

Abstract
Deficits of protein phosphatase-2A (PP2A) play a crucial role in tau hyperphosphorylation, amyloid overproduction, and synaptic suppression of Alzheimer's disease (AD), in which PP2A is inactivated by the endogenously increased inhibitory protein, namely inhibitor-2 of PP2A (I2PP2A). Therefore, in vivo silencing I2PP2A may rescue PP2A and mitigate AD neurodegeneration. By infusion of lentivirus-shRNA targeting I2PP2A (LV-siI2PP2A) into hippocampus and frontal cortex of 11-month-old tg2576 mice, we demonstrated that expression of LV-siI2PP2A decreased remarkably the elevated I2PP2A in both mRNA and protein levels. Simultaneously, the PP2A activity was restored with the mechanisms involving reduction of the inhibitory binding of I2PP2A to PP2A catalytic subunit (PP2AC), repression of the inhibitory Leu309-demethylation and elevation of PP2AC. Silencing I2PP2A induced a long-lasting attenuation of amyloidogenesis in tg2576 mice with inhibition of amyloid precursor protein hyperphosphorylation and β-secretase activity, whereas simultaneous inhibition of PP2A abolished the antiamyloidogenic effects of I2PP2A silencing. Finally, silencing I2PP2A could improve learning and memory of tg2576 mice with preservation of several memory-associated components. Our data reveal that targeting I2PP2A can efficiently rescue Aβ toxicities and improve the memory deficits in tg2576 mice, suggesting that I2PP2A could be a promising target for potential AD therapies.
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder, characterized pathologically with brain accumulation of numerous neurofibrillary tangles and senile plaques.1 Since β-amyloid (Aβ) and hyperphosphorylated tau (p-tau) are respectively the core elements of the plaques and tangles, suppressing Aβ and p-tau accumulation could be a logic strategy for arresting AD pathologies.
Deficit of several protein phosphatases (PP), such as PP2A, PP2B, PP1, and PP5, has been identified in AD neurodegeneration, among them, PP2A is the most implicated: PP2A is the most active phosphatase in dephosphorylating the hyperphosphorylated tau proteins isolated from the AD brains and dephosphorylation restores the biological activity of tau proteins in promoting microtubule assembly and stabilizing microtubules;2 PP2A accounts for ~70% of the total tau phosphatase activity in human brain,3 and the activity of PP2A is significantly decreased in the brain of AD patients.4 Studies also show that inhibition of PP2A causes tau hyperphosphorylation and memory deficits in rats.5,6 In addition to tau proteins, an in vitro study showed that PP2A could also dephosphorylate amyloid precursor protein (APP) and therefore attenuate Aβ production.7 Furthermore, activation of PP2A promotes axonogenesis and facilitates synaptic plasticity.8 These data suggest that restoration of PP2A activity may rescue AD pathologies and the memory-associated synaptic deficits.
To date, the PP2A activator is very limited. It has been reported that ceramide can activate PP2A; however, the neurotoxicity of ceramide has greatly restricted its clinical application.9 Therefore, restitution of PP2A may represent a new strategy in arresting AD neurodegeneration. In the brain, the activity of PP2A is downregulated by its constitutive inhibitory proteins, namely inhibitor 1 (I1PP2A) and inhibitor 2 of PP2A (I2PP2A),10,11 both of which inhibit noncompetitively PP2A, including PP2AC monomer, dimeric, and trimeric complexes, with the Ki values in low nanomolar range.12 I2PP2A is a homologue of SET, which was first identified in a patient with acute leukemia.13 Later studies demonstrate that SET is a multifunctional protein widely expressed in human and mouse. SET can protect histones from acetylation, regulate G2/M transition, modulate HuR mRNA binding, or act as a transcription factor for P450c17 activation.14,15,16,17 In AD brains, the level of I2PP2A transcripts was upregulated and the elevated I2PP2A protein was coaccumulated with neurofibrillary tangles in the cytoplasm.18,19 A recent study demonstrate that upregulation of I2PP2A can reproduce AD-like histopathologies and memory deficit in rats.20 These data suggest that the elevated I2PP2A plays a crucial role in AD neurodegeneration with mechanisms involving PP2A inhibition, therefore, downregulation of I2PP2A may rescue PP2A and therefore mitigate AD neurodegeneration.
We tested this idea in the present study. We found that silencing I2PP2A by in vivo lentiviral vector delivery of shRNA targeting I2PP2A remarkably attenuated amyloidogenesis by activation of PP2A in tg2576 mice, a recognized mouse model of AD. We also observed that silencing I2PP2A improved learning and memory of the mice with preservation of several memory-associated components. We propose that in vivo targeting I2PP2A is a promising strategy in mitigating AD pathologies and memory deficits.
Results
Level of I2PP2A increases age dependently in hippocampus and frontal cortex of tg2576 mice with a correlated inhibition of PP2A
Elevation of I2PP2A with the inactivation of PP2A has been detected in the AD brains.18 To testify whether tg2576 is a suitable model for the current study, we first measured the alterations of I2PP2A and PP2A in the mice. An age-associated elevation of I2PP2A (Supplementary Figure S1a–c) with inactivation of PP2A (Supplementary Figure S1d) was observed from 6 to 23 months in both hippocampus and frontal cortex of the tg2576 mice. By Pearson analysis, a negative correlation between I2PP2A level and PP2A activity was shown (Supplementary Figure S1e,f). We also analyzed the protein level of I2PP2A in hippocampus of the age-matched wild-type littermates, but the increase was only detected at 23 months old (Supplementary Figure S1a,b). Normally, I2PP2A is mainly located in the nuclei, but it is largely translocated into the cytoplasm in AD brains.18 We therefore measured the cellular localization of I2PP2A. Compared with the age-matched control mice, a cytoplasmic translocation of I2PP2A was shown in the hippocampus of tg2576 mice measured at ~12.5 months (Supplementary Figure S2). These data indicate that tg2576 can serve as a perfect model for I2PP2A and PP2A studies, and the elevated/cytoplasmic translocated I2PP2A may underlie the inactivation of PP2A in the mice.
Expression of LV-siI2PP2A downregulates I2PP2A in hippocampus and the frontal cortex of tg2576 mice with stimulation of PP2A activity
To silence I2PP2A in vivo, we infused the virus expressing an eGFP-labeled LV-siI2PP2A or the scrambled control (LV-ssiI2PP2A) into the hippocampus (CA3 and CA1) and frontal cortex of tg2576 mice at ~11 months old and analyzed the mice at 4 or 6 weeks after injection. Robust expression of the LV-siI2PP2A with a significant reduction of I2PP2A protein and the transcript was detected in hippocampus and frontal cortex by direct fluorescence imaging (Figure 1a; for lower magnification, see Supplementary Figure S3), immunohistochemistry (Figure 1b), Western blot (Figure 1c), and RT-PCR assays (Figure 1d). Expression of LV-siI2PP2A restored or further stimulated PP2A activity in cortex and hippocampus of tg2576 mice (Figure 2a) with increase of total level of PP2AC, decrease of binding of I2PP2A to PP2AC (Figure 2b,c), and decrease of the inhibitory demethylation of PP2AC (Figure 2d,e), all of which may underlie the activation of PP2A. We also measured the level of PP2AA and PP2AB, the regulatory subunits, but no significant change was detected (Figure 2d). To further verify the factors leading to the reduced PP2AC demethylation, we measured methyltransferase (PPMT) and methylesterase (PME) that regulate the methylation level of PP2A. We found that protein level of PPMT significantly increased while the PME was not changed by I2PP2A silencing (Figure 2f,g), suggesting an enhanced methylation of PP2A by silencing I2PP2A.
