

ABSTRACT
Streptococcus pneumoniae is a common human nasopharyngeal commensal colonizing 10% to 40% of healthy individuals, depending on age. Despite a low invasive disease rate, widespread carriage ensures that infection occurs often enough to make S. pneumoniae a leading bacterial cause of respiratory disease worldwide. However, the mechanisms behind transition from asymptomatic colonization to dissemination and disease in otherwise sterile sites remain poorly understood but are epidemiologically strongly linked to infection with respiratory viruses. In this report, we show that infection with influenza A virus and treatment with the resulting host signals (febrile-range temperatures, norepinephrine, extracytoplasmic ATP, and increased nutrient availability) induce the release of bacteria from biofilms in a newly developed biofilm model on live epithelial cells both in vitro and during in vivo colonization. These dispersed bacteria have distinct phenotypic properties different from those of both biofilm and broth-grown, planktonic bacteria, with the dispersed population showing differential virulence gene expression characteristics resulting in a significantly increased ability to disseminate and cause infection of otherwise sterile sites, such as the middle ear, lungs, and bloodstream. The results offer novel and important insights into the role of interkingdom signaling between microbe and host during biofilm dispersion and transition to acute disease.
IMPORTANCE
This report addresses the mechanisms involved in transition from pneumococcal asymptomatic colonization to disease. In this study, we determined that changes in the nasopharyngeal environment result in the release of bacteria from colonizing biofilms with a gene expression and virulence phenotype different not only from that of colonizing biofilm bacteria but also from that of the broth-grown planktonic bacteria commonly used for pathogenesis studies. The work importantly also identifies specific host factors responsible for the release of bacteria and their changed phenotype. We show that these interkingdom signals are recognized by bacteria and are induced by influenza virus infection, which is epidemiologically strongly associated with transition to secondary pneumococcal disease. As virus infection is a common inducer of transition to disease among species occupying the nasopharynx, the results of this study may provide a basis for better understanding of the signals involved in the transition from colonization to disease in the human nasopharynx.
Introduction
Nasopharyngeal colonization with Streptococcus pneumoniae (pneumococcus) is a frequent finding in healthy individuals and begins within the first few weeks to months of life. By the age of 2 years, >95% of children have been colonized with individual serotypes that persist for weeks or months (1), and at any given time, 25% to 40% of children under the age of 5 years (2, 3) and 8% to 15% of adults are colonized (4, 5). Longitudinal studies have demonstrated that nasopharyngeal colonization is a necessary—but not sufficient—step in the pathogenesis of pneumococcal disease (6, 7). Nevertheless, despite a low attack rate of between 3 and 34 cases of invasive pneumococcal disease per 100,000 acquisitions (8, 9), colonization is widespread enough that disease burden caused by the pneumococcus remains a leading cause of acute otitis media (AOM), pneumonia, sepsis, meningitis, and death in children and in elderly and immunocompromised individuals worldwide (10).
We and others have demonstrated that asymptomatic pneumococcal colonization in the nasopharynx predominantly occurs within complex multicellular biofilm communities (11, 12), and we have recently shown that key features of the nasopharyngeal environment, such as interaction with epithelial cells, a temperature of 34°C, and limited nutrient availability, are required in order for in vitro models to recapitulate the in vivo biofilm phenotype (12, 13). Pneumococci grown as biofilms in vitro are hyperadhesive and readily colonize the murine nasopharynx but show attenuated virulence in models of invasive disease (14). Pneumococci isolated from the bloodstream and sputum of patients and animals instead appear to exist exclusively as diplococci (15). Despite the high disease burden, little is known about how bacteria transition from asymptomatic carriage within colonizing biofilm communities to planktonic diplococci capable of causing invasive disease.
Clinical, epidemiological, and experimental data suggest that the transition from nasopharyngeal colonization to invasive disease is highly associated with preceding or concomitant virus infections (16–23). The implementation of more sensitive, PCR-based assays has documented viral infection in the nasopharyngeal secretions from approximately 90% of children with AOM (24). Viral circulation has been associated with invasive pneumococcal disease in both children (25) and adults (26, 27), where bacterial strains detected in middle ear fluids, lungs, or blood of patients with acute disease are genetically identical to those isolated from the nasopharynx. Investigations in infant mice colonized with S. pneumoniae support this hypothesis, demonstrating that secondary infection with influenza A virus (IAV) increases the nasopharyngeal pneumococcal burden and leads to subsequent dissemination to the lower respiratory tract (28). This is supported by a recent survey of children that demonstrated that viral infection is temporally associated with an approximately 15-fold increase in the number of pneumococci detected in nasal cultures (29), suggesting that virus infections augment growth of bacteria or increased dissociation from the nasopharyngeal tissue or both.
It is clear that multiple mechanisms of viral and bacterial synergism exist. Viral presence can predispose the respiratory niche to bacterial invasion by increasing bacterial adhesion receptors, damaging the integrity of the epithelial barrier, and inducing a decrease in mucocilliary clearance as well as virus-induced immunosuppression (reviewed in reference 23). This is accompanied by activation of the sympathetic nervous system and increases in proinflammatory cytokines (30). The localized inflammatory response is characterized by injury of host epithelial cells resulting in the release of ATP and nutrients such as glucose and sialic acid in secretions (6) and may be manifested in the host by whole-body hyperthermia (fever). Importantly, environmental conditions are known to influence the development of biofilm communities and play a critical role in the precise mobilization of virulence determinants (14, 31). However, while dysregulated control of nasopharyngeal bacterial commensals has been hypothesized to be a likely part of this transition to disease, few studies have investigated this aspect.
The purpose of the present study was therefore to specifically investigate the signals and events that influence pneumococcal variation, promoting biofilm dispersal and egress and turning a passive nasal “commensal” into a destructive opportunistic pathogen. During asymptomatic carriage, biofilms can be detected over the respiratory epithelium that can persist for weeks to months with limited damage to the host. However, in laboratory assays, planktonic pneumococci rapidly invade and kill human respiratory epithelial cells (HRECs) (32) and no S. pneumoniae biofilm model has yet been developed where pneumococci are able to coexist with living cultures of human respiratory cells for more than 24 h (12, 33). We therefore developed a novel biofilm model on a live epithelial substratum to allow the study of host-pathogen interactions over time in vitro. Using this model and in vivo colonization in animals, we show that transition from colonization to disease is the result of changes in the colonizing environment induced by virus infection or other events leading to cell damage, diminished immunity, or increased inflammation in the airways. These changes lead to release of host molecules that can act as interkingdom signals to induce active dispersal of bacteria from colonizing biofilms, resulting in a released population with an increased ability to disseminate to and establish infection in otherwise sterile sites.
RESULTS
Development of a static biofilm model with living cultures of HRECs.
To study the role of virus infection in vitro, we needed to develop a model system to allow biofilm formation on live epithelial cells for prolonged periods of time. This was accomplished by first forming pneumococcal biofilms over prefixed NCI-H292 HRECs for 48 h at the nasopharyngeal temperature of 34°C (34), as described previously (12, 13), and then transferring these bacterial biofilm cells to live HRECs. We used both the common D39 Avery strain and the EF3030 clinical otitis media isolate that makes more dense biofilms in vitro, colonizes the nasopharynx more effectively than D39, but is less virulent both in the lungs and the bloodstream. Biofilm biomass increased steadily during the first 48 h of biofilm formation over prefixed HRECs for both strains and remained stable over the next 48 h following transfer to live HRECs (Fig. 1A). After transfer of biofilm bacteria from prefixed epithelia to live epithelia, biofilms regained biofilm-specific antimicrobial resistance within 24 h (Fig. 1B) and could be maintained on the live epithelium without substantial damage to or death of epithelial cells for up to 72 h (see Fig. 3E). Confocal imaging showed that at 48 h after biofilm transplantation to live HRECs, the biofilm covers nearly the entire surface of the infected epithelia, with no bacteria internalized into the live cells (Fig. 1C and D). Membrane and nucleus staining confirmed that biofilm-associated HRECs remained morphologically similar to noninfected cells (Fig. 1D).
FIG 1 .

