-like XP_003753270 and rat HMGB1 NP_037095, respectively. Although we have found a human hmgb1 pseudogene located in chromosome 3 analyzed by 1000 genome browser (GenBankTM accession number NG_000897.5, www.ncbi.nlm.nih.gov), it cannot preclude the possibility of human retaining two paralogs during the evolution. The same case appears in mouse, in which dozens of hmgb1 pseudogenes locate in chromosomes 2, 4, 6, 11, 13, and X (data not shown). Similarly, the possibility of the two paralogs in chicken cannot be ruled out. A phylogenetic tree constructed using the PHYML implementation of Maximum-Likelihood demonstrated that gHMGB1b clustered with human HMGB1 (Fig. 1), suggesting that the two homologs might share some similar characteristics in primary structure or even in physiological roles.
FIGURE 1.
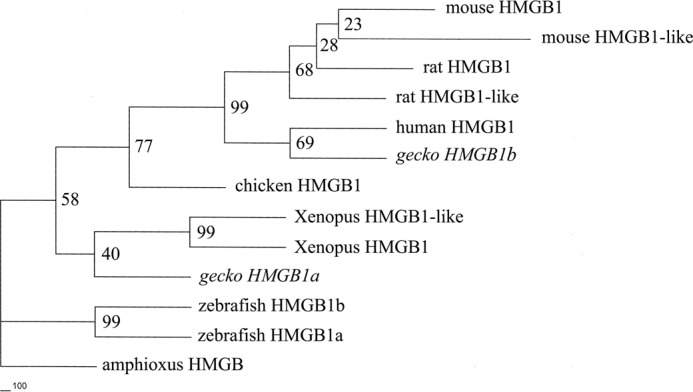
Phylogenetic tree of gHMGB1 paralogs and those of other representative vertebrates constructed by the neighbor joining method within the package PHYLIP 3.5c. Bootstrap majority consensus values on 1000 replicates are indicated at each branch point in percent. Amphioxus HMGB was designated as outgroup. Sequences obtained from GenBankTM are as follows: zebrafish HMGB1a (NM_199555); zebrafish HMGB1b (NM_001099251); Xenopus HMGB1 (U21933); Xenopus HMGB1-like (BC054148); chicken HMGB1 (NP_990233); rat HMGB1 (NP_037095); rat HMGB1-like (XP_003753270); mouse HMGB1 (NP_034569); mouse HMGB1-like (XP_889413); and human HMGB1 (NP_002119).
Differential Responses of gHMGB1a and gHMGB1b to the Spinal Cord Trauma and Inflammatory Challenge
Both Northern blotting and real time PCR were conducted to elucidate a basic expression pattern of the two gHMGB1 paralogs. As shown in Fig. 2, A and B, all samples revealed two transcripts, similar to the HMGB1 signals obtained in canine tissues of about 1.3 and 2.4 kb (54). The more sensitive real time PCR technique showed that gHMGB1a was ubiquitously expressed in all tissues examined but with low abundance in the kidney and liver (Fig. 2C). For gHMGB1b, the ovary displayed strong transcripts of 1.3 and 2.4 kb, with weak expression in the other tissues (Fig. 2B).
FIGURE 2.

Transcriptional expression of gHMGB1 paralogs in tissues and differential responses to injury and infectious challenges. A and B, Northern blotting analysis of gHMGB1a and gHMGB1b transcripts in different tissues. C, quantitative real time PCR analysis of gHMGB1a transcripts in different gecko tissues. Gecko EF-1α was used for normalization. D and E, quantitative RT-PCR amplification of gHMGB1a and gHMGB1b in the spinal cord from L13 to the 6th caudal vertebra following tail amputation at 0 day (con), 1 and 3 days (d) and 1 and 2 weeks (w). F and G, quantitative RT-PCR amplification of gHMGB1a and gHMGB1b in the different tissues after injection of LPS (7.5 mg/kg body weight) intraperitoneally. Quantities were normalized to endogenous EF-1α expression. Error bars represent the standard deviation (*, p < 0.01).
To understand the potential roles of gHMGB1 two paralogs, which might be involved in differential responses to the different stimuli, we established spinal cord trauma and lipopolysaccharide (LPS) injection models to determine the transcriptional expression changes of gHMGB1a and gHMGB1b. Real time PCR detection showed that the abundance of gHMGB1a mRNA in the injury site of the spinal cord displayed no obvious changes at 1 and 3 days and 1 and 2 weeks after tail amputation (Fig. 2D). In contrast, gHMGB1b transcription in spinal cord significantly increased at 3 days after amputation and decreased to the control level at 1 week. At 2 weeks after amputation, the expression of gHMGB1b increased again (Fig. 2E).
LPS dissolved in normal saline was injected intraperitoneally (7.5 mg/kg body weight). Samples were analyzed for 1 day (n = 15), 2 days (n = 15), and 3 days (n = 15) after injection. Tissues, including brain, spinal cord, spleen, and lung, were selected for analysis. Lung was chosen for its association with high level expression of HMGB1 after inflammation or acute injury (23, 55), and kidney and liver were excluded for the low abundance of HMGB1 (Fig. 2, A and B). Real time PCR revealed that gHMGB1a transcripts in brain decreased at 3 days after injection. However, its expression markedly increased at 2 and 3 days in spleen (Fig. 2F), an organ that contains quite a number of macrophages involved in the induction and maintenance of both innate and acquired immune defense (56). Comparatively, gHMGB1b transcripts demonstrated no obvious changes in all tissues examined (Fig. 2G).
These results indicated that gHMGB1a was probably involved in the response of external inflammatory stimuli, and gHMGB1b was probably involved in the regulation of spinal cord regeneration following injury.
gHMGB1a and gHMGB1b Colocalized with Neuron and Microglia
In CNS, HMGB1 displays a complex temporal and spatial distribution pattern and is implicated in facilitating neurite outgrowth and cell migration (57, 58). HMGB1 also serves to induce neuroinflammation after injury, such as lesions in the spinal cord and brain (16, 18, 59). To elucidate whether neuron and microglia, a type of glial cells that are the resident macrophages of the brain and spinal cord, have participated in the physiological processes through activation of one or both HMGB1 paralog signalings, we first succeeded in preparation of the recombinant proteins by eukaryotic expression in 293T cells and then of the polyclonal antibodies (Fig. 3, A and B). We detected the cellular localization of gHMGB1a and gHMGB1b after spinal cord injury at 3 days, using double immunofluorescent staining with combinations of antibodies, namely gHMGB1a/NF200 or gHMGB1b/NF200 for neurons and gHMGB1a/CD11b or gHMGB1b/CD11b for microglias. The results indicated that gHMGB1a was ubiquitously distributed in the gray and white matter, displaying colocalization with both neurons and microglias (Fig. 3, C and D). In comparison, the injury-induced gHMGB1b expression was mostly distributed in the gray matter, with colocalization with both neurons and microglias (Fig. 3, C and D). The results demonstrated that both neurons and microglias of the gecko could produce two HMGB1 paralogs, although only gHMGB1b exhibited marked responses to spinal cord injury.