Figure 1.
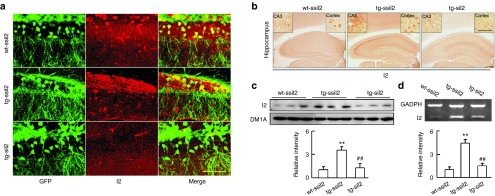
Expression of LV-siI2PP2A downregulates I2PP2A (I2). Virus expressing eGFP-labeled LV-I2PP2A (lentivirus) shRNA (siI2) or the scrambled control (ssiI2) (2 × 109 TU/ml) were injected into the hippocampal CA3 and CA1 and the frontal cortex of tg2576 mice (tg-ssiI2 and tg-siI2) or the wild type littermates (wt-ssiI2) at ~11 months old under a stereotaxic apparatus (2 µl per site). At ~12.5 months, the mice were killed for the following measurements. (a) The representative images show expression of LV-siI2 (green, direct fluorescent image) and the silencing efficacy of I2 (red, with I2 antibody) at hippocampal dentate gyrus (DG) of ~12.5-month mice (n = 3). Scale bars: 100 µm. (b) Level of I2 protein increased in cortex and hippocampus of tg2576 mice compared with the wt littermates, and expression of siI2 reduced the I2 signal measured by immunohistochemistry staining using I2 antibody (the inserts show enlarged images, n = 3). (c) Level of I2 protein in hippocampus of tg2576 mice increased ~3.5-fold of the control level and expression of LV-siI2 reduced I2 protein almost to the normal level measured by Western blotting (n = 3–5). (d) Level of I2 mRNA in hippocampus of tg2576 mice increased ~4.5-fold of the control level and expression of LV-siI2 partially reduced I2 mRNA level measured by RT-PCR (n = 3–5). **P < 0.01 versus wt-ssiI2; ##P < 0.01 versus tg-ssiI2 (mean ± SD).
Figure 2.
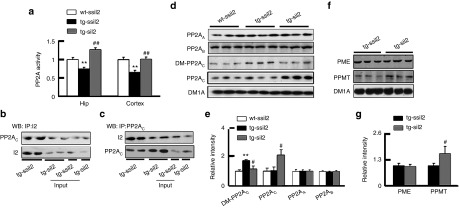
Silencing I2PP2A activates PP2A by multiple mechanisms. The mice were treated as described in Figure 1. (a) The activity of PP2A in hippocampus and cortex extracts of ~12.5-month tg2576 mice decreased and silencing I2PP2A (I2) by expression of LV-siI2 restored or further stimulated PP2A activity, measured by using a Serine/Threonine Phosphatase Assay Kit (n = 3–5). (b–c) Silencing I2 reduced the inhibitory binding of I2 with PP2AC in hippocampus of tg2576 mice, measured by immunoprecipitation (IP) and Western blot (WB) using anti-I2 or anti-PP2AC antibodies as indicated (n = 3). (d–e) Level of Leu309-demethylated PP2AC (DM-PP2AC) increased in hippocampus of tg2576 mice, and silencing I2 reduced the inhibitory demethylation of PP2AC with a significantly increased total level of PP2AC, whereas the protein levels PP2AA and PP2AB were not changed, measured by Western blotting (n = 3~5). (f–g) Silencing I2 increased protein level of PP2A methyltransferase (PPMT) with no change of PP2A methylesterase (PME) in hippocampus of tg2576 mice measured by Western blotting (n = 3–5). **P < 0.01 versus wt-ssiI2; #P < 0.05; ##P < 0.01 versus tg-ssiI2 (mean ± SD).
Silencing I2PP2A induces a long-lasting attenuation of Aβ toxicities in tg2576 mice
By Western blot and ELISA assay, we observed that levels of Aβ40 (17788 ± 2788 pg/mg protein) and Aβ42 (4950 ± 234 pg/mg protein) increased in ~12.5-month tg2576 mice compared with the age-matched controls (Aβ40, 31 ± 3 pg/mg protein; Aβ42, 5.1 ± 1 pg/mg protein), while silencing I2PP2A reduced the Aβ level (Aβ40, 4945 ± 1670 pg/mg protein; Aβ42, 4117 ± 197 pg/mg protein) (Figure 3a,b). An increased tau phosphorylation at multiple AD-related sites was also detected in ~12.5-month tg2576 mice, and silencing I2PP2A attenuated tau hyperphosphorylation at pS214, pS396 and tau-1 epitope (Figure 3c,d). The modified Bielschowsky's silver staining data showed that the expression of LV-siI2PP2A significantly decreased tau and Aβ aggregation (Figure 3e).
Figure 3.

Silencing I2PP2A induces a long-lasting attenuation of amyloidogenesis in tg2576 mice. The mice were treated as described in Figure 1. (a) The representative Western blot show an increased Aβ level in hippocampal extracts of ~12.5-month tg2576 mice infused with the scrambled siI2PP2A (tg-ssiI2) compared with the age-matched wild-type littermates (wt-ssiI2), and silencing I2 by LV-siI2PP2A (tg-siI2) reduced Aβ level compared with the tg-ssiI2 controls (n = 3). (b) ELISA data show increased levels of Aβ40 and Aβ42 in hippocampal extracts of tg2576 mice and the reduction of Aβ by silencing I2. The experiments were repeated for at least three times with triplicates. (c–d) An increased tau phosphorylation at multiple AD-related sites in tg2576 mice and the attenuation by I2 knockdown detected by Western blotting (note that tau-1 reacts with the unphosphorylated tau, therefore, an increased immunoreaction indicates reduced tau phosphorylation) (n = 3–5). (e) The representative silver staining images show an enhanced intracellular accumulation of the argyrophilic substances in hippocampus and the cortex of tg2576 mice, and the reduced accumulation by LV-siI2PP2A. Scale bars: 100 µm (n = 3). (f–h) At 18 months, LV-siI2 infused (at ~11 months) tg2576 mice still show lower levels of total and insoluble Aβ in hippocampus than the LV-ssiI2-infused control mice. The ELISA assay was repeated at least three times in triplicates. (i–k) The representative images and the quantitative analyses show increase of plaque load in hippocampus and cortex of ~23-month tg2576 mice immunostained with Aβ antibody, and attenuation of by I2 knockdown measured at the virus injection sites. Scale bars: 100 µm (n = 3). **P < 0.01 versus wt-ssiI2; #P < 0.05; ##P < 0.01 versus tg-ssiI2 (mean ± SD).