Establishment of a pneumococcal biofilm model on live epithelial cells. S. pneumoniae D39 or EF3030 bacteria were seeded on paraformaldehyde-fixed HRECs. At 48 h, biofilms were transplanted to live HRECs. (A and B) Biofilm biomass (A) and sensitivity to gentamicin (500 µg/ml) (B) were measured by viable counts over time. Green lines indicate bacterial biofilms on fixed epithelia, and blue lines represent the bacteria after transplantation to live epithelial cells. (C) Representative three-dimensional (3D) reconstruction of confocal image of a D39 biofilm at 48 h posttransplantation onto live HREC layers. Bacteria are stained red using Baclight Red, and epithelia were stained with 488-conjugated concanavalin A (green) and their nuclei with DAPI (blue). The numbers represent distances in micrometers. (D) Representative image from the midcell confocal section view of a D39 biofilm sample after 48 h on live HRECs showing no bacterial invasion. HRECs were stained with anti-secretory component (SC) antibody (red), nuclei were stained with DAPI (blue), and pneumococci were stained with anti-PspA antibody (green).
FIG 3 .

Characterization of pneumococcal populations for gene regulation, adherence, invasion, toxicity, and cytokine induction from exposed HRECs. Biofilm and dispersed and planktonic pneumococcal populations show distinct gene expression profiles of key virulence genes (P < 0.05 for all comparisons using 2-way analysis of variance [ANOVA]) (A) and differences in transparent- and opaque-phase states (*** = P < 0.001 using 1-way ANOVA). (C to F) The populations further differed in their ability to adhere to (C), invade (D), and kill (E) HRECs (** = P < 0.01 for all comparisons using 1-way ANOVA) and induce different levels of key cytokines involved in proinflammatory responses from the exposed HRECs (* = P < 0.05 using 2-way ANOVA) (F). Each experiment was repeated at least three times in duplicate. ns, nonsignificant.
Signals promoting biofilm dispersal in vitro.
Considering the clear association between respiratory virus infection and the development of pneumococcal disease, we used our epithelial biofilm models to investigate several potential signals for biofilm dispersal. We first investigated the impact of IAV infection on biofilm homeostasis using live epithelial cells as a substratum. At 24 h after IAV infection of HRECs, no notable cytopathic changes were found compared with control cells. At 48 to 72 h after virus infection, rounding of the cells, large nuclei, and prominent nucleoli were observed and several cells revealed cellular fusion, a phenomenon that became more manifest over the next 2 days, leading to cell detachment (data not shown). However, IAV infection of HRECs was accompanied by significantly increased ATP concentrations in cell culture supernatant already at 24 h postinoculation (Fig. 2A). This time point was used for the bacterial experiments, as host signals were induced without potential release of bacteria due to shedding of cells taking place.
FIG 2 .
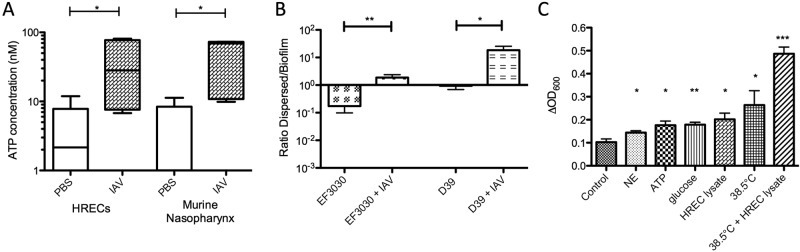
Dispersion of biofilm bacteria by virus infection and host factors. (A) The ATP concentrations in the supernatant of HRECs infected with influenza A virus (IAV) or in the nasopharyngeal lavage fluid recovered from mice 24 h after IAV infection were determined. (B) The impact of IAV infection on the ratio of bacteria in the supernatant (dispersed) to epithelium-associated bacteria (biofilm) 48 h after biofilm transplant to live HRECs was measured. (C) The change in the optical density at 600 nm (ΔOD600) of biofilm supernatant was measured 2 h after application of the indicated stimuli (NE = norepinephrine). The change in optical density correlated well with the number of CFU obtained from bacterial growth of the same supernatants. For each analysis, three independent assays were performed in duplicate. Statistical analysis was performed using Student’s t test (* = P < 0.05, ** = P < 0.01, *** = P < 0.001).
To test the impact of IAV infection on pneumococcal biofilm homeostasis, we transferred preformed biofilms made from both EF3030 and D39 pneumococci to a live epithelium and allowed 24 h for the biofilm to stabilize and reorganize before infecting the epithelium underlying the pneumococcal biofilm with IAV or buffer. At 24 h after IAV infection, the total bacterial load in each well was the same as the load in wells where the cells were not infected with virus; however, approximately 10-fold-more bacteria were present in the supernatant than in the biofilm communities attached to the virus-infected epithelium, shown by a significantly increased ratio of dispersed bacteria to biofilm bacteria (Fig. 2B).
To test the direct effect of host agents potentially associated with virus infection on biofilm integrity, application of host agents was performed on biofilms formed on fixed epithelia to avoid any potential involvement of host cell-mediated responses in biofilm dispersion. Exogenous application of norepinephrine (NE), ATP, glucose, or a complete HREC cell lysate (representing cell damage) added to 48-h biofilms induced a significant and rapid egress of cells from surface-attached biofilm communities to the supernatant, where smaller aggregates (predominantly diplococci) were observed microscopically. This increase of bacteria in the supernatant was not merely a result of outgrowth of bacteria already in the supernatant, as aliquots of supernatant removed from the biofilm culture and treated identically to the biofilm did not result in similar increases in bacterial numbers. Similarly, treatment with medium containing 2 mM of the potential dispersant calcium did not produce any detectable egress of bacteria from the biofilm (not shown). This indicated that NE, ATP, glucose, and cell lysate induced active biofilm dispersal and release of diplococci into the supernatant. Furthermore, temperature variation (simulating fever) of biofilms formed on fixed epithelia produced similar results, with the greatest effects seen at a temperature of 38.5°C applied in combination with a complete HREC cell lysate, which would mimic the host response to a febrile, symptomatic IAV infection (Fig. 2C). Combined, these results show that specific host molecules and environmental changes associated with virus infection are sensed by biofilm bacteria and activate biofilm dispersal.
Dispersed bacteria represent a distinct population of bacteria different from biofilm and broth-grown, planktonic cells.
Previous work has demonstrated that pneumococci grown in biofilms significantly downregulate a number of important virulence genes compared with broth-grown bacteria (14). To better understand the phenotype of biofilm-dispersed bacteria, we compared pneumococcal gene expression of the bacteria actively dispersed from the biofilm on fixed epithelia in response to 2 h at increased temperature (37°C) with both planktonic, broth-grown cultures and biofilm bacteria grown on fixed epithelial cells for 48 h. Consistent with previous reports (13, 14), we found that genes involved in competence were upregulated during biofilm formation (P < 0.05 compared with the dispersed and planktonic populations), while genes involved in virulence, such as the cps cassette for capsule production, and genes encoding pneumolysin, the adhesin PavA, and the licD2 locus involved in promoting the opaque phenotype were significantly downregulated during biofilm growth compared with both dispersed and planktonic, broth-grown bacteria (Fig. 3A). Of special interest was that dispersed bacteria not only showed significantly higher expression of these genes than biofilm bacteria but also showed significantly higher expression of lytA, licD2, and pavA (P < 0.05 for all genes) than broth-grown, planktonic controls (Fig. 3A), indicating that planktonic bacteria originating from broth cultures and from biofilm release are phenotypically distinct.
S. pneumoniae undergoes phase variation, alternating between a transparent phase with low capsule expression and high cell wall teichoic acid expression and an opaque phase with high capsule expression and low teichoic acid expression (35). During nasopharyngeal colonization, the transparent phase predominates, allowing increased adherence to host cells (36). In contrast, bacteria isolated from sites of acute pneumococcal disease show an almost exclusively opaque phenotype. To test whether the increased expression of licD2 seen in the dispersed population corresponded with an increase in the opaque phenotype, we used trans-oblique illumination to determine the phase of all three subpopulations. In both the EF3030 and D39 strains, colonies grown planktonically in broth were predominantly opaque, while colonies isolated from biofilms and grown on plates were predominantly transparent. Cells actively dispersed from biofilms showed consistently higher percentages of opaque colonies compared with either the broth-grown planktonic cultures (P < 0.05 for D39 pneumococci) or their biofilm counterparts (P < 0.01 for both strains; Fig. 3B).