FIGURE 3.

Distribution of gHMGB1a and gHMGB1b in the spinal cord. A, preparation of gHMGB1a and gHMGB1b recombinant proteins by eukaryotic expression in 293T cells. B, gHMGB1a and gHMGB1b polyclonal antibodies react specifically with spinal cord tissue by Western blot analysis. C, colocalization of gHMGB1a and gHMGB1b with NF-positive cells (arrowhead). D, colocalization of gHMGB1a and gHMGB1b with CD11b-positive cells (arrowhead). Lane M indicates protein marker. Rectangle indicates region magnified below. Scale bars, 100 μm.
Intracellular gHMGB1a and gHMGB1b Induced Differential Inflammatory Responses
HMGB1 was found to induce the synthesis of various inflammatory cytokines, including TNF-α and IL-1α by macrophages (60, 61). To gain insight into the gHMGB1a and gHMGB1b functions on mediating the release of the inflammatory cytokines in microglial cells, we overexpressed the plasmids of pEGFP-gHMGB1a or pEGFP-gHMGB1b into murine macrophage RAW 264.7 cells for 24 and 48 h, respectively. Results showed that gHMGB1a is localized in the cytoplasm and nucleus, whereas gHMGB1b is primarily localized in the nucleus of macrophages (Fig. 4B). The positive cells were selected by flow cytometry and subjected to transcriptional analysis (Fig. 4C). The expressions of TNF-α and IL-1α were significantly up-regulated after transfection with pEGFP-gHMGB1a for 48 h, and they were slightly up-regulated or unchanged after transfection with pEGFP-gHMGB1b (Fig. 4, D and E). Interestingly, there is only one amino acid difference in the first 20-residue fragment of the B-box between gHMGB1a and gHMGB1b (Fig. 4A), which has been found to affect the inflammatory activity (32). We subsequently exchanged the distinct amino acid residue by mutation (Lys ↔ Glu), and we examined the transcription of proinflammatory cytokines by transfection of mutated gHMGB1a (gHMGB1ma, Lys → Glu) or mutated gHMGB1b (gHMGB1mb, Glu → Lys) in RAW 264.7 cells. The potential of mediating the release of TNF-α and IL-1α by intracellular gHMGB1a and gHMGB1b has accordingly been exchanged. gHMGB1mb was capable of up-regulating the transcripts of IL-1α, but the role of gHMGB1ma was reversed (Fig. 4, F and G). The results revealed that the intracellular gHMGB1a and gHMGB1b induced differential inflammatory responses, and gHMGB1a was the main inflammatory mediator.
FIGURE 4.

Production of inflammatory cytokines in macrophage RAW 264.7 cells transfected with pEGFP-gHMGB1a, pEGFP-gHMGB1b, and mutation plasmids. A, alignment of first 20 amino acids from B-box domain of gHMGB1 paralogs, human and mouse HMGB1, showing an amino acid difference in gHMGB1b. B, overexpression of pEGFP-N1, pEGFP-gHMGB1a, pEGFP-gHMGB1b, mutation plasmids (Lys ↔ Glu) pEGFP-gHMGB1ma and pEGFP-gHMGB1mb in RAW 264.7 cells for 48 h. C, transfected cells were selected by flow cytometry. D and E, transcriptional analysis of TNF-α and IL-1α in macrophages following transfected with pEGFP-gHMGB1a and pEGFP-gHMGB1b. F and G, transcriptional analysis of TNF-α and IL-1α in macrophages following transfected with pEGFP-gHMGB1ma and pEGFP-gHMGB1mb. Control means cells transfected by pEGFP-N1. Quantities were normalized to endogenous GAPDH expression. Error bars in D–G represent the standard deviation (*, p < 0.01). Scale bars, 30 μm.
Involvements of Intracellular gHMGB1 Two Paralogs in the Apoptosis of Neurons
HMGB1 was found to activate not only macrophages but also neurons (23, 42). A plethora of studies have revealed that HMGB1 plays double-edged roles in neural development and neurodegeneration (59). Therefore, we transfected the plasmids of gHMGB1a and gHMGB1b into PC12 and SH-SY5Y cell lines to examine their effects on neurites. Results indicated that the two paralogs were incapable of promoting neurites outgrowth and elongation (data not shown), quite distinct from mammalian HMGB1 (57, 58). However, apoptosis was induced by overexpression of gHMGB1a, gHMGB1b, or their mutants for 72 h (Fig. 5, A and B). Annexin-V staining also demonstrated a marked correlation with apoptotic transfected cells (Fig. 5, C–Q). These indicated that both intracellular gHMGB1a and gHMGB1b could induce the neuronal apoptosis.
FIGURE 5.

Effects of intracellular gHMGB1 paralogs on the neurons. A, Western blot analysis of cleaved caspase3 in SH-SY5Y cells after transfection with pEGFP-gHMGB1a, pEGFP-gHMGB1b, pEGFP-gHMGB1ma, and pEGFP-gHMGB1mb for 72 h. Control means cells transfected with pEGFP-N1; B, statistics of A, and quantities were normalized to endogenous β-actin. Error bars in B represent the standard deviation (*, p < 0.01); C–Q, immunohistochemistry showing colocalization of transfected cells with annexin-V. Scale bars, 40 μm.
Extracellular gHMGB1 Two Paralogs Do Not Facilitate the Release of Inflammatory Cytokines
Previous studies have indicated that immune cells and damaged or necrotic cells can release HMGB1 into the extracellular milieu, where it triggers inflammatory responses by acting on various immune cells (33). To examine whether the extracellular gHMGB1 paralogs of regenerative gecko, especially for the significantly up-regulated gHMGB1b in the traumatic spinal cord, enacted the analogical inflammation-mediating function, RAW 264.7 cells were cultured with 0, 1, 5, or 10 μg/ml recombinant proteins for 24 h. The transcriptional expression levels of TNF-α and IL-1α exhibited no changes in macrophages (Fig. 6, A and B). We further conducted the parallel experiments by applying commercial hHMGB1 or mHMGB1 recombinant protein prepared by the same protocol. Assays by two-site sandwich ELISA showed that the recombinant proteins of gHMGB1 two paralogs, as well as their mutants, did not elicit the synthesis and secretion of TNF-α and IL-1α (Fig. 6, C–F), whereas mHMGB1 recombinant protein and commercial hHMGB1 significantly triggered the production of the two inflammatory cytokines (Fig. 6, C–F).
