

Abstract
HIV-associated neurologic disease continues to be a significant complication in the era of highly active antiretroviral therapy. A substantial subset of the HIV-infected population shows impaired neuropsychological performance as a result of HIV-mediated neuroinflammation and eventual central nervous system (CNS) injury. CNS compartmentalization of HIV, coupled with the evolution of genetically isolated populations in the CNS, is responsible for poor prognosis in patients with AIDS, warranting further investigation and possible additions to the current therapeutic strategy. This chapter reviews key advances in the field of neuropathogenesis and studies that have highlighted how molecular diversity within the HIV genome may impact HIV-associated neurologic disease. We also discuss the possible functional implications of genetic variation within the viral promoter and possibly other regions of the viral genome, especially in the cells of monocyte–macrophage lineage, which are arguably key cellular players in HIV-associated CNS disease.
1. INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) infects the central nervous system (CNS) initiating a cascade of neuroinflammation and eventually CNS injury. Despite the success of highly active antiretroviral therapy (HAART), neurocognitive impairment (NCI) continues to affect a significant proportion of infected patients. Although the incidence of HIV-1-associated dementia (HAD) has decreased, the overall prevalence of HIV-1-associated neurological disorders (HAND) has increased in the HAART era, primarily because the incidence of subtle forms of HIV-1-associated cognitive impairment has increased. In resource-limited settings, especially in the developing world, poor access to antiretroviral medication results in a much more severe prognosis for HIV-related CNS complications in late-stage HIV infection. HIV enters the nervous system within the first few weeks after initial systemic infection (Pilcher et al., 2001; Schacker, Collier, Hughes, Shea, & Corey, 1996), initiating a cascade of neuroinflammation and eventual CNS invasion and subsequent injury. CNS compartmentalization, including the cerebrospinal fluid (CSF), of HIV species may begin within the first year of infection. Thus, the CNS may be a potential independent site of HIV replication. Genetic variation within the HIV genome and associated selective pressures may lead to an increase in the prevalence of specialized variants that find a niche and begin evolving in the early stages of the disease. This review discusses the key features of HAND, the implications of the molecular and genetic diversity of the HIV-1 genome for HIV disease, and the importance of cells of the monocyte–macrophage lineage in the overall neuropathogenesis of HIV-1.
2. OVERVIEW OF HIV-1 CNS PATHOGENESIS
Entry of HIV-1 into the brain results in a chain of events leading to CNS disease and neurologic impairment. The virus must first circumvent the blood–brain barrier (BBB), a selectively permeable barrier separating the CNS from the peripheral circulation (Fig. 6.1). One route of entry into the CNS involves transit of HIV-1 across the BBB by means of infected cells trafficking from the periphery into the brain. This “Trojan horse” method of entry likely involves infected circulating monocytes carrying HIV-1 into the brain in the form of integrated provirus or infectious viral particles (Haase, 1986). Alternatively, HIV may also traffic into the CNS by lymphocytes that harbor viruses that replicate in macrophages, or as a cell-free virus entering through the endothelial cells or across cells of the choroid plexus (Collman et al., 1992; Spudich & Gonzalez-Scarano, 2012). Broad systemic infection and immune system activation may exacerbate this process, when infected (Hickey, 1999) and possibly uninfected cells within the CNS release chemotactic mediators into circulation, thereby drawing more activated cells harboring HIV-1 into the brain. This process may establish a positive feedback mechanism of viral entry and subsequent neuroinflammation (Fontaine, Poudrier, & Roger, 2011; Liu, Tang, McArthur, Scott, & Gartner, 2000; Yadav & Collman, 2009). HIV-1 infection of cells of the monocyte–macrophage lineage also induces increased expression of adhesion molecules on vascular endothelial cells, facilitating HIV-1 transit across the BBB (Blodget et al., 2012; Nottet et al., 1996; Rappaport et al., 1999). Infected macrophages induce greater expression of the adhesion molecules E-selectin and vascular cell adhesion molecule-1 (VCAM-1) on the surface of brain endothelial cells than do uninfected macrophages, suggesting that immune cell activation of monocytic cells following HIV-1 infection of the CNS likely plays a key role in facilitating transendothelial migration across the BBB (Miller et al., 2012; Nottet et al., 1996; Persidsky et al., 1997; Rappaport et al., 1999). Cells of the monocyte–macrophage lineage are the only cells in the CNS that routinely are shown to express HIV RNA or protein, although other cell types, such as astrocytes, have been shown to harbor HIV sequences but do not show a robust expression of HIV RNA or proteins (Spudich & Gonzalez-Scarano, 2012; Wiley, Schrier, Nelson, Lampert, & Oldstone, 1986). Among the different macrophage subtypes, perivascular macrophages are highly infected in the brains of HIV-1-infected individuals (Kim et al., 2006). Initially, it was thought that perivascular macrophages could not contribute to the long-term presence of HIV-1 in the brain owing to their rapid turnover rate, but reports have suggested that this cell population can harbor virus for long periods and can, therefore, serve as a reservoir for HIV-1, as previously reviewed (Spudich & Gonzalez-Scarano, 2012).
Figure 6.1.

Model of HIV trafficking across the BBB and its impact on the CNS. (1) Infected, activated monocytes transport HIV across the BBB through a “Trojan Horse” model, where they differentiate into perivascular macrophages. Infected perivascular macrophages then result in production of HIV within the CNS. (2) To a lesser extent, infected CD4+T cells also serve to carry HIV across the BBB and may also contribute to HIV pools within the CNS. (3) HIV produced in the CNS can result in infection of resident microglial cells. (4) The presence of multinucleate giant cells is an important feature of HIV-related brain pathology; these are produced as a result of cell-to-cell fusion. (5) Astrocytes are known to harbor HIV, but it is well established that they do not result in a productive infection. (6) The viral evolution and adaptation within the CNS adversely affect the physiology of neurons via a variety of mediators including ROS, nitrous oxide (NO), MMPs, and viral proteins that exhibit neurotoxic properties resulting in compromised neurologic functions.
Neurologic disease rarely manifests prior to the onset of immune system dysfunction (McArthur et al., 1997), and patients who do not show early signs of neurologic impairment typically progress through the asymptomatic stage of infection without experiencing a decline in neurologic status (Gannon, Khan, & Kolson, 2011; Selnes et al., 1990). The disconnect between initial infection of the CNS and the presentation of associated neurologic impairment may be explained by an initial immune system clearance of the virus followed by a reseeding of the CNS by HIV-1 at later time points in disease after the immune system has been functionally degraded. This is supported by evidence of increased levels of HIV-1-specific immunoglobulins within the CSF, intrathecally produced anti-HIV-1 antibodies, and increased numbers of HIV-1-specific CD8+ cytotoxic T lymphocytes (CTLs) (Krebs, Ross, McAllister, & Wigdahl, 2000). Recently, using the simian immunodeficiency virus model, it was shown that the intrathecal immune responses correlate inversely with the macrophage-tropic strains in the CNS (Selnes et al., 1990). An alternative explanation for the late onset of neurologic disease could be the selective infection of the CNS with a less neurovirulent strain of HIV-1, which results in a less cytotoxic but more chronic dissemination of the virus within the CNS (Krebs et al., 2000). Further explanation for the late-stage onset of neurological impairment comes from phylogenetic studies of HIV-1 gp160 sequences isolated from multiple tissue compartments as well as multiple compartments within the brain of HIV-1-infected patients (Liu et al., 2000). Recently, it was shown that HIV-1 R5 envelope (Env) sequences evolve with an increased positive charge and that this R5 subset evolves independently from highly macrophage-tropic variants with low-charge gp120s (Gonzalez-Perez et al., 2012). This study and others (Duncan & Sattentau, 2011; Peters, Duenas-Decamp, Sullivan, & Clapham, 2007) highlight the observations that HIV-1 R5 Envs evolve with very distinct properties at different sites in the body and are driven by powerful tissue-specific evolutionary pressures. Additional variables such as the effects of aging on brain, long-term CNS toxicity of HAART, and the impact of drugs of abuse need to be evaluated in detail to provide us with a conclusive model of these correlates of HIV disease.
Others have suggested that infected lymphocytes may be involved in the “Trojan horse” entry of HIV-1 into the CNS (Sloand et al., 1992; Weidenheim, Epshteyn, & Lyman, 1993). Similar to the case with cells of the monocyte–macrophage lineage, HIV-1 infection of CD4+ T cells results in upregulation of cellular adhesion molecules, including leukocyte function antigen-1 and very late antigen-4, which interact with vascular endothelial cell ligands intercellular adhesion molecule-1 (ICAM-1) and VCAM-1, thereby facilitating binding of lymphocytes to the endothelial cell surface (Sloand et al., 1992; Weidenheim et al., 1993). Lymphocytes may also secrete specific enzymes that degrade the basement membrane of endothelial cells, allowing the migration of HIV-1-infected T cells across the BBB (Sloand et al., 1992). However, owing to the relatively low frequency of lymphocytes infiltrating into the CNS, their contribution to HIV-1 trafficking to the brain and subsequent establishment and maintenance of infection remain a topic of debate.
3. CLINICAL DIAGNOSES OF HIV-1 CNS INFECTION
HIV-1 infection of the CNS can result in numerous motor and cognitive deficiencies (Cosenza, Zhao, Si, & Lee, 2002; Robertson, Liner, & Heaton, 2009; Wiley et al., 1986; Williams et al., 2001). Two distinctly recognizable conditions resulting from HIV-1 infection of the CNS are HAD and the less severe, subsyndromic condition called minor cognitive motor disorder (MCMD) (Cherner et al., 2002; Gartner, 2000; McArthur et al., 2003; Minagar et al., 2008; Williams & Hickey, 2002). Patients with either of these conditions are classified collectively as having NCI. Prior to the introduction of HAART in the industrialized world, ~20–30% of HIV-1-infected individuals developed HAD (Childs et al., 1999). The onset of HAD corresponds with high plasma viral loads, and although a reduced incidence of HAD has been observed with patients on HAART, the longer life expectancy of HIV patients has increased the prevalence of the disease (Childs et al., 1999). With the widespread use of HAART, MCMD has become more common (Cherner et al., 2002; McArthur et al., 2003). In the HAART era, it is estimated that ~10% of HIV-infected adults develop HAD; however, MCMD may be several times more common, involving as many as 30% of the HIV-infected population (Cherner et al., 2002; Sacktor et al., 2002). Furthermore, the clinical presentation of MCMD has been associated with neuropathological changes characteristic of HIV encephalitis, and MCMD is associated with a worse overall prognostic outlook (Cherner et al., 2002; McArthur et al., 2003; Mothobi & Brew, 2012; Sacktor et al., 2002). One means of explaining the development of MCMD is that the low-level viral replication associated with successful HAART regimens may lead to slowly progressing neurodegeneration. This is consistent with the longer life spans of patients receiving HAART, and possibly with the inability of certain antiretroviral drugs to effectively penetrate into the brain (Letendre et al., 2004). A recent comparative study (Heaton et al., 2011) of HIV-associated NCI before and after the introduction of HAART concludes that although HAD (the most severe form of HAND) appears to be less common in the HAART era, the long-term benefits of therapy with respect to milder forms of HAND remain in question because their prevalence appears to be increasing (Heaton et al., 2011; McArthur & Brew, 2010). To achieve an optimal comparison of HAND in a longitudinal framework would require development of more consistent clinical definitions of neurologic disease, clinical predictors, and a better-defined characterization of comorbid conditions. Study and compilation of all these variables would be greatly facilitated by developing larger and more representative HIV/AIDS cohorts. Nevertheless, we can safely conclude that the beneficial effects of HAART on neurological complications associated with HIV have been less than complete and that continuous efforts are needed to improve prognosis. Additional variables that need more precise definition in relation to neurologic complications of HIV include drugs of abuse, age, viral strains, genetic variation within the HIV genome during the course of disease, and timing of HAART initiation.
HAD cannot be defined in terms of a single disease entity but must rather be characterized in terms of a broad collection of symptoms encompassing cognitive, motor, and behavioral deficiencies corresponding to the presence of actively replicating HIV-1 within the CNS. HAD is a subcortical dementia, which differentiates it from dementia induced by Alzheimer’s disease, that presents clinically as a progressive decline in neurocognitive function (Kolson, Lavi, & Gonzalez-Scarano, 1998; Wendelken & Valcour, 2012). It may also include loss of memory, diminished ability to concentrate, psychomotor retardation, and frequent headache. Early in the course, HAD patients typically experience complications involving mental slowing, impaired motor control and lack of coordination, and behavioral alterations such as apathy, social withdrawal, and personality changes (Price, 1994). In late-stage disease, HAD patients exhibit severe, clinically recognizable cognitive, motor, and behavior deficits. Severe cases of HAD, which have become increasingly rare with the advent of effective combination antiretroviral therapeutics, may manifest with almost absolute mutism, incontinence, and severe, debilitating dementia (del Palacio, Alvarez, & Munoz-Fernandez, 2012; Price, 1994).
Clinical assessment of HIV-1-associated CNS disease requires surrogate biomarkers because brain and spinal cord are relatively inaccessible. HIV-1 RNA measurement in the CSF is one of the most practical means of examining CNS viral load (Marra, Maxwell, Collier, Robertson, & Imrie, 2007; Spudich et al., 2005). CSF markers of immune activation and inflammation have also been used as indicators of disease activity, including CCL2/monocyte chemotactic protein-1 (MCP1) and CXCL10/IP10 (chemokines that facilitate ingress of macrophages and lymphocytes across the BBB) (Chang, Ernst, St Hillaire, & Conant, 2004; Conant et al., 1998), β2-microglobulin and neopterin (Brew et al., 1992; Brew, Dunbar, Pemberton, & Kaldor, 1996; Enting et al., 2000), quinolic acid (Heyes et al., 2001), arachidonic acid metabolites (Genis et al., 1992), and oxidative stress markers (Schifitto et al., 2009) (Fig. 6.2). Reduced levels of N-acetylaspartate, which indicate decreased neuronal function, and elevated levels of choline, which indicate inflammation and membrane turnover, can also be utilized for overt HAD assessment using magnetic resonance spectroscopy (Chang, 1995; Meyerhoff et al., 1994).
Figure 6.2.
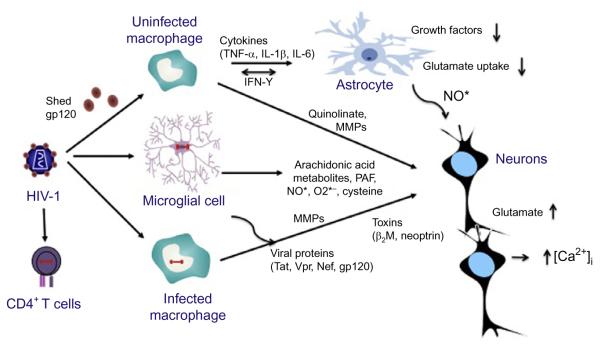
Potential pathways and mediators of CNS damage. Subsequent to neuroinvasion, HIV-1-infected perivascular macrophages and brain microglial cells are likely to be the major producers of infectious virus and neurotoxic cellular and viral proteins such as gp120, Tat, and Nef. The extent of CNS dysfunction observed during HIV-1 infection is likely due to both host and viral factors. CNS damage may occur through increased viral replication within the CNS, production of viral neurotoxic proteins, and release of toxins including NO, TNF-α, and quinolinic acid, all of which target neurons, astrocytes, endothelial cells, and oligodendrocytes (not shown).
Several pathologies are associated with HAD (Rosenblum, 1990), including white matter pallor, multinucleated-cell encephalitis (associated with the multinucleated giant cells, or MNGCs, or syncytia), and vacuolar myelopathy (Price, 1994). MNGCs are observed in only about half of all HAD patients on postmortem examination (Kato, Hirano, Llena, & Dembitzer, 1987; Wiley & Achim, 1994), and they are comprised of resident CNS mononuclear phagocytes often concentrated around blood vessels (Price, 1994). The presence of MNGCs in infected patients indicates active HIV-1 replication within the CNS (Budka, 1991; Budka et al., 1987). MNGC encephalitis occurs in the subcortical regions of the brain and is often associated with the presence of gliosis and white matter pallor (Chrysikopoulos, Press, Grafe, Hesselink, & Wiley, 1990; Epstein et al., 1984). In addition to these diagnostic approaches, functional magnetic resonance imaging and diffusion tensor imaging have been utilized to assess the changes in brain hemodynamics including alterations in cerebral blood flow, blood oxygen level dependence, and white matter morphometric changes (Ances et al., 2010; Spudich & Gonzalez-Scarano, 2012).
The onset and progression of HIV-1-associated neurodegeneration and subsequent decline in cognitive ability are likely dependent on multiple host and viral factors, some yet to be characterized (Fig. 6.3). This chapter reviews the relationship between genetic variation within the HIV-1 genome and host and the onset and progression of HAND.
Figure 6.3.

The dynamic interplay of multiple viral and host factors contributes to both the onset and severity of HIV-1-associated neurocognitive impairment. An individual’s HAART (highly active antiretroviral drug) and/or drugs of abuse status, as well as the presence of comorbidities resulting from opportunistic pathogens, greatly influence overall disease course and the establishment of central nervous system (CNS) pathology. One major complicating factor with respect to HIV-1-induced neurodegeneration is the ability of HIV-1 to adapt and evolve specific genetic variants in response to host immune pressures and subsequently to increase in its capacity to replicate within specific tissue compartments such as the CNS. This ability of HIV to evolve within the CNS eventually manifests as a variety of neurologic symptoms including asymptomatic neurocognitive impairment (ANI), mild neurocognitive disorder (MND), and HIV-associated dementia.
4. THE EVOLUTION AND ADAPTATION OF HIV-1 AND THE ESTABLISHMENT OF MOLECULAR DIVERSITY
HIV-1 molecular (and consequent phenotypic) diversity is present at the population level as well as among infected individuals. The rapidly evolving nature of HIV-1 results from several factors, including the error-prone nature of reverse transcriptase, selective pressures from the host and from antiretroviral therapy, replication dynamics, and genomic recombination. HIV-1 molecular heterogeneity is most often characterized within the context of specific point mutations, which present as synonymous (nonamino acid altering) and nonsynonymous (amino acid altering) variations (Rambaut, Posada, Crandall, & Holmes, 2004). However, this genetic diversity may also manifest as polymorphic nucleotides throughout the viral genome resulting from insertion, deletion, and recombinatorial events occurring within a viral population (Rambaut et al., 2004). The end consequence is altered viral protein structure and function, as well as changes within noncoding nucleic acid sequences, such as the viral promoter, the long terminal repeat (LTR), which are critical viral components that may dramatically alter the course of viral gene expression.
The clinical trajectory of HIV disease is generally well characterized. Substantial variability occurs with respect to the rate of disease progression among infected individuals; however, the role host factors play in HIV disease progression cannot be discounted (Lackner, Lederman, & Rodriguez, 2012; Lemey et al., 2007). The asymptomatic phase of infection is highly variable and can range from several months to more than 20 years (Lemey et al., 2007). This differential rate of disease progression likely results from the dynamic relationship between virus and host and from specific selective pressures placed upon the virus within a given host (Lackner et al., 2012; Lemey et al., 2007). External factors such as other exogenously acquired infectious diseases may also affect disease progression. Following HIV-1 infection, both a humoral and a cell-mediated immune response are mounted by the host to combat the virus, but without avail, both are eventually defeated as HIV evolves and adapts, enabling it to efficiently replicate (Frost et al., 2005; Richman, Wrin, Little, & Petropoulos, 2003). This process ultimately leads to the destruction of the host’s immune system and the establishment of multiple opportunistic diseases, which define the clinical progression to AIDS (Frost et al., 2005; Richman et al., 2003). The humoral immune response involves the production of neutralizing antibodies, which exert strong selective pressure on the HIV envelope gene (env) but do not effectively control viral replication (Cecilia, Kleeberger, Munoz, Giorgi, & Zolla-Pazner, 1999). Recent structural studies of the HIV-1 Env proteins have fueled interest in rational antibody design using candidate-induced antibodies to Env, as previously reviewed (Bonsignori et al., 2012; Walker & Burton, 2010). The CD8+ T-cell response likely serves in a protective capacity during HIV-1 infection, and evidence suggests that at least partial control of virus replication in vivo can be associated with the appearance of CTLs (Koup et al., 1994) and that the rate of disease progression is critically dependent on HLA class I alleles (Carrington, Dean, Martin, & O’Brien, 1999; Trachtenberg et al., 2003). It has been suggested that although the majority of the killing of HIV-1-infected cells results from the CTL response, small differences in CTL killing efficiency may be clinically relevant and may correspond with altered disease course (Asquith, Edwards, Lipsitch, & McLean, 2006).
HIV-1 possesses an enormous potential for evolutionary change, a consequence which is loss of the host’s immune response to effectively control viral replication (Lemey et al., 2007). A genetically diverse viral population exhibiting a high rate of mutation and recombination, in concert with rapid replication dynamics, facilitates the propagation of infection to a large population of HIV-1-susceptible cells and enables the virus to readily adapt to constantly changing physiological conditions within each individual host (Lemey et al., 2007). Multiple amino acid alterations have been shown to occur within the hypervariable region of HIV-1 Env, changes that allow the virus to evade the host’s humoral immune response but that do not negatively impact viral entry into target cells (Frost et al., 2005). HIV-1-specific CD8+ T cells more efficiently target other regions of the virus such as gag and nef (Addo et al., 2003; Cao, McNevin, Malhotra, & McElrath, 2003; Lichterfeld et al., 2004), especially early in infection. Although viral evolution facilitates the establishment of escape variants capable of evading the CTL response, studies have suggested that evolution of these escape (or partial escape) mutants during chronic HIV-1 infection may occur with, however, an associated loss in replicative fitness (Ganusov et al., 2011; Leslie et al., 2004; Lewis, Dagarag, Khan, Ali, & Yang, 2012; Martinez-Picado et al., 2006).
The HIV-1-specific immune response is becoming increasingly understood. The effect of viral evolution and establishment of genetic diversity on the course and outcome of HIV-1-associated immune and nervous system disease, however, remain unresolved. One issue that is generally accepted is the critical nature of viral evolution with respect to our understanding of the dynamic relationship between virus and host during chronic or persistent infection. Consistent patterns of HIV-1 evolution have been observed throughout the course of infection (Shankarappa et al., 1999); however, investigations of viral diversity and mean divergence from the founder strain of HIV-1 within patients with differing rates of disease progression often conflict with one another (Ganeshan, Dickover, Korber, Bryson, & Wolinsky, 1997). Studies aimed at distinguishing between adaptive and selective neutral mutations have led some to believe that delayed disease progression may be associated with increased positive selection of sites within and accelerated adaptation rates of HIV-1 Env (Ross & Rodrigo, 2002; Williamson, 2003). It remains a topic of debate, however, whether viral adaptation results from, or is the consequence of, differential rates of HIV-1-associated disease progression (Lemey et al., 2007). Moreover, the dynamics of antiretroviral drugs also determine the path of HIV evolution and play a role in therapy outcome. Mathematical simulation studies have established that simulation of clinical trials with new and untested HIV treatment protocols could be used as a potent tool in selecting novel antiretroviral combinations (Hill, Rosenbloom, & Nowak, 2012; Rosenbloom, Hill, Rabi, Siliciano, & Nowak, 2012). Although this simulation approach may have biases toward success and may be limited with respect to the incorporation of all possible sequence variants that would render the compound combination inferior in clinical trials, the simulation can still be used as a preliminary step to strengthen a proposed antiretroviral combination prior to testing in preliminary efficacy analyses.
Studies analyzing the ratio of synonymous (nonamino acid changing) to nonsynonymous (amino acid changing) mutations within HIV-1 have provided insight into the mechanisms governing viral adaptation and the establishment of genetic diversity (Seo, Thorne, Hasegawa, & Kishino, 2002). Fluctuations in the rate of synonymous substitutions have been suggested to reflect changes in mutation rates, whereas nonsynonymous substitution rates may also be affected by changes in selective pressure and effective population size (Lemey et al., 2007). Studies of HIV-1 evolution have been based on the assumption that the rate of synonymous changes is relatively constant among HIV-1-infected patients, an assumption that has been questioned (Ganeshan et al., 1997; Lemey et al., 2007). The rate of synonymous change depends on multiple factors including viral generation time, which may vary considerably among individuals (Lemey et al., 2007). In addition, it does not take into account the potential impact of synonymous changes on cis-acting effects between interactions of the genome with virion components that may alter the overall virion maturation and infectivity. Viral replication rates may depend on the activation state of the host’s immune system (Silvestri & Feinberg, 2003), the physiological environment (Martinez-Picado et al., 2006), and the environmental conditions existing within latently infected cell populations (Kelly, 1996; Kelly & Morrow, 2003). An investigation utilizing a new computational technique to estimate absolute rates of synonymous and nonsynonymous mutations and characterize how these rates change over time has suggested that the trajectory of HIV-1-associated disease progression among infected individuals may be predicted by the rate of synonymous mutations and that nonsynonymous mutations evolve as a consequence of differential antibody selective pressure. This approach builds on previous relaxed-clock methodology (Drummond et al., 2006), and by comparing evolutionary rates for specific branches within HIV-1 phylogenies, potentially biasing effects of deleterious polymorphisms are corrected for (Lemey et al., 2007). Using this method, a previously unidentified association between the rate of silent HIV-1 evolution and the rate of disease progression has been discovered, demonstrating that host immune mechanisms associated with HIV-1 pathogenesis may also play a role in modulating viral replication and ultimately place restrictions on HIV-1 evolution (Lemey et al., 2007). Most investigations that have compared the rate of nonsynonymous to synonymous changes have concluded that evolution of pol, env, or nef in brain isolates is adaptive in nature (Gray et al., 2011; Huang, Alter, & Wooley, 2002; Spudich & Gonzalez-Scarano, 2012). The challenge now is to understand the selective pressures driving these adaptive changes. To this end, understanding the specific features of the immune response within the CNS during this adaptation period will be important.
5. MOLECULAR DIVERSITY OF HIV-1 ENV AND NEUROLOGIC DISEASE
HIV-1 Env exists as a trimer in the virion and includes the surface glycoprotein transmembrane subunits gp120 and gp41. Initial viral attachment and subsequent entry into host cells are catalyzed by a high-affinity interaction between gp120 and the host cellular surface antigen CD4. This interaction results in gp120 undergoing a conformational change, exposing the coreceptor-binding site (Doms, 2000). The dynamic interaction between CD4-bound gp120 and the coreceptor then results in additional conformational changes culminating in the structural rearrangement of gp41 and facilitation of virus fusion and entry (Doms, 2000). The chemokine receptor CCR5 is the primary HIV-1 coreceptor utilized by the virus for infection of monocytic phagocytes, and viruses containing CCR5-utilizing envelopes (R5) are the predominant HIV-1 variants isolated from infected brain (Albright et al., 1999; Gorry et al., 2002; Smit et al., 2001). Brain microglia and tissue macrophages are known to express lower levels of CD4 and CCR5 on their surfaces than do peripheral blood CD4+ T cells (Lee, Sharron, Montaner, Weissman, & Doms, 1999; Lewin et al., 1996; Wang et al., 2002). CD4 and CCR5 by HIV-1 are interdependent on one another, with one becoming more critical as the other becomes limiting (Doms & Moore, 2000). HIV Env proteins exhibiting increased tropism for cells of the monocyte–macrophage lineage also possess the ability to utilize low levels of both CD4 and CCR5 for fusion and entry, suggesting that decreased dependence on these surface molecules may represent an adaptation preferentially favoring viral replication within the CNS (Gorry et al., 2002; Gray et al., 2005; Martín, LaBranche, & González-Scarano, 2001; Peters et al., 2004; Thomas et al., 2007). However, the mechanistic details concerning how HIV-1 acquires this capacity to enter cells with limiting receptor and coreceptor levels are unclear. However, many of these studies focused on studying isolates derived from postmortem tissues and, therefore, would reflect an end-stage disease phenotype. Moreover, the CNS environment is that of an “immunological privileged” site with a low penetration of antibodies, thus allowing propagation of sequence configurations that would promote neutralization. In fact, isolates from brain are often sensitive to neutralizing antisera (Dunfee et al., 2007; Martin-Garcia, Cocklin, Chaiken, & Gonzalez-Scarano, 2005; Spudich & Gonzalez-Scarano, 2012).
The amino acid sequence of HIV Env determines its affinity for CD4 binding, and sequence alterations within the envelope protein may affect cellular tropism of HIV-1 by changing coreceptor utilization (Hoffman, Stephens, Narayan, & Doms, 1998; Wang et al., 1998). Efficient infection of the brain by HIV-1 is predicated on macrophage tropism, with the majority of viruses isolated from the brain of HIV-1-infected patients representing viruses that preferentially utilize CCR5 for attachment and entry into host cells (Albright et al., 1999; Gorry et al., 2001; Reddy et al., 1996). Studies have suggested that viruses isolated from the brain of HIV-1-infected patients may have higher affinity for CCR5 than do CCR5-utilizing viruses isolated from other tissue compartments, indicating a decreased dependence on CD4 expression by target cells (Gorry et al., 2002; Martín et al., 2001; Shieh, Martin, Baltuch, Malim, & Gonzalez-Scarano, 2000). The chemokine receptor CCR3 has also been implicated in the infection of microglia, and it has been shown that targeting either CCR5 or CCR3 protected both microglia and monocyte-derived macrophages (MDMs) from many strains of HIV-1 (Agrawal et al., 2009; He et al., 1997; Martin-Garcia, Cao, Varela-Rohena, Plassmeyer, & Gonzalez-Scarano, 2006). Additionally, CXCR4 (X4) utilizing viruses and dual-tropic (X4R5) viruses are rarely found in the brain, despite reports suggesting their ability to induce neuronal damage (Chan et al., 1999; Gorry et al., 2001; Reddy et al., 1996).
Studies have demonstrated that brain-derived HIV-1 sequences from HAD patients differ from those isolated from non-HAD patients, suggesting that specific HIV envelope sequences may be associated with clinical onset of dementia (Power et al., 1994; van Marle & Power, 2005). Furthermore, comparative molecular and biological analyses of HIV-1 isolates derived from both HAD and non-HAD patients have suggested that differences in viral tropism can discriminate between patients with and without dementia, indicating that neurotropic HIV-1 variants evolve independently within the brain and contribute to neuropathogenesis (Smit et al., 2001). Several studies have demonstrated that HIV-1 strains derived from patients with HAD differ from those derived from patients without dementia, primarily within the V1 and V3 region of gp120. Variation within the V1 region, in addition to the V2 region, has been associated with altered replication efficiency in macrophages (Toohey, Wehrly, Nishio, Perryman, & Chesebro, 1995), and the V3 region of Env has been implicated as a primary determinant of macrophage tropism and subsequent cytopathicity (Chesebro, Wehrly, Nishio, & Perryman, 1992; Hwang, Boyle, Lyerly, & Cullen, 1991; Korber, MacInnes, Smith, & Myers, 1994; Rossi et al., 2008). Single amino acid alterations within this region have been shown to change viral tropism and entry (McKnight et al., 1995). In addition, association has been made between specific V3 sequences and HAD (Chang et al., 1998; Korber et al., 1994; Power et al., 1994). HAD patients exhibit impaired serological responses against CCR5-dependent HIV-1 strains and increased molecular diversity within the V3 region of Env, suggesting the emergence of viral mutants that may preferentially infect the brain and mediate neurodegeneration (Van Marle et al., 2002). One specific Env variant present in the CD4-binding site of gp120, N283, has been identified at high frequency in brain-derived viruses of HAD patients (Dunfee et al., 2006). N283 has been shown to increase the affinity of gp120 for CD4 by decreasing the dissociation rate between these two molecules, thus enabling HIV-1 to utilize lower levels of CD4 for binding and entry and subsequently enhancing viral replication within macrophages and microglia (Dunfee et al., 2006). The N283 HIV-1 Env variant is found significantly more often in brain-derived Envs from HAD patients (41%) compared with non-HAD patients (8%), suggesting that this macrophage-tropic HIV-1 variant may be specifically associated with neurologic disease (Dunfee et al., 2006). Studies have shown that brain-derived HIV-1 isolates differ from those typically found in systemic circulation (Epstein et al., 1991; Korber, Kunstman, et al., 1994; Wong et al., 1997), and comparisons between peripheral blood-derived viruses and those derived from multiple brain compartments in the same patient have suggested segregated evolution of viral strains present within differing brain regions (Chang et al., 1998). Several studies have shown that HIV-1 proteins may be directly neurotoxic (Dahiya, Nonnemacher, & Wigdahl, 2012; Li et al., 2012; Liu et al., 2000; Meucci et al., 1998; Muller, Schroder, Ushijima, Dapper, & Bormann, 1992) (also see sections below). Individual HIV-1 isolates have been shown to exhibit differential induction of neuronal apoptosis, and this induction is independent of viral replication capacity (Ohagen et al., 1999). Recently, HIV-1 infection was shown to upregulate cathepsin B in macrophages and to reduce cystatin–cathepsin interactions, eventually leading to neuronal apoptosis (Rodriguez-Franco et al., 2012). Also, it was shown that gp120 induced caspase-3-dependent neuronal apoptosis by enhancing A-type transient outward K+ currents via CXCR4-protein kinase C signaling (Xu et al., 2011). The V3 region of the HIV-1 Env, in addition to conferring increased tropism and subsequent infection of macrophages and microglial cells (Strizki et al., 1996), has been shown to impact the release of neurotoxic molecules following infection of macrophages (Cunningham et al., 1997; Kaul, Garden, & Lipton, 2001; Khanna et al., 2000; Power et al., 1998) and may, itself, be neurotoxic (Pattarini, Pittaluga, & Raiteri, 1998).
6. GENETIC DIVERSITY WITHIN HIV-1 Tat, Vpr, AND THE LTR, AND ITS CONTRIBUTION TO THE ONSET AND SEVERITY OF HIV-1-ASSOCIATED NEUROLOGICAL SYSTEM DISEASE
6.1. HIV-1 transactivator protein Tat
HIV-1 transcription involves an early, Tat-independent and a late, Tat-dependent phase, and transactivation of the viral genome is a critical step in the viral replication cycle, as previously reviewed (Dahiya et al., 2012; Li et al., 2012). The presence of Tat has been shown to increase LTR-mediated transcriptional activity by several hundredfold, and in the absence of Tat, viral replication falls to nearly undetectable levels (Doppler, Schalasta, Amtmann, & Sauer, 1992; Green, Ishino, & Loewenstein, 1989; Rice & Mathews, 1988). Tat is a unique transcription factor in that it binds to the “UCU” bulge of the transactivation response element (TAR), a cis-acting RNA enhancer element contained within the 5′ end of all viral transcripts (Brady & Kashanchi, 2005; Rappaport et al., 1999). The interaction of HIV-1 Tat with TAR RNA increases viral transcription and elongation (Raha, Cheng, & Green, 2005; Selby, Bain, Luciw, & Peterlin, 1989). Specifically, HIV-1 Tat is known to promote the binding of P-TEFb (cyclin T1 and cdk9) to the TAR region located within the viral promoter, immediately downstream of the transcriptional initiation site, and the interaction of Tat with P-TEFb and the TAR element results in hyperphosphorylation of the C-terminal domain and subsequent increased processivity of RNA polymerase II (pol II) (Raha et al., 2005; Zhou et al., 2000). The Tat-PTEFb crystal structure has shown that Tat forms extensive contacts with both the CycT1 and Cdk9 subunits in P-TEFb, resulting in a conformational change and constitutive activation of the enzyme (Tahirov et al., 2010). HIV-1 Tat may also be involved with the formation of the transcriptional preinitiation complex (Dahiya et al., 2012; Raha et al., 2005).
In addition to the HIV-1 LTR, Tat is known to upregulate several other viral as well as cellular genes. Within the CNS, Tat has been shown to stimulate HIV-1 LTR-mediated viral gene expression in the absence of TAR (Taylor & Khalili, 1994), an activity that may result from its ability to enhance the activity of cytokines such as tumor necrosis factor-α (TNF-α) (Sawaya et al., 1998). TNF-α also has the ability to activate the HIV-1 LTR via activation of cytoplasmic nuclear factor kappa B (NF-κB) (Nabel, Rice, Knipe, & Baltimore, 1988; Sawaya et al., 1998), and this positive feedback mechanism may lead to constitutive TNF-α and HIV-1 Tat synthesis by infected glial and microglial cell populations within the brain, ultimately resulting in paracrine dysregulation and damage to neighboring neurons and astrocytes. Similarly, evidence generated from stable expression studies has indicated that HIV-1 Tat may inhibit TNF-α-induced repression of TNF receptor p55, thereby resulting in the amplification of TNF-α activity (Chiao et al., 2001).
HIV-1 Tat can also be secreted from infected cells, including infected macrophages, microglia, and astrocytes and may consequently be taken up by neighboring, uninfected cells (Ensoli et al., 1993; Verhoef, Klein, & Berkhout, 1996). Tat protein has been detected within the brain of infected individuals, and the uptake of Tat by CNS cells has toxic consequences, resulting in large part from neuronal apoptosis (Hudson et al., 2000; Nath et al., 1996). Extracellular Tat can enter neurons via endocytosis through interaction with the low-density lipoprotein receptor-related protein present on the neuronal surface (Vendeville et al., 2004). Recently, it was shown that Tat could bind to the promoters of the phosphatase and the tensin homologue and protein phosphatase 2A (PP2A), eventually resulting in apoptosis of HIV-1-infected CD4+ T cells (Kim, Kukkonen, Gupta, & Aldovini, 2010). Furthermore, Tat has been shown to be transported along anatomical pathways within the brain, indicating that the neurotoxic effects of HIV-1 Tat may occur in regions far removed from the site of active infection (Bruce-Keller et al., 2003). Interestingly, secreted or extracellular Tat has been shown to function as a specific CXCR4 antagonist, selecting against X4-utilizing viruses, and thereby greatly influencing the development and progression of HIV-1 disease (Xiao et al., 2000), specifically within the CNS where R5 viruses are thought to play the predominant role in pathogenesis.
The neuropathologic properties associated with HIV-1 Tat stem from its ability to either directly or indirectly induce apoptosis, upregulate cytokines and chemokines, and interact with matrix metalloproteinases (MMPs). The ability of HIV-1 Tat to upregulate TNF-α and interleukin-1β (IL-1β) has been associated with increased expression of cell adhesion molecules on endothelial cells. Likewise, Tat-induced upregulation of MCP-1 has been shown to exacerbate neuroinvasion, facilitating the loss of BBB integrity, a pathological hallmark of late-stage HAND (Avison et al., 2004; Mayne et al., 1998; Nath, Conant, Chen, Scott, & Major, 1999). Although HIV-1 Tat and various MMPs, including MMP-1, -2, and -9, are known to be independently cytotoxic to cells within the CNS, studies have suggested that the dynamic interaction between Tat and MMPs may be neuroprotective (Johnston et al., 2001; Zhang et al., 2003). Specifically, MMP-1 has been shown to selectively cleave HIV-1 Tat and thereby neutralize its neurotoxic potential (Rumbaugh et al., 2006).
The molecular diversity of HIV-1 Tat protein isolated from brains of patients infected with different HIV-1 clades has been examined, as previously reviewed (Li et al., 2012). Studies examining Tat proteins representative of HIV-1 subtypes B, C, and BF recombinants have demonstrated important structural and functional differences (Siddappa et al., 2006; Turk et al., 2006). BF recombinant HIV-1 isolates, from Argentina, appear to have a replicative advantage over subtype B isolates, possibly owing to the differential ability of Tat to interact with the LTR, and subtype C Tat protein has been shown to be more highly ordered than subtype B Tat. In addition, subtype C Tat protein has been demonstrated to be consistently inferior to subtype B Tat in biological assays with respect to its ability to promote viral proliferation, induce TNF-α and IL-6 expression, and upregulate chemokine coreceptor expression (Siddappa et al., 2006). However, studies have also shown that HIV-1 subtype C Tat exhibits greater transcriptional activity in the Jurkat CD4+ T-cell line when compared with subtypes B and E and that this higher level of transactivation is not LTR sequence dependent but results from variations in the C Tat sequence at amino acid residues 57 (Arg in B and E and Ser in C) and 63 (Glu in B, E, and C), which are within and close to the basic domain, respectively (Kurosu et al., 2002). Phylogenetic analyses of Tat sequences from patients with and without HAD have shown clustering of sequences with respect to clinical diagnosis of neurological impairment as well as tissue of origin (Bratanich et al., 1998; Mayne et al., 1998). Nonsynonymous versus synonymous mutation rates among brain-derived Tat sequences isolated from patients with NCI were shown to be significantly greater than those isolated from patients without clinical evidence of neurologic disease (Bratanich et al., 1998). Collectively, these studies suggest that differing selective pressures act on individual HIV-1 genes within the CNS and that these differing pressures may influence both the development and subsequent severity of NCI. Comparisons of matched brain- and spleen-derived Tat sequences have suggested that greater sequence homology exists among brain-derived Tat clones than what is observed between brain- and spleen-derived clones (Mayne et al., 1998). Additionally, significant sequence heterogeneity exists within brain-derived Tat in domains associated with viral replication and intracellular transport (Mayne et al., 1998). Importantly, HIV-1 Tat derived from HAD patients has been associated with greater neuronal death both in vitro and in vivo compared with Tat from non-HAD patients, and this characteristic has been attributed, in part, to enhanced MMP-2 expression induced by brain-derived HIV-1 Tat (Johnston et al., 2001). Interestingly, however, these same brain-derived Tat isolates also appear to be limited in their ability to enhance viral gene expression despite their increased activation of host transcriptional machinery (Silva et al., 2003). However, one must remember that these viral gene activation studies were performed with a viral regulatory region that was derived from a non-CNS tissue source and may, therefore, not be naturally compatible with respect to optimal LTR activation by a Tat protein selected for CNS replication. This is particularly important because previous studies (Burdo, Gartner, Mauger, & Wigdahl, 2004; Hogan, Nonnemacher, Krebs, Henderson, & Wigdahl, 2003; Hogan, Stauff, et al., 2003) demonstrated that LTRs derived from the CNS are likely to be structurally and functionally different from LTRs derived from other tissue sources and that colinear Tat and LTR combinations may result in more efficient LTR activation (Li et al., 2011). Nonetheless, taken together, these reports suggest that genetic diversity of HIV-1 Tat very likely contributes to the establishment and severity of HAND.
6.2. HIV-1 Vpr
Viral protein r (Vpr) is a 96-amino acid accessory protein that is packaged into the HIV-1 virion via its association with the p6 domain of HIV-1 Gag (Emerman & Malim, 1998). Vpr is a multifunctional protein affecting both early and late stages of the HIV-1 viral life cycle and is associated with the nuclear localization and import of the HIV-1 preintegration complex (Lu, Spearman, & Ratner, 1993; Mahalingam, Collman, Patel, Monken, & Srinivasan, 1995; Mahalingam, Khan, Jabbar, et al., 1995; Mahalingam, Khan, Murali, et al., 1995). Lacking a true nuclear localization sequence, Vpr is known to localize to the nucleus when expressed in vitro (Lu et al., 1993; Mahalingam, Khan, Jabbar, et al., 1995; Mahalingam, Khan, Murali, et al., 1995). Therefore, Vpr likely facilitates nuclear localization via its interaction with cellular proteins involved in nuclear import, possibly karyopherin α and β (Lu et al., 1993; Mahalingam, Collman, et al., 1995; Mahalingam, Khan, Jabbar, et al., 1995; Mahalingam, Khan, Murali, et al., 1995). Vpr has been suggested by several studies to induce cell cycle arrest in HIV-1-infected cells, with Vpr-expressing cells accumulating in the G2 phase (Ayyavoo et al., 1997; Jowett et al., 1995). The efficacy of Vpr-induced transactivation of the LTR has been shown to correlate with the induction of G2 arrest in host cells (Ardon et al., 2006; DeHart et al., 2007; Zimmerman et al., 2004). Interestingly, cell cycle arrest of infected cells has been shown to increase HIV-1 LTR transcriptional activity, independent of Vpr (Cohen, Dehni, Sodroski, & Haseltine, 1990). Vpr has also been shown by several studies to transactivate the HIV-1 LTR by a variety of mechanisms (Cohen et al., 1990; Goh et al., 1998; McAllister et al., 2000; Sawaya et al., 1999). Vpr has been shown to activate the LTR via interaction with HIV-1 Tat and to indirectly increase LTR activity through its interaction with the transcription factor p300 (Felzien et al., 1998; Sawaya et al., 1999). Studies have suggested that Vpr may be involved in ternary complexes with Sp and the LTR, and investigations have indicated that Vpr is able to directly interact with the HIV-1 LTR via its binding to C/EBP-binding sites I and II (Burdo, Gartner, et al., 2004; Hogan, Nonnemacher, et al., 2003).
Studies of Vpr binding to HIV-1 LTR sequences encompassing the ATF/CREB, C/EBP site I, and the promoter-distal NF-κB site have suggested that the Vpr preferentially interacts with sequences spanning C/EBP site I and the adjacent NF-κB-binding site (Burdo, Nonnemacher, et al., 2004). This result in addition to the established proximity of the these two binding sites indicates that Vpr and NF-κB binding may be mutually exclusive; however, the downstream NF-κB element would still be available for binding independent of Vpr interactions upstream (Burdo, Gartner, et al., 2004). Importantly, it has been demonstrated that HIV-1 Vpr induces IL-8 production in monocytes through activation of both NF-κB and NF-IL-6 (C/EBP), and elevated levels of IL-8 are thought to be responsible for certain clinical manifestations observed among AIDS patients throughout the course of disease (Roux, Alfieri, Hrimech, Cohen, & Tanner, 2000). This finding suggests that HIV-1 LTR activity is likely influenced by a complex and dynamic balance between Vpr and members of the C/EBP transcription factor family, as well as NF-κB isoforms (Burdo, Nonnemacher, et al., 2004). Additionally, both Vpr and C/EBP factors are known to be required for efficient HIV-1 replication within cells of myeloid lineage. Studies have suggested that Vpr-regulated promoter activation may be enhanced as a result of increased binding of NF-κB and C/EBP factors to their respective binding sites (Roux et al., 2000), and other studies have suggested that Vpr may also mediate promoter activity via direct binding to C/EBP sites and other adjacent binding sequences (Burdo, Nonnemacher, et al., 2004; Hogan, Nonnemacher, et al., 2003). Evidence supports the concept that sequence-dependent interactions between Vpr and C/EBP site I may occur in the context of neurologic disease. Electrophoretic mobility shift (EMS) analyses have revealed a direct association between Vpr and HIV-1 LTR sequences, which include C/EBP site I, the promoter-distal NF-κB site, and the upstream ATF–CREB-binding site (Burdo, Nonnemacher, et al., 2004; Hogan, Nonnemacher, et al., 2003). This relationship was shown to be sequence-specific with respect to C/EBP site I (Burdo, Nonnemacher, et al., 2004). The 3T C/EBP-binding site variant, described earlier, which binds C/EBP factors with low relative affinity, has also been shown to be the C/EBP site I variant that binds Vpr with the highest relative affinity (Hogan, Nonnemacher, et al., 2003). Importantly, the affinity of C/EBP-binding sites for Vpr is associated with HAD, with high-affinity sites being more prevalent in HAD patients (Burdo, Nonnemacher, et al., 2004).
Cell types within the CNS that are capable of supporting productive HIV-1 infection are limited to macrophages and microglia; however, neuropathological abnormalities associated with cognitive impairment (MCMD and HAD) are thought to result, in large part, from neuronal dropout and apoptosis of neurons (Gelbard et al., 1995; Ohagen et al., 1999; Petito, 1995). Astrocytes do not support a high-level productive HIV-1 replication, potentially involving a defect in Rev function (Gorry et al., 1999; Messam & Major, 2000; Neumann et al., 1995; Tornatore, Chandra, Berger, & Major, 1994). Therefore, an indirect mechanism leading to apoptosis of neurons may exist, and HIV-1 Vpr is one of the viral gene products implicated in this process. Recently, it was shown that the effect of Vpr on neuronal death is in part via released proinflammatory factors. In this study, supernatants from Vpr-deleted HIV-1 mutant-infected MDMs contained lower concentrations of IL-1β, IL-8, and TNF-α and showed reduced neurotoxicity compared with wild-type HIV-1-infected MDM supernatants (Guha et al., 2012).
Importantly, free Vpr has been identified in the serum of HIV-1-infected patients and in the CSF of HIV/AIDS patients with neurological disease (Levy, Refaeli, & Weiner, 1995). Studies have linked Vpr to the induction of apoptosis of T cells, and although the HIV-1 envelope glycoprotein gp120 and Tat regulatory protein have been most commonly associated with cellular death during HIV-1 infection, virion-encapsulated Vpr may also be involved in CNS cell death in vivo. Experiments using extracellular Vpr have demonstrated that Vpr is able to bind promonocytic and lymphoid cells and increase permissiveness to HIV-1 replication (Levy et al., 1995). Extracellular Vpr has been shown to associate directly with the plasmalemma of cultured rat hippocampal neurons and causes a large inward sodium current, depolarization, and cell death (Levy et al., 1995; Piller, Jans, Gage, & Jans, 1998). In addition, HIV-1 Vpr has been shown to potently induce apoptosis both in the undifferentiated neuronal precursor cell line NT-2 and in mature human neurons (Patel, Mukhtar, & Pomerantz, 2000). Thus, based on the cytotoxic and neurotoxic effects of extracellular Vpr, one may postulate that cell-free Vpr likely contributes to cellular depletion within lymphoid, peripheral blood, and CNS tissue compartments. Furthermore, the fact that extracellular Vpr is present in the serum and CSF of AIDS patients with neurologic disease suggests that extracellular Vpr may play a significant role in AIDS pathology and HIV-1-associated neurologic complications (Levy et al., 1995; Piller et al., 1998). Moreover, Vpr has been implicated in modulating the host glucocorticoid receptor to affect transcription from the LTR as well as other host genes (Refaeli, Levy, & Weiner, 1995). Vpr has also been shown to transactivate promoters containing glucocorticoid-responsive elements. This modulation is most likely via direct interaction with the glucocorticoid receptor, with Vpr acting as a coactivator of glucocorticoid receptor (Kino et al., 1999). Vpr has also been shown to induce oxidative stress in microglial cells via the hypoxia-inducible factor pathway (Deshmane et al., 2009) and has been shown to interact with ANT, PP2A, and HAX-1, which have been shown to play important roles in pathways that culminate in neuronal degeneration (Na et al., 2011; Zhao, Li, & Bukrinsky, 2011). Nonetheless, extensive studies related to the role Vpr plays in HIV pathogenesis have established it as a crucial accessory protein with a multitude of functions spread across different stages of the viral life cycle.
6.3. HIV-1 LTR activity within cells of the monocyte–macrophage lineage
The HIV-1 LTR is approximately 640 bp long and consists of the U3, R, and U5 segmented regions. The U3 region is further divided into the modulatory, enhancer, and core regions, which facilitate the interaction of both viral and cellular proteins involved with regulating viral gene expression (Cullen, 1991; Pereira, Bentley, Peeters, Churchill, & Deacon, 2000). With HIV-1 subtype B, the core region contains the TATAA box and a GC-rich sequence, which facilitates binding of members of the Sp family of transcription factors. The TATAA box binds TBP (TATAA-binding protein), in addition to other cellular proteins involved with the pol II transcriptional complex (Jones & Peterlin, 1994). The enhancer element is located immediately upstream of the core region and is associated primarily with the presence of two 10-bp NF-κB-binding sites (Nabel & Baltimore, 1987). The modulatory region, which consists of sequences located upstream of the NF-κB-binding sites, contains numerous transcription factor-binding sites specific for factors including C/EBP, ATF/CREB, LEF-1, NF-AT, and many others (Krebs et al., 2000). Studies of the modulatory region have revealed that this region is rich in cis-acting-binding elements, which serve to both repress and activate the HIV-1 LTR (Pereira et al., 2000). Furthermore, the interaction of viral proteins, specifically Vpr and Tat, with the LTR provides an additional element of complexity to the regulation of viral gene expression.
One of the primary regulators of HIV-1 LTR activity in all susceptible host cell populations, including cells of the monocyte–macrophage lineage, is NF-κB (Asin, Bren, Carmona, Solan, & Paya, 2001). Several studies have been aimed at determining the dependence of NF-κB family members with respect to transcriptional activation of the HIV-1 LTR in T cells and its subsequent effect on reactivation of HIV-1 from latency (Chen, Feinberg, & Baltimore, 1997; Folks et al., 1986; Ross, Buckler-White, Rabson, Englund, & Martin, 1991). Depending on the type of T cell examined, and differences in the experimental approaches employed, results from these studies have been conflicting. Generally, HIV-1 LTR NF-κB-binding sites are indispensable with respect to viral replication in CD4+ T-cell lines (Alcami et al., 1995; Chen et al., 1997). Studies involving human monocytic cells and transformed human monocyte and macrophage cell lines have centered, in large part, on determining how monocytic differentiation affects HIV-1 expression and how HIV-1 infection results in NF-κB activation (Griffin, Leung, Folks, Kunkel, & Nabel, 1989; Raziuddin et al., 1991; Schuitemaker et al., 1992). Interestingly, differentiated macrophages already contain a constitutive nuclear pool of NF-κB, and what role this preexisting pool of NF-κB plays with respect to modulating HIV-1 gene expression remains unclear (Asin et al., 2001). However, studies have suggested that preexisting NF-κB heterodimers within the nuclei of these cells play a role in transcriptional initiation following infection by HIV-1 (Asin et al., 2001). Overall, studies have concluded that NF-κB cis-acting elements within the HIV-1 LTR are critical for efficient LTR activity and subsequent viral gene expression both within CD4+ T cells and in cells of monocyte–macrophage lineage.
The importance of NF-κB can also be observed in the difference in LTR activity of different HIV-1 subtypes. HIV-1 subtype B is the predominant subtype in North America and in Europe, whereas subtypes C and E are most prevalent in other parts of the world, including east Asia, Thailand, India, and southern Africa (Janssens, Buve, & Nkengasong, 1997; Novitsky et al., 1999; Ping et al., 1999). One of the most striking differences between HIV-1 subtype C and other HIV-1 subtypes, such as B and E, resides in the LTR. Subtype C viruses have been shown to contain three NF-κB-binding sites within the enhancer element of the LTR, whereas subtypes B and E have only two and one, respectively (Gao et al., 1996; Kurosu et al., 2001; Montano et al., 2000). The functional consequence of this difference was revealed by transient transfection assay in HeLa cells, which showed that subtype C LTRs have higher promoter/enhancer activity compared with subtypes B and E. Subtype C does appear to be transmitted more efficiently than other subtypes (Essex, 1999), and specific genetic biological differences such as increased NF-κB-binding sites may play a role in this increased transmission efficiency.
Investigations of the transcriptional regulation of the HIV-1 LTR in cells of the monocyte–macrophage lineage have also focused on the role of C/EBP and Sp transcription factors with respect to regulation of viral gene expression within the CNS. C/EBP factors have been shown to be critically involved in the regulation of monocyte-specific gene expression (Matsusaka et al., 1993; Pope et al., 1994; Tanaka et al., 1995). C/EBPβ has been shown to bind at least two sites within the HIV-1 LTR and has been demonstrated to activate viral transcription in transient expression analyses (Ross et al., 2001; Tesmer, Rajadhyaksha, Babin, & Bina, 1993). Studies have revealed that at least one intact C/EBP site is required for HIV-1 replication in monocytic cells; however, this is not required for replication in T cells (Henderson & Calame, 1997; Henderson, Connor, & Calame, 1996; Henderson, Zou, & Calame, 1995).
7. SEQUENCE VARIATION OF SPECIFIC TRANSCRIPTION FACTOR-BINDING SITES WITHIN THE HIV-1 LTR AND ITS CORRELATION WITH NERVOUS SYSTEM DISEASE
The relationship between LTR genetic diversity and HIV-1 disease is complex. Several reports have suggested that LTR sequence variation may alter promoter activity in varying cell types (Henderson & Calame, 1997; Henderson et al., 1996, 1995; Krebs, Mehrens, Pomeroy, Goodenow, & Wigdahl, 1998; McAllister et al., 2000). Numerous investigations have reported sequence variation within LTRs isolated from infected patients (Hogan, Nonnemacher, et al., 2003; Hogan, Stauff, et al., 2003; Michael, D’Arcy, Ehrenberg, & Redfield, 1994; Nonnemacher, Irish, Liu, Mauger, & Wigdahl, 2004; Ross et al., 2001). Comparative analysis of LTRs isolated from both peripheral blood mononuclear cells and brain across a population of individuals has also revealed compartmentalization of the specific LTR variants and showed that LTRs isolated from the CNS are more closely related to previously characterized brain-derived LTRs than to LTRs isolated from other physiological compartments (Hogan, Nonnemacher, et al., 2003; Hogan, Stauff, et al., 2003; Michael et al., 1994; Nonnemacher et al., 2004; Ross et al., 2001). These naturally occurring sequence alterations within the LTR, which appear to arise as a result of tissue-specific selective pressures, may have a profound impact on the ability of the LTR to support HIV-1 infection by differentially modulating the ability of critical transcription factors to bind the LTR (Hogan, Stauff, et al., 2003). Although several studies have suggested that there is no correlation between LTR sequence variation and altered viral tropism and replication (Pomerantz, Feinberg, Andino, & Baltimore, 1991; Schuitemaker et al., 1993; Velpandi, Nagashunmugam, Otsuka, Cartas, & Srinivasan, 1992), studies involving the analysis of two different LTR variants have demonstrated that increased LTR activity based on transient expression studies corresponds to increased viral replication (Golub, Li, & Volsky, 1991; McAllister et al., 2000). Studies utilizing an HIV-1 LAI infectious molecular clone revealed that when the native high-affinity NF-κB-proximal Sp-binding site III was replaced with a low-affinity site, replication within Jurkat CD4+ T cells was markedly decreased, whereas little effect on replication was observed within U-937 monocytic cells (McAllister et al., 2000). These results were consistent with previously published results (Zeichner, Hirka, Andrews, & Alwine, 1992; Zeichner, Kim, & Alwine, 1991a, 1991b), demonstrating that mutations introduced into the HIV-1 LTR that resulted in altered transient expression activity also resulted in similar alteration in viral replication potential when the same mutations were placed into the context of a replication-competent virus. Transient transfection analyses of HIV-1 LAI LTR-luciferase constructs in the Jurkat T-cell line have suggested that the large reduction in viral replication within these cells, caused by low-affinity Sp-binding site III variants, may be the result of reduced basal, Vpr, and Tat-mediated LTR activity (McAllister et al., 2000). When examined within the context of an HIV-1 YU-2 LTR-luciferase construct, the naturally low-affinity Sp-binding site III was replaced with a high-affinity site, which resulted in increased basal YU-2 LTR activity in Jurkat T cells and reduced LTR activity in U-937 monocytic cells (McAllister et al., 2000). In addition, LTRs derived from HIV-1-infected patients have been shown to differentially regulate transient expression in a cell type-specific manner, a finding that is reinforced by studies involving cell type-specific reporter gene expression directed by a brain-derived LTR in transgenic mouse CNS tissue (Corboy, Buzy, Zink, & Clements, 1992; McAllister et al., 2000; Michael et al., 1994).
Several investigations have compared LTR sequences from HIV-1-infected long-term nonprogressors (LTNPs) and rapid progressors, and in each of these studies, no direct relationship could be established between LTR sequence variation and disease progression (Zhang et al., 1997). Furthermore, transient expression analyses performed in both cell lines and primary monocytes demonstrated no simple correlation between promoter length and rapidity of disease course (Zhang et al., 1997). However, two LTNPs were shown to harbor virus that exhibited what could be defined as a defective LTR. G to A hypermutations were observed throughout the promoter region of LTRs derived from one LTNP (Zhang et al., 1997), and another LTNP was shown to harbor virus with multiple insertions and deletions across the LTR (Rousseau, Abrams, Lee, Urbano, & King, 1997), both indicating defects within the 5′ LTR structure and suggesting that impaired functionality of the HIV-1 LTR may correspond to long-term nonprogression in a subset of HIV-1-infected patients. Other reports have postulated that LTRs with increased activity may correlate with greater viral infectivity and propagation throughout high-risk populations. HIV-1 subtype C LTRs, which have been show to contain three NF-κB-binding sites, exhibit enhanced LTR activation when compared with LTRs containing only one or two of these sites. Additionally, studies have shown that subtype C viruses also produce increased levels of p24, thus indicating greater replication rates than viruses representative of other HIV-1 subtypes (Naghavi, Schwartz, Sonnerborg, & Vahlne, 1999).
Studies have demonstrated that specific HIV-1 LTR C/EBP-binding site sequence configurations may be preferentially compartmentalized in the brain of infected patients and that these LTR variants exhibit enhanced LTR-mediated transcriptional activity (Hogan, Stauff, et al., 2003; Ross et al., 2001). Transient expression studies have suggested that an NF-κB-proximal C/EBP site I that binds C/EBP factors with high relative affinity results in increased basal as well as IL-6-induced LTR activity (Ross et al., 2001). Investigations have revealed that specific HIV-1 LTR C/EBP configurations preferentially encountered in the brain exhibit enhanced LTR-specific activity (Ross et al., 2001). A high relative affinity 6G C/EBP site I (T to G change at nucleotide position 6) was commonly found in brain-derived LTRs but was infrequently encountered in peripheral blood-derived LTRs, as demonstrated by analyses of variations at each nucleotide position within C/EBP site I (Hogan, Stauff, et al., 2003). A differential level of conservation was also observed at C/EBP site II. Analyses of overall conservation of each site demonstrated that C/EBP site II was highly conserved in LTRs derived from brain and less conserved among those derived from the peripheral blood compartment. Overall, these studies demonstrated that brain-derived LTRs contain two high relative affinity C/EBP-binding sites and suggest that these sites may play a particular role in LTR-directed transcription with respect to CNS disease.
Studies have also suggested a direct correlation between specific C/EBP sequence variants and HAD. Sequence analysis of C/EBP-binding sites I and II using peripheral blood-derived HIV-1 LTR sequences was reported in three studies (Estable et al., 1996; Kirchhoff, Greenough, Hamacher, Sullivan, & Desrosiers, 1997; Michael et al., 1994). Because these published reports used different classification systems to describe disease severity, LTRs were designated as belonging to one of three groups prior to analysis. HIV-1 LTR sequences from asymptomatic patients with nonprogressing or stage I disease were assigned to disease severity group 1 (DSG 1) (Hogan, Stauff, et al., 2003). DSG 2 was comprised of patients characterized as having slow-progressing, stage II or III disease (Hogan, Stauff, et al., 2003). DSG 3 consisted of patients who were originally classified as having progressing, stage IV HIV-1 disease (Hogan, Stauff, et al., 2003). At C/EBP-binding site I, a 3T (C to T change at nucleotide position 3) configuration was observed at low prevalence within LTRs isolated from the peripheral blood of HIV-1-infected patients early in disease, and at relatively high prevalence from patients with late-stage disease (Hogan, Stauff, et al., 2003). The prevalence of the 3T C/EBP site I variant was not identified among LTRs from DSG1 patients and increased from approximately 8% of all DSG 2 LTRs to nearly 50% of all DSG 3 LTRs (Hogan, Stauff, et al., 2003). Within C/EBP site II, the consensus B (conB) configuration increased significantly throughout disease progression, whereas the prevalence of the 6G and 4C (T to C change at nucleotide position 4) variants decreased (Hogan, Stauff, et al., 2003). The conB configuration at C/EBP site II increased in prevalence from approximately 24% of all DSG 1 LTRs to approximately 93% of all DSG 3 LTRs. Conversely, the 4C C/EBP site II sequence variant decreased from approximately 28% in DSG 1 to approximately 8% in DSG 2 and was completely absent among DSG 3 LTRs (Hogan, Stauff, et al., 2003). Likewise, the 6G C/EBP site sequence variant decreased in prevalence from nearly 35% in DSG 1 to approximately 1% in DSG 3 LTRs. Interestingly, in this as well as similar studies, described in more detail below, involving analysis of Sp transcription factor-binding sites I, II, and III, both NF-κB-binding sites I and II were shown to be highly conserved in the conB configuration throughout disease progression (Hogan, Stauff, et al., 2003; Nonnemacher et al., 2004).
With respect to the impact of the genetic variants in C/EBP sites I and II and CNS disease, the 3T C/EBP site I variant was also observed in 25% of all brain-derived LTRs from patients diagnosed with dementia but was absent in brain-derived LTRs from patients without dementia (Hogan, Stauff, et al., 2003). The 3T C/EBP site I sequence configuration has been shown to have low relative affinity for C/EBP factors (Burdo, Gartner, et al., 2004; Hogan, Stauff, et al., 2003). Taken together, these results suggest that the 3T C/EBP site I configuration may provide a valuable tool in evaluating the likelihood of HIV-1-infected patients developing HAD. Similar to the observations made concerning the 3T C/EBP site I variant, the 6G and 4C C/EBP site II variants were observed in approximately 10% and 7% of HAD patients, respectively, and neither the 6G nor the 4C variant was found in LTRs derived from patients without dementia (Hogan, Stauff, et al., 2003). One study examining the regional distribution of HIV-1 LTRs containing the 6G and 4C C/EBP site II sequence variants within the brains of patients with HAD revealed statistically significant differences, with the high-affinity 6G C/EBP site II accumulating in the midfrontal gyrus and the low-affinity 4C C/EBP site II accumulating in the cerebellum (Burdo, Gartner, et al., 2004). These observations are consistent with reports of viral replication rates within these neuroanatomical regions, with the midfrontal gyrus representing a neuroanatomical region known to exhibit high-level HIV-1 replication (Glass, Fedor, Wesselingh, & McArthur, 1995), whereas viral gene expression within the cerebellum has been shown to occur at very low levels in patients with HAD (Burdo, Gartner, et al., 2004). This suggests that the presence of the 4C C/EBP site II variant within the cerebellum may represent a means by which HIV-1 maintains a silent genome and establishes a latent viral reservoir in the brain.
The three Sp-binding sites that comprise the remaining sequences within the core region of the HIV-1 LTR are also very important to HIV-1 basal and Tat-mediated transactivation (McAllister et al., 2000). Mutation of these binding sites diminishes both basal promoter activity and viral replication (although not in all cell types) (J.J. McAllister and B. Wigdahl, unpublished observation). Specific sequence variations in the NF-κB-proximal Sp site III present within the brain-derived HIV-1 variant, YU-2, result in a failure to interact efficiently with members of the Sp transcription factor family (McAllister et al., 2000) (unpublished observation). This result, combined with the observation that the ratio of Sp1:Sp3 factor binding to Sp site III is increased during monocytic differentiation, suggests that HIV-1 replication within cells of monocyte–macrophage lineage in the brain may be impacted by changes in Sp factor expression that accompany monocytic differentiation as well as alterations of the functional interactions between Sp factors and the NF-κB proximal, G/C-rich Sp-binding site (McAllister et al., 2000) (unpublished observation). Sequence variation within the Sp-binding sites and altered Sp factor recruitment may also impact the ability of HIV-1 Vpr to interact with and subsequently upregulate LTR activity (McAllister et al., 2000 and unpublished observations). Studies aimed at characterizing specific Sp-binding site sequence variants within 348 peripheral blood-derived HIV-1 LTRs isolated from patients with disease ranging in severity from DSG 1 to DSG 3 have demonstrated the presence of a low-affinity 5T (C to T change at nucleotide position 5) variant in Sp site III (Nonnemacher et al., 2004). The 5T Sp site III was shown to increase in prevalence throughout disease progression, with approximately 60% of all DSG 3 LTRs (Nonnemacher et al., 2004). Similar to the 3T C/EBP site I variant, the 5T sequence configuration results in substantially decreased binding affinity for Sp transcription factors, as demonstrated by both EMS (Nonnemacher et al., 2004) and surface plasmon resonance analyses (Nonnemacher and Wigdahl, unpublished observations). Interestingly, when the 3T C/EBP site I containing LTRs were examined for the presence of the 5T Sp site III sequence variant, an absolute correlation was observed with respect to DSG3. Of nine DSG2 LTRs that contained the 3T C/EBP site I sequence variant, six also contained the 5T Sp site III variant; and of 44 DSG3 LTRs that contained the 3T C/EBP site I sequence variant, all 44 also contained the 5T Sp site III variant (unpublished data). Importantly, the 5T Sp site III variant was also observed in 16% of all brain-derived LTRs from patients diagnosed with dementia but was absent in brain-derived LTRs from patients without dementia (unpublished observations). Taking into consideration that decreased binding of C/EBP and Sp factors corresponds with impaired HIV-1 LTR activity, the sequence variation observed within this region is almost certainly relevant to viral pathogenesis and may also influence both the course and severity of immunologic as well neurologic HIV-1 disease progression.
8. HOST GENETIC DETERMINANTS OF HIV-1 INFECTION AND CNS DISEASE
The course of HIV-1 infection is determined by a complex and dynamic interplay between viral and host factors. The very existence of LTNPs, in addition to individuals who have been exposed to HIV but remain uninfected, strongly suggests the existence of predisposing factors that may represent major determinants of clinical disease outcome. Post-seroconversion, progression to AIDS may take as little as 2 years in some individuals, while others may remain symptom-free for more than a decade. To this end, several studies have determined that the marked heterogeneity among infected individuals is governed, at least in part, by host genetic variants that serve to modulate virus replication and antiviral immunity (Carrington & O’Brien, 2003; Fauci, 2003; O’Brien & Nelson, 2004). The most widely recognized host genetic variant to be associated with HIV-1 disease progression is the 32-bp deletion in the coding region of the CCR5 gene (CCR5-Δ32). CCR5-Δ32 is one of the most significant host polymorphism with respect to HIV infection, and it has been shown to effectively block HIV-1 infection in homozygous individuals and to significantly retard disease progression and development of AIDS in heterozygotes (Carrington et al., 1997; Dean et al., 1996; Liu et al., 1996; O’Brien & Nelson, 2004; Samson et al., 1996). Subsequent to the discovery of CCR5-Δ 32, 13 additional host polymorphisms have been identified, and these genetic variants differ widely with respect to their influence on HIV-1-associated disease progression and development of AIDS-defining illnesses (O’Brien & Nelson, 2004). Here, we restrict our discussion to CCR5-Δ32 and CCR2-V64I, which have been linked to HIV-1 neuropathogenesis.
Chemokines and chemokine receptors play a critical role in the pathogenesis and transmission of HIV-1 (Michael, 1999; Paxton & Kang, 1998). The chemokine receptor CCR5 is utilized by R5 strains of HIV-1 to gain entry into host cells, particularly those of the monocyte–macrophage lineage, cells that have been shown to be intricately involved in HAND (Alkhatib et al., 1996; Berger et al., 1998; Choe et al., 1996; Roos et al., 1992). Several single-nucleotide polymorphisms identified within the regulatory region of CCR5 have been implicated in modulating the rate of HIV-1 disease progression (Kostrikis et al., 1999; Martin et al., 1998). The CCR5-Δ32 gene variant has been shown to result in the production of aberrant CCR5 protein and is known to provide considerable protection against HIV-1 infection in individuals homozygous for the mutation and to result in slowed disease progression in heterozygous patients infected with HIV-1 (Barroga et al., 2000; Liu et al., 1996; Singh et al., 2003). Chemokines and their cognate receptors are also expressed within the brain (Glabinski et al., 1995), and CCR5 has been shown to be the primary coreceptor utilized by brain-derived HIV-1 strains isolated from HAD patients (Albright et al., 1999; Boven, van der Bruggen, van Asbeck, Marx, & Nottet, 1999). This, in addition to resident brain microglia and macrophages being the primary cells supporting replication of R5 HIV-1 strains within the CNS, leads to the conclusion that any host genetic variant that may inhibit or at least decrease the ability of HIV-1 to infect these cell types could have a profound effect on the neuropathogenesis of HIV-1 infection and subsequent neurocognitive abnormalities.
The chemokine receptor CCR2 has been shown to function as a minor coreceptor for HIV-1, and its natural ligand is MCP-1, a β-chemokine, which has been linked to neuropsychological impairment (Conant et al., 1998; Weiss, Cuff, & Berman, 1999). A genetic variant within the promoter region of MCP-1 (−2518-G/A polymorphism) has been associated with increased HIV-1 disease progression and development of HAD (Singh et al., 2004), and homozygosity for an MCP-1 2578-G allele has been associated with a 50% decrease in the risk of becoming infected with HIV-1 (Gonzalez et al., 2002). A specific polymorphism (G to A change at nucleotide position 190) in the coding region of the CCR2 gene results in the expression of isoleucine in place of valine at amino acid position 64 (V64I), and this nonsynonymous mutation has been linked with impaired progression of HIV-1 disease in adults (Kostrikis et al., 1998; van Rij et al., 1998); however, this association has been contested by other studies (Michael et al., 1997; Mulherin et al., 2003). In a study investigating development of neurologic complications in 121 HIV-1 patients, the CCR2-V64I polymorphism correlated with more rapid progression to neuropsychological impairment (Singh et al., 2004). An observation that was further confirmed among individuals possessing the CCR5-wt/wt genotype suggested that heterozygosity for the CCR5-Δ32 genotype could not be used to explain the differential rate of progression to neurologic disease in this patient cohort (Singh et al., 2004). Interestingly, although the CCR2-V64I allele was associated with more rapid onset of neuropsychological impairment, it was shown not to have an impact on the overall rate of HIV-1-associated disease progression (Singh et al., 2004). Additionally, the CCR2-64 genotype did not correspond to differential HIV-1 plasma viral load or to CD4+ T-cell count, suggesting that this genotype is not directly involved with viral entry or replication but with the host inflammatory response mediated by the binding of MCP-1 to the CCR2 receptor within the CNS (Singh et al., 2004). Moreover, no association was observed between the genotype of CCR2 and HIV disease progression or therapeutic response (Philpott et al., 2004), indicating that HAART therapy benefits may overshadow the advantages imparted by the V64I polymorphism.
More recently, to investigate the virus–host interactions and their role in HIV-1 pathogenesis, genome-wide association studies (GWAS) have been conducted (Table 6.1) that utilized unbiased searches at the genome-wide level to identify yet unidentified genetic factors and cellular pathways associated with HIV-1 infection and these studies have been previously reviewed (van Manen, van ‘t Wout, & Schuitemaker, 2012). These association studies were greatly facilitated by a number of clinical HIV-1 disease cohorts that have been established over the years. An alternative approach of sequencing the whole exome that selectively sequences the coding regions can prove to be more cost-effective, but the caveat is that many trait-related single-nucleotide polymorphisms fall in intergenic and noncoding regions (Hindorff et al., 2009; Manolio, 2010). Moreover, the gaps in our knowledge of the viral promoter, the LTR, can also be filled by application of comparative genomics and deep sequencing techniques to find key signatures within the LTR that associate with changes in viral pathogenesis and/or alterations in clinical measurements of disease severity such as viral load, CD4+ T-cell count, HAND, and/or other pathologies associated with HIV disease.
Table 6.1.
Genetic factors and cellular pathways associated with HIV-1 infection identified through genome-wide association studies (GWAS) on HIV pathogenesis
| Significant and confirmed | |||
|---|---|---|---|
|
| |||
| Phenotype | Association | Validation technique | Reference(s) |
|
| |||
| RNA VL set point |
HCP5 (rs2395029) |
Genotyping, Illumina’s HumanHap550 BeadChip |
Catano et al. (2008), Fellay et al. (2007), Stern et al. (2012), Telenti and Johnson (2012) |
|
| |||
| VL controllers | −35 HLAC (rs9264942) |
Genotyping, genotype– phenotype associations |
Catano et al. (2008), Fellay et al. (2007) |
|
|
|||
| HLA-B*5703 | IFN-γ ELISPOT, tetramer staining, Illumina HumanHap BeadChip |
Pereyra et al. (2010) | |
|
| |||
| Long-term nonprogression |
HCP5 (rs2395029) |
Meta-analysis using Infinium II HumanHap300 BeadChips |
Limou et al. (2009) |
|
|
|||
| CXCR6 (rs2234358) |
Illumina Infinium II HumanHap300 BeadChips |
Limou et al. (2010) | |
|
| |||
| Progression to AIDS |
PARD3B (rs11884476) |
Regression analysis | Troyer et al. (2011) |
|
| |||
| Significant | |||
|
| |||
| VL controllers | MICA (rs4418214) >300 SNPs in MHC |
Genotype association, regression modeling |
Pereyra et al. (2010) |
|
| |||
| Confirmed | |||
|
| |||
| CD4 T-cell decline |
ZNRD1 (rs9261174) |
Genotyping, shRNA gene knockdown |
Ballana et al. (2010), Fellay et al. (2007) |
|
| |||
| Progression to AIDS |
PROX1 (rs17762192) |
Meta-analysis; immunoblotting |
Herbeck et al. (2010) |
|
| |||
| Nevirapine tolerance |
CCHCR1 (rs1265112) |
Illumina HumanHap550v3 Genotyping BeadChip |
Chantarangsu et al. (2011), Lingappa et al. (2011) |
|
| |||
| Putative | |||
|
| |||
| Long-term nonprogression |
C6orf48 (rs9368699) |
Meta-analysis using Infinium II HumanHap300 |
Limou et al. (2009) |
| Progression to AIDS |
PRMT6 (rs4118325) |
Illumina HumanHap300 BeadChips |
Le Clerc et al. (2009) |
| SOX5 (rs1522232) |
Illumina HumanHap300 BeadChips |
||
| AGR3 (rs152363) |
Illumina’s Infinium HumanHap300 BeadChip |
van Manen et al. (2011) | |
|
| |||
|
In vitro replication in macrophages |
DYRK1A (rs12483205) |
Illumina 610 Quad BeadChip, in vitro HIV-1 replication assays |
Bol et al. (2011) |
| Motherto- child transmission |
HS3ST3A1 (rs8069770) |
Illumina’s HumanHap650Y Genotyping BeadChip |
Joubert et al. (2010) |
Significant and confirmed: p value for association less than 5× 10−8 and repeated independently to confirm the association with significance. Significant: meets current standard for genome-wide significance in GWAS (has a p value below 5 × 10−8); needs to be replicated independently. Confirmed: SNP tested to have association with a phenotype but failed to achieve a significant p value to have genome-wide significance (i.e., p value >5 × 10−8). Putative: p value >5 × 10−8 and yet to be confirmed by independent analysis.
Adapted from van Manen et al. (2012).
Nevertheless, the efforts in these large-scale approaches (Fellay, Shianna, Telenti, & Goldstein, 2010) have enabled studies to examine HIV pathogenesis at a more global level, but the inter-individual variability that plays an important role in disease progression remains to be explored. The shift to whole genome sequencing will further help in accelerating the effort to find signatures that can be potentially used for therapeutic intervention regardless of whether they are of cellular or viral in origin.
9. CONCLUSION
The onset of HIV-1 CNS disease is largely dependent on the trafficking of infected monocytic cells from the periphery across the BBB, where virus is subsequently disseminated to susceptible cell populations within the brain including microglia and perivascular macrophages. Phylogenetic studies have suggested that cells of the monocyte–macrophage lineage may become infected within the bone marrow, and molecular genetic analyses of HIV-1 LTR, Tat, and Env sequences have revealed that evolutionary events occur both within the periphery and in the CNS (Liu et al., 2000). Current evidence indicates that HIV-1 within the brain may adapt specifically to selective pressures present within that physiological compartment, conferring upon the virus the capacity to utilize lower levels of CD4 and CCR5 for binding and entry (Doms, 2000; Doms & Moore, 2000). This observation, combined with results suggesting the presence of greater molecular diversity among HIV-1 quasispecies isolated from HAD versus non-HAD patients (Power et al., 1994), supports the idea that HIV-1 becomes specifically conditioned for efficient replication within the CNS. To date, no specific brain-adapted, neurovirulent virus has been identified, but lack of these observations may be due to limited experimental capabilities and incomplete patient information. If a brain-specific virus exists, one might postulate that it would result in greater neurologic disease and more severe NCI of its host. However, this also has not been observed, perhaps owing in part to the suppressive effects of combination antiretroviral therapy on reseeding of the virus in the brain owing to effective control of the viral replication in the periphery or more effective penetration of antiretroviral drug combinations into the CNS resulting in improved intra-CNS control of viral gene expression and replication.
The past two decades have witnessed an extraordinary experimental effort centered on elucidating both the causes and effects of HIV-1 infection of the CNS. Through these efforts, a number of viral adaptations and evolutionary events have been observed. Genetic mutations present throughout the HIV-1 genome have been identified and correlated both with peripheral immune and/or CNS disease progression and severity (Burdo, Gartner, et al., 2004; Dunfee et al., 2006; Hogan, Stauff, et al., 2003; Johnston et al., 2001). Multiple amino acid variants have been identified within the hypervariable regions of the HIV-1 Env that facilitate viral evasion of the host’s adaptive immune response (Dunfee et al., 2006). Brain-derived HIV-1 Env sequences from HAD patients have been shown to differ from those of non-HAD patients, suggesting that specific HIV-1 Env sequences may correlate with neurotropism, neuroinvasion, CNS infection, and the subsequent onset of NCI (Van Marle et al., 2002). Investigations of the HIV-1 LTR within cells of the monocyte–macrophage lineage have focused heavily on the role of C/EBP and Sp transcription factor families. Specific C/EBP-binding site sequence variants contained within the LTR that are associated with increased LTR-mediated transcriptional activity have been identified within the brains of HAD patients (Burdo, Gartner, et al., 2004; Hogan, Stauff, et al., 2003; Ross et al., 2001). Likewise, sequence variation within Sp transcription factor-binding sites, which results in altered Sp factor recruitment and thus diminished viral replication, may also impact the capacity of HIV-1 Vpr to modulate LTR activity (McAllister et al., 2000; Nonnemacher et al., 2004). Analyses of HIV-1 Tat derived from patients with and without clinical indications of neurological impairment have revealed evidence of phylogenetic clustering with respect to neuropathological status and tissue of origin (Bratanich et al., 1998; Mayne et al., 1998). HIV-1 Tat isolated from the brain of HAD patients has been linked with increased neuronal death (Johnston et al., 2001), and evidence suggests that brain-derived HIV-1 Tat may be limited in its ability to upregulate viral gene expression (Silva et al., 2003).
The knowledge obtained by studying these individual viral evolutionary events has greatly enhanced our understanding of how HIV-1 establishes infection within the CNS and ultimately causes disease; however, the overall value of this information is limited in that the analysis of these individual mutations alone does not allow for a complete appreciation of how the virus is adapting to global environmental changes within a given host or host population. In order to gain an accurate understanding of why HIV-1 selects for specific genetic variants, one must take a step back and determine whether multiple molecular alterations may be coselected for within the same viral genome. For example, the 3T C/EBP site I-binding site sequence variant would, at first glance, seem deleterious to the virus in that it prevents binding of a transcription factor that has been shown to be critical for viral replication with cells of the monocyte lineage. The same can be said for Tat variants isolated from the brains of infected patients, which appear to be more toxic to neuronal cells and less capable of transactivating the HIV-1 LTR. However, on further examination, these and other seemingly nonadvantageous viral structural and functional changes begin to make sense within the bigger picture of HIV-1 adaptation to changing host conditions. Although the 3T C/EBP site I variant binds C/EBP factors with low relative affinity, it has been shown to bind Vpr with high relative affinity, and studies have shown that only one functional C/EBP site is required for viral replication within cells of the monocyte–macrophage lineage (Burdo, Nonnemacher, et al., 2004; Hogan, Nonnemacher, et al., 2003). Interestingly, in all HIV-1 LTRs identified as containing the 3T C/EBP site I configuration, C/EBP-binding site II has been identified as containing a conB sequence configuration, a high-affinity C/EBP-binding site. Why, then, would the virus choose to sacrifice C/EBP binding in exchange for Vpr binding to the LTR? The answer to that question is not completely known, but one explanation may be found in the fact that Vpr binding to the HIV-1 LTR has been correlated with HAD (Burdo, Gartner, et al., 2004; Hogan, Stauff, et al., 2003). Therefore, within the brain, the virus contains a Tat protein that is inefficient with respect to driving viral gene expression, a deficiency that may be compensated for by Vpr binding to and subsequently transactivating the HIV-1 LTR, a process that can occur only after the virus has mutated in such a way as to ameliorate binding of C/EBP factors to C/EBP site I, while maintaining binding integrity at C/EBP site II. A similar story can be told regarding the 5T Sp site III sequence variant. Although this theory has not yet been demonstrated experimentally, perhaps by preventing binding of Sp factors to Sp-binding site III the virus may facilitate greater binding of NF-κB, a transcription factor that has been shown to be absolutely required for viral replication within all HIV-1-susceptible host cell populations (McAllister et al., 2000; Nonnemacher et al., 2004).
Regional distribution analysis of HIV-1 LTR sequence variants has revealed a relationship between the accumulation patterns of specific C/EBP site II variants’ viral replication rates (Burdo, Gartner, et al., 2004). The 6G C/EBP site II sequence configuration was shown to localize in the midfrontal gyrus, an area known to support relatively high levels of viral gene expression, whereas the 4C C/EBP site II variant was shown to accumulate in the cerebellum, a region exhibiting low-level viral replication (Burdo, Gartner, et al., 2004). These results, which provide evidence for the regional distribution of specific HIV-1 LTR genotypes within the brains of HAD and non-HAD patients, are consistent with investigations suggesting the neuroanatomical regionalization of HIV-1 Env sequences. It would be of great value to determine whether multiple HIV-1 gene products such as Env, LTR, and Tat evolve independently of one another, thus making the regional distribution of specific sequence variants merely coincidental, or whether HIV-1 evolves in a coordinated manner. Coordinated selection of multiple genetic variants within differing genomic regions could explain, at least in part, the occurrence of particular sequence alterations that have been identified throughout the HIV-1 genome and that correspond with neurologic disease. The idea that multiple HIV-1 genes may adapt and evolve together in response to changing physiological conditions within specific tissue compartments, as well as microenvironments within those compartments, raises the possibility that specific brain-adapted HIV-1 envelopes correspond to specific brain-adapted Tat proteins, which in turn correspond to specific brain-adapted LTR sequences. Further investigation is required to substantiate this concept.
It may be useful to consider both the timing of HIV-1 entry into the CNS and the mechanisms by which it may distribute and accumulate within specific brain regions. As discussed previously, regionalization could very well be the result of brain-specific viral evolution whereby HIV-1 adapts to the particular physiological conditions present within varying CNS microenvironments. Another possibility is that, through the continual trafficking of HIV-1 to the CNS during the course of infection, only those viral species already genetically adapted for survival within particular brain regions remain viable, while all others die out. Another possible explanation of the region-specific distribution of HIV-1 within the brain is that trafficking of HIV-1 occurs primarily during late-stage disease and that viral evolutionary events occur prior to neuroinvasion within the peripheral blood and possibly the bone marrow. This latter possibility is, however, contradictory to evidence suggesting that the 6G and 4C C/EBP site II sequence configurations are found only in the peripheral blood in patients with early-stage disease but are present in the brain in HAD patients with late-stage disease. This finding would indicate that HIV-1 containing these LTR genotypes must invade the CNS during the early stages of disease.
One of the most profound advancements that might be realized by identifying and characterizing HIV-1 genetic mutations is their potential use as diagnostic markers of disease progression and as targets for preventive and therapeutic strategies. Several genetic mutations have been associated with varying degrees of disease severity, including the 3T C/EBP site I and 5T Sp site III variants. These markers of disease progression are currently being evaluated for their potential prognostic value (Burdo, Gartner, et al., 2004; Burdo, Nonnemacher, et al., 2004; Hogan, Nonnemacher, et al., 2003; Hogan, Stauff, et al., 2003; Nonnemacher et al., 2004). Studies are ongoing to develop an HIV-1 sequence database that will allow for the identification of additional sequence variants that may also serve as molecular markers of HIV-1 immune and nervous system disease. Developing an understanding of viral as well as host genetic heterogeneity may also prove useful in the prevention and treatment of HIV-1 infection and disease. This factor is being utilized in microbicide development, where the knowledge of how HIV-1 selectively adapts to more readily and efficiently infect and replicate within specific cellular compartments may generate effective pharmacologic barriers to viral entry and infection. Of course, enhancing our appreciation of how HIV-1 adapts for replication within the CNS will greatly facilitate the development of the next generation of antiretroviral therapeutics that can more effectively target compartmentalized virus within the brain and other tissue reservoirs.
It is clear that individual genetic mutations cannot be fully understood in isolation and that specific genomic mutations may very well be coselected for within specific viruses and viral quasispecies in response to local and global physiological changes within the host’s peripheral circulation as well as specific tissue compartments such as the brain. Based on the available data, there is little debate as to whether genetic diversity of HIV-1 contributes to the onset and severity of HIV-1-associated neurologic disease; however, further investigation is required to definitively determine the impact that the genomic heterogeneity of HIV-1, and that of the host, has on the development of neuropathology and the onset of NCI.
ACKNOWLEDGMENTS
This work was supported in part by funds from the Public Health Service, National Institutes of Health, through grants from the National Institute of Neurological Disorders and Stroke (NS32092 to B. W.) and the National Institute of Drug Abuse (DA19807 to B. W.). M. R. N. was also supported by faculty development funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease.
REFERENCES
- Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, et al. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. Journal of Virology. 2003;77:2081. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal L, Maxwell CR, Peters PJ, Clapham PR, Liu SM, Mackay CR, et al. Complexity in human immunodeficiency virus type 1 (HIV-1) co-receptor usage: Roles of CCR3 and CCR5 in HIV-1 infection of monocyte-derived macrophages and brain microglia. The Journal of General Virology. 2009;90:710. doi: 10.1099/vir.0.006205-0. [DOI] [PubMed] [Google Scholar]
- Albright AV, Shieh JT, Itoh T, Lee B, Pleasure D, O’Connor MJ, et al. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. Journal of Virology. 1999;73:205. doi: 10.1128/jvi.73.1.205-213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcami J, Lain de Lera T, Folgueira L, Pedraza MA, Jacque JM, Bachelerie F, et al. Absolute dependence on kappa B responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. The EMBO Journal. 1995;14:1552. doi: 10.1002/j.1460-2075.1995.tb07141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, et al. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Ances BM, Vaida F, Yeh MJ, Liang CL, Buxton RB, Letendre S, et al. HIV infection and aging independently affect brain function as measured by functional magnetic resonance imaging. The Journal of Infectious Diseases. 2010;201:336. doi: 10.1086/649899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardon O, Zimmerman ES, Andersen JL, DeHart JL, Blackett J, Planelles V. Induction of G2 arrest and binding to cyclophilin A are independent phenotypes of human immunodeficiency virus type 1 Vpr. Journal of Virology. 2006;80:3694. doi: 10.1128/JVI.80.8.3694-3700.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asin S, Bren GD, Carmona EM, Solan NJ, Paya CV. NF-kappaB cis-acting motifs of the human immunodeficiency virus (HIV) long terminal repeat regulate HIV transcription in human macrophages. Journal of Virology. 2001;75:11408. doi: 10.1128/JVI.75.23.11408-11416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asquith B, Edwards CT, Lipsitch M, McLean AR. Inefficient cytotoxic T lymphocyte-mediated killing of HIV-1-infected cells in vivo. PLoS Biology. 2006;4:e90. doi: 10.1371/journal.pbio.0040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avison MJ, Nath A, Greene-Avison R, Schmitt FA, Bales RA, Ethisham A, et al. Inflammatory changes and breakdown of microvascular integrity in early human immunodeficiency virus dementia. Journal of Neurovirology. 2004;10:223. doi: 10.1080/13550280490463532. [DOI] [PubMed] [Google Scholar]
- Ayyavoo V, Mahalingam S, Rafaeli Y, Kudchodkar S, Chang D, Nagashunmugam T, et al. HIV-1 viral protein R (Vpr) regulates viral replication and cellular proliferation in T cells and monocytoid cells in vitro. Journal of Leukocyte Biology. 1997;62:93. doi: 10.1002/jlb.62.1.93. [DOI] [PubMed] [Google Scholar]
- Ballana E, Senserrich J, Pauls E, Faner R, Mercader JM, Uyttebroeck F, et al. ZNRD1 (zinc ribbon domain-containing 1) is a host cellular factor that influences HIV-1 replication and disease progression. Clinical Infectious Diseases. 2010;50:1022. doi: 10.1086/651114. [DOI] [PubMed] [Google Scholar]
- Barroga CF, Raskino C, Fangon MC, Palumbo PE, Baker CJ, Englund JA, et al. The CCR5Delta32 allele slows disease progression of human immunodeficiency virus-1-infected children receiving antiretroviral treatment. The Journal of Infectious Diseases. 2000;182:413. doi: 10.1086/315704. [DOI] [PubMed] [Google Scholar]
- Berger EA, Doms RW, Fenyo EM, Korber BT, Littman DR, Moore JP, et al. A new classification for HIV-1. Nature. 1998;391:240. doi: 10.1038/34571. [DOI] [PubMed] [Google Scholar]
- Blodget E, Shen C, Aldrovandi G, Rollie A, Gupta SK, Stein JH, et al. Relationship between microbial translocation and endothelial function in HIV infected patients. PLoS One. 2012;7:e42624. doi: 10.1371/journal.pone.0042624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol SM, Moerland PD, Limou S, van Remmerden Y, Coulonges C, van Manen D, et al. Genome-wide association study identifies single nucleotide polymorphism in DYRK1A associated with replication of HIV-1 in monocyte-derived macrophages. PLoS One. 2011;6:e17190. doi: 10.1371/journal.pone.0017190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignori M, Alam SM, Liao HX, Verkoczy L, Tomaras GD, Haynes BF, et al. HIV-1 antibodies from infection and vaccination: Insights for guiding vaccine design. Trends in Microbiology. 2012;20:532–539. doi: 10.1016/j.tim.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boven LA, van der Bruggen T, van Asbeck BS, Marx JJ, Nottet HS. Potential role of CCR5 polymorphism in the development of AIDS dementia complex. FEMS Immunology and Medical Microbiology. 1999;26:243. doi: 10.1111/j.1574-695X.1999.tb01395.x. [DOI] [PubMed] [Google Scholar]
- Brady J, Kashanchi F. Tat gets the “green” light on transcription initiation. Retrovirology. 2005;2:69. doi: 10.1186/1742-4690-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratanich AC, Liu C, McArthur JC, Fudyk T, Glass JD, Mittoo S, et al. Brain-derived HIV-1 tat sequences from AIDS patients with dementia show increased molecular heterogeneity. Journal of Neurovirology. 1998;4:387. doi: 10.3109/13550289809114537. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Bhalla RB, Paul M, Sidtis JJ, Keilp JJ, Sadler AE, et al. Cerebrospinal fluid beta 2-microglobulin in patients with AIDS dementia complex: An expanded series including response to zidovudine treatment. AIDS. 1992;6:461. [PubMed] [Google Scholar]
- Brew BJ, Dunbar N, Pemberton L, Kaldor J. Predictive markers of AIDS dementia complex: CD4 cell count and cerebrospinal fluid concentrations of beta 2-microglobulin and neopterin. The Journal of Infectious Diseases. 1996;174:294. doi: 10.1093/infdis/174.2.294. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A. Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. The Journal of Neuroscience. 2003;23:8417. doi: 10.1523/JNEUROSCI.23-23-08417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budka H. Neuropathology of human immunodeficiency virus infection. Brain Pathology. 1991;1:163. doi: 10.1111/j.1750-3639.1991.tb00656.x. [DOI] [PubMed] [Google Scholar]
- Budka H, Costanzi G, Cristina S, Lechi A, Parravicini C, Trabattoni R, et al. Brain pathology induced by infection with the human immunodeficiency virus (HIV). A histological, immunocytochemical, and electron microscopical study of 100 autopsy cases. Acta Neuropathologica (Berlin) 1987;75:185. doi: 10.1007/BF00687080. [DOI] [PubMed] [Google Scholar]
- Burdo TH, Gartner S, Mauger D, Wigdahl B. Region-specific distribution of human immunodeficiency virus type 1 long terminal repeats containing specific configurations of CCAAT/enhancer-binding protein site II in brains derived from demented and nondemented patients. Journal of Neurovirology. 2004;10(Suppl. 1):7–14. doi: 10.1080/753312746. [DOI] [PubMed] [Google Scholar]
- Burdo TH, Nonnemacher M, Irish BP, Choi CH, Krebs FC, Gartner S, et al. High-affinity interaction between HIV-1 Vpr and specific sequences that span the C/EBP and adjacent NF-kappaB sites within the HIV-1 LTR correlate with HIV-1-associated dementia. DNA and Cell Biology. 2004;23:261. doi: 10.1089/104454904773819842. [DOI] [PubMed] [Google Scholar]
- Cao J, McNevin J, Malhotra U, McElrath MJ. Evolution of CD8+ T cell immunity and viral escape following acute HIV-1 infection. Journal of Immunology. 2003;171:3837. doi: 10.4049/jimmunol.171.7.3837. [DOI] [PubMed] [Google Scholar]
- Carrington M, Dean M, Martin MP, O’Brien SJ. Genetics of HIV-1 infection: Chemokine receptor CCR5 polymorphism and its consequences. Human Molecular Genetics. 1999;8:1939. doi: 10.1093/hmg/8.10.1939. [DOI] [PubMed] [Google Scholar]
- Carrington M, Kissner T, Gerrard B, Ivanov S, O’Brien SJ, Dean M. Novel alleles of the chemokine-receptor gene CCR5. American Journal of Human Genetics. 1997;61:1261. doi: 10.1086/301645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annual Review of Medicine. 2003;54:535. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- Catano G, Kulkarni H, He W, Marconi VC, Agan BK, Landrum M, et al. HIV-1 disease-influencing effects associated with ZNRD1, HCP5 and HLA-C alleles are attributable mainly to either HLA-A10 or HLA-B*57 alleles. PLoS One. 2008;3:e3636. doi: 10.1371/journal.pone.0003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecilia D, Kleeberger C, Munoz A, Giorgi JV, Zolla-Pazner S. A longitudinal study of neutralizing antibodies and disease progression in HIV-1-infected subjects. The Journal of Infectious Diseases. 1999;179:1365. doi: 10.1086/314773. [DOI] [PubMed] [Google Scholar]
- Chan SY, Speck RF, Power C, Gaffen SL, Chesebro B, Goldsmith MA. V3 recombinants indicate a central role for CCR5 as a coreceptor in tissue infection by human immunodeficiency virus type 1. Journal of Virology. 1999;73:2350. doi: 10.1128/jvi.73.3.2350-2358.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L. In vivo magnetic resonance spectroscopy in HIV and HIV-related brain diseases. Reviews in the Neurosciences. 1995;6:365. doi: 10.1515/revneuro.1995.6.4.365. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, St Hillaire C, Conant K. Antiretroviral treatment alters relationship between MCP-1 and neurometabolites in HIV patients. Antiviral Therapy. 2004;9:431. doi: 10.1177/135965350400900302. [DOI] [PubMed] [Google Scholar]
- Chang J, Jozwiak R, Wang B, Ng T, Ge YC, Bolton W, et al. Unique HIV type 1 V3 region sequences derived from six different regions of brain: Region-specific evolution within host-determined quasispecies. AIDS Research and Human Retroviruses. 1998;14:25. doi: 10.1089/aid.1998.14.25. [DOI] [PubMed] [Google Scholar]
- Chantarangsu S, Mushiroda T, Mahasirimongkol S, Kiertiburanakul S, Sungkanuparph S, Manosuthi W, et al. Genome-wide association study identifies variations in 6p21.3 associated with nevirapine-induced rash. Clinical Infectious Diseases. 2011;53:341. doi: 10.1093/cid/cir403. [DOI] [PubMed] [Google Scholar]
- Chen BK, Feinberg MB, Baltimore D. The kappaB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. Journal of Virology. 1997;71:5495. doi: 10.1128/jvi.71.7.5495-5504.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherner M, Masliah E, Ellis RJ, Marcotte TD, Moore DJ, Grant I, et al. Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology. 2002;59:1563. doi: 10.1212/01.wnl.0000034175.11956.79. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Wehrly K, Nishio J, Perryman S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: Definition of critical amino acids involved in cell tropism. Journal of Virology. 1992;66:6547. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao C, Bader T, Stenger JE, Baldwin W, Brady J, Barrett JC. HIV type 1 Tat inhibits tumor necrosis factor alpha-induced repression of tumor necrosis factor receptor p55 and amplifies tumor necrosis factor alpha activity in stably tat-transfected HeLa Cells. AIDS Research and Human Retroviruses. 2001;17:1125. doi: 10.1089/088922201316912736. [DOI] [PubMed] [Google Scholar]
- Childs EA, Lyles RH, Selnes OA, Chen B, Miller EN, Cohen BA, et al. Plasma viral load and CD4 lymphocytes predict HIV-associated dementia and sensory neuropathy. Neurology. 1999;52:607. doi: 10.1212/wnl.52.3.607. [DOI] [PubMed] [Google Scholar]
- Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- Chrysikopoulos HS, Press GA, Grafe MR, Hesselink JR, Wiley CA. Encephalitis caused by human immunodeficiency virus: CT and MR imaging manifestations with clinical and pathologic correlation. Radiology. 1990;175:185. doi: 10.1148/radiology.175.1.2315479. [DOI] [PubMed] [Google Scholar]
- Cohen EA, Dehni G, Sodroski JG, Haseltine WA. Human immunodeficiency virus vpr product is a virion-associated regulatory protein. Journal of Virology. 1990;64:3097. doi: 10.1128/jvi.64.6.3097-3099.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman R, Balliet JW, Gregory SA, Friedman H, Kolson DL, Nathanson N, et al. An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. Journal of Virology. 1992;66:7517. doi: 10.1128/jvi.66.12.7517-7521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3117. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corboy JR, Buzy JM, Zink MC, Clements JE. Expression directed from HIV long terminal repeats in the central nervous system of transgenic mice. Science. 1992;258:1804. doi: 10.1126/science.1465618. [DOI] [PubMed] [Google Scholar]
- Cosenza MA, Zhao ML, Si Q, Lee SC. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathology. 2002;12:442. doi: 10.1111/j.1750-3639.2002.tb00461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Regulation of gene expression in the human immunodeficiency virus type 1. Advances in Virus Research. 1991;40:1. doi: 10.1016/s0065-3527(08)60275-4. [DOI] [PubMed] [Google Scholar]
- Cunningham AL, Naif H, Saksena N, Lynch G, Chang J, Li S, et al. HIV infection of macrophages and pathogenesis of AIDS dementia complex: Interaction of the host cell and viral genotype. Journal of Leukocyte Biology. 1997;62:117. doi: 10.1002/jlb.62.1.117. [DOI] [PubMed] [Google Scholar]
- Dahiya S, Nonnemacher MR, Wigdahl B. Deployment of the human immunodeficiency virus type 1 protein arsenal: Combating the host to enhance viral transcription and providing targets for therapeutic development. The Journal of General Virology. 2012;93:1151. doi: 10.1099/vir.0.041186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multi-center AIDS cohort study, multicenter hemophilia cohort study, San Francisco city cohort, ALIVE study. Science. 1996;273:1856. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- DeHart JL, Zimmerman ES, Ardon O, Monteiro-Filho CM, Arganaraz ER, Planelles V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virology Journal. 2007;4:57. doi: 10.1186/1743-422X-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Palacio M, Alvarez S, Munoz-Fernandez MA. HIV-1 infection and neurocognitive impairment in the current era. Reviews in Medical Virology. 2012;22:33. doi: 10.1002/rmv.711. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Mukerjee R, Fan S, Del Valle L, Michiels C, Sweet T, et al. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxiainducible factor 1alpha expression. The Journal of Biological Chemistry. 2009;284:11364. doi: 10.1074/jbc.M809266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW. Beyond receptor expression: The influence of receptor conformation, density, and affinity in HIV-1 infection. Virology. 2000;276:229. doi: 10.1006/viro.2000.0612. [DOI] [PubMed] [Google Scholar]
- Doms RW, Moore JP. HIV-1 membrane fusion: Targets of opportunity. The Journal of Cell Biology. 2000;151:F9. doi: 10.1083/jcb.151.2.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler C, Schalasta G, Amtmann E, Sauer G. Binding of NF-kB to the HIV-1 LTR is not sufficient to induce HIV-1 LTR activity. AIDS Research and Human Retroviruses. 1992;8:245. doi: 10.1089/aid.1992.8.245. [DOI] [PubMed] [Google Scholar]
- Drummond NS, Vilar FJ, Naisbitt DJ, Hanson A, Woods A, Park BK, et al. Drug-specific T cells in an HIV-positive patient with nevirapine-induced hepatitis. Antiviral Therapy. 2006;11:393. [PubMed] [Google Scholar]
- Duncan CJ, Sattentau QJ. Viral determinants of HIV-1 macrophage tropism. Viruses. 2011;3:2255. doi: 10.3390/v3112255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunfee RL, Thomas ER, Gorry PR, Wang J, Taylor J, Kunstman K, et al. The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15160. doi: 10.1073/pnas.0605513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunfee RL, Thomas ER, Wang J, Kunstman K, Wolinsky SM, Gabuzda D. Loss of the N-linked glycosylation site at position 386 in the HIV envelope V4 region enhances macrophage tropism and is associated with dementia. Virology. 2007;367:222. doi: 10.1016/j.virol.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerman M, Malim MH. HIV-1 regulatory/accessory genes: Keys to unraveling viral and host cell biology. Science. 1998;280:1880. doi: 10.1126/science.280.5371.1880. [DOI] [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, et al. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. Journal of Virology. 1993;67:277. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enting RH, Foudraine NA, Lange JM, Jurriaans S, van der Poll T, Weverling GJ, et al. Cerebrospinal fluid beta2-microglobulin, monocyte chemotactic protein-1, and soluble tumour necrosis factor alpha receptors before and after treatment with lamivudine plus zidovudine or stavudine. Journal of Neuroimmunology. 2000;102:216. doi: 10.1016/s0165-5728(99)00219-2. [DOI] [PubMed] [Google Scholar]
- Epstein LG, Kuiken C, Blumberg BM, Hartman S, Sharer LR, Clement M, et al. HIV-1 V3 domain variation in brain and spleen of children with AIDS: Tissue-specific evolution within host-determined quasispecies. Virology. 1991;180:583. doi: 10.1016/0042-6822(91)90072-j. [DOI] [PubMed] [Google Scholar]
- Epstein LG, Sharer LR, Cho ES, Myenhofer M, Navia B, Price RW. HTLV-III/LAV-like retrovirus particles in the brains of patients with AIDS encephalopathy. AIDS Research. 1984;1:447. doi: 10.1089/aid.1.1983.1.447. [DOI] [PubMed] [Google Scholar]
- Essex M. Human immunodeficiency viruses in the developing world. Advances in Virus Research. 1999;53:71. doi: 10.1016/s0065-3527(08)60343-7. [DOI] [PubMed] [Google Scholar]
- Estable MC, Bell B, Merzouki A, Montaner JS, O’Shaughnessy MV, Sadowski IJ. Human immunodeficiency virus type 1 long terminal repeat variants from 42 patients representing all stages of infection display a wide range of sequence polymorphism and transcription activity. Journal of Virology. 1996;70:4053. doi: 10.1128/jvi.70.6.4053-4062.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauci AS. HIV and AIDS: 20 Years of science. Nature Medicine. 2003;9:839. doi: 10.1038/nm0703-839. [DOI] [PubMed] [Google Scholar]
- Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellay J, Shianna KV, Telenti A, Goldstein DB. Host genetics and HIV-1: The final phase? PLoS Pathogens. 2010;6:e1001033. doi: 10.1371/journal.ppat.1001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felzien LK, Woffendin C, Hottiger MO, Subbramanian RA, Cohen EA, Nabel GJ. HIV transcriptional activation by the accessory protein, VPR, is mediated by the p300 co-activator. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5281. doi: 10.1073/pnas.95.9.5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folks T, Powell DM, Lightfoote MM, Benn S, Martin MA, Fauci AS. Induction of HTLV-III/LAV from a nonvirus-producing T-cell line: Implications for latency. Science. 1986;231:600. doi: 10.1126/science.3003906. [DOI] [PubMed] [Google Scholar]
- Fontaine J, Poudrier J, Roger M. Short communication: Persistence of high blood levels of the chemokines CCL2, CCL19, and CCL20 during the course of HIV infection. AIDS Research and Human Retroviruses. 2011;27:655. doi: 10.1089/aid.2010.0261. [DOI] [PubMed] [Google Scholar]
- Frost SD, Wrin T, Smith DM, Kosakovsky Pond SL, Liu Y, Paxinos E, et al. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18514. doi: 10.1073/pnas.0504658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshan S, Dickover RE, Korber BT, Bryson YJ, Wolinsky SM. Human immunodeficiency virus type 1 genetic evolution in children with different rates of development of disease. Journal of Virology. 1997;71:663. doi: 10.1128/jvi.71.1.663-677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon P, Khan MZ, Kolson DL. Current understanding of HIV-associated neurocognitive disorders pathogenesis. Current Opinion in Neurology. 2011;24:275. doi: 10.1097/WCO.0b013e32834695fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganusov VV, Goonetilleke N, Liu MK, Ferrari G, Shaw GM, McMichael AJ, et al. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. Journal of Virology. 2011;85:10518. doi: 10.1128/JVI.00655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Robertson DL, Morrison SG, Hui H, Craig S, Decker J, et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. Journal of Virology. 1996;70:7013. doi: 10.1128/jvi.70.10.7013-7029.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner S. HIV infection and dementia. Science. 2000;287:602. doi: 10.1126/science.287.5453.602. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, James HJ, Sharer LR, Perry SW, Saito Y, Kazee AM, et al. Apoptotic neurons in brains from paediatric patients with HIV-1 encephalitis and progressive encephalopathy. Neuropathology and Applied Neurobiology. 1995;21:208. doi: 10.1111/j.1365-2990.1995.tb01052.x. [DOI] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: Implications for the neuropathogenesis of HIV disease. The Journal of Experimental Medicine. 1992;176:1703. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabinski AR, Tani M, Aras S, Stoler MH, Tuohy VK, Ransohoff RM. Regulation and function of central nervous system chemokines. International Journal of Developmental Neuroscience. 1995;13:153. doi: 10.1016/0736-5748(95)00017-b. [DOI] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: Correlations with dementia. Annals of Neurology. 1995;38:755. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, Nowak MA, et al. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nature Medicine. 1998;4:65. doi: 10.1038/nm0198-065. [DOI] [PubMed] [Google Scholar]
- Golub EI, Li GR, Volsky DJ. Induction of dormant HIV-1 by sodium butyrate: Involvement of the TATA box in the activation of the HIV-1 promoter. AIDS. 1991;5:663. [PubMed] [Google Scholar]
- Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, et al. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13795. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez MP, O’Connell O, Lin R, Sullivan WM, Bell J, Simmonds P, et al. Independent evolution of macrophage-tropism and increased charge between HIV-1 R5 envelopes present in brain and immune tissue. Retrovirology. 2012;9:20. doi: 10.1186/1742-4690-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, et al. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. Journal of Virology. 2001;75:10073. doi: 10.1128/JVI.75.21.10073-10089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry PR, Howard JL, Churchill MJ, Anderson JL, Cunningham A, Adrian D, et al. Diminished production of human immunodeficiency virus type 1 in astrocytes results from inefficient translation of gag, env, and nef mRNAs despite efficient expression of Tat and Rev. Journal of Virology. 1999;73:352. doi: 10.1128/jvi.73.1.352-361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry PR, Taylor J, Holm GH, Mehle A, Morgan T, Cayabyab M, et al. Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate. Journal of Virology. 2002;76:6277. doi: 10.1128/JVI.76.12.6277-6292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LR, Gabuzda D, Cowley D, Ellett A, Chiavaroli L, Wesselingh SL, et al. CD4 and MHC class 1 down-modulation activities of nef alleles from brain-and lymphoid tissue-derived primary HIV-1 isolates. Journal of Neurovirology. 2011;17:82. doi: 10.1007/s13365-010-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray L, Sterjovski J, Churchill M, Ellery P, Nasr N, Lewin SR, et al. Uncoupling coreceptor usage of human immunodeficiency virus type 1 (HIV-1) from macrophage tropism reveals biological properties of CCR5-restricted HIV-1 isolates from patients with acquired immunodeficiency syndrome. Virology. 2005;337:384. doi: 10.1016/j.virol.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Green M, Ishino M, Loewenstein PM. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress HIVLTR-driven gene expression. Cell. 1989;58:215. doi: 10.1016/0092-8674(89)90417-0. [DOI] [PubMed] [Google Scholar]
- Griffin GE, Leung K, Folks TM, Kunkel S, Nabel GJ. Activationof HIVgene expression during monocyte differentiation by induction of NF-kappa B. Nature. 1989;339:70. doi: 10.1038/339070a0. [DOI] [PubMed] [Google Scholar]
- Guha D, Nagilla P, Redinger C, Srinivasan A, Schatten GP, Ayyavoo V. Neuronal apoptosis by HIV-1 Vpr: Contribution of proinflammatory molecular networks from infected target cells. Journal of Neuroinflammation. 2012;9:138. doi: 10.1186/1742-2094-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase AT. Pathogenesis of lentivirus infections. Nature. 1986;322:130. doi: 10.1038/322130a0. [DOI] [PubMed] [Google Scholar]
- He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. Journal of Neurovirology. 2011;17:3. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson AJ, Calame KL. CCAAT/enhancer binding protein (C/EBP) sites are required for HIV-1 replication in primary macrophages but not CD4(+) T cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8714. doi: 10.1073/pnas.94.16.8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson AJ, Connor RI, Calame KL. C/EBP activators are required for HIV-1 replication and proviral induction in monocytic cell lines. Immunity. 1996;5:91. doi: 10.1016/s1074-7613(00)80313-1. [DOI] [PubMed] [Google Scholar]
- Henderson AJ, Zou X, Calame KL. C/EBP proteins activate transcription from the human immunodeficiency virus type 1 long terminal repeat in macrophages/monocytes. Journal of Virology. 1995;69:5337. doi: 10.1128/jvi.69.9.5337-5344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbeck JT, Gottlieb GS, Winkler CA, Nelson GW, An P, Maust BS, et al. Multistage genomewide association study identifies a locus at 1q41 associated with rate of HIV-1 disease progression to clinical AIDS. The Journal of Infectious Diseases. 2010;201:618. doi: 10.1086/649842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Ellis RJ, Ryan L, Childers ME, Grant I, Wolfson T, et al. Elevated cerebrospinal fluid quinolinic acid levels are associated with region-specific cerebral volume loss in HIV infection. Brain. 2001;124:1033. doi: 10.1093/brain/124.5.1033. [DOI] [PubMed] [Google Scholar]
- Hickey WF. Leukocyte traffic in the central nervous system: The participants and their roles. Seminars in Immunology. 1999;11:125. doi: 10.1006/smim.1999.0168. [DOI] [PubMed] [Google Scholar]
- Hill AL, Rosenbloom DI, Nowak MA. Evolutionary dynamics of HIV at multiple spatial and temporal scales. Journal of Molecular Medicine (Berlin, Germany) 2012;90:543. doi: 10.1007/s00109-012-0892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9362. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman TL, Stephens EB, Narayan O, Doms RW. HIV type I envelope determinants for use of the CCR2b, CCR3, STRL33, and APJ coreceptors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11360. doi: 10.1073/pnas.95.19.11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan TH, Nonnemacher MR, Krebs FC, Henderson A, Wigdahl B. HIV-1 Vpr binding to HIV-1 LTR C/EBP cis-acting elements and adjacent regions is sequence-specific. Biomedicine and Pharmacotherapy. 2003;57:41. doi: 10.1016/s0753-3322(02)00333-5. [DOI] [PubMed] [Google Scholar]
- Hogan TH, Stauff DL, Krebs FC, Gartner S, Quiterio SJ, Wigdahl B. Structural and functional evolution of human immunodeficiency virus type 1 long terminal repeat CCAAT/enhancer binding protein sites and their use as molecular markers for central nervous system disease progression. Journal of Neurovirology. 2003;9:55. doi: 10.1080/13550280390173292. [DOI] [PubMed] [Google Scholar]
- Huang KJ, Alter GM, Wooley DP. The reverse transcriptase sequence of human immunodeficiency virus type 1 is under positive evolutionary selection within the central nervous system. Journal of Neurovirology. 2002;8:281. doi: 10.1080/13550280290100716. [DOI] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, et al. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. Journal of Neurovirology. 2000;6:145. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- Janssens W, Buve A, Nkengasong JN. The puzzle of HIV-1 subtypes in Africa. AIDS. 1997;11:705. doi: 10.1097/00002030-199706000-00002. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, et al. HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Annals of Neurology. 2001;49:230. doi: 10.1002/1531-8249(20010201)49:2<230::aid-ana43>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Jones KA, Peterlin BM. Control of RNA initiation and elongation at the HIV-1 promoter. Annual Review of Biochemistry. 1994;63:717. doi: 10.1146/annurev.bi.63.070194.003441. [DOI] [PubMed] [Google Scholar]
- Joubert BR, Lange EM, Franceschini N, Mwapasa V, North KE, Meshnick SR. A whole genome association study of mother-to-child transmission of HIV in Malawi. Genome Medicine. 2010;2:17. doi: 10.1186/gm138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowett JB, Planelles V, Poon B, Shah NP, Chen ML, Chen IS. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 M phase of the cell cycle. Journal of Virology. 1995;69:6304. doi: 10.1128/jvi.69.10.6304-6313.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Hirano A, Llena JF, Dembitzer HM. Neuropathology of acquired immune deficiency syndrome (AIDS) in 53 autopsy cases with particular emphasis on microglial nodules and multinucleated giant cells. Acta Neuropathology (Berlin, Germany) 1987;73:287. doi: 10.1007/BF00686624. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kelly JK. Replication rate and evolution in the human immunodeficiency virus. Journal of Theoretical Biology. 1996;180:359. doi: 10.1006/jtbi.1996.0108. [DOI] [PubMed] [Google Scholar]
- Kelly NJ, Morrow CD. Yeast tRNA(Phe) expressed in human cells can be selected by HIV-1 for use as a reverse transcription primer. Virology. 2003;313:354. doi: 10.1016/s0042-6822(03)00243-5. [DOI] [PubMed] [Google Scholar]
- Khanna KV, Yu XF, Ford DH, Ratner L, Hildreth JK, Markham RB. Differences among HIV-1 variants in their ability to elicit secretion of TNF-alpha. Journal of Immunology. 2000;164:1408. doi: 10.4049/jimmunol.164.3.1408. [DOI] [PubMed] [Google Scholar]
- Kim WK, Alvarez X, Fisher J, Bronfin B, Westmoreland S, McLaurin J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. The American Journal of Pathology. 2006;168:822. doi: 10.2353/ajpath.2006.050215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Kukkonen S, Gupta S, Aldovini A. Association of Tat with promoters of PTEN and PP2A subunits is key to transcriptional activation of apoptotic pathways in HIV-infected CD4+ T cells. PLoS Pathogens. 2010;6:e1001103. doi: 10.1371/journal.ppat.1001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Gragerov A, Kopp JB, Stauber RH, Pavlakis GN, Chrousos GP. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. The Journal of Experimental Medicine. 1999;189:51. doi: 10.1084/jem.189.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F, Greenough TC, Hamacher M, Sullivan JL, Desrosiers RC. Activity of human immunodeficiency virus type 1 promoter/TAR regions and tat1 genes derived from individuals with different rates of disease progression. Virology. 1997;232:319. doi: 10.1006/viro.1997.8586. [DOI] [PubMed] [Google Scholar]
- Kolson DL, Lavi E, Gonzalez-Scarano F. The effects of human immunodeficiency virus in the central nervous system. Advances in Virus Research. 1998;50:1. doi: 10.1016/s0065-3527(08)60804-0. [DOI] [PubMed] [Google Scholar]
- Korber BT, Kunstman KJ, Patterson BK, Furtado M, McEvilly MM, Levy R, et al. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: Evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. Journal of Virology. 1994;68:7467. doi: 10.1128/jvi.68.11.7467-7481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber BT, MacInnes K, Smith RF, Myers G. Mutational trends in V3 loop protein sequences observed in different genetic lineages of human immunodeficiency virus type 1. Journal of Virology. 1994;68:6730. doi: 10.1128/jvi.68.10.6730-6744.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrikis LG, Huang Y, Moore JP, Wolinsky SM, Zhang L, Guo Y, et al. A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation. Nature Medicine. 1998;4:350. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- Kostrikis LG, Neumann AU, Thomson B, Korber BT, McHardy P, Karanicolas R, et al. A polymorphism in the regulatory region of the CC-chemokine receptor 5 gene influences perinatal transmission of human immunodeficiency virus type 1 to African-American infants. Journal of Virology. 1999;73:10264. doi: 10.1128/jvi.73.12.10264-10271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. Journal of Virology. 1994;68:4650. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs FC, Mehrens D, Pomeroy S, Goodenow MM, Wigdahl B. Human immunodeficiency virus type 1 long terminal repeat quasispecies differ in basal transcription and nuclear factor recruitment in human glial cells and lymphocytes. Journal of Biomedical Science. 1998;5:31. doi: 10.1007/BF02253354. [DOI] [PubMed] [Google Scholar]
- Krebs FC, Ross H, McAllister J, Wigdahl B. HIV-1-associated central nervous system dysfunction. Advances in Pharmacology. 2000;49:315. doi: 10.1016/s1054-3589(00)49031-9. [DOI] [PubMed] [Google Scholar]
- Kurosu T, Mukai T, Auwanit W, Ayuthaya PI, Saeng-Aroon S, Ikuta K. Variable sequences in the long terminal repeat and Its downstream region of some of HIV Type 1 CRF01_AE recently distributing among Thai carriers. AIDS Research and Human Retroviruses. 2001;17:863. doi: 10.1089/088922201750252061. [DOI] [PubMed] [Google Scholar]
- Kurosu T, Mukai T, Komoto S, Ibrahim MS, Li YG, Kobayashi T, et al. Human immunodeficiency virus type 1 subtype C exhibits higher transactivation activity of Tat than subtypes B and E. Microbiology and Immunology. 2002;46:787. doi: 10.1111/j.1348-0421.2002.tb02766.x. [DOI] [PubMed] [Google Scholar]
- Lackner AA, Lederman MM, Rodriguez B. HIV pathogenesis: The host. Cold Spring Harbor Perspectives in Medicine. 2012;2:a007005. doi: 10.1101/cshperspect.a007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clerc S, Limou S, Coulonges C, Carpentier W, Dina C, Taing L, et al. Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03) The Journal of Infectious Diseases. 2009;200:1194. doi: 10.1086/605892. [DOI] [PubMed] [Google Scholar]
- Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5215. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Kosakovsky Pond SL, Drummond AJ, Pybus OG, Shapiro B, Barroso H, et al. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Computational Biology. 2007;3:e29. doi: 10.1371/journal.pcbi.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nature Medicine. 2004;10:282. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- Letendre SL, McCutchan JA, Childers ME, Woods SP, Lazzaretto D, Heaton RK, et al. Enhancing antiretroviral therapy for human immunodeficiency virus cognitive disorders. Annals of Neurology. 2004;56:416. doi: 10.1002/ana.20198. [DOI] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, Weiner DB. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. Journal of Virology. 1995;69:1243. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin SR, Sonza S, Irving LB, McDonald CF, Mills J, Crowe SM. Surface CD4 is critical to in vitro HIV infection of human alveolar macrophages. AIDS Research and Human Retroviruses. 1996;12:877. doi: 10.1089/aid.1996.12.877. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Dagarag M, Khan B, Ali A, Yang OO. Partial escape of HIV-1 from cytotoxic T lymphocytes during chronic infection. Journal of Virology. 2012;86:7459. doi: 10.1128/JVI.06724-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Aiamkitsumrit B, Pirrone V, Nonnemacher MR, Wojno A, Passic S, et al. Development of co-selected single nucleotide polymorphisms in the viral promoter precedes the onset of human immunodeficiency virus type 1-associated neurocognitive impairment. Journal of Neurovirology. 2011;17:92. doi: 10.1007/s13365-010-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Dahiya S, Kortagere S, Aiamkitsumrit B, Cunningham D, Pirrone V, et al. Impact of Tat genetic variation on HIV-1 disease. Advances in Virology. 2012;2012:123605. doi: 10.1155/2012/123605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichterfeld M, Yu XG, Cohen D, Addo MM, Malenfant J, Perkins B, et al. HIV-1 Nef is preferentially recognized by CD8 T cells in primary HIV-1 infection despite a relatively high degree of genetic diversity. AIDS. 2004;18:1383. doi: 10.1097/01.aids.0000131329.51633.a3. [DOI] [PubMed] [Google Scholar]
- Limou S, Coulonges C, Herbeck JT, van Manen D, An P, Le Clerc S, et al. Multiple-cohort genetic association study reveals CXCR6 as a new chemokine receptor involved in long-term nonprogression to AIDS. The Journal of Infectious Diseases. 2010;202:908. doi: 10.1086/655782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limou S, Le Clerc S, Coulonges C, Carpentier W, Dina C, Delaneau O, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) The Journal of Infectious Diseases. 2009;199:419. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- Lingappa JR, Petrovski S, Kahle E, Fellay J, Shianna K, McElrath MJ, et al. Genomewide association study for determinants of HIV-1 acquisition and viral set point in HIV-1 serodiscordant couples with quantified virus exposure. PLoS One. 2011;6:e28632. doi: 10.1371/journal.pone.0028632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Liu Y, Tang XP, McArthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: Evidence for monocyte trafficking into brain. Journal of Neurovirology. 2000;6(Suppl. 1):S70. [PubMed] [Google Scholar]
- Lu YL, Spearman P, Ratner L. Human immunodeficiency virus type 1 viral protein R localization in infected cells and virions. Journal of Virology. 1993;67:6542. doi: 10.1128/jvi.67.11.6542-6550.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam S, Collman RG, Patel M, Monken CE, Srinivasan A. Functional analysis of HIV-1 Vpr: Identification of determinants essential for subcellular localization. Virology. 1995;212:331. doi: 10.1006/viro.1995.1490. [DOI] [PubMed] [Google Scholar]
- Mahalingam S, Khan SA, Jabbar MA, Monken CE, Collman RG, Srinivasan A. Identification of residues in the N-terminal acidic domain of HIV-1 Vpr essential for virion incorporation. Virology. 1995;207:297. doi: 10.1006/viro.1995.1081. [DOI] [PubMed] [Google Scholar]
- Mahalingam S, Khan SA, Murali R, Jabbar MA, Monken CE, Collman RG, et al. Mutagenesis of the putative alpha-helical domain of the Vpr protein of human immunodeficiency virus type 1: Effect on stability and virion incorporation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3794. doi: 10.1073/pnas.92.9.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA. Genomewide association studies and assessment of the risk of disease. The New England Journal of Medicine. 2010;363:166. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- Marra CM, Maxwell CL, Collier AC, Robertson KR, Imrie A. Interpreting cerebrospinal fluid pleocytosis in HIV in the era of potent antiretroviral therapy. BMC Infectious Diseases. 2007;7:37. doi: 10.1186/1471-2334-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MP, Dean M, Smith MW, Winkler C, Gerrard B, Michael NL, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science. 1998;282:1907. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- Martín J, LaBranche CC, González-Scarano F. Differential CD4/CCR5 utilization, gp120 conformation, and neutralization sensitivity between envelopes from a microglia-adapted human immunodeficiency virus type 1 and its parental isolate. Journal of Virology. 2001;75:3568. doi: 10.1128/JVI.75.8.3568-3580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. Journal of Virology. 2006;80:3617. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Garcia J, Cao W, Varela-Rohena A, Plassmeyer ML, Gonzalez-Scarano F. HIV-1 tropism for the central nervous system: Brain-derived envelope glycoproteins with lower CD4 dependence and reduced sensitivity to a fusion inhibitor. Virology. 2006;346:169. doi: 10.1016/j.virol.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Martin-Garcia J, Cocklin S, Chaiken IM, Gonzalez-Scarano F. Interaction with CD4 and antibodies to CD4-induced epitopes of the envelope gp120 from a micro-glial cell-adapted human immunodeficiency virus type 1 isolate. Journal of Virology. 2005;79:6703. doi: 10.1128/JVI.79.11.6703-6713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10193. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne M, Bratanich AC, Chen P, Rana F, Nath A, Power C. HIV-1 tat molecular diversity and induction of TNF-alpha: Implications for HIV-induced neurological disease. Neuroimmunomodulation. 1998;5:184. doi: 10.1159/000026336. [DOI] [PubMed] [Google Scholar]
- McAllister JJ, Phillips D, Millhouse S, Conner J, Hogan T, Ross HL, et al. Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: Evidence for cell type-specific gene regulation and viral replication. Virology. 2000;274:262. doi: 10.1006/viro.2000.0476. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ. HIV-associated neurocognitive disorders: Is there a hidden epidemic? AIDS. 2010;24:1367. doi: 10.1097/QAD.0b013e3283391d56. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, et al. Human immunodeficiency virus-associated dementia: An evolving disease. Journal of Neurovirology. 2003;9:205. doi: 10.1080/13550280390194109. [DOI] [PubMed] [Google Scholar]
- McArthur JC, McClernon DR, Cronin MF, Nance-Sproson TE, Saah AJ, St Clair M, et al. Relationship between human immunodeficiency virus-associated dementia and viral load in cerebrospinal fluid and brain. Annals of Neurology. 1997;42:689. doi: 10.1002/ana.410420504. [DOI] [PubMed] [Google Scholar]
- McKnight A, Weiss RA, Shotton C, Takeuchi Y, Hoshino H, Clapham PR. Change in tropism upon immune escape by human immunodeficiency virus. Journal of Virology. 1995;69:3167. doi: 10.1128/jvi.69.5.3167-3170.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messam CA, Major EO. Stages of restricted HIV-1 infection in astrocyte cultures derived from human fetal brain tissue. Journal of Neurovirology. 2000;6(Suppl. 1):S90. [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14500. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhoff DJ, MacKay S, Poole N, Dillon WP, Weiner MW, Fein G. N-acetylaspartate reductions measured by 1H MRSI in cognitively impaired HIV-seropositive individuals. Magnetic Resonance Imaging. 1994;12:653. doi: 10.1016/0730-725x(94)92460-0. [DOI] [PubMed] [Google Scholar]
- Michael NL. Host genetic influences on HIV-1 pathogenesis. Current Opinion in Immunology. 1999;11:466. doi: 10.1016/S0952-7915(99)80078-8. [DOI] [PubMed] [Google Scholar]
- Michael NL, Chang G, Louie LG, Mascola JR, Dondero D, Birx DL, et al. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nature Medicine. 1997;3:338. doi: 10.1038/nm0397-338. [DOI] [PubMed] [Google Scholar]
- Michael NL, D’Arcy L, Ehrenberg PK, Redfield RR. Naturally occurring genotypes of the human immunodeficiency virus type 1 long terminal repeat display a wide range of basal and Tat-induced transcriptional activities. Journal of Virology. 1994;68:3163. doi: 10.1128/jvi.68.5.3163-3174.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TI, Borkowsky W, DiMeglio LA, Dooley L, Geffner ME, Hazra R, et al. Metabolic abnormalities and viral replication are associated with biomarkers of vascular dysfunction in HIV-infected children. HIV Medicine. 2012;13:264. doi: 10.1111/j.1468-1293.2011.00970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagar A, Commins D, Alexander JS, Hoque R, Chiappelli F, Singer EJ, et al. NeuroAIDS: Characteristics and diagnosis of the neurological complications of AIDS. Molecular Diagnosis and Therapy. 2008;12:25. doi: 10.1007/BF03256266. [DOI] [PubMed] [Google Scholar]
- Montano MA, Nixon CP, Ndung’u T, Bussmann H, Novitsky VA, Dickman D, et al. Elevated tumor necrosis factor-alpha activation of human immunodeficiency virus type 1 subtype C in Southern Africa is associated with an NF-kappaB enhancer gain-of-function. The Journal of Infectious Diseases. 2000;181:76. doi: 10.1086/315185. [DOI] [PubMed] [Google Scholar]
- Mothobi NZ, Brew BJ. Neurocognitive dysfunction in the highly active anti-retroviral therapy era. Current Opinion in Infectious Diseases. 2012;25:4. doi: 10.1097/QCO.0b013e32834ef586. [DOI] [PubMed] [Google Scholar]
- Mulherin SA, O’Brien TR, Ioannidis JP, Goedert JJ, Buchbinder SP, Coutinho RA, et al. Effects of CCR5-Delta32 and CCR2-64I alleles on HIV-1 disease progression: The protection varies with duration of infection. AIDS. 2003;17:377. doi: 10.1097/01.aids.0000050783.28043.3e. [DOI] [PubMed] [Google Scholar]
- Muller WE, Schroder HC, Ushijima H, Dapper J, Bormann J. gp120 of HIV-1 induces apoptosis in rat cortical cell cultures: Prevention by memantine. European Journal of Pharmacology. 1992;226:209. doi: 10.1016/0922-4106(92)90063-2. [DOI] [PubMed] [Google Scholar]
- Na H, Acharjee S, Jones G, Vivithanaporn P, Noorbakhsh F, McFarlane N, et al. Interactions between human immunodeficiency virus (HIV)-1 Vpr expression and innate immunity influence neurovirulence. Retrovirology. 2011;8:44. doi: 10.1186/1742-4690-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- Nabel GJ, Rice SA, Knipe DM, Baltimore D. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science. 1988;239:1299. doi: 10.1126/science.2830675. [DOI] [PubMed] [Google Scholar]
- Naghavi MH, Schwartz S, Sonnerborg A, Vahlne A. Long terminal repeat promoter/enhancer activity of different subtypes of HIV type 1. AIDS Research and Human Retroviruses. 1999;15:1293. doi: 10.1089/088922299310197. [DOI] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. The Journal of Biological Chemistry. 1999;274:17098. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. Journal of Virology. 1996;70:1475. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Felber BK, Kleinschmidt A, Froese B, Erfle V, Pavlakis GN, et al. Restriction of human immunodeficiency virus type 1 production in a human astrocytoma cell line is associated with a cellular block in Rev function. Journal of Virology. 1995;69:2159. doi: 10.1128/jvi.69.4.2159-2167.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonnemacher MR, Irish BP, Liu Y, Mauger D, Wigdahl B. Specific sequence configurations of HIV-1 LTR G/C box array result in altered recruitment of Sp isoforms and correlate with disease progression. Journal of Neuroimmunology. 2004;157:39. doi: 10.1016/j.jneuroim.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Nottet HS, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, et al. Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. Journal of Immunology. 1996;156:1284. [PubMed] [Google Scholar]
- Novitsky VA, Montano MA, McLane MF, Renjifo B, Vannberg F, Foley BT, et al. Molecular cloning and phylogenetic analysis of human immunodeficiency virus type 1 subtype C: A set of 23 full-length clones from Botswana. Journal of Virology. 1999;73:4427. doi: 10.1128/jvi.73.5.4427-4432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien SJ, Nelson GW. Human genes that limit AIDS. Nature Genetics. 2004;36:565. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- Ohagen A, Ghosh S, He J, Huang K, Chen Y, Yuan M, et al. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: Evidence for a role of the envelope. Journal of Virology. 1999;73:897. doi: 10.1128/jvi.73.2.897-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CA, Mukhtar M, Pomerantz RJ. Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. Journal of Virology. 2000;74:9717. doi: 10.1128/jvi.74.20.9717-9726.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattarini R, Pittaluga A, Raiteri M. The human immunodeficiency virus-1 envelope protein gp120 binds through its V3 sequence to the glycine site of N-methyl-D-aspartate receptors mediating noradrenaline release in the hippocampus. Neuroscience. 1998;87:147. doi: 10.1016/s0306-4522(98)00125-0. [DOI] [PubMed] [Google Scholar]
- Paxton WA, Kang S. Chemokine receptor allelic polymorphisms: Relationships to HIV resistance and disease progression. Seminars in Immunology. 1998;10:187. doi: 10.1006/smim.1998.0132. [DOI] [PubMed] [Google Scholar]
- Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Research. 2000;28:663. doi: 10.1093/nar/28.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, et al. A model for monocyte migration through the blood–brain barrier during HIV-1 encephalitis. Journal of Immunology. 1997;158:3499. [PubMed] [Google Scholar]
- Peters PJ, Bhattacharya J, Hibbitts S, Dittmar MT, Simmons G, Bell J, et al. Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. Journal of Virology. 2004;78:6915. doi: 10.1128/JVI.78.13.6915-6926.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Duenas-Decamp MJ, Sullivan WM, Clapham PR. Variation of macrophage tropism among HIV-1 R5 envelopes in brain and other tissues. Journal of Neuroimmune Pharmacology. 2007;2:32. doi: 10.1007/s11481-006-9042-2. [DOI] [PubMed] [Google Scholar]
- Petito CK. Mechanisms of cell death in brains of patients with AIDS. Journal of Neuropathology and Experimental Neurology. 1995;54:404. [PubMed] [Google Scholar]
- Philpott S, Burger H, Tarwater PM, Lu M, Gange SJ, Anastos K, et al. CCR2 genotype and disease progression in a treated population of HIV type 1-infected women. Clinical Infectious Diseases. 2004;39:861. doi: 10.1086/423386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilcher CD, Shugars DC, Fiscus SA, Miller WC, Menezes P, Giner J, et al. HIV in body fluids during primary HIV infection: Implications for pathogenesis, treatment and public health. AIDS. 2001;15:837. doi: 10.1097/00002030-200105040-00004. [DOI] [PubMed] [Google Scholar]
- Piller SC, Jans P, Gage PW, Jans DA. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: Implications for AIDS pathology. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4595. doi: 10.1073/pnas.95.8.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping LH, Nelson JA, Hoffman IF, Schock J, Lamers SL, Goodman M, et al. Characterization of V3 sequence heterogeneity in subtype C human immuno-deficiency virus type 1 isolates from Malawi: Underrepresentation of X4 variants. Journal of Virology. 1999;73:6271. doi: 10.1128/jvi.73.8.6271-6281.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RJ, Feinberg MB, Andino R, Baltimore D. The long terminal repeat is not a major determinant of the cellular tropism of human immunodeficiency virus type 1. Journal of Virology. 1991;65:1041. doi: 10.1128/jvi.65.2.1041-1045.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope M, Betjes MG, Romani N, Hirmand H, Cameron PU, Hoffman L, et al. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell. 1994;78:389. doi: 10.1016/0092-8674(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Power C, McArthur JC, Johnson RT, Griffin DE, Glass JD, Perryman S, et al. Demented and nondemented patients with AIDS differ in brain-derived human immunodeficiency virus type 1 envelope sequences. Journal of Virology. 1994;68:4643. doi: 10.1128/jvi.68.7.4643-4649.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C, McArthur JC, Nath A, Wehrly K, Mayne M, Nishio J, et al. Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. Journal of Virology. 1998;72:9045. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RW. Understanding the AIDS dementia complex (ADC). The challenge of HIV and its effects on the central nervous system. Research Publications—Association for Research in Nervous and Mental Disease. 1994;72:1. [PubMed] [Google Scholar]
- Raha T, Cheng SW, Green MR. HIV-1 Tat stimulates transcription complex assembly through recruitment of TBP in the absence of TAFs. PLoS Biology. 2005;3:e44. doi: 10.1371/journal.pbio.0030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Posada D, Crandall KA, Holmes EC. The causes and consequences of HIV evolution. Nature Reviews. Genetics. 2004;5:52. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, et al. Molecular pathway involved in HIV-1-induced CNS pathology: Role of viral regulatory protein, Tat. Journal of Leukocyte Biology. 1999;65:458. doi: 10.1002/jlb.65.4.458. [DOI] [PubMed] [Google Scholar]
- Raziuddin, Mikovits JA, Calvert I, Ghosh S, Kung HF, Ruscetti FW. Negative regulation of human immunodeficiency virus type 1 expression in monocytes: Role of the 65-kDa plus 50-kDa NF-kappa B dimer. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:9426. doi: 10.1073/pnas.88.21.9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RT, Achim CL, Sirko DA, Tehranchi S, Kraus FG, Wong-Staal F, et al. Sequence analysis of the V3 loop in brain and spleen of patients with HIV encephalitis. AIDS Research and Human Retroviruses. 1996;12:477. doi: 10.1089/aid.1996.12.477. [DOI] [PubMed] [Google Scholar]
- Refaeli Y, Levy DN, Weiner DB. The glucocorticoid receptor type II complex is a target of the HIV-1 vpr gene product. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3621. doi: 10.1073/pnas.92.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AP, Mathews MB. Transcriptional but not translational regulation of HIV-1 by the tat gene product. Nature. 1988;332:551. doi: 10.1038/332551a0. [DOI] [PubMed] [Google Scholar]
- Richman DD, Wrin T, Little SJ, Petropoulos CJ. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4144. doi: 10.1073/pnas.0630530100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K, Liner J, Heaton R. Neuropsychological assessment of HIV-infected populations in international settings. Neuropsychology Review. 2009;19:232. doi: 10.1007/s11065-009-9096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Franco EJ, Cantres-Rosario YM, Plaud-Valentin M, Romeu R, Rodriguez Y, Skolasky R, et al. Dysregulation of macrophage-secreted cathepsin B contributes to HIV-1-linked neuronal apoptosis. PLoS One. 2012;7:e36571. doi: 10.1371/journal.pone.0036571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos MT, Lange JM, de Goede RE, Coutinho RA, Schellekens PT, Miedema F, et al. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. The Journal of Infectious Diseases. 1992;165:427. doi: 10.1093/infdis/165.3.427. [DOI] [PubMed] [Google Scholar]
- Rosenbloom DI, Hill AL, Rabi SA, Siliciano RF, Nowak MA. Anti-retroviral dynamics determines HIV evolution and predicts therapy outcome. Nat Med. 2012;18:1378–1385. doi: 10.1038/nm.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum MK. Infection of the central nervous system by the human immuno-deficiency virus type 1. Morphology and relation to syndromes of progressive encephalopathy and myelopathy in patients with AIDS. Pathology Annual. 1990;25(Pt. 1):117. [PubMed] [Google Scholar]
- Ross EK, Buckler-White AJ, Rabson AB, Englund G, Martin MA. Contribution of NF-kappa B and Sp1 binding motifs to the replicative capacity of human immunodeficiency virus type 1: Distinct patterns of viral growth are determined by T-cell types. Journal of Virology. 1991;65:4350. doi: 10.1128/jvi.65.8.4350-4358.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross HL, Gartner S, McArthur JC, Corboy JR, McAllister JJ, Millhouse S, et al. HIV-1 LTR C/EBP binding site sequence configurations preferentially encountered in brain lead to enhanced C/EBP factor binding and increased LTR-specific activity. Journal of Neurovirology. 2001;7:235. doi: 10.1080/13550280152403281. [DOI] [PubMed] [Google Scholar]
- Ross HA, Rodrigo AG. Immune-mediated positive selection drives human immunodeficiency virus type 1 molecular variation and predicts disease duration. Journal of Virology. 2002;76:11715. doi: 10.1128/JVI.76.22.11715-11720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Querido B, Nimmagadda M, Cocklin S, Navas-Martin S, Martin-Garcia J. The V1-V3 region of a brain-derived HIV-1 envelope glycoprotein determines macrophage tropism, low CD4 dependence, increased fusogenicity and altered sensitivity to entry inhibitors. Retrovirology. 2008;5:89. doi: 10.1186/1742-4690-5-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau C, Abrams E, Lee M, Urbano R, King MC. Long terminal repeat and nef gene variants of human immunodeficiency virus type 1 in perinatally infected long-term survivors and rapid progressors. AIDS Research and Human Retroviruses. 1997;13:1611. doi: 10.1089/aid.1997.13.1611. [DOI] [PubMed] [Google Scholar]
- Roux P, Alfieri C, Hrimech M, Cohen EA, Tanner JE. Activation of transcription factors NF-kappaB and NF-IL-6 by human immunodeficiency virus type 1 protein R (Vpr) induces interleukin-8 expression. Journal of Virology. 2000;74:4658. doi: 10.1128/jvi.74.10.4658-4665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh J, Turchan-Cholewo J, Galey D, St Hillaire C, Anderson C, Conant K, et al. Interaction of HIV Tat and matrix metalloproteinase in HIV neuropathogenesis: A new host defense mechanism. The FASEB Journal. 2006;20:1736. doi: 10.1096/fj.05-5619fje. [DOI] [PubMed] [Google Scholar]
- Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. Journal of Neurovirology. 2002;8:136. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Sawaya BE, Khalili K, Rappaport J, Serio D, Chen W, Srinivasan A, et al. Suppression of HIV-1 transcription and replication by a Vpr mutant. Gene Therapy. 1999;6:947. doi: 10.1038/sj.gt.3300907. [DOI] [PubMed] [Google Scholar]
- Sawaya BE, Thatikunta P, Denisova L, Brady J, Khalili K, Amini S. Regulation of TNFalpha and TGFbeta-1 gene transcription by HIV-1 Tat in CNS cells. Journal of Neuroimmunology. 1998;87:33. doi: 10.1016/s0165-5728(98)00044-7. [DOI] [PubMed] [Google Scholar]
- Schacker T, Collier AC, Hughes J, Shea T, Corey L. Clinical and epidemiologic features of primary HIV infection. Annals of Internal Medicine. 1996;125:257. doi: 10.7326/0003-4819-125-4-199608150-00001. [DOI] [PubMed] [Google Scholar]
- Schifitto G, Yiannoutsos CT, Ernst T, Navia BA, Nath A, Sacktor N, et al. Selegiline and oxidative stress in HIV-associated cognitive impairment. Neurology. 2009;73:1975. doi: 10.1212/WNL.0b013e3181c51a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuitemaker H, Groenink M, Meyaard L, Kootstra NA, Fouchier RA, Gruters RA, et al. Early replication steps but not cell type-specific signalling of the viral long terminal repeat determine HIV-1 monocytotropism. AIDS Research and Human Retroviruses. 1993;9:669. doi: 10.1089/aid.1993.9.669. [DOI] [PubMed] [Google Scholar]
- Schuitemaker H, Kootstra NA, Koppelman MH, Bruisten SM, Huisman HG, Tersmette M, et al. Proliferation-dependent HIV-1 infection of monocytes occurs during differentiation into macrophages. The Journal of Clinical Investigation. 1992;89:1154. doi: 10.1172/JCI115697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby MJ, Bain ES, Luciw PA, Peterlin BM. Structure, sequence, and position of the stem-loop in tar determine transcriptional elongation by tat through the HIV-1 long terminal repeat. Genes and Development. 1989;3:547. doi: 10.1101/gad.3.4.547. [DOI] [PubMed] [Google Scholar]
- Selnes OA, Miller E, McArthur J, Gordon B, Munoz A, Sheridan K, et al. HIV-1 infection: No evidence of cognitive decline during the asymptomatic stages. The multicenter AIDS cohort study. Neurology. 1990;40:204. doi: 10.1212/wnl.40.2.204. [DOI] [PubMed] [Google Scholar]
- Seo TK, Thorne JL, Hasegawa M, Kishino H. A viral sampling design for testing the molecular clock and for estimating evolutionary rates and divergence times. Bioinformatics. 2002;18:115. doi: 10.1093/bioinformatics/18.1.115. [DOI] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. Journal of Virology. 1999;73:10489. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JT, Martin J, Baltuch G, Malim MH, Gonzalez-Scarano F. Determinants of syncytium formation in microglia by human immunodeficiency virus type 1: Role of the V1/V2 domains. Journal of Virology. 2000;74:693. doi: 10.1128/jvi.74.2.693-701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddappa NB, Venkatramanan M, Venkatesh P, Janki MV, Jayasuryan N, Desai A, et al. Transactivation and signaling functions of Tat are not correlated: Biological and immunological characterization of HIV-1 subtype-C Tat protein. Retrovirology. 2006;3:53. doi: 10.1186/1742-4690-3-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva C, Zhang K, Tsutsui S, Holden JK, Gill MJ, Power C. Growth hormone prevents human immunodeficiency virus-induced neuronal p53 expression. Annals of Neurology. 2003;54:605. doi: 10.1002/ana.10729. [DOI] [PubMed] [Google Scholar]
- Silvestri G, Feinberg MB. Turnover of lymphocytes and conceptual paradigms in HIV infection. The Journal of Clinical Investigation. 2003;112:821. doi: 10.1172/JCI19799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Barroga CF, Hughes MD, Chen J, Raskino C, McKinney RE, et al. Genetic influence of CCR5, CCR2, and SDF1 variants on human immunodeficiency virus 1 (HIV-1)-related disease progression and neurological impairment, in children with symptomatic HIV-1 infection. The Journal of Infectious Diseases. 2003;188:1461. doi: 10.1086/379038. [DOI] [PubMed] [Google Scholar]
- Singh KK, Ellis RJ, Marquie-Beck J, Letendre S, Heaton RK, Grant I, et al. CCR2 polymorphisms affect neuropsychological impairment in HIV-1-infected adults. Journal of Neuroimmunology. 2004;157:185. doi: 10.1016/j.jneuroim.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Sloand EM, Klein HG, Banks SM, Vareldzis B, Merritt S, Pierce P. Epidemiology of thrombocytopenia in HIV infection. European Journal of Haematology. 1992;48:168. doi: 10.1111/j.1600-0609.1992.tb00591.x. [DOI] [PubMed] [Google Scholar]
- Smit TK, Wang B, Ng T, Osborne R, Brew B, Saksena NK. Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): Evidence for neurotropic HIV variants. Virology. 2001;279:509. doi: 10.1006/viro.2000.0681. [DOI] [PubMed] [Google Scholar]
- Spudich S, Gonzalez-Scarano F. HIV-1-related central nervous system disease: Current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harbor Perspectives in Medicine. 2012;2:a007120. doi: 10.1101/cshperspect.a007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich SS, Nilsson AC, Lollo ND, Liegler TJ, Petropoulos CJ, Deeks SG, et al. Cerebrospinal fluid HIV infection and pleocytosis: Relation to systemic infection and antiretroviral treatment. BMC Infectious Diseases. 2005;5:98. doi: 10.1186/1471-2334-5-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M, Czaja K, Rauch A, Rickenbach M, Gunthard HF, Battegay M, et al. HLA-Bw4 identifies a population of HIV-infected patients with an increased capacity to control viral replication after structured treatment interruption. HIV Medicine. 2012;13:589. doi: 10.1111/j.1468-1293.2012.01019.x. [DOI] [PubMed] [Google Scholar]
- Strizki JM, Albright AV, Sheng H, O’Connor M, Perrin L, Gonzalez-Scarano F. Infection of primary human microglia and monocyte-derived macrophages with human immunodeficiency virus type 1 isolates: Evidence of differential tropism. Journal of Virology. 1996;70:7654. doi: 10.1128/jvi.70.11.7654-7662.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, et al. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Khalili K. Activation of HIV-1 transcription by Tat in cells derived from the CNS: Evidence for the participation of NF-kappa B—A review. Advances in Neuroimmunology. 1994;4:291. doi: 10.1016/s0960-5428(06)80270-6. [DOI] [PubMed] [Google Scholar]
- Telenti A, Johnson WE. Host genes important to HIV replication and evolution. Cold Spring Harbor Perspectives in Medicine. 2012;2:a007203. doi: 10.1101/cshperspect.a007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer VM, Rajadhyaksha A, Babin J, Bina M. NF-IL6-mediated transcriptional activation of the long terminal repeat of the human immunodeficiency virus type 1. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7298. doi: 10.1073/pnas.90.15.7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ER, Dunfee RL, Stanton J, Bogdan D, Taylor J, Kunstman K, et al. Macrophage entry mediated by HIV Envs from brain and lymphoid tissues is determined by the capacity to use low CD4 levels and overall efficiency of fusion. Virology. 2007;360:105. doi: 10.1016/j.virol.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toohey K, Wehrly K, Nishio J, Perryman S, Chesebro B. Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology. 1995;213:70. doi: 10.1006/viro.1995.1547. [DOI] [PubMed] [Google Scholar]
- Tornatore C, Chandra R, Berger JR, Major EO. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology. 1994;44:481. doi: 10.1212/wnl.44.3_part_1.481. [DOI] [PubMed] [Google Scholar]
- Trachtenberg E, Korber B, Sollars C, Kepler TB, Hraber PT, Hayes E, et al. Advantage of rare HLA supertype in HIV disease progression. Nature Medicine. 2003;9:928. doi: 10.1038/nm893. [DOI] [PubMed] [Google Scholar]
- Troyer JL, Nelson GW, Lautenberger JA, Chinn L, McIntosh C, Johnson RC, et al. Genome-wide association study implicates PARD3B-based AIDS restriction. The Journal of Infectious Diseases. 2011;203:1491. doi: 10.1093/infdis/jir046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk G, Carobene M, Monczor A, Rubio AE, Gomez-Carrillo M, Salomon H. Higher transactivation activity associated with LTR and Tat elements from HIV-1 BF intersubtype recombinant variants. Retrovirology. 2006;3:14. doi: 10.1186/1742-4690-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Manen D, Delaneau O, Kootstra NA, Boeser-Nunnink BD, Limou S, Bol SM, et al. Genome-wide association scan in HIV-1-infected individuals identifying variants influencing disease course. PLoS One. 2011;6:e22208. doi: 10.1371/journal.pone.0022208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Manen D, van ‘t Wout AB, Schuitemaker H. Genome-wide association studies on HIV susceptibility, pathogenesis and pharmacogenomics. Retrovirology. 2012a;9:70. doi: 10.1186/1742-4690-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G, Power C. Human immunodeficiency virus type 1 genetic diversity in the nervous system: Evolutionary epiphenomenon or disease determinant? Journal of Neurovirology. 2005;11:107. doi: 10.1080/13550280590922838. [DOI] [PubMed] [Google Scholar]
- Van Marle G, Rourke SB, Zhang K, Silva C, Ethier J, Gill MJ, et al. HIV dementia patients exhibit reduced viral neutralization and increased envelope sequence diversity in blood and brain. AIDS. 2002;16:1905. doi: 10.1097/00002030-200209270-00007. [DOI] [PubMed] [Google Scholar]
- van Rij RP, de Roda Husman AM, Brouwer M, Goudsmit J, Coutinho RA, Schuitemaker H. Role of CCR2 genotype in the clinical course of syncytium-inducing (SI) or non-SI human immunodeficiency virus type 1 infection and in the time to conversion to SI virus variants. The Journal of Infectious Diseases. 1998;178:1806. doi: 10.1086/314522. [DOI] [PubMed] [Google Scholar]
- Velpandi A, Nagashunmugam T, Otsuka T, Cartas M, Srinivasan A. Structure-function studies of HIV-1: Influence of long terminal repeat U3 region sequences on virus production. DNA and Cell Biology. 1992;11:369. doi: 10.1089/dna.1992.11.369. [DOI] [PubMed] [Google Scholar]
- Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Molecular Biology of the Cell. 2004;15:2347. doi: 10.1091/mbc.E03-12-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoef K, Klein A, Berkhout B. Paracrine activation of the HIV-1 LTR promoter by the viral Tat protein is mechanistically similar to trans-activation within a cell. Virology. 1996;225:316. doi: 10.1006/viro.1996.0606. [DOI] [PubMed] [Google Scholar]
- Walker LM, Burton DR. Rational antibody-based HIV-1 vaccine design: Current approaches and future directions. Current Opinion in Immunology. 2010;22:358. doi: 10.1016/j.coi.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZX, Berson JF, Zhang TY, Cen YH, Sun Y, Sharron M, et al. CXCR4 sequences involved in coreceptor determination of human immunodeficiency virus type-1 tropism. Unmasking of activity with M-tropic Env glycoproteins. The Journal of Biological Chemistry. 1998;273:15007–15015. doi: 10.1074/jbc.273.24.15007. [DOI] [PubMed] [Google Scholar]
- Wang J, Crawford K, Yuan M, Wang H, Gorry PR, Gabuzda D. Regulation of CC chemokine receptor 5 and CD4 expression and human immunodeficiency virus type 1 replication in human macrophages and microglia by T helper type 2 cytokines. The Journal of Infectious Diseases. 2002;185:885. doi: 10.1086/339522. [DOI] [PubMed] [Google Scholar]
- Weidenheim KM, Epshteyn I, Lyman WD. Immunocytochemical identification of T-cells in HIV-1 encephalitis: Implications for pathogenesis of CNS disease. Modern Pathology. 1993;6:167. [PubMed] [Google Scholar]
- Weiss JM, Cuff CA, Berman JW. TGF-beta downmodulates cytokine-induced monocyte chemoattractant protein (MCP)-1 expression in human endothelial cells. A putative role for TGF-beta in the modulation of TNF receptor expression. Endothelium. 1999;6:291. doi: 10.3109/10623329909078496. [DOI] [PubMed] [Google Scholar]
- Wendelken LA, Valcour V. Impact of HIV and aging on neuropsychological function. Journal of Neurovirology. 2012;18:256. doi: 10.1007/s13365-012-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CA, Achim C. Human immunodeficiency virus encephalitis is the pathological correlate of dementia in acquired immunodeficiency syndrome. Annals of Neurology. 1994;36:673. doi: 10.1002/ana.410360422. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:7089. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: Implications for the neuropathogenesis of AIDS. The Journal of Experimental Medicine. 2001;193:905. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annual Review of Neuroscience. 2002;25:537. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- Williamson S. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Molecular Biology and Evolution. 2003;20:1318. doi: 10.1093/molbev/msg144. [DOI] [PubMed] [Google Scholar]
- Wong JK, Ignacio CC, Torriani F, Havlir D, Fitch NJ, Richman DD. In vivo compartmentalization of human immunodeficiency virus: Evidence from the examination of pol sequences from autopsy tissues. Journal of Virology. 1997;71:2059. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, et al. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11466. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Liu J, Chen L, Liang S, Fujii N, Tamamura H, et al. HIV-1 gp120 enhances outward potassium current via CXCR4 and cAMP-dependent protein kinase A signaling in cultured rat microglia. Glia. 2011;59:997. doi: 10.1002/glia.21171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav A, Collman RG. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. Journal of Neuroimmune Pharmacology. 2009;4:430. doi: 10.1007/s11481-009-9174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeichner SL, Hirka G, Andrews PW, Alwine JC. Differentiation-dependent human immunodeficiency virus long terminal repeat regulatory elements active in human teratocarcinoma cells. Journal of Virology. 1992;66:2268. doi: 10.1128/jvi.66.4.2268-2273.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeichner SL, Kim JY, Alwine JC. Analysis of the human immunodeficiency virus long terminal repeat by in vitro transcription competition and linker scanning mutagenesis. Gene Expression. 1991a;1:15. [PMC free article] [PubMed] [Google Scholar]
- Zeichner SL, Kim JY, Alwine JC. Linker-scanning mutational analysis of the transcriptional activity of the human immunodeficiency virus type 1 long terminal repeat. Journal of Virology. 1991b;65:2436. doi: 10.1128/jvi.65.5.2436-2444.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Huang Y, Yuan H, Chen BK, Ip J, Ho DD. Genotypic and phenotypic characterization of long terminal repeat sequences from long-term survivors of human immunodeficiency virus type 1 infection. Journal of Virology. 1997;71:5608. doi: 10.1128/jvi.71.7.5608-5613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nature Neuroscience. 2003;6:1064. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- Zhao RY, Li G, Bukrinsky MI. Vpr-host interactions during HIV-1 viral life cycle. Journal of Neuroimmune Pharmacology. 2011;6:216. doi: 10.1007/s11481-011-9261-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Halanski MA, Radonovich MF, Kashanchi F, Peng J, Price DH, et al. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Molecular and Cellular Biology. 2000;20:5077. doi: 10.1128/mcb.20.14.5077-5086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman ES, Chen J, Andersen JL, Ardon O, Dehart JL, Blackett J, et al. Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Molecular and Cellular Biology. 2004;24:9286. doi: 10.1128/MCB.24.21.9286-9294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]