The variation in opacity and expression of virulence determinants correlated well with adherence, invasion, and toxicity to HRECs. Both EF3030 and D39 biofilm bacteria showed increased adherence to HRECs compared with dispersed or broth-grown bacteria (P < 0.01 compared with both dispersed and planktonic pneumococci for EF3030; Fig. 3C). Furthermore, dispersed bacteria showed significantly higher internalization than both planktonic and biofilm pneumococci (P < 0.05 for both strains and both comparisons; Fig. 3D), with the more virulent D39 strain showing higher levels of internalization than the EF3030 strain. The internalization translated into toxicity of dispersed pneumococci for HRECs over the first 18 h that was rapid and significantly higher than that of planktonic, broth-grown bacteria and dramatically higher than that of the biofilms from which they were liberated over the entire time course of the experiment (Fig. 3E). The more virulent phenotype in vitro for dispersed cells was supported by inflammatory cytokine levels that were significantly higher than those seen upon exposure to their biofilm counterparts or to broth-grown, planktonic bacteria (Fig. 3F). These results confirmed the expression data indicating that the dispersed bacterial population was distinct not only from the biofilm bacteria but also from conventional, broth-grown bacteria showing less adherence, more invasion, and higher toxicity to HRECs.
In vivo phenotype of planktonic, dispersed, and biofilm populations in a murine colonization model.
Having used in vitro models to confirm the enhanced toxicity and inflammatory potential of bacterial cells dispersed from mature biofilms, we next tested their in vivo phenotype using established mouse models for pneumococcal colonization and disease. Intranasal inoculation of the EF3030 strain showed that all three populations colonized the nasopharyngeal tissue, but with a distinct propensity for dispersed bacteria to colonize less tightly, as ~12.5% of total nasal colonization was recovered in the lavage fluid compared with 1.7% for broth-grown and 0.15% for biofilm bacteria (P < 0.05 for both comparisons; Fig. 4A). In analogy, dispersed EF3030 bacteria inoculated in the nares of mice disseminated to the lungs in large numbers and grew dramatically, whereas neither planktonic nor biofilm pneumococci showed a significant bacterial presence in the lungs (P < 0.001; Fig. 4A). Furthermore, EF3030 (a middle ear clinical isolate) was detected in the middle ears. All populations migrated to the middle ears; however, dispersed bacteria showed a bacterial burden that was approximately 2 log10 higher (P < 0.05) than that seen with the other bacterial populations (Fig. 4A).
FIG 4 .

Colonization and dissemination of pneumococcal populations after intranasal inoculation, aspiration inoculation, or intraperitoneal inoculation. (A and B) Individual 6-week-old female BALB/cByJ mice were inoculated with biofilm (designated “B”) or dispersed (designated “D”) or broth-grown, planktonic (designated “P”) populations of EF3030 (A) and D39 (B) pneumococci intranasally without anesthesia to determine nasopharyngeal colonization and dissemination from the nasopharynx to the lungs and middle ears. Additionally, the bacterial burden in nasopharyngeal (NP) lavage fluid was determined. (C and D) Mice were furthermore inoculated with EF3030 (C) and D39 (D) pneumococci intranasally after anesthesia in an aspiration pneumonia model, and the bacterial burden in the lung tissue and dissemination to the bloodstream were determined by plate counts. (E and F) Finally, each bacterial population of EF3030 (E) and D39 (F) pneumococci was injected intraperitoneally to determine the respective levels of virulence upon reaching the vascular compartment. For both nonanesthetized and anesthetized intranasal challenges, nasal lavage fluids and all tissues were collected at 48 h postinfection. For intraperitoneal challenge, samples were collected at 24 h postinfection. Each dot in the graphs represents an individual mouse. An “X” represents a mouse that became moribund and required euthanasia before the end of the experiment. Each experiment included at least 6 mice. Statistical analysis was performed using one-way ANOVA (* = P < 0.05, ** = P < 0.01, and *** = P < 0.001, indicating significant differences between the populations).
A histological examination of the nasal epithelium, middle ears, and lungs of mice 7 days after intranasal colonization showed that colonization with dispersed bacteria led to pronounced denudation of the nasal epithelium despite similar levels of tissue colonization of the populations (Fig. 5, row 1). Furthermore, we found large inflammatory infiltrates in the middle ear cavity and lungs. In contrast, colonization with biofilm bacteria showed the presence of shorter, intact cilia in the lining of the nasal epithelium, with no inflammatory infiltration of the lung or middle ears, which correlated well with the low bacterial load in these tissues. Inoculation with broth-grown planktonic bacteria led to a mixed phenotype, with localized areas of epithelial denudation in the nares, minimal inflammatory presence in the middle ear, and small areas of perivascular inflammation in the lungs (Fig. 5, rows 1 to 3).
FIG 5 .

Histological representation of mouse tissues infected with various pneumococcal populations. Representative histological images of nasal epithelium, middle ear space, and lungs 7 days following nonanesthetized intranasal challenge (rows 1 to 3) with PBS (mock-infected control), biofilm, or dispersed or planktonic populations. Row 4 shows histological sections of the lungs 48 h following intratracheal aspiration of these same bacterial populations. Tissues were stained with hematoxylin and eosin and examined microscopically at magnifications of ×400 for row 1 and ×200 for rows 2 to 4. The sections are labeled with relevant structures (LI, leukocyte infiltrate; TM, tympanic membrane; SM, stapes muscle; ME, middle ear cavity; Spn, S. pneumoniae).
Colonization experiments with the D39 mouse-adapted strain showed mostly similar results. Biofilm pneumococci showed a distinct tissue tropism for the nasopharyngeal tissue, colonizing at a higher bacterial burden at 48 h compared with both dispersed and broth-grown populations (P < 0.05; Fig. 4B). All populations were detected in equal amounts in the nasal lavage fluid (Fig. 4B). However, as dispersed D39 pneumococci were present at a lower burden in the nasal tissue, their presence in the lavage fluid represented a significantly greater fraction of the total bacterial population in the nasopharynx, with ~10% of bacteria loosely associated with the tissue compared with the biofilm or planktonic populations, where ~0.1% to 1% of colonizing bacteria were recovered in the lavage fluid. Following intranasal inoculation, only one of the mice inoculated with the planktonic population and one of the mice inoculated with the biofilm population had detectable bacteria in the lungs after 48 h (Fig. 4B), whereas dispersed bacteria rapidly migrated to the lungs and established a productive infection. Interestingly, no detectable amounts of D39 bacteria of any population were recovered from the middle ears (Fig. 4B). Histological analysis of the nasal epithelium and lungs of D39-inoculated mice showed results very similar to those described above for EF3030 (not shown), with the exception that none of the populations were identified in the middle ears.
In general, the results confirm that the phenotype of the dispersed bacteria showed that they were more toxic and had a propensity to disseminate from the nasopharynx to the lungs and middle ears at a level that was higher than that of their broth-grown planktonic counterparts and much higher than that of the biofilm bacteria.
In vivo phenotype of planktonic, dispersed, and biofilm populations in a murine pneumonia model.
As intranasal inoculation resulted in higher levels of dissemination to sterile sites such as the lungs by the dispersed phenotype, we wanted to address the virulence potential in the lungs by inoculating equal numbers of all of the bacterial populations through aspiration. Mice given EF3030 biofilm bacteria showed low levels of bacteria in the lungs after 48 h (Fig. 4C) that induced only minimal recruitment of inflammatory cells with localized areas of perivascular inflammation (Fig. 5, row 4) and resulted in no dissemination to the bloodstream. Mice given D39 biofilm bacteria showed complete or partial clearance of bacteria by 48 h, with only 3 of the challenged mice showing dissemination with low bacterial numbers to the bloodstream (Fig. 4D). In contrast, challenge with cells dispersed from the same biofilms by exposure to 37°C for 2 h led to high bacterial burdens in the lungs and 3/6 mice challenged with EF3030 and all mice challenged with D39 produced hematogenous dissemination.