We detected an age-dependent Aβ accumulation measured at 6, 12, 18, and 23 months in tg2576 mice (Supplementary Figure S4). Previous study demonstrated that the lentiviral shRNA-mediated gene silencing could be long lasting or even be transferred to the next generation,21 therefore, we further measured the long-term effects of I2PP2A silencing on Aβ accumulation. To this end, the tg2576 mice were infused with siI2PP2A at ~11 months and examined at 18 months. As shown in Figure 3f–h, both total and insoluble Aβ were still lower in siI2PP2A-infused tg-mice (total Aβ40, 18088 ± 712 pg/mg protein, Aβ42, 5914 ± 204 pg/mg protein; insoluble Aβ40, 17902 ± 624 pg/mg protein, Aβ42, 5811 ± 213 pg/mg protein) than in the tg-ssiI2PP2A mice (total Aβ40, 26600 ± 600 pg/mg protein, Aβ42, 6857 ± 196 pg/mg protein; insoluble Aβ40, 26022 ± 768 pg/mg protein, Aβ42, 6800 ± 112 pg/mg protein). At 23 months, both density and the intensity of Aβ-positive plaques were lower in siI2PP2A-expressing mice than in the age-matched ssiI2PP2A-expressing control mice (Figure 3i–k). Of note, the impact of I2PP2A silencing on Aβ accumulation seems to be correlated with the expression of I2PP2A, as the expression of siI2PP2A was widely spread in the hippocampus, leading to the attenuation of Aβ accumulation in whole hippocampus (CA1, CA3, and DG) (Figure 3i), whereas the expression of siI2PP2A was only localized in the injected area of the cortex, resulting in no changes of the Aβ pathology in the adjacent regions (Supplementary Figure S5).
Given the established role of neuroinflammatory processes in AD and the role of PP2A in stimulating astrocytes,22 we detected the inflammatory response to the LV-siRNA treatment in 12.5-month tg2576 mice by measuring the expression levels of glial fibrillary acidic protein (GFAP, a marker of astrocytes), and Iba-1 (a marker of microglia). No significant difference of GFAP and Iba-1 was observed between siI2PP2A-infused and the ssiI2PP2A-infused control mice (Supplementary Figure S6).
These data together indicate that in vivo targeting I2PP2A by lentiviral vector delivery of siI2PP2A not only mitigates tau pathologies but also induces a long-lasting attenuation of amyloidogenesis.
Activation of PP2A mediates the siI2PP2A-induced attenuation of Aβ accumulation with mechanisms involving APP dephosphorylation and β-secretase inhibition
To verify the role of PP2A in siI2PP2A-induced attenuation of amyloidogenesis, we infused OA into the hippocampal CA3 and CA1 regions of tg2576 mice for 48 hours to inhibit PP2A after expression of siI2PP2A for 6 weeks. Compared with the tg-mice injected with siI2PP2A alone (Aβ40, 4916 ± 451 pg/mg protein, Aβ42, 4317 ± 202 pg/mg protein), simultaneous inhibition of PP2A by OA abolished the siI2PP2A-induced reduction of Aβ40 (12368 ± 1116 pg/mg protein) and Aβ42 (4803 ± 180 pg/mg protein) in hippocampus (Figure 4a,b). We further confirmed the role of PP2A in attenuating amyloidogenesis in N2a/APP cells. As shown in Figure 4c,d, inhibition of PP2A by OA increased Aβ level, whereas activation of PP2A with C6-ceramide (DES) attenuated the OA-induced Aβ overproduction. Furthermore, simultaneous inhibition of PP2A by expression of siPP2AC abolished the siI2PP2A-induced reduction of Aβ (Figure 4e,f). These data strongly suggest that the activation of PP2A mediates the siI2PP2A-induced attenuation of Aβ accumulation.
Figure 4.

Simultaneous inhibition of PP2A abolishes the siI2PP2A-induced attenuation of Aβ accumulation. (a) The mice were first injected as described in Figure 1. After ~1.5 months, okadaic acid (OA) was infused into the hippocampal CA3 and CA1 (0.2 µmol/l, 2 µl per site), and inhibition of PP2A activity by OA was detected in hippocampal extracts by using a Serine/Threonine Phosphatase Assay Kit (n = 3–5 in triplicates). (b) The mice were treated as described in panel a. Levels of Aβ were reduced by expression of LV-siI2 in 12.5-month tg2576 mice, while simultaneous inhibition of PP2A by OA reversed partially the siI2-induced reduction of Aβ levels measured by ELISA assay (n = 3–5 in triplicates). (c–d) N2a/APP cells treated with 5 nmol/l OA for 24 hours increased Aβ levels, whereas PP2A activator C6-ceramide (DES) (7 nmol/l) treatment attenuated the OA-induced Aβ production. The proteins were isolated by native Tris-tricine gradient gel electrophoresis (8–12%) and probed by antibody 6E10. The experiment was repeated for three times. (e) Silencing I2 increased PP2AC in N2a/APP cells, whereas simultaneous expression of siPP2AC for 24 hours downregulated the siI2-induced overexpression of PP2AC measured by Western blotting. The experiment was repeated at least three times. (f) Silencing I2 reduced Aβ levels, whereas simultaneous downregulation of PP2AC by siPP2AC partially reversed the siI2-induced Aβ reduction in the culture medium of N2a/APP cells measured by ELISA. The experiment was repeated at least three times with triplicates.
*P < 0.05 versus ssiI2; **P < 0.01 versus tg-ssiI2 or ssiI2 or Ctr; #P < 0.05; ##P < 0.01 versus tg-siI2 or siI2 or OA (mean ± SD).
It has been well characterized that PP2A is the most implicated phosphatase in dephosphorylating tau proteins; however, the role of PP2A in Aβ production is not fully illustrated. We therefore studied how PP2A activation may attenuate Aβ production. First, we measured the level of APP phosphorylation at Thr668 (pAPP), because the phosphorylation of APP at this site has been reported to facilitate Aβ production.23,24 Compared with the age-matched wild-type littermate, a significant increase of APP phosphorylation was observed in tg2576 mice (Figure 5a). Expression of siI2PP2A-inhibited APP phosphorylation (Figure 5a), which was abolished by simultaneous inhibition of PP2A through hippocampal infusion of OA (Figure 5b). To further verify the role of PP2A in APP phosphorylation, we treated N2a/APP cells with OA alone or together with DES. As shown in Figure 5c, activation of PP2A by DES not only decreased the endogenous pAPP, but also attenuated the OA-induced APP phosphorylation. Furthermore, overexpression of wild-type PP2AC (wtPP2AC) decreased pAPP, whereas downregulation of PP2A by siPP2AC partially reversed the siI2PP2A-induced reduction of APP phosphorylation (Figure 5d,e). These data suggest that restitution of PP2A may attenuate Aβ generation through dephosphorylating APP.
Figure 5.

