

Abstract
Thrombotic thrombocytopenic purpura (TTP) is an episodic microangiopathic coagulopathy characterized by profound thrombocytopenia and hemolytic anemia. Neurovascular complications, including stroke and TIA, occur commonly upon initial TTP presentation, and the underlying etiology of such neurologic manifestations is usually hinted by the concomitant hematologic abnormalities.
PEARL
Thrombotic thrombocytopenic purpura (TTP) is an episodic microangiopathic coagulopathy characterized by profound thrombocytopenia and hemolytic anemia. Neurovascular complications, including stroke and TIA, occur commonly upon initial TTP presentation, and the underlying etiology of such neurologic manifestations is usually hinted by the concomitant hematologic abnormalities.
OY-STER
TTP is rarely suspected in the setting of a normal or near-normal platelet count, but neurologic manifestations can precede the onset of striking hematologic abnormalities in TTP. Recognizing this atypical TTP presentation has major therapeutic implications. Atypical TTP may be the underlying stroke etiology in young and middle-aged women without cardiovascular risk factors and subtle or no hematologic abnormalities.
CASE REPORT
A 29-year-old previously healthy, right-handed Hispanic woman presented with 30 minutes of sudden-onset left-sided hemianesthesia, left tongue numbness, dysarthria, and generalized weakness. She had never experienced similar symptoms but complained of sharp left anterior chest pain over the course of the week prior to presentation. She had 2 children and no history of obstetric complications. She denied tobacco or drug use and used alcohol occasionally. Her only medication was norgestimate-ethinyl estradiol oral contraceptive pills. She had no family history of stroke or hematologic disorders. On examination, she appeared anxious but alert and oriented. Her general examination was unremarkable, except for tachycardia (144/min) and hypertension (152/77 mm Hg). Her neurologic examination was significant for subtle left lower facial weakness and left hemisensory loss to pinprick that spared the face but affected the arm, torso, and leg. Her NIH Stroke Scale (NIHSS) score was 2 due to sensory loss. Her initial head CT and CT angiogram were unremarkable. MRI of the brain with diffusion-weighted imaging revealed multifocal areas of acute infarction in the right insular cortex, right medial temporal lobe, and medial right occipital cortex (figure). IV thrombolysis was not offered as the neurologic deficit was minimal and improved rapidly. Her blood count showed leukocytes 8.6 × 103 cells/mm3, hemoglobin 12.2 g/dL, hematocrit 34.9%, and platelets 125 × 103 cells/mm3. Her serum electrolytes, creatinine, blood glucose, thyroid-stimulating hormone, liver function tests urinalysis, lipid panel, and glycosylated hemoglobin A1c were within normal limits. Her urine toxicologic screen was negative. She had an elevated erythrocyte sedimentation rate of 74 mm/h, C-reactive protein of 3.58 mg/dL, and troponin of 0.07 U/L. Her chest x-ray was normal and her ECG was significant for sinus tachycardia. During hospitalization, transthoracic and transesophageal echocardiograms showed no abnormalities. There were no notable events on telemetry. Autoimmune and hypercoagulability workup was relevant only for antinuclear antibody titer of 1:160 in a speckled pattern. Complement, protein S activity, lupus anticoagulant, antiphospholipid antibodies, anti-β2 glycoprotein antibodies, homocysteine, antithrombin III activity, factor VIII activity, factor V Leiden, and prothrombin G20210A mutation were normal or negative. An autoimmune panel including anti-Smith, Jo, myeloperoxidase, proteinase 3, ribonucleoprotein, SSA, SSB, heparin-induced platelet antibodies, and direct antiglobulin tests was also negative. Bacterial blood cultures, syphilis rapid plasma reagin titer, HIV antibodies, and hepatitis serologies were negative. Platelet aggregation studies were normal. She was diagnosed with a cryptogenic stroke, although her presentation suggested an embolic mechanism with possible prothrombotic or vasculitic etiologies. Discontinuation of oral contraception was recommended and the patient was discharged on acetylsalicylic acid 325 mg per day and rosuvastatin 20 mg per day, with plan for Holter monitoring, repeat hypercoagulability workup, and consideration for cerebral angiogram as an outpatient.
Figure. Brain MRI upon initial presentation with left-sided hemianesthesia, left tongue numbness, and dysarthria.
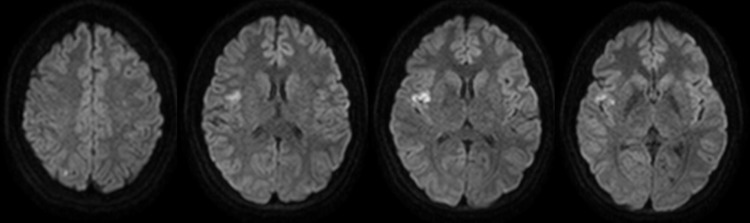
Acute ischemic brain infarct is demonstrated by sagittal views of diffusion-weighted MRI sequence corresponding to a conspicuous area of restricted diffusion in the right insular cortex. Punctate foci of restricted diffusion were also seen in the right medial temporal lobe and right occipital cortex.
Two months later, she presented to our service with acute-onset right-hand numbness, hemiparesis, and transient aphasia for 45 minutes that resolved spontaneously. She had mild weakness to hand grip, slow finger tapping, and mildly decreased sensation to light touch on the right hand. Her neurologic examination was otherwise intact, with an NIHSS of 1, for sensory loss. Repeat head CT was negative. MRI of the brain showed no evidence of new acute ischemic lesions. This time, her platelet count was significantly low at 19 × 103 cells/mm3 and her hemoglobin was 9 g/dL. A peripheral blood smear revealed the presence of schistocytes, slight polychromasia, vacuolated polymorphonuclear cells, reactive lymphocytes, and large platelets. She had an absolute reticulocyte count of 219 × 103 cells/mm3 (normal 32–147 × 103 cells/mm3), haptoglobin 5 mg/dL (normal 16–200 mg/dL), and lactate dehydrogenase 485 U/L (normal 135–214 U/L). An undetectable activity level of the metalloproteinase ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif 13) (normal ≥67%) and increased ADAMTS13 inhibitor level to 1.6 units (normal ≤0.4 units) further supported the diagnosis of TTP. She was started on prednisone 100 mg orally every day and therapeutic plasma exchange (TPE) with subsequent correction of the thrombocytopenia. A rapid hematologic relapse within 15 days prompted 4 pulses of rituximab 375 mg/m2 IV every week and initiation of cyclosporine 100 mg PO twice a day. Cyclosporine was stopped 2 months later due to side effects. Her platelet count stabilized after 4 months of treatment with prednisone and TPE 3 times per week. A slow prednisone taper was then started and TPE was discontinued soon after. She has remained neurologically stable.
DISCUSSION
Our patient is of particular interest because her presentation with a cryptogenic stroke is evidence that neurovascular complications of TTP may develop before the onset of alarming hematologic abnormalities. TTP has been defined in clinical trials by the presence of thrombocytopenia and microangiopathic hemolytic anemia.1 Indeed, 95% of patients with newly diagnosed TTP have platelets lower than 60 × 103 cells/mm3. Yet a landmark analysis conducted years before an effective treatment was available already identified that some patients may present in an atypical fashion, with neurologic manifestations that precede the recognition of TTP.2 Recent reports of patients with TTP-related stroke and normal or near-normal platelets support that this atypical form of TTP may not be rare.3–7 The table summarizes the 8 cases of atypical TTP presenting with stroke available in the literature. All patients are young or middle-aged women without cardiovascular risk factors. Most of them had subtle hematologic abnormalities and only 2 had normal levels of both platelets and hemoglobin. The vascular distribution of the infarcts corresponds predominantly to a distal microembolic pattern, although major branch occlusion may also occur. Of course, TTP may be expected as the underlying cause of stroke if there is prior history of TTP. However, our patient and some of those previously reported had no such history. In all cases, this situation delayed the identification of the etiology of the stroke and led to inadequate treatments. TTP can be a devastating condition, with mortality of nearly 90% if not treated promptly.2 The introduction of TPE for the treatment of TTP has significantly improved its prognosis, and has made the classic pentad of thrombocytopenia, hemolytic anemia, fever, and neurologic and renal complications less common in clinical practice.1 The neurovascular complications of TTP improve once TPE and adequate immunosuppression are instituted. Thus, recognition of atypical TTP as a potential cause of cryptogenic stroke has major therapeutic implications.
Table.
Clinical features of patients with acute stroke caused by atypical thrombotic thrombocytopenic purpura

TTP is caused by the inability of ADAMTS13 to cleave von Willebrand factor multimers. This induces systemic platelet aggregation, microvascular thrombosis, intravascular hemolysis from fibrin deposition, and mechanical trauma.8 Signs of end-organ damage such as brain infarction are seen once thrombocytopenia and anemia are evident. It is possible, however, that a subtle ADAMTS13 dysfunction causes widespread but subclinical end-organ damage that is clinically noticeable only by its dramatic effects on the function of the nervous system.9 On the other hand, local prothrombotic changes summated to a very incipient ADAMTS13 dysfunction may be significant enough to cause focal platelet aggregation, without permanently overcoming the systemic antithrombotic mechanisms.10 This could alternatively explain the presence of neurologic symptoms in the absence of thrombocytopenia or anemia. Regardless of whether the initial events in atypical TTP are local or systemic, endogenous antithrombotic mechanisms seem to activate following the initial end-organ insult and the progressive subclinical thrombocytopenia and hemolytic anemia. This is suggested by a characteristic lag between stroke presentation and the development of the typical hematologic features of TTP seen in all patients (table). The measurement of ADAMTS13 activity has been introduced into clinical practice to support the diagnosis of TTP and to monitor disease activity. Nevertheless, ADAMTS13 is usually not measured in patients with normal hematologic parameters. Our analysis shows that atypical TTP may be the underlying stroke etiology in young and middle-aged women without cardiovascular risk factors and subtle or no hematologic abnormalities. Thus, systematic evaluation of the value of measuring ADAMTS13 activity and other TTP diagnostic parameters in cryptogenic stroke may be indicated in this population.
Cryptogenic stroke may be caused by an atypical form of TTP in which thrombocytopenia and hemolytic anemia are absent or clinically mild. Atypical TTP should be suspected even in the absence of frank hematologic abnormalities in young or middle-aged women without cardiovascular risk factors presenting with cryptogenic stroke. Recognizing atypical TTP as a potential stroke etiology has major therapeutic implications.
AUTHOR CONTRIBUTIONS
Dr. Julio C. Rojas: study concept and design, acquisition of data, analysis and interpretation. Dr. Chirantan Banerjee: acquisition of data, analysis and interpretation. Dr. Fazeel Siddiqui: acquisition of data. Dr. Bardia Nourbakhsh: acquisition of data. Dr. Craig Powell: study concept and design, critical revision of the manuscript for important intellectual content and supervision.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura: Canadian Apheresis Study Group. N Engl J Med 1991;325:393–397 [DOI] [PubMed] [Google Scholar]
- 2.Amorosi EL, Ultmann JE. Thrombotic thrombocytopenic purpura: report of 16 cases and review of the literature. Medicine 1966;45:139–159 [Google Scholar]
- 3.Downes KA, Yomtovian R, Tsai HM, Silver B, Rutherford C, Sarode R. Relapsed thrombotic thrombocytopenic purpura presenting as an acute cerebrovascular accident. J Clin Apher 2004;19:86–89 [DOI] [PubMed] [Google Scholar]
- 4.Idowu M, Reddy P. Atypical thrombotic thrombocytopenic purpura in a middle-aged woman who presented with a recurrent stroke. Am J Hematol Epub 2012 Apr 26 [DOI] [PubMed]
- 5.O'Brien TE, Crum ED. Atypical presentations of thrombotic thrombocytopenic purpura. Int J Hematol 2002;76:471–473 [DOI] [PubMed] [Google Scholar]
- 6.Sevy A, Doche E, Squarcioni C, et al. Stroke in a young patient treated by alteplase heralding an acquired thrombotic thrombocytopenic purpura. J Clin Apher 2011;26:152–155 [DOI] [PubMed] [Google Scholar]
- 7.Tsai HM, Shulman K. Rituximab induces remission of cerebral ischemia caused by thrombotic thrombocytopenic purpura. Eur J Haematol 2003;70:183–185 [DOI] [PubMed] [Google Scholar]
- 8.Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009;24:47–52 [DOI] [PubMed] [Google Scholar]
- 9.Ono T, Mimuro J, Madoiwa S, et al. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood 2006;107:528–534 [DOI] [PubMed] [Google Scholar]
- 10.Amann K. ADAMTS13: more than just TMA and TTP. Nephrol Dial Transpl 2011;26:1761–1764 [DOI] [PubMed] [Google Scholar]