Histological examination of the lungs of mice directly infected with dispersed bacteria showed the greatest influx of inflammatory cells, with extensive vasculitis and perivascular, peribronchiolar, and interstitial inflammation along with areas of intra-alveolar hemorrhage and edema (Fig. 5, row 4). Exposure to broth-grown planktonic bacteria led to a significantly lower bacterial load in the lungs (Fig. 4D) that resulted in moderate inflammatory infiltration (Fig. 5) and limited hematogenous dissemination, with 1/6 mice showing bacteremia using the EF3030 strain and 4/6 using D39 bacteria (Fig. 4D).
Biofilm populations show attenuated virulence and biofilm-dispersed pneumococci are hypervirulent during bacteremia after intraperitoneal challenge.
To determine if the role played by dispersed bacteria was specific to tissue infections, we injected organisms directly into the peritoneum to bypass the respiratory tract. After injection of between 2 ×105 and 5 ×105 CFU/mouse of the various EF3030 populations, 2 mice infected with dispersed EF3030 became moribund before 24 h had passed and 5/6 mice were bacteremic despite the traditional view that EF3030 is a nonbacteremic strain (37). In contrast, all animals inoculated with planktonic or biofilm EF3030 cultures were healthy at the end of the experiment at 24 h and bacteria had been completely cleared (Fig. 4E).
Using the same inoculum of D39 bacteria, a strain that is classically more virulent in mouse septicemia, mice infected with dispersed D39 populations rapidly became moribund, with the first mouse sacrificed by 14 h postinfection and 6/9 mice requiring euthanasia before the end of the experiment at 24 h. This phenotype was much more virulent that that observed when broth-grown bacteria were used. Inoculation with these bacteria resulted in no moribund mice within the first 24 h, although the majority of the mice had the same bacterial burden in the blood as mice infected with dispersed bacteria. None of the biofilm-inoculated animals were moribund at 24 h, and these mice showed a significantly lower bacterial load than the two planktonic groups except for a significant bacterial load in 2/9 mice (Fig. 4E). These results suggest that the dispersed phenotype may play a critical role in the survival and growth of the pneumococcus during the development of acute infection in the vascular compartment.
Influenza virus infection and dispersing stimuli induce transition to disease in vivo.
Considering the high carriage rate of pneumococci, it is not unlikely that a substantial proportion of those who contract IAV are already asymptomatically colonized by S. pneumoniae. As colonization always precedes dissemination to other sites (38), changes in the upper respiratory tract resulting from IAV infection likely alter the colonizing microenvironment of the nasopharynx, affecting preexisting pneumococcal biofilms.
To assess the impact of IAV infection on asymptomatic pneumococcal colonization, we used the established carriage model described above. In the absence of virus, mice remain colonized with D39 or EF3030 for 1 to 3 weeks without infection of the lower respiratory tract or the development of bacteremia (data not shown). We induced stable asymptomatic carriage with either biofilm-grown EF3030 or D39. At 48 h postinoculation, mice were intranasally inoculated with 40 PFU of IAV. Infection with IAV confirmed the results of many human epidemiological studies and showed that the mice had a marked level of viral pneumonia on day 1 post-IAV inoculation, showing increased lethargy, huddling, and ruffled fur. We found that 24 h after IAV infection, both of the colonizing pneumococcal strains had successfully disseminated to both the lungs and middle ears, whereas the non-virus-infected group, inoculated with phosphate-buffered saline (PBS), showed no presence of bacteria in the lungs (Fig. 6A and B). Only a limited bacterial presence was observed for the EF3030 clinical otitis strain in the middle ear. To determine the fate of disseminated bacteria over time, we followed the mice over 5 days post-IAV inoculation. EF3030 bacteria continuously colonized the nasopharynx at a high level over all 5 days in both the presence and the absence of virus (Fig. 6A and C), but dissemination to the lungs increased over time to induce pneumonia and to the middle ears that persisted to induce acute otitis media only in the presence of IAV. The D39 strain, being a more invasive strain, decreased in colonization over time (Fig. 6B and D). And yet, following damage to the lungs from the viral pneumonia, D39 pneumococci that actively disseminated to the lung increased their bacterial burden over time and substantial bacterial pneumonia was seen on day 5 post-IAV infection (Fig. 6B and D). Although the D39 strain also disseminated to the middle ear by day 1, it was slowly cleared over time, suggesting that this strain is less otogenic, in agreement with the results shown in Fig. 4B.
FIG 6 .

Bacterial dissemination of mice stably colonized with pneumococci and challenged with influenza A virus. Bacterial burden in tissues and nasopharyngeal lavage fluid was measured for individual 6-week-old female BALB/cByJ mice colonized intranasally without anesthesia with EF3030 (A and C) or D39 (B and D) biofilm bacteria at 1 (A and B) or 5 (C and D) days postinfection with IAV. Each dot represents an individual mouse. Statistical analysis was performed using Student’s t test. * = P < 0.05, ** = P < 0.01, *** = P < 0.001.
To further explore the mechanism involved in transition from colonization to disease, we used this same colonization model to assess the effect of host-derived interkingdom signals seen during viral infection that were found to disperse pneumococcal biofilms in vitro as described above. Infection of mice with IAV induced a significant release of ATP in nasopharyngeal secretions, suggesting this to be an important host signaling molecule (Fig. 2A). As in our in vitro experiments, intranasal application of ATP, NE, or glucose or a brief (4-h) exposure of the mice to febrile-range hyperthermia (FRH) led to the dissemination of both EF3030 and D39 pneumococci from the nasopharynx. There was a moderate increase in the percentage of nasal bacteria found in the lavage fluid compared to the tissue-associated population. This was accompanied by the dissemination of bacteria to the middle ear or lungs within 24 h (Fig. 7). No such bacterial invasion of the lower respiratory tract was detected after intranasal administration of PBS. All host signals that induced dispersion effectively caused dissemination to the lungs for both strains. For dissemination to the middle ear, febrile-range hyperthermia and norepinephrine were especially effective in causing dissemination and ATP worked effectively also for the EF3030 otitis media isolate (Fig. 7A).
FIG 7 .

Bacterial dissemination of mice stably colonized with pneumococci and challenged with dispersants. The bacterial burden in tissues and nasopharyngeal (NP) lavage fluid was measured for individual 6-week-old female BALB/cByJ mice colonized intranasally without anesthesia with biofilm bacteria from (A) EF3030 or (B) D39 pneumococci for 48 h and then challenged intranasally with 20 µl PBS, 10 mM ATP, 100 nM norepinephrine (NE), or 1 M glucose (Glu) or subjected to 4 h of febrile-range hyperthermia (FRH). Each experiment represents at least three individual experiments with duplicate samples. Statistical analysis was performed using Student’s t test, comparing treatment conditions to PBS control. * = P < 0.05, ** = P < 0.01, *** = P < 0.001.
To confirm the crucial role of activating events leading to specific release of bacteria from the biofilms with increased virulence, we tested the in vivo phenotype of bacteria passively released from EF3030 and D39 biofilms (see Fig. S1 in the supplemental material). Released bacteria from EF3030 biofilm overgrowth inoculated intranasally showed no dissemination to the lungs and only a limited presence in the middle ears that was not significantly greater than that seen during challenge with intact biofilm bacteria as shown in Fig. 4. Similarly, intraperitoneal challenge of this same population resulted in complete clearance within 24 h. Together, these results demonstrate that activation of the biofilm with specific host signaling molecules is required to induce the liberation and dispersion of bacteria that can disseminate to normally sterile sites.
DISCUSSION
A current challenge in the field of pneumococcal pathogenesis is to understand how asymptomatic pneumococcal colonization progresses to dissemination, specific tissue damage, and disease or death of the host. This study therefore focused on better understanding of factors influencing S. pneumoniae biofilm dispersion and its relationship to the onset of pneumococcal disease. We demonstrated that pneumococcal biofilm dispersion is an active process induced by alterations of the nasopharyngeal environment. These data provide one potential explanation of the process by which colonizing pneumococcal biofilms disperse to become virulent pneumococci primed for invasive disease.