Activation of PP2A mediates the siI2PP2A-induced APP dephosphorylation at Thr668. (a) The mice were treated as described in Figure 1. The levels of total APP (tAPP) and the Thr668-phosphorylated APP (pAPP) were increased in hippocampal extracts of ~12.5 months tg2576 mice (tg-ssiI2) compared with the age-matched wild-type littermates (wt-ssiI2), both mice were transfected with the scrambled siI2PP2A; expression of siI2 (tg-siI2) decreased the phosphorylation level of APP compared with tg-ssiI2, measured by Western blotting (n = 6). (b) In tg2576 mice, simultaneous inhibition of PP2A by hippocampal (CA3 and CA1) infusion of okadaic acid (2 µl each) reversed the siI2PP2A-suppressed APP phosphorylation (n = 6). (c) In N2a/APP cells, inhibition of PP2A by OA increased APP phosphorylation compared with the control (Ctr), whereas simultaneous activation of PP2A by DES attenuated the OA-induced APP phosphorylation. The experiments were repeated two times in triplicates. (d) In N2a/APP cells, upregulation of PP2A by overexpression of wild type PP2AC (wtPP2AC) reduced the phosphorylation level of APP. The experiments were repeated two times in triplicates. (e) In N2a/APP cells, I2 knockdown by expression of siI2PP2A (siI2) reduced APP phosphorylation, while simultaneous inhibition of PP2A by expression of siPP2AC reversed the siI2-suppressed APP phosphorylation. The experiments were repeated at least for three times. *P < 0.05; **P < 0.01 versus wt-ssiI2 or Ctr or ssiI2; #P < 0.05 versus pcDNA; ##P < 0.01 vs tg-ssiI2 or tg-siI2 or siI2 or OA (mean ± SD).
Aβ production is also regulated by α-, β- and γ-secretases; therefore, we measured the alterations of the secretases. The activity assay showed that only β-secretase but not α- and γ-secretase was significantly activated in hippocampus and the cortex of ~12.5-month tg2576 mice, and silencing I2PP2A completely reversed the activity of β-secretase (Figure 6a,b). In addition, upregulation of β-secretase and the attenuation by siI2PP2A in hippocampus of 12.5-month tg2576 mice were also detected by Western blot (Figure 6c,d) and RT-PCR analyses (Figure 6e,f), whereas inhibition of PP2A by brain injection of OA abolished the siI2PP2A-induced inhibition of β-secretase in the mice (Figure 6g). In N2a/APP cells, inhibition of PP2A by OA activated β-secretase, whereas stimulation of PP2A by DES or overexpression of wtPP2AC inhibited β-secretase (Figure 6h,i). These data suggest that β-secretase inhibition is also involved in the siI2PP2A-induced attenuation of Aβ generation.
Figure 6.

Activation of PP2A mediates the siI2PP2A-induced inhibition of β-secretase. (a–b) The mice were treated as described in Figure 1, and then the activity of α-, β-, and γ-secretases in hippocampus and cortex extracts of ~12.5 months tg2576 mice and the wild-type littermates (wt) was measured. β-secretase was activated in tg2576 mice compared with the wt, and expression of LV-siI2PP2A (siI2) inhibited β-secretase. The experiments were repeated at least two times in triplicates. (c–d) The protein level of β-site APP-cleaving enzyme 1 (BACE1) was increased in hippocampal extracts of ~12.5 months tg2576 mice measured by Western blotting, and expression of LV-siI2PP2A reduced the protein level of BACE1 (n = 3–5). (e–f) The mRNA level of BACE1 was increased in hippocampal extracts of ~12.5 months tg2576 mice measured by RT-PCR, and expression of LV-siI2PP2A reduced the mRNA level of BACE1 (n = 3–5). (g) Simultaneous inhibition of PP2A by hippocampal infusion of OA (2 µl each at CA3 and CA1) for 48 hours abolished the LV-siI2-induced inhibition of β-secretase in ~12.5 months tg2576 mice after injection of LV-siI2 for 6 weeks (n = 3). (h) In N2a/APP cells, inhibition of PP2A by OA (5 nmol/l) for 24 hours activated β-secretase, whereas activation of PP2A by DES (7 nmol/l) for 24 hours inhibited β-secretase. The experiments were repeated at least three times in triplicates. (i) In N2a/APP cells, overexpression of wild-type PP2AC (wtPP2AC) for 24 hours inhibited β-secretase when compared with pcDNA vector transfected cells. The experiments were repeated at least three times in duplicates. *P < 0.05; **P < 0.01 versus wt-ssiI2 or Ctr or pcDNA; #P < 0.05; ##P < 0.01 versus tg-ssiI2 or tg-siI2 or OA (mean ± SD).
Silencing I2PP2A improves learning and memory with preservation of several memory-associated components
Tg2576 mice show learning and memory deficits at 12.5 months compared with the age-matched wild-type controls, and downregulation of I2PP2A remarkably improved the cognitive functions of these mice as measured by Morris water maze test at ~12 months (Figure 7a,b) and step-down avoidance test at ~12.5 months (Figure 7c,d). The improved cognitive function was still significant at 18 months measured by step-down avoidance test (Figure 7e,f). The role of PP2A activation in the improved memory was evaluated by hippocampal infusion of OA for 48 hours after expression of siI2PP2A for 6 weeks. We observed unexpectedly that simultaneous inhibition of PP2A by OA did not induce any obvious difference when compared with the control group (Figure 7g,h). These data suggest that silencing I2PP2A improves the learning and memory of tg2576 mice and this effect seems independent of PP2A activation.
Figure 7.

Silencing I2PP2A improves learning and memory independent of PP2A activation in tg2576 mice. (a) Tg2576 mice (tg) at ~12 months had spatial learning deficit compared with the wild-type littermates (wt), and silencing I2PP2A (I2) improved the learning ability measured by the latency to find the hidden platform during 7 days water maze training (n = 15–20 each group). (b) At ~12 months, the tg-mice showed spatial memory deficits and silencing I2 improved the memory ability measured by the time spent in each quadrant after removed platform at day 9 (OPP, opposite quadrant; AL, adjacent left quadrant; Targ, target quadrant; AR, adjacent right quadrant; n = 15–20 each group). (c–d) At ~12.5 months, the tg-mice showed deficits of short-term memory (STM) (measured at 15 minutes after training) and long-term memory (LTM) (measured at 24 hours after training) by step-down avoidance test, and knockdown I2 improved the memory (n = 15–20 for each group). (e–f) At ~18 months, the tg-mice showed deficits of STM and LTM measured by step-down avoidance test, and knockdown I2 improved the memory (n = 8–10 each group). (g–h) Simultaneous inhibition of PP2A by hippocampal (CA3 and CA1) infusion of OA after expression of LV-siI2PP2A for 6 weeks did not significantly affect the siI2-induced improvement of STM and LTM measured at ~12.5 months (n = 6–7 each group). *P < 0.05; **P < 0.01 versus wt-ssiI2; #P < 0.05; ##P < 0.01 versus tg-ssiI2 (mean ± SD).