FIGURE 6.

Effects of gHMGB1 paralogs recombinant proteins on the release of inflammatory cytokines. A and B, determination of TNF-α and IL-1α transcripts in macrophage RAW 264.7 cells after treatment with 0, 1, 5, and 10 μg/ml recombinant proteins prepared from gHMGB1a, gHMGB1b, gHMGB1ma and gHMGB1mb, for 24 h, respectively. C and D, ELISA of TNF-α from supernants (C) and pellets (D) of macrophages cultured with 1 μg/ml gHMGB1a, gHMGB1b, gHMGB1ma. and gHMGB1mb recombinant proteins for 24 h. E and F, ELISA of IL-1α from supernants (E) and pellets (F) of macrophages cultured with 1 μg/ml gHMGB1a, gHMGB1b, gHMGB1ma, and gHMGB1mb recombinant proteins for 24 h. G, ELISA of TNF-α from spinal cord segments proximal to amputation following injection of 5 μl of 300 μg/ml gHMGB1 recombinant proteins for 24, 48, and 72 h, respectively. H, ELISA of IL-1α from spinal cord segments proximal to amputation following injection of 5 μl of 300 μg/ml gHMGB1 recombinant proteins for 24, 48, and 72 h, respectively. A total of 5 μl of 300 μg/ml mHMGB1 recombinant proteins, commercial hHMGB1, or 5 μl of 200 μg/ml LPS was performed as positive control. Negative control means ELISA of cytokines immediately after amputation or injection with a dose of 5 μl of saline. Error bars in C–H represent the standard deviation (*. p < 0.01).
Following injection of 5 μl of 300 μg/ml gHMGB1 or mHMGB1 recombinant proteins, as well as equivalent commercial hHMGB1 into the spinal cord proximal to the injured site immediately after tail amputation, we collected 0.5-cm segments at 0–3 days, respectively, and we evaluated the expression levels of inflammatory cytokines. A dose of 5 μl of saline or 5 μl of 200 μg/ml LPS was performed as control. ELISAs displayed that TNF-α and IL-1α could be directly initiated by spinal cord amputation, reaching a basic amount of inflammatory cytokines. Injection of gHMGB1 recombinant proteins was unable to potentiate the release of TNF-α and IL-1α compared with saline control (Fig. 6, G and H). But injection of LPS, mHMGB1 recombinant protein, or commercial hHMGB1 significantly elicited the up-regulation of TNF-α and IL-1α in vivo (Fig. 6, G and H). Our results demonstrated that extracellular gHMGB1 paralogs did not enact inflammatory functions in the spontaneously regenerating spinal cord.
gHMGB1a and gHMGB1b Exhibit Interactions with RAGE but Not with TLR2 or TLR4 Receptor
Studies have shown that HMGB1 initiates the multiple intracellular signaling by interaction with RAGE, TLR2, TLR4, or other family members, leading to the activation of NFκB (28, 28). To shed light on the relationship of gHMGB1a and gHMGB1b with TLR2, TLR4, and RAGE receptors, we cultured macrophage RAW 264.7 or SH-SY5Y cells with 1 μg/ml recombinant gHMGB1a, gHMGB1b, or 0.1 m PBS, pH 7.4, respectively. The two cell lines expressed the three surface receptors. However, NFκB has not been activated by the gHMGB1 two paralogs (Fig. 7, A and B), in contrast to what has been done by mammalian HMGB1 (28, 29). We further carried out immunoprecipitation experiments using anti-TLR2, -TLR4, or -RAGE antibody, and then we detected the components of the TLR2-, TLR4-, and RAGE-associated complexes with anti-His antibody, to avoid cross-reacting with endogenous HMGB1 of the cultured cells. As shown in Fig. 7, C–E, gHMGB1a or gHMGB1b coprecipitated with RAGE, but not with TLR2 or TLR4 receptor. These results indicated that gHMGB1a or gHMGB1b in spontaneously regenerating spinal cord could not bind with cell surface receptors TLR2 or TLR4 to initiate the production of inflammatory cytokines.
FIGURE 7.

Binding assays of gHMGB1a and gHMGB1b with RAGE, TLR2, and TLR4 receptors. A, Western blot analysis of RAGE, TLR2, TLR4, and p65NFκB after macrophages and SH-SY5Y cells were cultured with 1 μg/ml gHMGB1a or gHMGB1b recombinant proteins for 24 h, respectively; B, statistical analysis of A; C–E, immunoprecipitation (IP) using anti-RAGE (C), -TLR2 (D), or -TLR4 antibody (E) and detection of the components of the RAGE- (C), TLR2- (D), or TLR4-associated complexes (D) with anti-His antibody.
gHMGB1a and gHMGB1b Promote the Oligodendrocyte Migration by Activation of Rac1/Cdc42 Signaling
In the CNS, extracellular HMGB1 is highly active in enhancing neurite outgrowth through interaction with RAGE to communicate with the GTPases Cdc42 and Rac (62–64). To verify the effects of gHMGB1 paralogs on the neurons, we cocultured SH-SY5Y cells with 1 μg/ml exogenous recombinant gHMGB1a or gHMGB1b and then assayed the neurite length. Statistical analysis displayed no significant differences between control and treatment groups. Also, the paralogs did not mediate neuronal apoptosis, which could be induced by endogenous overexpression (Fig. 8, A and B). Daston and Ratner (65) have ever described that HMGB1 was the early marker of oligodendrocytes in the developing rat spinal cord, and Fages et al. (57) found that HMGB1 was involved in the migration of neuroblastoma cells. Given such defined functions, we therefore assayed the migration of oligodendrocyte Gsn3 cells cocultured with 1 μg/ml exogenous recombinant gHMGB1a or gHMGB1b. Transwell experiments demonstrated that both recombinant gHMGB1a and gHMGB1b proteins were able to potentiate the cell migration (Fig. 8, C–E and H), and anti-gHMGB1a or anti-gHMGB1b antibody (1:50 dilution) was able to block the effect, accordingly (Fig. 8, F–H).
FIGURE 8.

Effects of gHMGB1 paralog recombinant proteins on the neurons and oligodendrocytes. A, Western blot analysis of cleaved caspase3 in the SH-SY5Y cells cultured with 1 μg/ml exogenous recombinant gHMGB1a, gHMGB1b, or mutant proteins. B, immunofluorescence of SH-SY5Y cells detected by NF-200 antibody, and analysis of the neurite length. C–G, assays of oligodendrocytes Gsn3 migration by transwell experiments after cells cultured with PBS, pH 7.4 (C), recombinant gHMGB1a (D), or gHMGB1b proteins (E) for 36 h; anti-gHMGB1a antibody (F) and anti-gHMGB1b antibody (G) were able to accordingly block the effects. H, statistical analysis of migrated cell numbers in C–G. Error bars in H represent the standard deviation (*, p < 0.01). Scale bars, 25 μm in B and 50 μm in C–G.
RAGE engagement by HMGB1 ligand activates various signaling pathways in a cell-specific manner, eventually impinging on canonical NFκB and (or) CREB with consequent regulation of migration. RAGE signaling-dependent regulation of cell motility also occurs independently of its effects on gene transcription via a Dia-1/Src/Ras/PI3K/RhoA/ROCK or a Dia-1/Rac1/Cdc42 pathway (66). To unveil the migration-associated signaling(s) activated by gHMGB1a/RAGE or gHMGB1b/RAGE interaction, oligodendrocyte Gsn3 cells were cocultured with 1 μg/ml exogenous recombinant gHMGB1a or gHMGB1b. Western blots revealed that oligodendrocytes expressed the RAGE receptor (Fig. 9A), rather than TLR2 and TLR4 receptors (data not shown). Transcriptional factors NFκB and its upstream regulators p-ERK1/2 have been shown to be activated, whereas CREB and other pathway molecules p-p38 and p-JNK have not (Fig. 9A). Rac1 and Cdc42, but not RhoA, were significantly activated to mediate oligodendrocyte migration (Fig. 9B). These results demonstrated that gHMGB1a and gHMGB1b could potentiate the oligodendrocyte migration by activation of both NFκB and Rac1/Cdc42 signaling, and gHMGB1b, the up-regulated protein in spontaneously regenerating spinal cord, was the critical mediator that was possibly implicated in the promoting spinal cord regeneration.
FIGURE 9.

Examination of HMGB1/RAGE signal pathway associated with oligodendrocyte migration. A, Western blot analysis of RAGE, p65NFκB, CREB, p-CREB, JNK, p-JNK, p38, p-p38, ERK1/2, and p-ERK1/2 implicated in the pathway activation. B, determination of Rac1/Cdc42/RhoA in signaling activation. Error bars in A and B represent the standard deviation (*, p < 0.01). Con, control.
DISCUSSION
Many conflicting lines of evidence have resulted in confusion and considerable debate regarding the detrimental versus beneficial role of inflammation, particularly after CNS injury (67). On the one hand, acute inflammatory response after spinal cord injury has been well documented to mediate the progressive tissue damage (68, 69); on the other hand, some observations indicate that inflammation can also be beneficial for injured CNS tissue by promoting clearance of cell debris and secretion of neurotrophic factors (70, 71). Nevertheless, it is generally acceptable that uncontrolled inflammation drives the progression of the CNS tissue damages, whereas HMGB1 protein, a self-derived immune activator, facilitates the vicious self-propagating inflammatory circle contributing to the pathophysiology (4, 72). In the condition of uncontrolled inflammatory reaction, neutrophils, monocytes/macrophages, and lymphocytes were rapidly recruited and activated, contributing to the expansion of the lesion size and series of pathological changes by releasing a spectrum of inflammatory cytokines or generation of free radicals such as NO (69, 73–76). Extracellular HMGB1 acts as ligand and sensor for innate immunity to promote recruitments of inflammatory cells and to elicit release of inflammatory cytokines, thus initiating a vicious circle between injured neurons and uncontrolled inflammation (13, 72, 77). In traumatic spinal cord of rat, HMGB1 was significantly up-regulated from 6 h, acting on the roles of the release of TNF-α, IL-1β, and IL-6 (15, 18). Monocytes cultured with HMGB1 release TNF-α, IL-1, IL-6, IL-8, and macrophage inflammatory protein-1 but not anti-inflammatory cytokines, such as IL-10 and IL-12 (23, 25). As such, HMGB1 has become a “necrotic marker” participating in the proinflammatory cascade of secondary damage after spinal cord injury (18, 42). In contrast to the failures of adult spinal cord regeneration in chicken and mammals, several vertebrates, including fish, amphibians, and reptiles, are capable of spontaneous regeneration following cord transection or tail amputation (38, 78). Although changes in regenerative ability resulted from combinations of several factors, such as multipotent neural progenitors, extraneuronal milieu, and cellular and molecular changes acquired during evolution, any valuable clues derived from the comparative studies might be beneficial for potential regeneration in mammals. Interestingly, injured spinal cords in regeneration-competent lower vertebrates do not lead to secondary tissue damage, which often occurs in chicken and mammals. HMGB1 has not been shown to be involved in the pathophysiological events, suggesting that there is close association between tightly regulated inflammatory responses by gHMGB1 paralogs and successful spinal cord regeneration.
Two paralogs of intracellular gHMGB1 were able to trigger the apoptosis of neurons, and how these intracellular proteins activated the apoptotic signaling has not been determined. Bioinformatic analysis has even revealed that overexpressed HMGB1 can influence cell fate, such as apoptosis or proliferation, through regulation of cell circle factors, including c-Myc, p53, pRb, Ras, etc. (79). The possibility of intracellular gHMGB1-mediated neuronal apoptosis may be caused by aberrant expression of these cell circle factors. The apoptosis of neurons, although much less than those of oligodendrocytes, has been assumed as an important process that affects the development of neuronal tissue damage following acute spinal cord injury (80–82). But it has been shown to be required for the successful regeneration of the amputated tail, including spinal cord (83). In Xenopus tadpole, inhibition of caspase-dependent apoptosis resulted in a failure to induce proliferation in the growth zone, a mispatterning of axons in the regenerate, and the appearance of ectopic otoliths in the neural tube (83). The requirement of apoptosis may be non-cell autonomous for some cells during regeneration (83, 84). Distinct with necrosis, cells undergoing programmed cell death induce negligible inflammation in the surrounding tissue, due to the retention of HMGB1 within the apoptotic cells (13, 85). In regenerating spinal cord of gecko, the neuronal apoptosis mediated by up-regulation of gHMGB1b following injury would therefore produce negligible inflammatory response. In addition, intracellular gHMGB1b in immune cells of spinal cord, as shown in cultured macrophage RAW 264.7 cells, triggered less release of inflammatory cytokines. Taken together, gHMGB1b elicits limited inflammatory responses in the injured spinal cord.
One of the pathological outcomes of acute and chronic SCI is demyelination of axons at traumatic site due to oligodendrocyte death through necrosis and/or apoptosis (82). Primary demyelination occurs within the 1st day, and the subsequent progression, which is independent of injury intensity, is attributed to the second injury (86). The poorly understood inflammatory mechanisms have contributed to the increased loss of myelin after SCI (86). It is defined that both gHMGB1 paralogs have not militated against integrity of myelin by eliciting enormous inflammatory cytokines compared with those of mammals. Instead, they have been shown to potentiate migration of oligodendrocytes, a prerequisite for normal myelination in the vertebrate CNS. The cellular events might be involved in the replenishment of necrotic and/or apoptotic oligodendrocytes suffering from direct insults or ensheathment of regenerating axons.
HMGB1 could interact with multiple cell surface receptors, including RAGE, TLR2, TLR4, TLR9, and Mac-1, etc. (6, 29, 87). Among them, RAGE, TLR2, and TLR4 pathway signalings have been strongly implicated in the pathogenesis of sterile injury by activation of NFκB (29, 33, 34). HMGB1/TLR4 signaling is mostly required for the activation of inflammatory cytokine release in macrophages, whereas HMGB1/TLR2 and HMGB1/RAGE are dispensable. Exposure of macrophages to HMGB1, for example, was able to induce TNF production by macrophages derived from TLR2 knock-out and RAGE knock-out in quantities comparable with wild-type macrophages, whereas TNF release was significantly inhibited in macrophages from TLR4 knock-out mice (34). Similar results could be observed for other HMGB1-induced cytokine mediators in these macrophages, demonstrating that the TLR4 knock-out macrophages also failed to produce MIP-2, IL-6, IL-1β, and IL-10, although these were produced normally by TLR2 and RAGE knock-out macrophages (34). Furthermore, HMGB1 activation of macrophages from double knock-out of TLR2 and RAGE led to significant stimulation of TNF release, but double knock-out of TLR4 and RAGE completely prevented TNF release in response to HMGB1 (34). So, it is understandable that two paralogs of gHMGB1 failed to initiate the release of inflammatory cytokines in macrophages due to undetectable interaction with TLR2 and TLR4. RAGE has been shown to be expressed on neurons, endothelial macrophages, oligodendrocytes, and immature dendritic cells (64, 88–92). RAGE signaling requires adaptor proteins, such as diaphanous-1, TIRAP, and MyD88. The structural properties of ligand, the cell type, and the number of RAGE molecules expressed on the cell surface may condition RAGE signaling (66). HMGB1/RAGE signaling has been documented to contribute to the outgrowth of the neurites (64, 88) and cell migration (57, 89). In this study, extracellular gHMGB1a and gHMGB1b failed to promote the neurite outgrowth, but it potentiated oligodendrocyte migration in a manner of activation of both NFκB and Rac1/Cdc42 signaling, suggesting a cell-specific HMGB1/RAGE activation mechanism.
Genome duplication is an evolutionary innovation to create functionally diverse proteins from a limited number of genes. These duplicated genes were assigned to execute specific functions to meet various physiological requirements, except those suffered from degeneration (37, 93). In the meantime, animals have evolved alternative switches to regulate these paralogs, and such cases were found in the regulation of Globin gene superfamily and other proteins (43, 94, 95). Both paralogs of gHMGB1 were found to be ubiquitously expressed and share similar binding patterns with cell surface receptors. However, traumatic spinal cord induces only gHMGB1b up-regulation, rather than gHMGB1a, which is induced in spleen and lungs by infectious signals and LPS, suggesting that established switch(es) might control spatial expression of the two paralogs. The exquisite regulatory strategy, by which gHMGB1b participates in spinal cord regeneration, could be readily explained by the need of repetitive repair of tail loss in complex natural environments.
In conclusion, as illustrated in Fig. 10, the spontaneously regenerating spinal cord switches on the up-regulation of gHMGB1b rather than gHMGB1a. The intracellular protein could not create a vicious circle of inflammation by negligible release of inflammatory cytokines in macrophages or by promoting neuronal apoptosis, nor could the extracellular protein activate inflammatory signaling by undetectable interaction with TRL2 and TLR4 receptors. Instead, gHMGB1b interacted with RAGE to facilitate oligodendrocytes migration by activation of both NFκB and Rac1/Cdc42 signaling. Such cellular events under tightly controlled inflammatory milieu might be implicated in the spinal cord regeneration.
FIGURE 10.

Illustration of gHMGB1 paralog functions on different cells of traumatic spinal cord. Red ellipsoids indicate gHMGB1a, and green ones indicate gHMGB1b.
Acknowledgment
We thank Professor Wei Qian for providing His tag antibody.
This work was supported by National Natural Science Foundation of China Grants 31071874 and 31171405, the Priority Academic Program Development of Jiangsu Higher Education Institutions, and Basic Research Program of Jiangsu Education Department Grant 10KJA180041.
- SCI
- spinal cord injury
- DAMP
- endogenous damage-associated molecular pattern
- TLR
- toll-like receptors
- RAGE
- receptor for advanced glycation end product
- gHMGB1
- gecko HMGB1
- mHMGB1
- mouse HMGB1
- Q-PCR
- quantitative PCR
- CREB
- cAMP-response element-binding protein
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Brambilla R., Bracchi-Ricard V., Hu W. H., Frydel B., Bramwell A., Karmally S., Green E. J., Bethea J. R. (2005) Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 202, 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Popovich P. G., Longbrake E. E. (2008) Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci. 9, 481–493 [DOI] [PubMed] [Google Scholar]
- 3. Hausmann O. N. (2003) Post-traumatic inflammation following spinal cord injury. Spinal Cord 41, 369–378 [DOI] [PubMed] [Google Scholar]
- 4. Gao H. M., Hong J. S. (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horner P. J., Gage F. H. (2000) Regenerating the damaged central nervous system. Nature 407, 963–970 [DOI] [PubMed] [Google Scholar]
- 6. Tian J., Avalos A. M., Mao S. Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., Hua J., An L. L., Audoly L., La Rosa G., Bierhaus A., Naworth P., Marshak-Rothstein A., Crow M. K., Fitzgerald K. A., Latz E., Kiener P. A., Coyle A. J. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 [DOI] [PubMed] [Google Scholar]
- 7. Gensel J. C., Kigerl K. A., Mandrekar-Colucci S. S., Gaudet A. D., Popovich P. G. (2012) Achieving CNS axon regeneration by manipulating convergent neuro-immune signaling. Cell Tissue Res. 349, 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kvarnhammar A. M., Cardell L. O. (2012) Pattern-recognition receptors in human eosinophils. Immunology 136, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kigerl K. A., Popovich P. G. (2009) Toll-like receptors in spinal cord injury. Curr. Top. Microbiol. Immunol. 336, 121–136 [DOI] [PubMed] [Google Scholar]
- 10. Rubartelli A., Lotze M. T. (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28, 429–436 [DOI] [PubMed] [Google Scholar]
- 11. Lotze M. T., Tracey K. J. (2005) High mobility group box 1 protein (HMGB1), nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342 [DOI] [PubMed] [Google Scholar]
- 12. Rauvala H., Rouhiainen A. (2007) RAGE as a receptor of HMGB1 (Amphoterin): roles in health and disease. Curr. Mol. Med. 7, 725–734 [DOI] [PubMed] [Google Scholar]
- 13. Scaffidi P., Misteli T., Bianchi M. E. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 [DOI] [PubMed] [Google Scholar]
- 14. Qiu J., Nishimura M., Wang Y., Sims J. R., Qiu S., Savitz S. I., Salomone S., Moskowitz M. A. (2008) Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 28, 927–938 [DOI] [PubMed] [Google Scholar]
- 15. Kawabata H., Setoguchi T., Yone K., Souda M., Yoshida H., Kawahara K., Maruyama I., Komiya S. (2010) High mobility group box 1 is up-regulated after spinal cord injury and is associated with neuronal cell apoptosis. Spine 35, 1109–1115 [DOI] [PubMed] [Google Scholar]
- 16. Kim J. B., Sig Choi J., Yu Y. M., Nam K., Piao C. S., Kim S. W., Lee M. H., Han P. L., Park J. S., Lee J. K. (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu K., Mori S., Takahashi H. K., Tomono Y., Wake H., Kanke T., Sato Y., Hiraga N., Adachi N., Yoshino T., Nishibori M. (2007) Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 21, 3904–3916 [DOI] [PubMed] [Google Scholar]
- 18. Chen K. B., Uchida K., Nakajima H., Yayama T., Hirai T., Rodriguez Guerrero A., Kobayashi S., Ma W. Y., Liu S. Y., Zhu P., Baba H. (2011) High-mobility group box-1 and its receptors contribute to proinflammatory response in the acute phase of spinal cord injury in rats. Spine 36, 2122–2129 [DOI] [PubMed] [Google Scholar]
- 19. Read C. M., Cary P. D., Crane-Robinson C., Driscoll P. C., Norman D. G. (1993) Solution structure of a DNA-binding domain from HMG1. Nucleic Acids Res. 21, 3427–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasheva E., Sarov M., Bidjekov K., Ugrinova I., Sarg B., Lindner H., Pashev I. G. (2004) In vitro acetylation of HMGB-1 and -2 proteins by CBP: the role of the acidic tail. Biochemistry 43, 2935–2940 [DOI] [PubMed] [Google Scholar]
- 21. Bustin M. (1999) Regulation of DNA-dependent activities by the functional motifs of the high mobility group chromosomal proteins. Mol. Cell. Biol. 19, 5237–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bianchi M. E., Beltrame M. (2000) Upwardly mobile proteins. The role of HMG proteins in chromatine structure, gene expression and neoplasia. EMBO Rep. 1, 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H., Bloom O., Zhang M., Vishnubhakat J. M., Ombrellino M., Che J., Frazier A., Yang H., Ivanova S., Borovikova L., Manogue K. R., Faist E., Abraham E., Andersson J., Andersson U., Molina P. E., Abumrad N. N., Sama A., Tracey K. J. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251 [DOI] [PubMed] [Google Scholar]
- 24. Yang H., Tracey K. J. (2010) Targeting HMGB1 in inflammation. Biochim. Biophys. Acta 1799, 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Andersson U., Harris H. E. (2010) The role of HMGB1 in the pathogenesis of rheumatic disease. Biochim. Biophys. Acta 1799, 141–148 [DOI] [PubMed] [Google Scholar]
- 26. Tsung A., Sahai R., Tanaka H., Nakao A., Fink M. P., Lotze M. T., Yang H., Li J., Tracey K. J., Geller D. A., Billiar T. R. (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201, 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taniguchi N., Kawahara K., Yone K., Hashiguchi T., Yamakuchi M., Goto M., Inoue K., Yamada S., Ijiri K., Matsunaga S., Nakajima T., Komiya S., Maruyama I. (2003) High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 48, 971–981 [DOI] [PubMed] [Google Scholar]
- 28. Park J. S., Svetkauskaite D., He Q., Kim J. Y., Strassheim D., Ishizaka A., Abraham E. (2004) Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377 [DOI] [PubMed] [Google Scholar]
- 29. Park J. S., Gamboni-Robertson F., He Q., Svetkauskaite D., Kim J. Y., Strassheim D., Sohn J. W., Yamada S., Maruyama I., Banerjee A., Ishizaka A., Abraham E. (2006) High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 290, C917–C924 [DOI] [PubMed] [Google Scholar]
- 30. Ozinsky A., Underhill D. M., Fontenot J. D., Hajjar A. M., Smith K. D., Wilson C. B., Schroeder L., Aderem A. (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 13766–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bianchi M. E., Manfredi A. A. (2007) High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 220, 35–46 [DOI] [PubMed] [Google Scholar]
- 32. Li J., Kokkola R., Tabibzadeh S., Yang R., Ochani M., Qiang X., Harris H. E., Czura C. J., Wang H., Ulloa L., Wang H., Warren H. S., Moldawer L. L., Fink M. P., Andersson U., Tracey K. J., Yang H. (2003) Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol. Med. 9, 37–45 [PMC free article] [PubMed] [Google Scholar]
- 33. Ulloa L., Messmer D. (2006) High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 17, 189–201 [DOI] [PubMed] [Google Scholar]
- 34. Yang H., Hreggvidsdottir H. S., Palmblad K., Wang H., Ochani M., Li J., Lu B., Chavan S., Rosas-Ballina M., Al-Abed Y., Akira S., Bierhaus A., Erlandsson-Harris H., Andersson U., Tracey K. J. (2010) A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U.S.A. 107, 11942–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moleri S., Cappellano G., Gaudenzi G., Cermenati S., Cotelli F., Horner D. S., Beltrame M. (2011) The HMGB protein gene family in zebrafish: Evolution and embryonic expression patterns. Gene Expr. Patterns 11, 3–11 [DOI] [PubMed] [Google Scholar]
- 36. Putnam N. H., Butts T., Ferrier D. E., Furlong R. F., Hellsten U., Kawashima T., Robinson-Rechavi M., Shoguchi E. (2008) The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071 [DOI] [PubMed] [Google Scholar]
- 37. Mazet F., Shimeld S. M. (2002) Gene duplication and divergence in the early evolution of vertebrates. Curr. Opin. Genet. Dev. 12, 393–396 [DOI] [PubMed] [Google Scholar]
- 38. Ferretti P., Zhang F., O'Neill P. (2003) Changes in spinal cord regenerative ability through phylogenesis and development: lessons to be learnt. Dev. Dyn. 226, 245–256 [DOI] [PubMed] [Google Scholar]
- 39. Wang Y., Wang R., Jiang S., Zhou W., Liu Y., Wang Y., Gu Q., Gu Y., Dong Y., Liu M., Gu X., Ding F., Gu X. (2011) Gecko CD59 is implicated in proximodistal identity during tail regeneration. Plos one 6, e17878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y., Gu Q., Dong Y., Zhou W., Song H., Liu Y., Liu M., Yuan Y., Ding F., Gu X., Wang Y. (2012) Inhibition of gecko GSK-3β promotes elongation of neurites and oligodendrocyte processes but decreases the proliferation of blastemal cells. J. Cell. Biochem. 113, 1842–1851 [DOI] [PubMed] [Google Scholar]
- 41. McLean K. E., Vickaryous M. K. (2011) A novel amniote model of epimorphic regeneration: the leopard gecko, Eublepharis macularius. BMC Dev. Biol. 16, 11–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tian L., Rauvala H., Gahmberg C. G. (2009) Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 30, 91–99 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y., Dong Y., Song H., Liu Y., Liu M., Yuan Y., Ding F., Gu X., Wang Y. (2012) Involvement of gecko SNAP25b in spinal cord regeneration by promoting outgrowth and elongation of neurites. Int. J. Biochem. Cell Biol. 44, 2288–2298 [DOI] [PubMed] [Google Scholar]
- 44. Hasegawa A., Iwasaka H., Hagiwara S., Noguchi T. (2011) Relationship between HMGB1 and tissue protective effects of HSP72 in a LPS-induced systemic inflammation model. J. Surg. Res. 169, 85–91 [DOI] [PubMed] [Google Scholar]
- 45. Liu Y., Ding F., Liu M., Jiang M., Yang H., Feng X., Gu X. (2006) EST-based identification of genes expressed in brain and spinal cord of Gekko japonicus, a species demonstrating intrinsic capacity of spinal cord regeneration. J. Mol. Neurosci. 29, 21–28 [DOI] [PubMed] [Google Scholar]
- 46. Rehm B. H. (2001) Bioinformatic tools for DNA/protein sequence analysis, functional assignment of genes and protein classification. Appl. Microbiol. Biotechnol. 57, 579–592 [DOI] [PubMed] [Google Scholar]
- 47. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 [DOI] [PubMed] [Google Scholar]
- 49. Liu M., Gu Y., Liu Y., Li J., He J., Lin S., Gu X. (2010) Establishment and characterization of two cell lines derived from primary cultures of Gekko japonicus cerebral cortex. Cell Biol. Int. 34, 153–161 [DOI] [PubMed] [Google Scholar]
- 50. Yu B., Qian T., Wang Y., Zhou S., Ding G., Ding F., Gu X. (2012) miR-182 inhibits Schwann cell proliferation and migration by targeting FGF9 and NTM, respectively, at an early stage following sciatic nerve injury. Nucleic Acids Res. 40, 10356–10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sessa L., Bianchi M. E. (2007) The evolution of high mobility group box (HMGB) chromatin proteins in multicellular animals. Gene 387, 133–140 [DOI] [PubMed] [Google Scholar]
- 52. Bianchi M. E., Beltrame M. (1998) Flexing DNA: HMG-box proteins and their partners. Am. J. Hum. Genet. 63, 1573–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yamada S., Maruyama I. (2007) HMGB1, a novel inflammatory cytokine. Clin. Chim. Acta 375, 36–42 [DOI] [PubMed] [Google Scholar]
- 54. Murua Escobar H., Meyer B., Richter A., Becker K., Flohr A. M., Bullerdiek J., Nolte I. (2003) Molecular characterization of the canine HMGB1. Cytogenet. Genome Res. 101, 33–38 [DOI] [PubMed] [Google Scholar]
- 55. Abraham E., Arcaroli J., Carmody A., Wang H., Tracey K. J. (2000) HMG-1 as a mediator of acute lung inflammation. J. Immunol. 165, 2950–2954 [DOI] [PubMed] [Google Scholar]
- 56. Wijburg O. L., Heemskerk M. H., Boog C. J., Van Rooijen N. (1997) Role of spleen macrophages in innate and acquired immune responses against mouse hepatitis virus strain A59. Immunology 92, 252–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fages C., Nolo R., Huttunen H. J., Eskelinen E., Rauvala H. (2000) Regulation of cell migration by amphoterin. J. Cell Sci. 113, 611–620 [DOI] [PubMed] [Google Scholar]
- 58. Merenmies J., Pihlaskari R., Laitinen J., Wartiovaara J., Rauvala H. (1991) 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J. Biol. Chem. 266, 16722–16729 [PubMed] [Google Scholar]
- 59. Fang P., Schachner M., Shen Y. Q. (2012) HMGB1 in development and diseases of the central nervous system. Mol. Neurobiol. 45, 499–506 [DOI] [PubMed] [Google Scholar]
- 60. Erlandsson Harris H., Andersson U. (2004) Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 34, 1503–1512 [DOI] [PubMed] [Google Scholar]
- 61. Andersson U., Wang H., Palmblad K., Aveberger A. C., Bloom O., Erlandsson-Harris H., Janson A., Kokkola R., Zhang M., Yang H., Tracey K. J. (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192, 565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hori O., Brett J., Slattery T., Cao R., Zhang J., Chen J. X., Nagashima M., Lundh E. R., Vijay S., Nitecki D. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J. Biol. Chem. 270, 25752–25761 [DOI] [PubMed] [Google Scholar]
- 63. Rauvala H., Pihlaskari R. (1987) Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J. Biol. Chem. 262, 16625–16635 [PubMed] [Google Scholar]
- 64. Huttunen H. J., Kuja-Panula J., Sorci G., Agneletti A. L., Donato R., Rauvala H. (2000) Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 275, 40096–40105 [DOI] [PubMed] [Google Scholar]
- 65. Daston M. M., Ratner N. (1994) Amphoterin (P30, HMG-1) and RIP are early markers of oligodendrocytes in the developing rat spinal cord. J. Neurocytol. 23, 323–332 [DOI] [PubMed] [Google Scholar]
- 66. Sorci G., Riuzzi F., Giambanco I., Donato R. (2013) RAGE in tissue homeostasis, repair, and regeneration. Biochim. Biophys. Acta 1833, 101–109 [DOI] [PubMed] [Google Scholar]
- 67. David S., Kroner A. (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388–399 [DOI] [PubMed] [Google Scholar]
- 68. Fitch M. T., Doller C., Combs C. K., Landreth G. E., Silver J. (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J. Neurosci. 19, 8182–8198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fleming J. C., Norenberg M. D., Ramsay D. A., Dekaban G. A., Marcillo A. E., Saenz A. D., Pasquale-Styles M., Dietrich W. D., Weaver L. C. (2006) The cellular inflammatory response in human spinal cords after injury. Brain 129, 3249–3269 [DOI] [PubMed] [Google Scholar]
- 70. Schwartz M., Kipnis J. (2004) A common vaccine for fighting neurodegenerative disorders: recharging immunity for homeostasis. Trends Pharmacol. Sci. 25, 407–412 [DOI] [PubMed] [Google Scholar]
- 71. Ding Y. M., Jaumotte J. D., Signore A. P., Zigmond M. J. (2004) Effects of 6-hydroxydopamine on primary cultures of substantia nigra: specific damage to dopamine neurons and the impact of glial cell line-derived neurotrophic factor. J. Neurochem. 89, 776–787 [DOI] [PubMed] [Google Scholar]
- 72. Yanai H., Ban T., Taniguchi T. (2012) High-mobility group box family of proteins: ligand and sensor for innate immunity. Trends Immunol. 33, 633–640 [DOI] [PubMed] [Google Scholar]
- 73. Nesic O., Xu G. Y., McAdoo D., High K. W., Hulsebosch C., Perez-Pol R. (2001) IL-1 receptor antagonist prevents apoptosis and caspase-3 activation after spinal cord injury. J. Neurotrauma 18, 947–956 [DOI] [PubMed] [Google Scholar]
- 74. Ferguson A. R., Christensen R. N., Gensel J. C., Miller B. A., Sun F., Beattie E. C., Bresnahan J. C., Beattie M. S. (2008) Cell death after spinal cord injury is exacerbated by rapid TNF α-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J. Neurosci. 28, 11391–11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Probert L., Eugster H. P., Akassoglou K., Bauer J., Frei K., Lassmann H., Fontana A. (2000) TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain 123, 2005–2019 [DOI] [PubMed] [Google Scholar]
- 76. Lu J., Ashwell K. W., Waite P. (2000) Advances in secondary spinal cord injury: role of apoptosis. Spine 25, 1859–1866 [DOI] [PubMed] [Google Scholar]
- 77. Orlova V. V., Choi E. Y., Xie C., Chavakis E., Bierhaus A., Ihanus E., Ballantyne C. M., Gahmberg C. G., Bianchi M. E., Nawroth P. P., Chavakis T. (2007) A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 26, 1129–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sîrbulescu R. F., Zupanc G. K. (2011) Spinal cord repair in regeneration-competent vertebrates: adult teleost fish as a model system. Brain Res. Rev. 67, 73–93 [DOI] [PubMed] [Google Scholar]
- 79. Gong H., Zuliani P., Komuravelli A., Faeder J. R., Clarke E. M. (2010) Analysis and verification of the HMGB1 signaling pathway. BMC Bioinformatics 11, Suppl. 7, S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Crowe M. J., Bresnahan J. C., Shuman S. L., Masters J. N., Beattie M. S. (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 3, 73–76 [DOI] [PubMed] [Google Scholar]
- 81. Li G. L., Brodin G., Farooque M., Funa K., Holtz A., Wang W. L., Olsson Y. (1996) Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J. Neuropathol. Exp. Neurol. 55, 280–289 [DOI] [PubMed] [Google Scholar]
- 82. Mekhail M., Almazan G., Tabrizian M. (2012) Oligodendrocyte-protection and remyelination post-spinal cord injuries: a review. Prog. Neurobiol. 96, 322–339 [DOI] [PubMed] [Google Scholar]
- 83. Tseng A. S., Adams D. S., Qiu D., Koustubhan P., Levin M. (2007) Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev. Biol. 301, 62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kiba T. (2002) The role of the autonomic nervous system in liver regeneration and apoptosis-recent developments. Digestion 66, 79–88 [DOI] [PubMed] [Google Scholar]
- 85. Savill J., Dransfield I., Gregory C., Haslett C. (2002) A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2, 965–975 [DOI] [PubMed] [Google Scholar]
- 86. Blight A. R. (1985) Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent. Nerv. Syst. Trauma 2, 299–315 [DOI] [PubMed] [Google Scholar]
- 87. Chen G. Y., Tang J., Zheng P., Liu Y. (2009) CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huttunen H. J., Fages C., Rauvala H. (1999) Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 274, 19919–19924 [DOI] [PubMed] [Google Scholar]
- 89. Kuniyasu H., Chihara Y., Takahashi T. (2003) Co-expression of receptor for advanced glycation end products and the ligand amphoterin associates closely with metastasis of colorectal cancer. Oncol. Rep. 10, 445–448 [PubMed] [Google Scholar]
- 90. Kokkola R., Andersson A., Mullins G., Ostberg T., Treutiger C. J., Arnold B., Nawroth P., Andersson U., Harris R. A., Harris H. E. (2005) RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 61, 1–9 [DOI] [PubMed] [Google Scholar]
- 91. Dumitriu I. E., Baruah P., Valentinis B., Voll R. E., Herrmann M., Nawroth P. P., Arnold B., Bianchi M. E., Manfredi A. A., Rovere-Querini P. (2005) Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 174, 7506–7515 [DOI] [PubMed] [Google Scholar]
- 92. Qin J., Goswami R., Dawson S., Dawson G. (2008) Expression of the receptor for advanced glycation end products in oligodendrocytes in response to oxidative stress. J. Neurosci. Res. 86, 2414–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Volff J. N. (2005) Genome evolution and biodiversity in teleost fish. Heredity 94, 280–294 [DOI] [PubMed] [Google Scholar]
- 94. Hoffmann F. G., Opazo J. C., Storz J. F. (2012) Whole-genome duplications spurred the functional diversification of the globin gene superfamily in vertebrates. Mol. Biol. Evol. 29, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Johansson J. U., Ericsson J., Janson J., Beraki S., Stanić D., Mandic S. A., Wikström M. A., Hökfelt T., Ogren S. O., Rozell B., Berggren P. O., Bark C. (2008) An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS Genet. 4, e1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]