Biofilm formation during nasopharyngeal colonization provides pneumococci with a protected sanctuary for sessile cells and a platform for person-to-person transmission and is a prerequisite for the development of invasive disease. However, recent work has shown that large bacterial aggregates are more susceptible to complement-mediated opsonophagocytosis within the vascular compartment (39) and that in vitro-grown biofilms are attenuated for virulence (14). Our results demonstrating that biofilms grown on epithelial cells downregulate important virulence factors, are predominantly in the transparent phase, induce lower cytokine responses in epithelial cells, and are noninvasive and nontoxic to HRECs support these observations. Likewise, our in vivo models suggest that the formation of biofilm structures within the nasopharynx allows increased bacterial adherence and persistence while ensuring the rapid clearance from any normally sterile areas into which they may gain entry such as the lung or vascular compartment, possibly through microaspiration.
Biofilms formed during nasopharyngeal colonization must balance attachment, growth, and eventual dispersion processes within a dynamic nasopharyngeal environment. In both bacteria and fungi, the nutritional status of the environment dictates biofilm dispersal. In general, increased nutrient availability is associated with inhibiting biofilm formation, increasing detachment, and triggering dispersion, and it is possible that other host signals are involved in this regulation (40, 41). Here, we found that pneumococcal cells dispersed from biofilms in response to hyperthermia treatment were more inflammatory and more invasive in both in vitro and in vivo assays, while passive overgrowth and seeding of biofilm bacteria into the supernatant did not result in any phenotypic traits shared with their actively dispersed counterparts but did result in traits similar to those seen with the biofilm parent cultures. Phenotypic analysis showed that heat-dispersed cells were predominantly opaque, with increased capsule production and markedly higher expression of a number of virulence factors required for invasive disease. Trappetti et al. found that opaque broth-grown bacteria, but not transparent-biofilm-derived pneumococci, were able to translocate from the nasopharynx to the lungs and brain of mice (42). These findings support previous reports that human convalescent sera from individuals recovered from pneumococcal pneumonia primarily recognize bacterial proteins in planktonic versus biofilm pneumococci (43). However, given the largely artificial phenotype of planktonic broth-grown bacteria, in aggregate these data suggest that phenotypically altered cells dispersed from biofilms in response to specific environmental cues, and not colonizing biofilm bacteria or cells passively released during normal growth, are more likely to represent the etiologic agents of acute disease.
It is becoming increasingly evident that cell-cell signaling inside the nasopharynx plays a key role in commensal survival, pathogen colonization, and host defense. An important component is interkingdom signaling, or the recognition of eukaryotic signaling molecules by commensal bacteria and pathogens. Changes in the nasopharyngeal microenvironment, including nutrient availability, temperature, ion concentrations, and host signaling molecules capable of triggering biofilm dispersion, can be the result of viruses joining the microbial microflora as a prelude to secondary bacterial infections of the respiratory tract (44). Fever often peaks within 24 h of viral infection onset and is accompanied by other physical signs, including cough, sore throat, headache, and myalgia, and correlates with a significant release, either in nasopharyngeal fluid or in plasma, of a number of cytokines and chemokines (45). IAV infection also activates the sympathetic nervous system, resulting in the release of granules containing neurotransmitters, including the classic stress hormone NE and ATP (30, 46, 47), and ATP may also be released through virus-induced cell damage. A growing body of evidence recognizes both ATP and NE as important interkingdom signaling molecules during a number of different host-pathogen interactions (48). Release of sympathetic neurotransmitters occurs nonsynaptically, and NE diffuses widely in tissues with a half-life on the order of 7 h (49). Likewise, rapid release of extracellular ATP into the airway surface liquid and local microenvironment from distressed or injured eukaryotic cells due to pathogens or other etiological factors functions as an important “danger signal,” activating host inflammatory and immune responses (50, 51). Both low levels of NE and high extracellular concentrations of ATP exhibit activating effects on leukocytes (52, 53). In addition, high extracellular ATP concentrations induce rapid increases in monolayer permeability of pulmonary epithelial and endothelial cells, allowing increased transmigration of immune cells to an area of acute infection (52, 54).
As indicated by our results, the changes of the microenvironment induced by these agents have the paradoxical effect of increasing bacterial dissemination into normally sterile areas. Induction of a febrile response after virus infection correlated with increased concentrations of ATP in the nasopharyngeal lavage fluid and resulted in dispersion of bacteria from bacterial biofilm communities both in vitro and in vivo. Surprisingly, even a limited period of whole-body hyperthermia or a single intranasal application of glucose, NE, or ATP applied to healthy mice with stable asymptomatic nasopharyngeal colonization induced the development of pneumococcal pneumonia and otitis media within 24 h. Although the mechanisms of pneumococcal sensing of host factors are not yet understood, this is an exciting focus for future studies. These data suggest that interkingdom signaling, through the molecules identified in this study as well as other potential host factors and mechanisms, allows communication between respiratory microbes and their hosts, leading bacteria to activate their virulence genes and turning a passive commensal into a destructive pathogen.
Clinical findings support these observations, as an increasing body of data points to enhanced levels of ATP in airway secretions of patients with airway damage and respiratory diseases (reviewed in reference 52) where secondary pneumococcal infections are significantly more common. Therefore, massive dispersion under favorable growth conditions may reflect a bacterial survival strategy whereby S. pneumoniae benefits from the biofilm mode of growth under restrictive environmental conditions imposed by a healthy host but abandons this mode in response to interkingdom signaling and microenvironmental changes that suggest a state of decreased host immunity.
Temperature is another critical and ubiquitous environmental signal that governs the development and virulence of diverse microbial pathogens, including viruses, parasites, and bacteria (55–58). Microbial survival is frequently contingent on initiating appropriate responses to environmental temperature cues—often signaling successful infection of the host. In the host, febrile-range hyperthermia (FRH) enhances serum cytokine levels and other modulators of the acute inflammatory response (59). However, increased levels of tumor necrosis factor alpha (TNF-α) and other immediate proinflammatory cytokines induced both by fever-range hyperthermia and by bacteria actively dispersed from the commensal nasopharyngeal biofilms could be undesirable due to their role in lung injury. In the setting of acute lung injury or infection, FRH augments the development of central pathophysiologic features of respiratory disease, including neutrophil accumulation, loss of endothelial and epithelial barrier function, and epithelial injury (60). Adult respiratory distress syndrome occurs in one-fourth of humans with heat stroke (61), demonstrating that exposure to hyperthermia may in and of itself be sufficient to activate pathways involved in lung injury. Unlike other organisms, such as Streptococcus pyogenes, which produce a variety of tissue-damaging substances, the pneumococcus produces relatively few toxins, with pneumolysin being the principal one. Instead, the pneumococcus causes an intense inflammatory response, and in most organs—including the lungs, middle ear, and meninges—this inflammatory response constitutes the disease.
While animal and cell culture studies clearly establish the importance of fever management, the outcomes of clinical studies addressing this issue are less clear (62). Clinical studies suggest that the effects of fever depend primarily on the severity of the underlying illness and the timing of antipyretic therapy (62). Chiappini and colleagues recently found that parental administration of antipyretics given to children with upper respiratory tract infections as soon as symptoms manifested resulted in a significant decrease in the clinical signs of respiratory infection, including nasal discharge and nontransparent tympanic membranes (63). However, many other studies have found that antipyretic treatment can have detrimental effects, increasing the length of time to recovery in viral illnesses (64) and decreasing survival during sepsis (reviewed in reference 62). Together, these observations suggest that FRH may play an important role in the initial pathogenesis of acute lung injury complicating respiratory infections, including secondary bacterial pneumonia, but may also confer a protective benefit in systemic disease.
In conclusion, we have proposed a novel model for understanding the mechanisms underlying pneumococcal and viral interactions in the respiratory tract. Our findings suggest that synergistic interactions may occur that influence and disturb the natural equilibrium of the microbiota in the nasopharyngeal niche, prompting physiologic changes that lead to the conversion of Streptococcus pneumoniae from a passive colonizing agent to a virulent pathogen primed for invasion. These phenotypically modified dispersed bacteria are primed to take advantage of the virus-induced changes in airway function and epithelial damage to the respiratory tract that prime the upper airway and lung for subsequent bacterial infection. Pneumococcal disease may therefore be the result of both variation on the bacterial level and capitalization on opportunities presented by the host. The insight gained into the pathogenesis of the interaction between influenza virus and pneumococcus may serve as a starting point for understanding the mechanisms underlying the processes seen with other commensal bacteria that have both colonizing and invasive phenotypes.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, Buffalo, NY, and the Veterans Affairs Medical Center, Buffalo, NY. All bacterial inoculations and treatments were performed under conditions designed to minimize any potential suffering of the animals.