To further explore the mechanisms that may underlie the improved learning and memory of the mice, we measured the levels of several memory-related proteins. Levels of NR2A/B, c-fos, total cAMP response element-binding protein (CREB), and the phosphorylated form at Ser133 (pCREB) were decreased in ~12.5-month tg2576 mice compared with the wild-type littermates, and silencing I2PP2A increased the levels of NR2A/B, synaptophysin (Syt), c-fos, tCREB, and pCREB (Figure 8a,b). The level of synapsin-1 (Syn-1) increased in ~12.5-month tg2576 mice, and silencing I2PP2A did not obviously affect the levels, and no difference was observed for PSD95 (Figure 8a,b). We also found that the dendrite branches, density of dendritic spine and the mushroom-like spines decreased significantly in ~12.5-month tg2576 mice, which were partially restored by silencing I2PP2A (Figure 8c–h).
Figure 8.

Silencing I2PP2A preserves several memory-associated components. (a–b) Levels of NR2A/B, c-fos, CREB, and pCREB were reduced in ~12.5 months tg2576 mice, whereas knockdown I2PP2A (I2) restored the levels of the memory-associated proteins analyzed by Western blot (n = 3–5). (c–e) The representative images showing decreased dendrite branch numbers in hippocampal DG region of ~12.5-month tg2576 mice, and preservation of the dendrite complexity by knockdown I2. (c) Dendritic arbors shown by Golgi stain. (d) Reconstruction of the dendritic arbors. (e) Quantitative analysis of dendrite branch numbers. (f–h) The total spine number and the mushroom-shaped spines were decreased in ~12.5-month tg2576 mice, and knockdown I2 preserved spine plasticity analyzed by Golgi stain and NeuronStudio system. Scale bars: 100 µm. *P < 0.05; **P < 0.01 versus wt-ssiI2; #P < 0.05; ##P < 0.01 versus tg-ssiI2 (mean ± SD).
These data together indicate that downregulation of I2PP2A may improve learning and memory by preservation of several memory-associated components in tg2576 mice.
Since the attenuation effect of siI2PP2A in pathologies and memory deficits was still significant at 18 months of the mice, we detected I2PP2A level and PP2A activity at this age. We found that decrease of I2PP2A level (Supplementary Figure S7) and preservation of PP2A activity (Supplementary Figure S8) were still significant at 18 months. These data further confirm the role of I2PP2A and PP2A in the observed attenuation effects.
Discussion
PP2A is the most active phosphatase in dephosphorylating the hyperphosphorylated tau isolated from the AD brains,2 in which PP2A activity is significantly inhibited.4 PP2A also plays a role in Aβ production7 and synaptic functions.8,25 I2PP2A is a constitutive protein inhibitor of PP2A.10,11 Its level is elevated in the AD brains,18 and upregulation of I2PP2A reproduces AD-like histopathology and memory deficit in rats.20 These studies indicate that silencing I2PP2A may activate PP2A and therefore improve the pathologies and cognitive functions of AD. In the present study, we found an age-dependent elevation of I2PP2A with a subsequent inactivation of PP2A in tg2576 mice, in which tau hyperphosphorylation, Aβ accumulation and memory deficit were reported26,27 and confirmed in the current study. Expression of LV-siI2PP2A downregulated I2PP2A and activated PP2A with a simultaneous attenuation of tau hyperphosphorylation, Aβ accumulation and memory deficits. We also demonstrate that PP2A activation is essential in attenuating amyloidosis by silencing I2PP2A, whereas the improved cognitive function seems independent of PP2A activation but involves preservation of several synapse/memory-associated components. These findings indicate that targeting I2PP2A by LV-shRNA could be a promising strategy in rescuing AD, and the mechanistic studies provide new insights on how I2PP2A regulates PP2A, and how PP2A mediates the effects of I2PP2A on amyloid metabolisms and the cognition-related machineries.
Though it is currently not fully understood how I2PP2A may interact with PP2A and therefore inhibit its activity, it is known that the biological activity of PP2A is regulated by the total level of PP2A catalytic subunit (PP2AC) (the activity-executing subunit), and its inhibitory phosphorylation at Tyr307 and demethylation at Leu309.28,29,30 Our data showed that silencing I2PP2A stimulated PP2A activity through mechanisms involving increase of protein level of PP2AC and decrease of Leu309-demethylation, and reduction of the binding level of PP2AC with I2PP2A. In line with our results, a recent study also reported that silencing I2PP2A results in activation of PP2A due to reduced binding of PP2AC with I2PP2A.31 Our results show that I2PP2A regulates PP2A activity by multiple pathways, involving gene expression, direct interaction and modulating the upstream regulatory factors, such as methyltransferase. Of note, we also noticed that downregulation of I2PP2A at hippocampus not only restored but even further stimulated PP2A activity. Given that the activity of PP2A is significantly decreased in the AD brains, silencing I2PP2A could be a plausible strategy to rescue PP2A.
It is well documented that PP2A is the most implicated phosphatase in dephosphorylating the hyperphosphorylated tau,2 whereas much less is known regarding the role of PP2A in Aβ-related amyloidosis. Aβ is produced from APP by β- and γ-secretase cleavage pathway.32 Under physiological conditions, APP is largely metabolized through α-secretase pathway, by which only low level of Aβ is produced. Many factors, such as activation of β- or/and γ-secretase, can stimulate Aβ production.33,34 In vivo studies strongly suggest that APP phosphorylation facilitates Aβ generation through β-secretase pathway, as in the AD brains and its transgenic mouse models, all Aβ plaques contain pT668-APP or β-secretase, and the production of Aβ is significantly reduced when Thr668-phosphorylation is either abolished by mutation or inhibited with kinase inhibitors.23,24 On the other hand, a recent in vitro study indicates that Thr668-phosphorylation of APP does not affect the C-terminal γ-secretase cleavage.35 Our current data show that the activation of PP2A by silencing I2PP2A decreases APP phosphorylation with a remarkable reduction of Aβ, whereas simultaneous inhibition of PP2A increases APP phosphorylation with accumulation of Aβ we also demonstrate that silencing I2PP2A suppresses β-secretase without affecting γ-secretase. These data together indicate that phosphorylation of APP at Thr668 may facilitate the β-secretase but not γ-secretase cleavage of APP to increase Aβ generation. Since OA may also inhibit PP1, PP4 and PP5,36 we used different approaches, including expression of wtPP2AC or siPP2AC plasmids and specific PP2A activity assay to confirm the role of PP2A in mediating the effects of I2PP2A silencing. These data strongly suggest that stimulation of PP2A by silencing I2PP2A can efficiently inhibit amyloidogenesis through inhibiting APP phosphorylation and β-secretase activity.
We also noticed that the effects of I2PP2A inhibition on tau dephosphorylation were not as prominent as expected in tg2576 mice. It could be because I2PP2A inhibition affects other protein phosphatases (such as PP2B, PP1, and PP5), and as well as tau kinases, which also affect tau phosphorylation. Given the more pronounced effects of siI2PP2A on Aβ deposition and memory impairment than tau phosphorylation, it implies that Aβ may play a more critical role than tau hyperphosphorylation in tg2576 mice. We also observed that the effects of I2PP2A inhibition were much more drastic on Aβ40 than Aβ42 levels. Of note, silencing I2PP2A did not affect γ-secretase, whereas it reduced phosphorylation of APP at Thr668. Consistent with our results, previous studies also observed that phosphorylation of APP led to a more prominent elevation of Aβ40 than Aβ42.37,38 On the basis of these data, we propose that the phosphorylation status of APP at Thr668 may change the conformation of APP and therefore differentially affect its cleavage at the C-terminal.
The tg2576 mice used in the present study show memory deficits at ~12 months old onward, which is consistent with the previous reports.26,27 We therefore injected the mice at 11 months and did the behavioral tests at 12–12.5 months. We found that downregulation of I2PP2A remarkably improved the learning and memory with attenuation of tau and Aβ pathologies, and more impressively, the improvement was still significant when the mice were reared to old ages. The long-term efficiency of lentiviral vector-mediated gene targeting was also observed in previous studies.21 Unexpectedly, we observed that the cognitive improvement by I2PP2A silencing seemed independent of PP2A activation. We found that I2PP2A knockdown increased the levels of NR2A/B, c-fos, synaptophysin, total, and the phosphorylated CREB. Since I2PP2A can also regulate gene replication,39 core histone acetylation,17,40 and protein transcription;41 these functions of I2PP2A may contribute to the alterations of the observed synapse- or memory-associated proteins, which deserves further studies. In addition, I2PP2A silencing also increased the dendritic complexity with increased spine density and mushroom-like spines. The role of neuronal dendrites is to receive and process synaptic inputs; a recent study demonstrates that rapid spine stabilization and synaptic enhancement play an important role at the onset of behavioral learning.42 We believe that the influence of I2PP2A on the memory-associated proteins and/or dendritic morphology at least contribute to the improved synaptic plasticity and cognition.
Taken together, we demonstrate here that silencing the elevated I2PP2A by in vivo lentiviral vector delivery attenuates tau pathologies and amyloidogenesis by restoration of PP2A activity in tg2576 mice; it also improves learning and memory with the mechanisms involving preservation of several memory-associated components. Therefore, I2PP2A could be a promising target for AD.
Materials and Methods
Production and brain infusion of lentiviral constructs. The I2PP2A-shRNA sequence was selected by referring a previous study,43 and it was cloned into LentiLox 3.7. The eGFP sequence was driven by a cytomegalovirus promoter and terminated using polyadenylation signal in the 3′ long terminal repeat. The third generation packaging systems was used for lentiviral production. These vectors include: pMDLg/pRRE (gag/pol elements), pRSV-REV, and pMD.G (env elements). The eGFP-labeled lentiviral I2PP2A-shRNA (LV-siI2PP2A) and the scrambled control (LV-ssiI2PP2A) were constructed according to the standard procedure. The recombinant lentivirus was produced by transient transfection of HEK293T cells using the calcium phosphate method; the virus was harvested at 48 and 72 hours post-transfection and purified by centrifugation at 4 °C. The titer of the virus was 2 × 109 TU/ml. The in vivo knock-down efficiency was measured by immunofluorescence staining, Western blotting, and RT-PCR after injection of LV-siI2PP2A into the mouse brains.
For brain injections, ~11-month-old tg2576 mice harboring the human amyloid precursor protein 695 with Swedish double mutation (APP695; K670N/M671L, Jackson Lab) were positioned respectively in a stereotaxic instrument, then 2 µl LV-siI2PP2A (tg-siI2PP2A) or LV-ssiI2PP2A (tg-ssiI2PP2A) were unilaterally injected into the hippocampus CA3 (AP -2.0, ML -1.5, DV -2.0), CA1 (AP -2.0, ML -1.8, DV -1.1) and the frontal cortex (AP -1.0, ML -2.0, DV -1.5) at a rate of 0.50 µl/min. To receive a comparable result, the age-matched wild-type littermates also received infusion of ssiI2 (wt-ssiI2PP2A). The syringe was left in place for ~3 minutes before being withdrawn. The genotype of the mice was assessed by PCR from tail clip digestions. All mice were kept at 24 ± 2 °C on daily 12 hours light-dark cycles with ad libitum access to food and water. The triple sites injection did not significantly increase the death rate or change the normal activity of the mice compared with the noninjected controls. The expression of the lentiviral vectors in hippocampus was detected in CA3, CA1, and DG regions, whereas the expression in the frontal cortex was only detected in the injected site. Therefore, we used the whole hippocampus and the injected site of the cortex, respectively, for the biochemical measurements. The animal experiments were carried out according to the “Policies on the Use of Animals and Humans in Neuroscience Research” approved by the Society for Neuroscience in 1995; and supervised by Animal Administration and Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology.
Behavioral procedures. Four weeks after brain infusion of the lentiviral vectors, the spatial learning and memory were assessed by Morris water maze test.44 For spatial learning, mice were trained in water maze to find a hidden platform for 7 consecutive days, three trials per day with a 30-second interval from 14:00 to 20:00 pm. On each trial, the mice started from one of the four quadrants facing the wall of the pool and ended when the animal climbed on the platform. If the mice did not locate the platform in 60 seconds, they were guided to the platform. The swimming path and the time used to find the platform (latency) or to pass through the previous platform quadrant were recorded each day by a video camera fixed to the ceiling, 1.5 meters from the water surface. The camera was connected to a digital-tracking device attached to an IBM computer. The spatial memory was tested 2 days (day 9) after the last training. The longer a mouse stayed in the previous platform-located quadrant, the better it scored the spatial memory.
The step-down avoidance test was performed 7 days after the Morris water maze test at ~12.5 months or when the mice were reared to ~18 months by following a previous procedure.45 Briefly, the mouse was kept in the cage for 3 minutes to adapt to the environment before experiments, then the mice received training by delivering electrical shock (36 v) for 3 minutes (the mice will step up onto the platform). The short-term memory (STM) and long-term memory (LTM) were tested respectively at 15 minutes and 24 hours after the training by measuring the step-down latency and the errors made in 3 minutes.
Cell culture and transfection. N2a cells with stable expression of APP695 (N2a/APP) (kind gift of Dr. Xu, Burnham Institute, SD) were cultured in a 1:1 mixture of DMEM and OPTI-MEM supplemented with 15% FBS and 100 U/ml penicillin/100 mg/ml streptomycin in a humidified atmosphere of 5% CO2 in air at 37 °C. The cells were plated onto six-well plates overnight and the plasmids were transfected the next day using Lipofectamine 2000 according to the manufacturer's instruction (Invitrogen, CA).
Real time PCR. Total RNA (3 μg in 25 μl), isolated using Trizol (Invitrogen, CA), was reversely transcribed and the produced cDNA (1 μl) was measured by real time PCR. The primers for I2PP2A: 5′-CTTCAACTCTGGTCAAATAATGCA-3′, 5′-GAACAAAAATATAACAAACTCCGC-3′ for β-site APP cleaving enzyme 1 (BACE1): 5′-CGG-GAGTGGTATTATGAAGTG-3′, 5′-AGGATGGTGATGCGGAAG-3′ and for glyceraldehyde phosphate dehydrogenase (GAPDH): 5′-GAAGGTGAAGGTCGGAGTC-3′, 5′-GAAGATGGTGATGGGATTTC-3′.
Western blot. The hippocampi were rapidly removed and homogenized at 4 °C using a Teflon glass homogenizer in 50 mmol/l Tris-HCl, pH 7.4, 150 mmol/l NaCl, 10 mmol/l NaF, 1 mmol/l Na3VO4, 5 mmol/l EDTA, 2 mmol/l benzamidine, and 1 mmol/l phenylmethylsulfonyl fluoride. The extract was mixed with sample buffer (3:1, v/v) containing 200 mmol/l Tris-HCl, pH 7.6, 8% SDS, 40% glycerol, 40 mmol/l dithiothreitol, boiled for 10 minutes, and then centrifuged at 12,000g for 10 minutes at 25 °C. The supernatant was stored at –80 °C for Western blot analysis. The protein concentration in the supernatant was estimated by bicinchoninic acid (BCA) kit according to manufacturer's instructions. For general protein analyses, 10% SDS-polyacrylamide gel electrophoresis was used to isolate the protein and nitrocellulose membrane was used for transfer (10 mA for 2 hours followed by 276 mA for 1 hour). For analyzing Aβ, the proteins were separated on 8–12% Tris-tricine gradient gel and transferred to 0.2 µm PVDF membranes (150 mA for 14 hours followed by 600 mA for 1 hour). The membranes were blocked with 5% nonfat milk dissolved in TBS Tween-20 (50 mmol/l Tris HCl, pH 7.6, 150 mmol/l NaCl, 0.2% Tween-20) for 1 hour and probed with primary antibody at 4 °C for overnight. Then, the blots were incubated with antimouse or anti-rabbit IgG conjugated to horseradish peroxidase (1:5,000) for 1 hour at 37 °C, and visualized with enhanced chemiluminescence. The blots were quantitatively analyzed by Kodak Digital Science 1D software (Eastman Kodak, New Haven, CT). The antibody used in the current study is listed in Supplementary Table S1.
ELISA, immunocytochemistry and quantitative analysis. The Aβ levels were measured using Signal Select β-Amyloid ELISA Kits (Biosource, NY). Immunohistochemical studies were performed as described previously,46 and immunofluorescence imaging was carried out using a laser scanning confocal microscope (Olympus FV500, Tokyo, Japan). The quantitative analysis of Aβ deposits was performed as described by Oddo et al.47 Briefly, brain sections were stained with monoclonal anti-β-amyloid antibody, which detects Aβ40 and Aβ42. The images of stained sections were captured using an Olympus Provis AX80 microscope (B&B Microscopes, Philadelphia, PA) and imported into Image-Pro Plus 6.2 software (Media Cybernetics, Bethesda, MD). The area covered with Aβ deposits in the virus-injected regions of the hippocampus or cortex was measured. Three to five coronal sections were analyzed per mouse, and the average ratios were used to calculate group means.
PP2A and secretase activity assay. PP2A activity was measured using a Serine/Threonine Phosphatase Assay Kit by following the manufacturer's instructions, which can specifically measure PP2A but not the other protein phosphatases through selection of specific substrate and alternative buffer systems (Promega, MA). The absorbance was read at 600 nm (BioTek Instruments, VT). The activity of α-, β- or γ-secretase was estimated using α-, β- or γ-Secretase Activity Kits by following the manufacturer's instructions (R&D Systems, MN) and the absorbance was read at excitation 355 nm and emission 510 nm (BioTek Instruments, VT).
Golgi impregnation and dendritic morphology analysis. For Golgi stain, the mice were anesthetized and then perfused transcardially with 4% paraformaldehyde, and brain tissue was processed as described.48 Individual sections were incubated overnight at room temperature in water solution of 3.5% K2Cr2O7 and 0.4% OsO4. The sections were then sandwiched in two glass slides and incubated in 1% AgNO3(aq) for 5 hours at room temperature in dark. Then the slide assemblies were dismantled in water and the sections were mounted on gel-coated slides (0.5% porcine gelatin), dehydrated in a series of graded ethanol rinses, cleared with Histoclear, and cover slipped with cytoseal. The images were taken using Olympus BX60 (Tokyo, Japan).
To analyze the dendritic morphology, Z-stacks (step size 1 µm) from 5 to 7 neurons were generated using a confocal microscope (LSM510, Zeiss) in bright-field mode (20×objective) and reconstructed in ImagePro in combination with the NeuroDraw toolbox for each animal. Total number of branch points was analyzed for every neuron.
In addition, the spine density was determined in two segments of dendrites at a distance of 90–110 µm (proximal) and 190–210 µm (distal) from the soma. To acquire images for spine analysis, the dendritic segments were imaged under bright-field illumination on a Zeiss Axioimager microscope with a 63× oil immersion objective, and spine morphology was analyzed according to a previously reported method,49 which does not assess spine density in a three dimensional manner but focuses on spines paralleled to the plane of section. Although the method may underestimate the total number of spines, it facilitates a direct comparison of treatment groups when they are analyzed in an identical manner. Image J software was used to calculate linear spine density,50 which was presented as the number of spines per 10 mm of dendrite length. On the basis of morphology, spines were classified into the following categories: (i) thin: spines with a long neck and a visible small head; (ii) mushroom: big spines with a well-defined neck and a very voluminous head; and (iii) stubby: very short spines without a distinguishable neck and stubby appearance. The ratio of mushroom spine was calculated. Data from 5 to 7 neurons were averaged per animal and used in further statistical analysis.
Statistical analysis. Data were analyzed with SPSS 12.0 statistical software. The one-way analysis of variance procedure followed by least significant difference post hoc tests was used to determine the statistical significance of the means.
SUPPLEMENTARY MATERIAL Figure S1. The age-dependent alterations of I2PP2A and PP2A activity in tg2576 mice and the wild-type littermates. Figure S2. Alteration of the cytoplasmic translocation of I2PP2A in the hippocampus of 12.5-month tg2576 mice. Figure S3. The direct fluorescence images after hippocampal (CA1 and CA3) and frontal cortex infusion of LV-siI2PP2A for 6 weeks. Figure S4. An age-dependent alteration of Aβ accumulation in tg2576 mice. Figure S5. The representative images show plaque load in the cortex of ~23 months tg2576 mice adjacent to the injection site. Figure S6. Measurement of glial fibrillary acidic protein (GFAP, a marker of astrocytes) and Iba-1 (a marker of microglia) by Western blotting. Figure S7. Alterations of I2PP2A in hippocampus of tg2576 mice measured at 18 months after infused with LV-siI2PP2A or the scrambled control at 11 months. Figure S8. Alteration of PP2A activity in hippocampus of tg2576 mice measured at 18 months after infused with LV-siI2PP2A or the scrambled control at 11 months. Table S1. Antibodies employed in the study.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81270418 and 91132305), Alzheimer's Association (IIRG-09-133433), and FIRCA grant (1R03 TW008744-01A1). The authors declare no conflict of interest.
Supplementary Material
References
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–4860. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, et al. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience. 2003;118:1175–1182. doi: 10.1016/s0306-4522(02)00697-8. [DOI] [PubMed] [Google Scholar]
- Tian Q, Lin ZQ, Wang XC, Chen J, Wang Q, Gong CX, et al. Injection of okadaic acid into the meynert nucleus basalis of rat brain induces decreased acetylcholine level and spatial memory deficit. Neuroscience. 2004;126:277–284. doi: 10.1016/j.neuroscience.2004.03.037. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, Dayal S, et al. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci. 2007;27:2751–2759. doi: 10.1523/JNEUROSCI.3316-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LQ, Zheng HY, Peng CX, Liu D, Li HL, Wang Q, et al. Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2. J Neurosci. 2010;30:3839–3848. doi: 10.1523/JNEUROSCI.5174-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry. 1995;34:1988–1996. doi: 10.1021/bi00006a020. [DOI] [PubMed] [Google Scholar]
- Li M, Damuni Z. I1PP2A and I2PP2A. Two potent protein phosphatase 2A-specific inhibitor proteins. Methods Mol Biol. 1998;93:59–66. doi: 10.1385/0-89603-468-2:59. [DOI] [PubMed] [Google Scholar]
- Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3' half to different genes: characterization of the set gene. Mol Cell Biol. 1992;12:3346–3355. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CM, Gallouzi IE, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol. 2000;151:1–14. doi: 10.1083/jcb.151.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela N, Rodriguez-Vilarrupla A, Estanyol JM, Diaz C, Pujol MJ, Agell N, et al. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J Biol Chem. 2003;278:1158–1164. doi: 10.1074/jbc.M207497200. [DOI] [PubMed] [Google Scholar]
- Compagnone NA, Zhang P, Vigne JL, Mellon SH. Novel role for the nuclear phosphoprotein SET in transcriptional activation of P450c17 and initiation of neurosteroidogenesis. Mol Endocrinol. 2000;14:875–888. doi: 10.1210/mend.14.6.0469. [DOI] [PubMed] [Google Scholar]
- Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104:119–130. doi: 10.1016/s0092-8674(01)00196-9. [DOI] [PubMed] [Google Scholar]
- Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. 2005;166:1761–1771. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 2005;579:363–372. doi: 10.1016/j.febslet.2004.11.097. [DOI] [PubMed] [Google Scholar]
- Wang X, Blanchard J, Kohlbrenner E, Clement N, Linden RM, Radu A, et al. The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 2010;24:4420–4432. doi: 10.1096/fj.10-158477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen PI, Koponen JK, Kärkkäinen AM, Malm TM, Pulkkinen KH, Koistinaho J, et al. Stable RNA interference: comparison of U6 and H1 promoters in endothelial cells and in mouse brain. J Gene Med. 2006;8:433–441. doi: 10.1002/jgm.860. [DOI] [PubMed] [Google Scholar]
- Liu XP, Zheng HY, Qu M, Zhang Y, Cao FY, Wang Q, et al. Upregulation of astrocytes protein phosphatase-2A stimulates astrocytes migration via inhibiting p38 MAPK in tg2576 mice. Glia. 2012;60:1279–1288. doi: 10.1002/glia.22347. [DOI] [PubMed] [Google Scholar]
- Shin RW, Ogino K, Shimabuku A, Taki T, Nakashima H, Ishihara T, et al. Amyloid precursor protein cytoplasmic domain with phospho-Thr668 accumulates in Alzheimer's disease and its transgenic models: a role to mediate interaction of Abeta and tau. Acta Neuropathol. 2007;113:627–636. doi: 10.1007/s00401-007-0211-z. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenceau A, Billard JM, Haditsch U, Mansuy IM, Dutar P. Different phosphatase-dependent mechanisms mediate long-term depression and depotentiation of long-term potentiation in mouse hippocampal CA1 area. Eur J Neurosci. 2003;18:1279–1285. doi: 10.1046/j.1460-9568.2003.02831.x. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, et al. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004;63:1080–1091. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- Sontag JM, Nunbhakdi-Craig V, Mitterhuber M, Ogris E, Sontag E. Regulation of protein phosphatase 2A methylation by LCMT1 and PME-1 plays a critical role in differentiation of neuroblastoma cells. J Neurochem. 2010;115:1455–1465. doi: 10.1111/j.1471-4159.2010.07049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud L, Chen S, Liu F, Li B, Khatoon S, Grundke-Iqbal I, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett. 2011;585:2653–2659. doi: 10.1016/j.febslet.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Luo X, Yan R. Inhibition of BACE1 for therapeutic use in Alzheimer's disease. Int J Clin Exp Pathol. 2010;3:618–628. [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- Matsushima T, Saito Y, Elliott JI, Iijima-Ando K, Nishimura M, Kimura N, et al. Membrane-microdomain localization of amyloid ß-precursor protein (APP) C-terminal fragments is regulated by phosphorylation of the cytoplasmic Thr668 residue. J Biol Chem. 2012;287:19715–19724. doi: 10.1074/jbc.M111.334847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Su Y, Li B, Zhou Y, Ryder J, Gonzalez-DeWhitt P, et al. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547:193–196. doi: 10.1016/s0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- Ryoo SR, Cho HJ, Lee HW, Jeong HK, Radnaabazar C, Kim YS, et al. Dual-specificity tyrosine (Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: evidence for a functional link between Down syndrome and Alzheimer's disease. 2008;104:1333–1344. doi: 10.1111/j.1471-4159.2007.05075.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Nagata K, Ui M, Hanaoka F. Template activating factor I, a novel host factor required to stimulate the adenovirus core DNA replication. J Biol Chem. 1993;268:10582–10587. [PubMed] [Google Scholar]
- Kutney SN, Hong R, Macfarlan T, Chakravarti D. A signaling role of histone-binding proteins and INHAT subunits pp32 and Set/TAF-Ibeta in integrating chromatin hypoacetylation and transcriptional repression. J Biol Chem. 2004;279:30850–30855. doi: 10.1074/jbc.M404969200. [DOI] [PubMed] [Google Scholar]
- Okuwaki M, Nagata K. Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J Biol Chem. 1998;273:34511–34518. doi: 10.1074/jbc.273.51.34511. [DOI] [PubMed] [Google Scholar]
- Roberts TF, Tschida KA, Klein ME, Mooney R. Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature. 2010;463:948–952. doi: 10.1038/nature08759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster JP, Leeuwen Iv, Scheres N, Anthony EC, Hordijk PL. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 2007;26:336–345. doi: 10.1038/sj.emboj.7601518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GP, Wei W, Zhou X, Zhang Y, Shi HH, