

Abstract
Evidence is growing that vascular risk factors (VRFs) for Alzheimer’s disease (AD) affect cerebral hemodynamics to launch a cascade of cellular and molecular changes that initiate cognitive deficits and eventual progression of AD. Neuroimaging studies have reported VRFs for AD to be accurate predictors of cognitive decline and dementia. In regions that participate in higher cognitive function, middle temporal, posterior cingulate, inferior parietal and precuneus regions, and neuroimaging studies indicate an association involving VRFs, cerebral hypoperfusion, and cognitive decline in elderly individuals who develop AD. The VRF can be present in cognitively intact individuals for decades before mild cognitive deficits or neuropathological signs are manifested. In that sense, they may be “ticking time bombs” before cognitive function is demolished. Preventive intervention of modifiable VRF may delay or block progression of AD. Intervention could target cerebral blood flow (CBF), since most VRFs act to lower CBF in aging individuals by promoting cerebrovascular dysfunction.
Keywords: vascular risk factors, Alzheimer’s, cerebral blood flow, hypoperfusion, aging, neuroimaging
Introduction
Blood flow in mammals is essential for life. No blood flow, no life. Paradoxically, blood flow can continue even when there is no life, as it happens when brain death occurs. Medical science argues that brain death is human death, because there is not only permanent cessation of all electrical brain activity but also irreversible loss of brainstem function where only artificial life support can keep the heart beating.
However, there is a level of blood flow decrease that gradually deprives the cognitive function and conscious awareness but allows quasielectrical brain activity, heartbeat, and respiration to continue functioning. Can this be considered life? Here, the key words are “level of blood flow decrease,” because when this is defined, it will determine the likely outlook for how brain cells that regulate cognition will either cope to stay functional or not. The level of blood flow decrease is measured by the amount of arterial blood perfusing, an area of neural tissue within a given time. 1
Normal resting cerebral blood flow (CBF) in adult human cortex is about 60 to 70 mL/100 g/min. Regionally, gray matter flow averages about 70 mL/100 g/min, and white matter is about 20 mL/100 g/min. It is important to note that these flow values normally decline slowly with advancing age by an estimated rate of 0.5% per year. 2 Consequently, from the age of 20 to 65, normal CBF drops 15% to 20%. 2–4 As previous studies have shown, 5–8 this 15% to 20% normal drop in CBF at an elderly age does not necessarily imply that cognitive function will be compromised. The reason is that adequate CBF is available to supply neuronal demand for energy metabolism and activity from neurovascular coupling and autoregulatory mechanisms.
However, this age-related decline in CBF can become critical to neuronal survival if an additional hemodynamic burden further lowers CBF levels, at which point, neuronal demand for energy nutrients may not be met by supply or compensated by neurovascular coupling. This issue is discussed later.
Brain blood flow rates that mildly decline to 50 mL/100 g/min can inhibit the synthesis of many proteins 9 and with further declines to 30 mL/100 g/min, neurologic symptoms and impaired neurotransmission will appear. 10 When CBF falls to 20 mL/100 g/min, electrical failure or irreversible neuronal damage can result within minutes. 11,12
Neuronal Responses to Reduced Cerebral Perfusion
Heiss 10 described 3 stages of reduced blood flow to the brain that can impact neuronal metabolism and anatomic structure (1) ischemia, (2) penumbra, and (3) hypoperfusion. Using this construct, neuroimaging techniques can detect, localize, and quantitate 3 neuronal states generated by vascular pathology affecting CBF hemodynamics, ischemic neurons with blood flow values below 12 mL/100 g/min, penumbral neurons with flows at 12 to 22 mL/100 g/min, and hypoperfused neurons with flow values exceeding 22 mL/100 g/min but still below normal threshold 10,13 (Figure 1).
Figure 1.

Four basic neuronal states generated by vascular pathology that reduces cerebral blood flow (CBF) supply and alters normal hemodynamic response to local or global ischemia. A, Hypoperfused neurons with blood flow values exceeding 22 mL/100 g/min but still below normal blood flow for gray matter; (B) penumbral neurons with blood flows ranging at 12 to 22 mL/100 g/min; (C) ischemic neurons with blood flow values under 12 mL/100 g/min. Very few, if any, structural changes are seen in hypoperfused neurons but their function may undergo progressive impairment. Penumbral neurons, also referred to as moderate ischemia, generally display mild structural changes and moderate functional deficits but are still capable of recovery assuming appropriate intervention is applied. Ischemic neurons can quickly reach the point of no return where the cell becomes unable to recover its normal morphology and function even if all processes leading to death are interventionally arrested. Acute ischemic neurons typically show cytoplasmic eosinophilia, dispersion of Nissl substance, nuclear shrinking (pyknosis), and nuclear displacement to the periphery. When ischemic neurons are chronically injured, as in neurodegeneration, no eosinophilia is seen and instead, the cell body shrinks and becomes more angular, synaptic damage can be seen, and axonal neurofibrillary tangles (NFTs) may dominate. D, Neuronal death is the state where permanent and complete arrest of energy metabolism occurs.
Nontraumatic ischemic neurons are commonly produced by an embolic stroke, where anatomic and functional changes generally occur relatively quickly. The anatomic changes consist of cytoplasmic swelling secondary to sodium influx and failure of membrane ionic regulation. 14 This is followed by characteristic eosinophilia, severe loss of the endoplasmic reticulum and Golgi apparatus, chromatin clumping, nuclear shrinking with peripheral shifting, and disappearance of axonal microtubules, which generate synaptic dystrophy and neurotransmission failure (Figure 1). Depending on its location and severity, a focal embolic insult to the brain will destablize ischemic neurons by inducing hypoxia, glutamate excitotoxicity, a massive and disruptive intracellular rise of Ca2+, mitochondrial depolarization, formation of oxygen and nitric oxide free radicals, and impairment of adenosine triphosphate (ATP) synthesis in fuel cells.
The biochemical cascade associated with anatomic changes in ischemic neurons and glial cells is the product of irreversible oxygen–glucose deprivation and ATP production failure that may have been initiated as a neurometabolic energy crisis in penumbral and hypoperfused neurons. 15–17 The ischemic neurons weathering these functional-anatomic changes are unlikely to recover spontaneously or with any intervention and at some point, will degenerate into an agonal state prior to death (Figure 1).
When CBF falls subnormally in brain tissue adjacent to an infarct but stays above the 20-mL threshold that tissue and its neuronal population are theoretically salvageable provided that appropriate intervention is applied. 18,19 The salvageable tissue is known as the ischemic penumbra. 11,20 The ischemic penumbra is a moderately hypoperfused zone peripheral to the infarct core, where neurons are functionally impaired but show no gross morphologic abnormalities 10,20 (Figure 1). In his classic article describing the ischemic penumbra, Astrup and his colleagues 11 recognized 2 stages where penumbral neurons can experience functional impairment and irreparable degenerative damage, usually with the former stage preceding the latter (Figure 1).
Penumbral neurons may remain viable for several hours, 19 several weeks, 13 or even beyond a year 21 as long as arterial collaterals supplied by the unaffected branches of the occluded vascular tree remain patent. The arterial collateral blood flow in the penumbral zone may be adequate to sustain tissue viability but generally not sufficient to support cognitive or neurological function. 11
Penumbral neurons are destined to follow a pathway irreversible to degeneration unless successful rescue by intervention is applied. 22
For this reason, the penumbral tissue territory on the rim of an occlusive infarct has become an important target for therapy, because it offers a window of opportunity to salvage these neurons and reverse the dysfunctional effect they have on the system. Consequently, if penumbral neurons can be rescued from their limited arterial blood supply, a reversal of lost sensory-motor deficits may result. This is the main objective of thrombolytic agents used in postischemic stroke. Thrombolytic agents such as recombinant tissue plasminogen activator work to reduce the occlusive insoluble fibrin clot and increase penumbral arterial blood flow. 23
Extending this principle, some studies have reported that restoring to normal decreased CBF supplying penumbral or hypoperfused neurons that contribute to cognitive deficits allow the impaired cognitive function to return to normal. 24–28
Hypoperfused neurons are generally not found in the ischemic or penumbral zones of an infarct; and although they are usually structurally intact, they can be functionally impaired, particularly among the elderly patients, to the point where cognitive symptoms can be detected. This functional and growing impairment of chronically hypoperfused brain neurons is the basis for the vascular hypothesis of Alzheimer’s disease (AD). 17,29
Despite its clinical importance, relatively little research has been done to study the pathophysiology of neurons following chronic brain hypoperfusion. Notwithstanding this lapse, a consistent rule has emerged from the tight coupling between cognitive function and CBF. The cognitive-CBF relationship can be appreciated by a Rotterdam study that examined 892 participants between the ages of 60 and 91 and reported that lower CBF was linked with deficient executive function, information processing speed, and global cognition. 28 This finding indicates that the influence of CBF on cognition is not incidental and may be an important step in better understanding the pathophysiology of AD.
For example, when CBF is temporarily decreased by occlusion of one or both carotid arteries in cognitively healthy humans or rats, the almost universal reaction is that some aspect of cognitive function will be diminished. 24,30,31 When a transient global cerebral hypoperfusion attack results in memory impairment, improvement in CBF reverses the memory impairment.
If the carotid artery occlusion is removed and CBF is restored to preocclusion levels (all other factors remaining constant), normal cognitive function will be restored. 25,30,31 This phenomenon is also observed when a transient global cerebral hypoperfusion attack results in memory impairment, improvement in CBF during clinical recovery reverses the memory impairment. 32
These simple experiments imply 2 things (1) sudden or acute drop in CBF can have a negative effect on cognitive function, especially at an advanced age and (2) timely restoration of CBF can reverse the loss of cognitive function. Having made these 2 assumptions, the next question should widen the reasoning process by asking whether CBF levels remain subnormal indefinitely, what further effects will it have on cognitive function? This question is dealt with in the following discussion.
Vascular Risk Factors and Critically Attained Threshold of Cerebral Hypoperfusion
Most neuroimaging studies of brain function rely on neurovascular coupling changes between regional brain metabolism and regional CBF (rCBF), 33–35 and useful information can be obtained when patients with mild cognitive impairment (MCI) or dementia are tested and compared with patients who are cognitively intact. The MCI is a diagnostic term developed to describe the prodromal stage of AD.
In 1993, we designed a series of experiments on young and old rats subjected to chronic brain hypoperfusion via carotid artery occlusion. 29 At various time intervals ranging from 1 to 6 months, spatial memory, histopathology, cellular, molecular, and neurophysiological changes were measured in these animals. 36–39 The experimental rat findings obtained, together with a careful review of the clinical literature, provided support to the premise that reduced blood flow to the brain seemed to ultimately induce cognitive dysfunction.
The aging rat findings intriguingly implied that a similar relationship involving the start of cerebral hypoperfusion and cognitive loss as seen in the rat experiments could occur in elderly persons. 40 However, although we knew the trigger for this process in aged rats involved blocking carotid artery blood flow to the brain, there still remained the question as to what factor or factors could lead to the cerebral hypoperfusion–cognitive dysfunction in humans. The answer came unexpectedly but not surprisingly in a landmark article published in 1997 by Hofman et al 41 showing in a prospective population-based study that carotid artery atherosclerosis was significantly associated with AD and with vascular dementia. We had known from our aging rat experiments that carotid artery occlusion not only reduced CBF but in time also resulted in cognitive loss, neuronal degeneration, and an assortment of cellular–molecular aberrations reminiscent of AD. 36–40
The study by Hofman et al 41 was strengthened by a report from Skoog et al 42 that appeared about the same time showing that a medical history of hypertension in elderly people 15 years before constituted a significant risk factor in the development of AD. Since it is well known that both atherosclerosis and hypertension are disorders known to reduce CBF by different physiological means during advanced aging, these 2 studies seemed complimentary. Moreover, the reports provided evidence for the missing link that we needed to propose the concept that vascular risk factors (VRFs) such as atherosclerosis and hypertension in aging individuals already burdened by age-related CBF decline could be the basis for triggering further cognitive impairment that would eventually evolve to AD. 40 Since 1997, both the Hofman and the Skoog studies have been amply confirmed and more than 2 dozen additional VRFs for AD have been added to the list including factors impairing cardiovascular, cerebrovascular, and peripheral vascular function. 43–48 Many of these VRFs are modifiable with proper intervention. 49
The Hofman and Skoog studies also led to our proposal in 1997 that AD can develop from 2 important events that occur during aging (1) normal age-related decline in CBF and (2) a further CBF drop when VRFs are acquired. These 2 events can lower blood flow to the brain to a critically attained threshold of cerebral hypoperfusion (CATCH) and induce cognitive dysfunction. 39
The CATCH is a progressive cerebrovascular insufficiency that with time will destabilize neurons, synapses, neurotransmission, and cognitive ability, eventually evolving into a severe neurodegenerative process characterized by the formation of amyloid β (Aβ) peptide-containing plaques, neurofibrillary tangles (NFTs), and amyloid angiopathy. 50 Clinical evidence suggests that 3 interactive factors are fundamental in the development of CATCH (1) duration of hypoperfusion, 30,42,51 (2) severity of hypoperfusion, 10,41,52,53 and (3) age of the patient. 3–4
If, for argument’s sake, AD can be regarded as a gigantic jigsaw puzzle with most of the pieces still missing, some key pieces of that puzzle, represented by the evidence gathered thus far pertaining to CBF changes, cognitive dysfunction, aging, VRFs, neurodegeneration, and dementia, offer a compelling partial image of that puzzle that may provide essential clues necessary for constructing a theoretical framework to decipher the rest of the AD puzzle (Figure 2).
Figure 2.
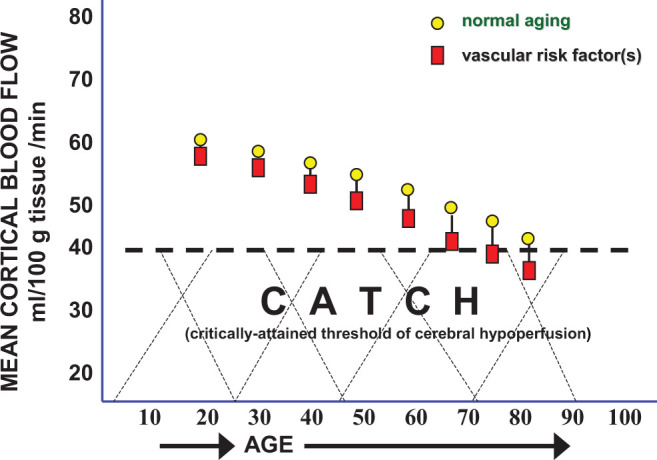
Normal resting cerebral blood flow (CBF) is approximately 65 mL/100g/min at a young age. The CBF normally declines 0.5% per year so from age 20 to 60, a 20% reduction in brain blood flow is normally expected (circles). In patients with mild cognitive impairment (MCI), CBF can further drop an additional 15% to 20%, but the cause for this CBF fall is unclear. Critically attained threshold of cerebral hypoperfusion (CATCH) argues that vascular risk factors in the presence of advanced aging can add another 15% reduction in CBF resulting in a brain metabolic energy crisis due to chronic, subnormal delivery of energy nutrients needed to sustain the high energy requirement of brain cell consumption.
Vascular Risk Factors and Neurometabolism
Many neuroimaging studies have shown agreement that hypoperfused neurons in elderly people may be one of the earliest markers of cognitive impairment preceding AD. 54,55 The patterns of hypoperfusion measured in people prior to AD appear to be independent of underlying cortical gray matter neurodegeneration. 54 In asymptomatic people at high risk of AD, brain hypoperfusion may exist before cerebral atrophy is present. 54–58
This finding suggests that neurodegeneration and the heavy accumulation of Aβ plaques and NFTs that define AD are produced after cerebral perfusion abnormalities.
Moreover, it has been reported that clinically asymptomatic patients and patients with cognitive impairment at autopsy showed similar amounts of Aβ plaques and NFTs deposits in regions thought to participate in higher cognitive function such as the middle frontal, middle temporal, inferior parietal, and precuneus regions. 59 The asymptomatic and cognitively impaired groups differed only while alive by a relatively higher level of rCBF in the asymptomatic individuals at the anterior and posterior cingulate and cuneus, when compared to the cognitively impaired group. 59
It should be noted that Aβ-containing plaques are also found in the brains of stable patients with MCI as well as in many cognitively normal individuals, and the accumulation of Aβ in brain does not correlate with cognitive deficits, neuronal loss, or severity of AD. 60–68 Conversely, it has been reported that mild and transient brain hypoperfusion increases Aβ levels profoundly and persistently by enhancing β-secretase protein expression (β-site amyloid precursor protein-cleaving enzyme 1 [BACE-1]). 69 The BACE-1 is the rate-limiting enzyme of Aβ, and its upregulation by brain hypoperfusion can promote Aβ overproduction. 70
The mechanism responsible for Aβ overproduction following brain hypoperfusion is unclear but may be related to reduced oxygen that is a potent regulator of BACE-1 levels and Aβ production. 71
Whether chronic brain hypoperfusion is the initiator of neurodegeneration and by-products such as Aβ and NFTs has not been established. For this reason, it is tempting and germane to consider, given the evidence available from neuroimaging studies, how VRFs may increase the odds of developing AD. Do VRFs upregulate the levels of Aβ and NFTs to promote AD or do they act principally to lower CBF to critical levels where the delivery of energy nutrients, particularly glucose and oxygen, threaten brain cell metabolism?
Studies reporting an association between cerebral perfusion deficits in people with cognitive impairment are supported by regional cerebral metabolic rate of glucose (rCMRglu) studies in patients with MCI. 56–57,72 There is general agreement that changes in blood flow appear to be accompanied by changes in glucose metabolism, 73–78 and this association can be tested using (18)F-fluorodeoxyglucose and positron emission tomography (FDG-PET) to detect CMRglu uptake in the brain in conjunction with arterial spin labeling (ASL) MRI, which measures CBF. 77 Neuroimaging studies combining FDG-PET for rCMRglu and ASL-MRI for CBF show a striking overlap of images in AD brain regions affected by reduced glucose uptake and low CBF. 78
Measuring CMRglu uptake in the brain of patients parallels CBF levels due to the tight coupling between rCBF and regional metabolism of glucose. 79,80 A deficient CMRglu uptake in the brain may also indicate synaptic dysfunction and reduced synaptic density. 81 The FDG-PET studies have generally shown a reduced glucose uptake in patients with MCI who later convert to AD. 56–57,72,82 In fact, asymptomatic individuals with hypoperfusion in the precuneus, posterior cingulate, lateral parietal cortex, and hippocampal region 83 or with a progression of reduced CMRglu uptake from the hippocampus to the parietotemporal and posterior cingulate cortices were later observed to convert to AD. 84
These FDG-PET findings point to an important clue that center on 3 main possibilities (1) the findings are spurious or coincidental; (2) the findings imply that neurodegenerative changes, such as accumulated Aβ plaques and NFTs, kill brain cells that take up glucose; and (3) the findings imply that the presence of cerebral hypopoperfusion, reflected by lowered CMRglu, delivers less glucose to the brain. The first possibility is unlikely, because the finding has been replicated many times by different international groups working independently. The second possibility could explain the lowered glucose uptake but only if neurodegenerative markers such as Aβ-containing plaques and NFTs are heavily present. However, by definition, MCI brains do not contain heavy neurodegenerative deposits or substantial neuronal loss or white–gray matter atrophy, and when they do, the patient no longer has MCI but rather AD. 85,86
The third possibility seems more likely to explain the reduced glucose uptake seen at the MCI stage even when cognitive dysfunction is very mildly affected. 84,87
Supporting this argument are 2 dozen VRFs for AD described so far, which appear to share 1 common factor, that is, to reduce brain perfusion or interfere with cerebral hemodynamics. 48,52,88–91 Some of these VRFs can be present in cognitively intact individuals for decades before mild cognitive deficits or neuropathological signs are manifested. 92–95 In that sense, they may be “ticking time bombs” before cognitive function is exploded unless preventive intervention is applied to those factors that are modifiable. 49 Intervention could target CBF, since there is growing evidence that most if not all VRFs act to lower CBF in aging individuals by inducing cerebrovascular dysfunction. 46,48,96–98 Altered brain hemodynamics during advanced aging can promote cerebral hypoperfusion to a critical level where abnormal delivery of high energy nutrients to brain cells results in mild cognitive deficits involving executive function, mental flexibility, psychomotor speed, and memory 40 (Figure 3). These mild cognitive changes can remain stable for a protracted period of time or, if CBF is allowed to worsen, progressively evolve to MCI and severe cognitive impairment 52,94 (Figure 3).
Figure 3.
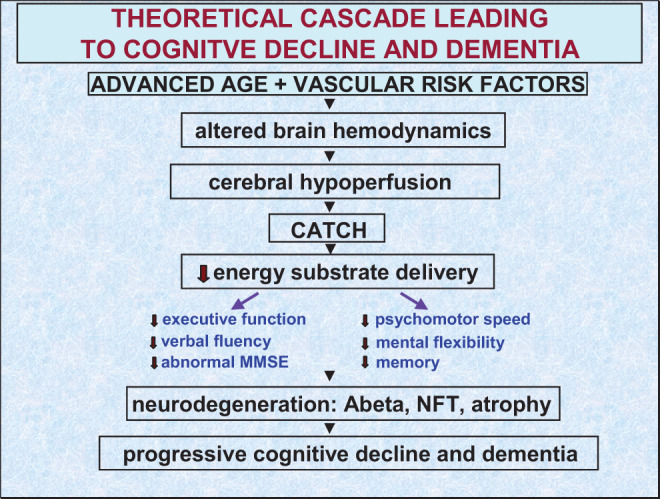
Theoretical cascade of events leading to cognitive failure and Alzheimer or vascular dementia. The cascade may begin with a double drop in cerebral blood flow (see Figure 2) during normal aging and in the presence of vascular risk factors. These 2 events are reported to alter brain hemodynamics giving rise to chronic cerebral hypoperfusion. With time, critically attained threshold of cerebral hypoperfusion (CATCH) is reached where brain cells, especially those sensitive to ischemia in the inferior temporal region, precuneus, and posterior cingulate cortex can no longer cope with reduced energy substrate delivery. The outcome of this process affects executive function, verbal fluency, psychomotor speed, and mental flexibility, which may progress to mild cognitive impairment, severe neurodegeneration, and dementia.
Longitudinal studies using FDG-PET in patients with MCI with a strong risk of conversion to AD were followed for an average of 3 years, and those patients who were shown to have reduced memory performance and decreased rCMRglu from the left temporoparietal area had a 90% rate of developing AD. 56 In 3 other studies, where patients with MCI were followed for 2 to 3 years, lower glucose uptake in the posterior cingulate cortex was a good marker in their eventual conversion to AD. 57,72,99
Although the CBF drop required to reach CATCH in cognitively intact individuals has only been implied, it is safe to say that blood flows falling below 40 mL/100 g/min in the parietal cortex, temporal cortex, and limbic system are consistent with cognitive deterioration in elderly individuals during MCI and supports the predicted conversion from unstable MCI to AD53,58 (Figure 2).
An interesting finding by Marshall et al 30 may shed some light on the critical level of CBF, where cognitive function is disturbed. These investigators reported using a balloon test to occlude the internal carotid artery temporarily in patients about to undergo carotid aneurysm surgery that revealed that a 30-minute occlusion lowering brain blood flow by about 15% from baseline resulted in a brief deterioration of a sustained attention task measuring alertness; if CBF further dropped another 15%, the sustained attention deficit persisted until the carotid artery occluder was removed. 30
There is a population of elderly individuals with VRFs of long duration who remain cognitively intact until death. 61–62 This finding suggests that it may be as important, if not more so, to study people who show resistance to dementia despite a neuropathologic or cerebrovascular picture that indicates high risk of AD. The answer could have far-reaching implications to a majority of elders with VRFs who will acquire dementia.
The collective findings reviewed here point to the conclusion that chronic lowering of CBF to a critical level (CATCH) may block cerebral autoregulation or neural signaling to arteriolar vasoregulation that normally counters the fallen cerebral perfusion. It is uncertain whether active intervention at this stage can reverse CATCH, but this is a crucial aspect of the AD puzzle that requires prompt research exploration.
Footnotes
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Buxton R. Introduction to Functional Magnetic Resonance Imaging: Principles and Techniques. Cambridge, England: Cambridge University Press; 2002. [Google Scholar]
- 2. Leenders KL, Perani D, Lammertsma AA, et al. Cerebral blood flow, blood volume and oxygen utilization. Normal values and effect of age. Brain. 2009;113(pt 1):27–47. [DOI] [PubMed] [Google Scholar]
- 3. Pantano P, Baron JC, Lebrun-Grandie P, Duquesnoy N, Bousser MG, Comar D. Regional cerebral blood flow and oxygen consumption in human aging. Stroke. 1994;15(4):635–641. [DOI] [PubMed] [Google Scholar]
- 4. Zou Q, Wu CW, Stein EA, Zang Y, Yang Y. Static and dynamic characteristics of cerebral blood flow during the resting state. Neuroimage. 2009;48(3):515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gur RC, Gur RE, Obrist WD, Skolnick BE, Reivich M. Age and regional cerebral blood flow at rest and during cognitive activity. Arch Gen Psychiatry. 1987;44(7):617–621. [DOI] [PubMed] [Google Scholar]
- 6. Creasey H, Rapoport SI. The aging human brain. Ann Neurol. 1985;17(1):2–10. [DOI] [PubMed] [Google Scholar]
- 7. Rapp PR, Amaral DG. Individual differences in the cognitive and neurobiological consequences of normal aging. Trends Neurosci. 1992;15(9):340–345. [DOI] [PubMed] [Google Scholar]
- 8. Grady CL, McIntosh AR, Bookstein F, Horwitz B, Rapoport SI, Haxby JV. Age-related changes in regional cerebral blood flow during working memory for faces. Neuroimage. 1998;8(4):409–425. [DOI] [PubMed] [Google Scholar]
- 9. Jacewicz M, Kiessling M, Pulsinelli WA. Selective gene expression in focal cerebral ischemia. J Cereb Blood Flow Metab. 1986;6(3):263–272. [DOI] [PubMed] [Google Scholar]
- 10. Heiss WD. Ischemic penumbra: evidence from functional imaging in man. J Cereb Blood Flow Metab. 2000;20(9):1276–1293. [DOI] [PubMed] [Google Scholar]
- 11. Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12(6):723–725. [DOI] [PubMed] [Google Scholar]
- 12. Branston NM, Strong AJ, Symon L. Extracellular potassium activity, evoked potential and tissue blood flow. Relationships during progressive ischaemia in baboon cerebral cortex. J Neurol Sci. 1977;32(3):305–321. [DOI] [PubMed] [Google Scholar]
- 13. Wise RJ, Bernardi S, Frackowiak RS, Legg NJ, Jones T. Serial observations on the pathophysiology of acute stroke. Brain. 1983;106(pt 1):197–222. [DOI] [PubMed] [Google Scholar]
- 14. Kalimo H, Olsson Y, Paljari L, Soderfeldt B. Structural changes in brain tissue under hypoxic-ischemic conditions. J Cereb Blood Flow Metab. 1982;2(suppl 1):S19–S22. [PubMed] [Google Scholar]
- 15. Cross JL, Meloni BP, Bakker AJ, Lee S, Knuckey NW. Modes of neuronal calcium entry and homeostasis following cerebral ischemia. Stroke Res Treat. 2010;2010:316862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–1568. [DOI] [PubMed] [Google Scholar]
- 17. de la Torre JC. The vascular hypothesis of Alzheimer's disease: bench to bedside and beyond. Neurodegener Dis. 2010;7(1-3):116–121. [DOI] [PubMed] [Google Scholar]
- 18. Lund CG, Aamodt AH, Russell D. Patient selection for intra-arterial cerebral revascularization in acute ischemic stroke. Acta Neurol Scand Suppl. 2013;(196):65–68. [DOI] [PubMed] [Google Scholar]
- 19. Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14(5):497–500. [DOI] [PubMed] [Google Scholar]
- 20. Baron JC. Mapping the ischaemic penumbra with PET: a new approach. Brain. 2001;124(pt 1):2–4. [DOI] [PubMed] [Google Scholar]
- 21. Brumm KP, Perthen JE, Liu TT, Haist F, Ayalon L, Love T. An arterial spin labeling investigation of cerebral blood flow deficits in chronic stroke survivors. Neuroimage. 2010;51(3):995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher M, Bastan B. Identifying and utilizing the ischemic penumbra. Neurology. 2012;79(13 suppl 1):S79–S85. [DOI] [PubMed] [Google Scholar]
- 23. Devos D, Sevin M, De Gaalon S, Lintia-Gaultier A, Guillon B. Management of ischemic stroke in the hyperacute phase. Panminerva Med. 2013;55(1):59–78. [PubMed] [Google Scholar]
- 24. de la Torre JC, Fortin T, Park GA, Pappas BA, Richard MT. Brain blood flow restoration ‘rescues' chronically damaged rat CA1 neurons. Brain Res. 1993;623(1):6–15. [DOI] [PubMed] [Google Scholar]
- 25. Borroni B, Tiberio G, Bonardelli S, et al. Is mild vascular cognitive impairment reversible? Evidence from a study on the effect of carotid endarterectomy. Neurol Res. 2004;26(5):594–597. [DOI] [PubMed] [Google Scholar]
- 26. Carnevale D, Mascio G, D'Andrea I, et al. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension. 2012;60(1):188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldsmith HS, Wu W, Zhong J, Edgar M. Omental transposition to the brain as a surgical method for treating Alzheimer's disease. Neurol Res. 2003;25(6):625–634. [DOI] [PubMed] [Google Scholar]
- 28. Poels MM, Ikram MA, Vernooij MW, et al. Total cerebral blood flow in relation to cognitive function: the Rotterdam Scan Study. J Cereb Blood Flow Metab. 2008;28(10):1652–1655. [DOI] [PubMed] [Google Scholar]
- 29. de la Torre JC, Mussivand T. Can disturbed brain microcirculation cause Alzheimer's disease? Neurol Res. 1993;15(3):146–153. [DOI] [PubMed] [Google Scholar]
- 30. Marshall RS, Lazar RM, Pile-Spellman J, et al. Recovery of brain function during induced cerebral hypoperfusion. Brain. 2001;124(pt 6):1208–1217. [DOI] [PubMed] [Google Scholar]
- 31. Duan W, Chun-Qing Z, Zheng J, Gui L, Huang HQ, Chen KN. Relief of carotid stenosis improves impaired cognition in a rat model of chronic cerebral hypoperfusion. Acta Neurobiol Exp (Wars). 2011;71(2):233–243. [DOI] [PubMed] [Google Scholar]
- 32. Yamane Y, Ishii K, Shimizu K, et al. Global cerebral hypoperfusion in a patient with transient global amnesia. J Comput Assist Tomogr. 2008;32(3):415–417. [DOI] [PubMed] [Google Scholar]
- 33. Roy CS, Sherrington CS. On the regulation of the blood-supply of the brain. J Physiol. 1890;11(1-2):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ingvar DH, Risberg J. Influence of mental activity upon regional cerebral blood flow in man. A preliminary study. Acta Neurol Scand Suppl. 1965;41(suppl 14):183–186. [DOI] [PubMed] [Google Scholar]
- 35. Chen Y, Wolk DA, Reddin JS, et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology. 2011;77(22):1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de la Torre JC, Cada A, Nelson N, Davis G, Sutherland RJ, Gonzalez-Lima F. Reduced cytochrome oxidase and memory dysfunction after chronic brain ischemia in aged rats. Neurosci Lett. 1997;223(3):165–168. [DOI] [PubMed] [Google Scholar]
- 37. de la Torre JC, Fortin T, Park GA, et al. Chronic cerebrovascular insufficiency induces dementia-like deficits in aged rats. Brain Res. 1992;582(2):186–195. [DOI] [PubMed] [Google Scholar]
- 38. de la Torre JC, Fortin T, Park GA, et al. Aged but not young rats develop metabolic, memory deficits after chronic brain ischemia. Neurol Res. 1992;14(suppl 2):177–180. [DOI] [PubMed] [Google Scholar]
- 39. de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol Aging. 2000;21(2):331–342. [DOI] [PubMed] [Google Scholar]
- 40. de la Torre JC. Hemodynamics of deformed microvessels in Alzheimer's disease brain. Ann N Y Acad Sci. 1997;826:75–91. [DOI] [PubMed] [Google Scholar]
- 41. Hofman A, Ott A, Breteler MM, et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam study. Lancet. 1997;349(9046):151–154. [DOI] [PubMed] [Google Scholar]
- 42. Skoog I, Lernfelt B, Landahl S, et al. 15-Year longitudinal study of blood pressure and dementia. Lancet. 1996;347(9009):1141–1145. [DOI] [PubMed] [Google Scholar]
- 43. Yarchoan M, Xie SX, Kling MA, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain. 2012;135(pt 12):3749–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goldstein FC, Levey AI, Steenland NK. High blood pressure and cognitive decline in mild cognitive impairment. J Am Geriatr Soc. 2013;61(1):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luzzi S, Vella L, Bartolini M, Provinciali L, Silvestrini M. Atherosclerosis in the evolution of Alzheimer's disease: can treatment reduce cognitive decline? J Alzheimers Dis. 2010;20(3):893–901. [DOI] [PubMed] [Google Scholar]
- 46. Orsucci D, Mancuso M, Ienco EC, Simoncini C, Siciliano G, Bonuccelli U. Vascular factors and mitochondrial dysfunction: a central role in the pathogenesis of Alzheimer's disease. Curr Neurovasc Res. 2013;10(1):76–80. [DOI] [PubMed] [Google Scholar]
- 47. Polidori MC, Pientka L, Mecocci P. A review of the major vascular risk factors related to Alzheimer's disease. J Alzheimers Dis. 2012;32(3):521–530. [DOI] [PubMed] [Google Scholar]
- 48. de la Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3(3):184–190. [DOI] [PubMed] [Google Scholar]
- 49. Pase MP. Modifiable vascular markers for cognitive decline and dementia: the importance of arterial aging and hemodynamic factors. J Alzheimers Dis. 2012;32(3):653–663. [DOI] [PubMed] [Google Scholar]
- 50. de la Torre JC, Stefano GB. Evidence that Alzheimer's disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev. 2000;34(3):119–136. [DOI] [PubMed] [Google Scholar]
- 51. Nitrini R, Buchpiguel CA, Caramelli P, et al. SPECT in Alzheimer's disease: features associated with bilateral parietotemporal hypoperfusion. Acta Neurol Scand. 2000;101(3):172–176. [DOI] [PubMed] [Google Scholar]
- 52. de la Torre JC. Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer's disease. J Alzheimers Dis. 2012;32(3):553–567. [DOI] [PubMed] [Google Scholar]
- 53. Ishii K, Sasaki M, Yamaji S, Sakamoto S, Maeda K. Cerebral blood flow changes in the primary motor and premotor cortices during hyperventilation. Ann Nucl Med. 1998;12(1):29–33. [DOI] [PubMed] [Google Scholar]
- 54. Johnson NA, Jahng GH, Weiner MW, et al. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology. 2005;234(3):851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alsop DC, Dai W, Grossman M, Detre JA. Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer's disease. J Alzheimers Dis. 2010;20(3):871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arnaiz E, Jelic V, Almkvist O, et al. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport. 2001;12(4):851–855. [DOI] [PubMed] [Google Scholar]
- 57. Chetelat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology. 2003;60(8):1374–1377. [DOI] [PubMed] [Google Scholar]
- 58. Huang C, Wahlund LO, Almkvist O, et al. Voxel- and VOI-based analysis of SPECT CBF in relation to clinical and psychological heterogeneity of mild cognitive impairment. Neuroimage. 2003;19(3):1137–1144. [DOI] [PubMed] [Google Scholar]
- 59. Codispoti KE, Beason-Held LL, Kraut MA, et al. Longitudinal brain activity changes in asymptomatic Alzheimer disease. Brain Behav. 2012;2(3):221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arriagada PV, Growdon JH, Hedley-White ET, Hyman B. Neurofibrillar tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 pt 1):631–639. [DOI] [PubMed] [Google Scholar]
- 61. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. [DOI] [PubMed] [Google Scholar]
- 62. Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30(7):1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gomez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41(1):17–24. [DOI] [PubMed] [Google Scholar]
- 64. Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol. 1999;58(4):376–388. [DOI] [PubMed] [Google Scholar]
- 65. Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31(8):1275–1283. [DOI] [PubMed] [Google Scholar]
- 66. Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–1500. [DOI] [PubMed] [Google Scholar]
- 67. Rodrigue KM, Kennedy KM, Park DC. Beta-amyloid deposition and the aging brain. Neuropsychol Rev. 2009;19(4):436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Neuropathology Group. Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet. 2001;357(9251):169–175. [DOI] [PubMed] [Google Scholar]
- 69. Velliquette RA, O’Connor T, Vassar R. Energy inhibition elevates beta secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J Neurosci. 2005;25(47):10874–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sun X, Bromley-Brits K, Song W. Regulation of β-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer's disease. J Neurochem. 2012;120(suppl 1):62–70. [DOI] [PubMed] [Google Scholar]
- 71. Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid-{beta}. Am J Pathol. 2010;177(1):300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Drzezga A, Lautenschlager N, Siebner H, et al. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer's disease: a PET follow-up study. Eur J Nucl Med Mol Imaging. 2003;30(8):1104–1113. [DOI] [PubMed] [Google Scholar]
- 73. Shaffer JL, Petrella JR, Sheldon FC, et al. Alzheimer’s Disease Neuroimaging Initiative. Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology. 2013;266(2):583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Meier-Ruge WA, Bertoni-Freddari C. Pathogenesis of decreased glucose turnover and oxidative phosphorylation in ischemic and trauma-induced dementia of the Alzheimer type. Ann N Y Acad Sci. 1997;826:229–241. [DOI] [PubMed] [Google Scholar]
- 75. Toussaint PJ, Perlbarg V, Bellec P, et al. Benali, for the Alzheimer's Disease Neuroimaging Initiative. Resting state FDG-PET functional connectivity as an early biomarker of Alzheimer's disease using conjoint univariate and independent component analyses. Neuroimage. 2012;63(2):936–946. [DOI] [PubMed] [Google Scholar]
- 76. Küntzelmann A, Guenther T, Haberkorn U, et al. Impaired cerebral glucose metabolism in prodromal Alzheimer's disease differs by regional intensity normalization. Neurosci Lett. 2013;534:12–17. [DOI] [PubMed] [Google Scholar]
- 77. Wolk DA, Detre JA. Arterial spin labeling MRI: an emerging biomarker for Alzheimer's disease and other neurodegenerative conditions. Curr Opin Neurol. 2012;25(4):421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Musiek ES, Chen Y, Korczykowski M, et al. Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer’s disease. Alzheimers Dement. 2012;8(1):51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Paulson OB, Hasselbalch SG, Rostrup E, Knudsen GM, Pelligrino D. Cerebral blood flow response to functional activation. J Cereb Blood Flow Metab. 2010;30(1):2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Buxton RB, Frank LR. A model for the coupling between cerebral blood flow and oxygen metabolism during neural stimulation. J Cereb Blood Flow Metab. 1997;17(1):64–72. [DOI] [PubMed] [Google Scholar]
- 81. Rocher AB, Chapon F, Blaizot X, Baron JC, Chavoix C. Resting-state brain glucose utilization as measured by PET is directly related to regional synaptophysin levels: a study in baboons. Neuroimage. 2004;20(3):1894–1898. [DOI] [PubMed] [Google Scholar]
- 82. Mosconi L, Tsui WH, De Santi S, et al. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology. 2005;64(11):1860–1867. [DOI] [PubMed] [Google Scholar]
- 83. Okonkwo OC, Xu G, Oh JM, et al. Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer's disease [published online December 12, 2012]. Cereb Cortex. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mosconi L, Mistur R, Switalski R, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2009;36(5):811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Haroutunian V, Hoffman LB, Beeri MS. Is there a neuropathology difference between mild cognitive impairment and dementia? Dialogues Clin Neurosci. 2009;11(12):171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wolf H, Hensel A, Kruggel F, et al. Structural correlates of mild cognitive impairment. Neurobiol Aging. 2004;25(7):913–924. [DOI] [PubMed] [Google Scholar]
- 87. Ruitenberg A, den Heijer T, Bakker SL, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005;57(6):789–794. [DOI] [PubMed] [Google Scholar]
- 88. de la Torre JC. Critical threshold cerebral hypoperfusion causes Alzheimer's disease. Acta Neuropathol. 1999;98(1):1–8. [DOI] [PubMed] [Google Scholar]
- 89. Mazza M, Marano G, Traversi G, Bria P, Mazza S. Primary cerebral blood flow deficiency and Alzheimer’s disease: shadows and lights. J Alzheimers Dis. 2011;23(3):375–389. [DOI] [PubMed] [Google Scholar]
- 90. Roher AE, Debbins JP, Malek-Ahmadi M, et al. Cerebral blood flow in Alzheimer's disease. Vasc Health Risk Manag. 2012;8:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kalaria RN, Akinyemi R, Ihara M. Does vascular pathology contribute to Alzheimer changes? J Neurol Sci. 2012;322(1-2):141–147. [DOI] [PubMed] [Google Scholar]
- 92. Park KW, Yoon HJ, Kang DY, Kim BC, Kim S, Kim JW. Regional cerebral blood flow differences in patients with mild cognitive impairment between those who did and did not develop Alzheimer's disease. Psychiatry Res. 2012;203(2-3):201–206. [DOI] [PubMed] [Google Scholar]
- 93. Chao LL, Pa J, Duarte A, et al. Patterns of cerebral hypoperfusion in amnestic and dysexecutive MCI. Alzheimer Dis Assoc Disord. 2009;23(3):245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Waldstein SR, Rice SC, Thayer JF, Najjar SS, Scuteri A, Zonderman AB. Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension. 2008;51(1):99–104. [DOI] [PubMed] [Google Scholar]
- 95. Inoue K, Meguro K, Akanuma K, Meguro M, Yamaguchi S, Fukuda H. Impaired memory and executive function associated with decreased medial temporal and prefrontal blood flow in Clinical Dementia Rating 0.5 status: the Osaki-Tajiri project. Psychogeriatrics. 2012;12(1):27–33. [DOI] [PubMed] [Google Scholar]
- 96. Clerici F, Caracciolo B, Cova I, et al. Does vascular burden contribute to the progression of mild cognitive impairment to dementia? Dement Geriatr Cogn Disord. 2012;34(3-4):235–243. [DOI] [PubMed] [Google Scholar]
- 97. Silvestrini M, Viticchi G, Altamura C, Luzzi S, Balucani C, Vernieri F. Cerebrovascular assessment for the risk prediction of Alzheimer's disease. J Alzheimers Dis. 2012;32(3):689–698. [DOI] [PubMed] [Google Scholar]
- 98. Richard F, Pasquier F. Can the treatment of vascular risk factors slow cognitive decline in Alzheimer's disease patients? J Alzheimers Dis. 2012;32(3):765–772. [DOI] [PubMed] [Google Scholar]
- 99. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42(1):85–94. [DOI] [PubMed] [Google Scholar]