Reagents.
Cell culture reagents were from Invitrogen, Carlsbad, CA. Bacterial and cell culture media and reagents were from VWR Inc., Radnor, PA. Chemically defined bacterial growth medium (CDM) was obtained from JRH Biosciences, Lexera, KS. Sheep blood was purchased from BioLink, Inc., Liverpool, NY. All remaining reagents were purchased from Sigma-Aldrich, St. Louis, MO.
Cells and bacterial and virus strains.
NCI-H292 bronchial carcinoma cells (ATCC CCL-1848), referred to here as HRECs, and MDCK cells (ATCC CCL-34) were grown on various surfaces as described previously (65). Pneumococcal strains were grown in a synthetic medium (CDM) as described previously (66). The study used serotype 19F otitis media isolate EF3030 (67) and classical serotype 2 Avery strain D39 (68). IAV strain A/PR8/34 (H3N2) (ATCC VR-777) was used, and titers were determined by plaque assays (69). Adherence and invasion assays were performed as described previously (70).
Determination of ATP release from virus-infected epithelia.
HRECs grown to confluence in 24-well tissue culture plates were infected with 1 × 105 PFU/ml IAV that were allowed to adsorb to the cells for 1 h, after which the virus was removed and 2 ml of fresh cell culture media with 2% fetal bovine serum (FBS) and 1 µg/ml CPCK trypsin (tosyl phenylalanyl chloromethyl ketone-treated trypsin) were added. At 24 h following IAV infection, the cell culture medium was changed to serum-free medium. After 2 h, supernatant was removed from wells and subjected to sterile filtering. Separately, nasopharyngeal lavage fluid from mice infected intranasally for 24 h with 40 PFU of IAV was collected. Levels of ATP release were determined using an ATP determination kit (Invitrogen) according to the manufacturer’s instructions and a Synergy 2 plate reader (Bio-Tek).
Cytokine release after bacterial stimulation of HRECs.
For stimulation experiments, confluent HREC layers were washed twice with serum-free media and then stimulated with pneumococcal populations prepared as described above in serum-free, RPMI 1640 medium. Cultures were incubated for 4 h at 34°C in 5% CO2. After incubation, culture supernatants were collected, centrifuged at 10,000 × g for 5 min, and stored at −80°C. Cytokine contents in the supernatants were determined by a sandwich enzyme-linked immunosorbent assay (ELISA) (Signosis) following the manufacturer’s protocols.
Static biofilm model on prefixed epithelia.
Static pneumococcal biofilms on prefixed epithelia were produced from CDM-grown pneumococci seeded onto confluent HRECs that were prefixed in 4% paraformaldehyde at 34°C as described previously (12). For our live epithelial-biofilm model, 48-h biofilms grown on prefixed epithelia were washed and resuspended by gentle pipetting in fresh antibiotic-free RPMI 1640 media with 2% FBS and then transplanted onto live HREC cultures grown to confluence in 24-well plates. Biofilms were maintained for up to 72 h, with changes of culture media every 4 h. For IAV infection, 24 h after biofilm transplantation, epithelial biofilm cultures were incubated with viruses at a multiplicity of infection of one virus/cell for 1 h. Virus was removed, and fresh media were added as indicated above for ATP determinations.
Confocal microscopy.
Biofilms were prepared on live epithelia as indicated above. Infected cell layers were then fixed in 4% paraformaldehyde. To identify internalized bacteria, cells were washed with PBS and permeabilized with 3% bovine serum albumin–0.1% Triton X-100–PBS, bacteria were labeled with anti-PspA antibody (gift from David Briles, UAB, Birmingham, AL) (1:50 dilution), and cells were labeled with anti-secretory component antibody (1:200 dilution). DAPI (4[prime],6-diamidino-2-phenylindole) (5 µg/ml) was also added. Biofilm bacteria, which could not be washed extensively, were visualized with BacLight Red (Molecular Probes), and epithelial cells were stained with 488-conjugated concanavalin A and DAPI.
Mouse sepsis, pneumonia, and nasopharyngeal colonization model.
For nasopharyngeal colonization experiments, BALB/cByJ mice (Jackson Laboratories, Bar Harbor, ME) were intranasally colonized with 5 × 106 CFU of various bacterial populations prepared in a 20-µl volume as described above and previously (13). Pneumonia was induced using the same inoculum pipetted into the nares of isofluorane-anesthetized mice. Sepsis was induced by intraperitoneal injection with 1 × 105 CFU bacteria. Mice were monitored six times daily for determination of illness and mortality according to indications correlating to huddling, ruffled fur, lethargy, and body temperature. Mice found to be moribund were euthanized. Nasopharyngeal lavage fluid and tissue, lung, and blood samples were collected, and bacterial burden was determined as described previously (66). Tissue homogenate, lavage fluid, and blood were sonicated to ensure dissociation of bacterial aggregates and then serially diluted on tryptic soy agar (TSA)–5% blood agar plates. Histopathological assessment was performed on slides stained with hematoxylin and eosin (H&E) prepared by the University at Buffalo Histology Core Facility.
Where indicated, mice were subjected to febrile-range hyperthermia for 4 h as described previously (59) or treated intranasally with a single dose of 10 mM ATP, 100 nM norepinephrine, or 1 M glucose in a 20-µl volume on day 2 after colonization.
RNA isolation and quantitative RT-PCR (qRT-PCR).
RNA was isolated from PBS-washed CDM-grown planktonic and biofilm samples as well as from dispersed biofilm bacteria. Bacterial pellets were resuspended in 0.5 ml of 0.9% NaCl, 1 ml of RNAprotect (Qiagen, Valencia, CA) was added, and the mixture was incubated at room temperature for 5 min. Cells were then pelleted at 9,000 × g for 2 min at room temperature, and RNA was purified using Qiashredder columns and an RNeasy minikit as described previously (66). Three independent biological samples were collected for each population, and the relative levels of gene expression were analyzed by using the 2–ΔΔCT method (71). The reference gene was 16S, and the primers spanning 100-bp-to-150-bp segments are reported in Table S1 in the supplemental material.
Statistical analysis.
The data were analyzed for statistical significance by a two-tailed Student’s t test or analysis of variance (ANOVA) with correction for multiple groups using Prism 5 software (GraphPad, La Jolla, CA). A P value of <0.05 was considered significant.
SUPPLEMENTAL MATERIAL
Colonization and dissemination of bacteria passively released from pneumococcal biofilms. Data represent tissue tropism of bacteria passively released from EF3030 (A) and D39 (B) biofilms during normal growth after 48 h. Mice were challenged via nonanesthetized intranasal (Colonization) and intraperitoneal (Sepsis) inoculation. For intranasal challenges, nasal lavage fluids and the respective tissues were collected at 48 h postinfection. For intraperitoneal challenge, samples were collected at 24 h postinfection. Bacterial burden was determined by viable counts of the respective fluid or tissue homogenate samples. Statistical analysis of this data, using Student’s t tests, showed no significant differences between bacterial burdens of biofilm bacteria and those reported in the legend to Fig. 4. Download
Oligonucleotides used in qRT-PCR analysis.
ACKNOWLEDGMENTS
The studies were funded by the Department of Microbiology and Immunology, School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY (A.P.H.).
We thank Ryan Reddinger for technical assistance with the mouse experiments.
Footnotes
Citation Marks LR, Davidson BA, Knight PR, Hakansson AP. 2013. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4(4):e00438-13. doi:10.1128/mBio.00438-13
REFERENCES
- 1. Gray BM, Converse GM, III, Dillon HC. 1980. Epidemiologic studies of Streptococcus pneumoniae in infants: acquisition, carriage, and infection during the first 24 months of life. J. Infect. Dis. 142:923–933 [DOI] [PubMed] [Google Scholar]
- 2. Korona-Glowniak I, Malm A. 2012. Characteristics of Streptococcus pneumoniae strains colonizing upper respiratory tract of healthy preschool children in Poland. ScientificWorldJournal 2012:732901. 10.1100/2012/732901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang SS, Hinrichsen VL, Stevenson AE, Rifas-Shiman SL, Kleinman K, Pelton SI, Lipsitch M, Hanage WP, Lee GM, Finkelstein JA. 2009. Continued impact of pneumococcal conjugate vaccine on carriage in young children. Pediatrics 124:e1–e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hussain M, Melegaro A, Pebody RG, George R, Edmunds WJ, Talukdar R, Martin SA, Efstratiou A, Miller E. 2005. A longitudinal household study of Streptococcus pneumoniae nasopharyngeal carriage in a UK setting. Epidemiol. Infect. 133:891–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Millar EV, Watt JP, Bronsdon MA, Dallas J, Reid R, Santosham M, O’Brien KL. 2008. Indirect effect of 7-valent pneumococcal conjugate vaccine on pneumococcal colonization among unvaccinated household members. Clin. Infect. Dis. 47:989–996 [DOI] [PubMed] [Google Scholar]
- 6. Weiser JN. 2010. The pneumococcus: why a commensal misbehaves. J. Mol. Med. 88:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simell B, Auranen K, Käyhty H, Goldblatt D, Dagan R, O’Brien KL, Pneumococcal Carriage Group 2012. The fundamental link between pneumococcal carriage and disease. Expert Rev. Vaccines 11:841–855 [DOI] [PubMed] [Google Scholar]
- 8. Sleeman KL, Griffiths D, Shackley F, Diggle L, Gupta S, Maiden MC, Moxon ER, Crook DW, Peto TE. 2006. Capsular serotype-specific attack rates and duration of carriage of Streptococcus pneumoniae in a population of children. J. Infect. Dis. 194:682–688 [DOI] [PubMed] [Google Scholar]
- 9. Anonymous. 2008. 23-valent pneumococcal polysaccharide vaccine. WHO position paper. Wkly. Epidemiol. Rec. 83:373–384 [PubMed] [Google Scholar]
- 10. Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, Jha P, Campbell H, Walker CF, Cibulskis R, Eisele T, Liu L, Mathers C, Child Health Epidemiology Reference Group of WHO and UNICEF 2010. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375:1969–1987 [DOI] [PubMed] [Google Scholar]
- 11. Muñoz-Elías EJ, Marcano J, Camilli A. 2008. Isolation of Streptococcus pneumoniae biofilm mutants and their characterization during nasopharyngeal colonization. Infect. Immun. 76:5049–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marks LR, Parameswaran GI, Hakansson AP. 2012. Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect. Immun. 80:2744–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marks LR, Reddinger RM, Hakansson AP. 2012. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. mBio 3:e00200-12. 10.1128/mBio.00200-12.PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanchez CJ, Kumar N, Lizcano A, Shivshankar P, Dunning Hotopp JC, Jorgensen JH, Tettelin H, Orihuela CJ. 2011. Streptococcus pneumoniae in biofilms are unable to cause invasive disease due to altered virulence determinant production. PLoS One 6:e28738. 10.1371/journal.pone.0028738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomasz A, Jamieson JD, Ottolenghi E. 1964. The fine structure of Diplococcus pneumoniae. J. Cell Biol. 22:453–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim PE, Musher DM, Glezen WP, Rodriguez-Barradas MC, Nahm WK, Wright CE. 1996. Association of invasive pneumococcal disease with season, atmospheric conditions, air pollution, and the isolation of respiratory viruses. Clin. Infect. Dis. 22:100–106 [DOI] [PubMed] [Google Scholar]
- 17. Launes C, de-Sevilla MF, Selva L, Garcia-Garcia JJ, Pallares R, Muñoz-Almagro C. 2012. Viral coinfection in children less than five years old with invasive pneumococcal disease. Pediatr. Infect. Dis. J. 31:650–653 [DOI] [PubMed] [Google Scholar]
- 18. Chertow DS, Memoli MJ. 2013. Bacterial coinfection in influenza: a grand rounds review. JAMA 309:275–282 [DOI] [PubMed] [Google Scholar]
- 19. Chonmaitree T, Howie VM, Truant AL. 1986. Presence of respiratory viruses in middle ear fluids and nasal wash specimens from children with acute otitis media. Pediatrics 77:698–702 [PubMed] [Google Scholar]
- 20. Henderson FW, Collier AM, Sanyal MA, Watkins JM, Fairclough DL, Clyde WA, Jr, Denny FW. 1982. A longitudinal study of respiratory viruses and bacteria in the etiology of acute otitis media with effusion. N, Engl. J. Med. 306:1377–1383 [DOI] [PubMed] [Google Scholar]
- 21. Pettigrew MM, Gent JF, Pyles RB, Miller AL, Nokso-Koivisto J, Chonmaitree T. 2011. Viral-bacterial interactions and risk of acute otitis media complicating upper respiratory tract infection. J. Clin. Microbiol. 49:3750–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCullers JA. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19:571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bakaletz LO. 2010. Immunopathogenesis of polymicrobial otitis media. J. Leukoc. Biol. 87:213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heikkinen T, Ruohola A, Waris M, Ruuskanen O. 2001. Respiratory viruses in nasopharyngeal specimens from children with acute otitis media, abstr 176. 4th Extraordinary Int. Symp. Recent Adv. Otitis Media. [Google Scholar]
- 25. Peltola V, Heikkinen T, Ruuskanen O, Jartti T, Hovi T, Kilpi T, Vainionpää R. 2011. Temporal association between rhinovirus circulation in the community and invasive pneumococcal disease in children. Pediatr. Infect. Dis. J. 30:456–461 [DOI] [PubMed] [Google Scholar]
- 26. Jansen AG, Sanders EA, Van DER Ende DA, van Loon AM, Hoes AW, Hak E. 2008. Invasive pneumococcal and meningococcal disease: association with influenza virus and respiratory syncytial virus activity? Epidemiol. Infect. 136:1448–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou H, Haber M, Ray S, Farley MM, Panozzo CA, Klugman KP. 2012. Invasive pneumococcal pneumonia and respiratory virus co-infections. Emerg. Infect. Dis. 18:294–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diavatopoulos DA, Short KR, Price JT, Wilksch JJ, Brown LE, Briles DE, Strugnell RA, Wijburg OL. 2010. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J. 24:1789–1798 [DOI] [PubMed] [Google Scholar]
- 29. Vu HT, Yoshida LM, Suzuki M, Nguyen HA, Nguyen CD, Nguyen AT, Oishi K, Yamamoto T, Watanabe K, Vu TD. 2011. Association between nasopharyngeal load of Streptococcus pneumoniae, viral coinfection, and radiologically confirmed pneumonia in Vietnamese children. Pediatr. Infect. Dis. J. 30:11–18 [DOI] [PubMed] [Google Scholar]
- 30. Grebe KM, Takeda K, Hickman HD, Bailey AL, Bailey AM, Embry AC, Bennink JR. 2010. Cutting edge: sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J. Immunol. 184:540–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allegrucci M, Hu FZ, Shen K, Hayes J, Ehrlich GD, Post JC, Sauer K. 2006. Phenotypic characterization of Streptococcus pneumoniae biofilm development. J. Bacteriol. 188:2325–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Håkansson A, Carlstedt I, Davies J, Mossberg AK, Sabharwal H, Svanborg C. 1996. Aspects on the interaction of Streptococcus pneumoniae and Haemophilus influenzae with human respiratory tract mucosa. Am. J. Respir. Crit. Care Med. 154:S187–S191 [DOI] [PubMed] [Google Scholar]
- 33. Vidal JE, Howery KE, Ludewick HP, Nava P, Klugman KP. 2013. Quorum sensing systems LuxS/AI-2 and com regulate Streptococcus pneumoniae biofilms in a bioreactor with living cultures of human respiratory cells. Infect. Immun. 81:1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keck T, Leiacker R, Riechelmann H, Rettinger G. 2000. Temperature profile in the nasal cavity. Laryngoscope 110:651–654 [DOI] [PubMed] [Google Scholar]
- 35. Kim JO, Weiser JN. 1998. Association of intrastrain phase variation in quantity of capsular polysaccharide and teichoic acid with the virulence of Streptococcus pneumoniae. J. Infect. Dis. 177:368–377 [DOI] [PubMed] [Google Scholar]
- 36. Cundell D, Masure HR, Tuomanen EI. 1995. The molecular basis of pneumococcal infection: a hypothesis. Clin. Infect. Dis. 21(Suppl. 3):S204–S211 [DOI] [PubMed] [Google Scholar]
- 37. van Ginkel FW, McGhee JR, Watt JM, Campos-Torres A, Parish LA, Briles DE. 2003. Pneumococcal carriage results in ganglioside-mediated olfactory tissue infection. Proc. Natl. Acad. Sci. U. S. A. 100:14363–14367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301 [DOI] [PubMed] [Google Scholar]
- 39. Dalia AB, Weiser JN. 2011. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe 10:486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sauer K, Cullen MC, Rickard AH, Zeef LA, Davies DG, Gilbert P. 2004. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 186:7312–7326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sellam A, Al-Niemi T, McInnerney K, Brumfield S, Nantel A, Suci PA. 2009. A Candida albicans early stage biofilm detachment event in rich medium. BMC Microbiol. 9:25. 10.1186/1471-2180-9-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trappetti C, Ogunniyi AD, Oggioni MR, Paton JC. 2011. Extracellular matrix formation enhances the ability of Streptococcus pneumoniae to cause invasive disease. PLoS One 6:e19844. 10.1371/journal.pone.0019844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanchez CJ, Hurtgen BJ, Lizcano A, Shivshankar P, Cole GT, Orihuela CJ. 2011. Biofilm and planktonic pneumococci demonstrate disparate immunoreactivity to human convalescent sera. BMC Microbiol. 11:245. 10.1186/1471-2180-11-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. 2013. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 9:e1003057. 10.1371/journal.ppat.1003057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. 2001. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J. Med. Virol. 64:262–268 [DOI] [PubMed] [Google Scholar]
- 46. Dunn AJ, Powell ML, Meitin C, Small PA. 1989. Virus infection as a stressor: influenza virus elevates plasma concentrations of corticosterone, and brain concentrations of MHPG and tryptophan. Physiol. Behav. 45:591–594 [DOI] [PubMed] [Google Scholar]
- 47. Peiris JS, Hui KP, Yen HL. 2010. Host response to influenza virus: protection versus immunopathology. Curr. Opin. Immunol. 22:475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pacheco AR, Sperandio V. 2009. Inter-kingdom signaling: chemical language between bacteria and host. Curr. Opin. Microbiol. 12:192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sautel M, Sacquet J, Vincent M, Sassard J. 1988. NE turnover in genetically hypertensive rats of Lyon strain. II. Peripheral organs. Am. J. Physiol. 255 (Pt 2):H736–H741 [DOI] [PubMed] [Google Scholar]
- 50. Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC. 2006. Physiological regulation of ATP release at the apical surface of human airway epithelia. J. Biol. Chem. 281:22992–23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, Ryffel B, Couillin I. 2010. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 182:774–783 [DOI] [PubMed] [Google Scholar]
- 52. Burnstock G, Brouns I, Adriaensen D, Timmermans JP. 2012. Purinergic signaling in the airways. Pharmacol. Rev. 64:834–868 [DOI] [PubMed] [Google Scholar]
- 53. Ricci A, Bronzetti E, Conterno A, Greco S, Mulatero P, Schena M, Schiavone D, Tayebati SK, Veglio F, Amenta F. 1999. alpha1-Adrenergic receptor subtypes in human peripheral blood lymphocytes. Hypertension 33:708–712 [DOI] [PubMed] [Google Scholar]
- 54. McClenahan D, Hillenbrand K, Kapur A, Carlton D, Czuprynski C. 2009. Effects of extracellular ATP on bovine lung endothelial and epithelial cell monolayer morphologies, apoptoses, and permeabilities. Clin. Vaccine Immunol. 16:43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hatta M, Hatta Y, Kim JH, Watanabe S, Shinya K, Nguyen T, Lien PS, Le QM, Kawaoka Y. 2007. Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog. 3:1374–1379. 10.1371/journal.ppat.0030133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pandya U, Allen CA, Watson DA, Niesel DW. 2005. Global profiling of Streptococcus pneumoniae gene expression at different growth temperatures. Gene 360:45–54 [DOI] [PubMed] [Google Scholar]
- 57. Klein BS, Tebbets B. 2007. Dimorphism and virulence in fungi. Curr. Opin. Microbiol. 10:314–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Siau A, Silvie O, Franetich JF, Yalaoui S, Marinach C, Hannoun L, van Gemert GJ, Luty AJ, Bischoff E, David PH, Snounou G, Vaquero C, Froissard P, Mazier D. 2008. Temperature shift and host cell contact up-regulate sporozoite expression of Plasmodium falciparum genes involved in hepatocyte infection. PLoS Pathog. 4:e1000121. 10.1371/journal.ppat.1000121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ostberg JR, Taylor SL, Baumann H, Repasky EA. 2000. Regulatory effects of fever-range whole-body hyperthermia on the LPS-induced acute inflammatory response. J. Leukoc. Biol. 68:815–820 [PubMed] [Google Scholar]
- 60. Tomashefski JF. 2000. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 21:435–466 [DOI] [PubMed] [Google Scholar]
- 61. el-Kassimi FA, Al-Mashhadani S, Abdullah AK, Akhtar J. 1986. Adult respiratory distress syndrome and disseminated intravascular coagulation complicating heat stroke. Chest 90:571–574 [DOI] [PubMed] [Google Scholar]
- 62. Hasday JD, Fairchild KD, Shanholtz C. 2000. The role of fever in the infected host. Microbes Infect. 2:1891–1904 [DOI] [PubMed] [Google Scholar]
- 63. Chiappini E, Parretti A, Becherucci P, Pierattelli M, Bonsignori F, Galli L, de Martino M. 2012. Parental and medical knowledge and management of fever in Italian pre-school children. BMC Pediatr. 12:97. 10.1186/1471-2431-12-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Graham NM, Burrell CJ, Douglas RM, Debelle P, Davies L. 1990. Adverse effects of aspirin, acetaminophen, and ibuprofen on immune function, viral shedding, and clinical status in rhinovirus-infected volunteers. J. Infect. Dis. 162:1277–1282 [DOI] [PubMed] [Google Scholar]
- 65. van Schilfgaarde M, van Alphen L, Eijk P, Everts V, Dankert J. 1995. Paracytosis of Haemophilus influenzae through cell layers of NCI-H292 lung epithelial cells. Infect. Immun. 63:4729–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tyx RE, Roche-Hakansson H, Hakansson AP. 2011. Role of dihydrolipoamide dehydrogenase in regulation of raffinose transport in Streptococcus pneumoniae. J. Bacteriol. 193:3512–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Andersson B, Dahmén J, Frejd T, Leffler H, Magnusson G, Noori G, Edén CS. 1983. Identification of an active disaccharide unit of a glycoconjugate receptor for pneumococci attaching to human pharyngeal epithelial cells. J. Exp. Med. 158:559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Avery OT, MacLeod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a dexoxyribonuceic acid fraction isolated from pneumococcus type III. J. Exp. Med. 79:137–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. 2000. B-1 and β-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. 192:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paterson GK, Nieminen L, Jefferies JM, Mitchell TJ. 2008. PclA, a pneumococcal collagen-like protein with selected strain distribution, contributes to adherence and invasion of host cells. FEMS Microbiol. Lett. 285:170–176 [DOI] [PubMed] [Google Scholar]
- 71. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Colonization and dissemination of bacteria passively released from pneumococcal biofilms. Data represent tissue tropism of bacteria passively released from EF3030 (A) and D39 (B) biofilms during normal growth after 48 h. Mice were challenged via nonanesthetized intranasal (Colonization) and intraperitoneal (Sepsis) inoculation. For intranasal challenges, nasal lavage fluids and the respective tissues were collected at 48 h postinfection. For intraperitoneal challenge, samples were collected at 24 h postinfection. Bacterial burden was determined by viable counts of the respective fluid or tissue homogenate samples. Statistical analysis of this data, using Student’s t tests, showed no significant differences between bacterial burdens of biofilm bacteria and those reported in the legend to Fig. 4. Download
Oligonucleotides used in qRT-PCR analysis.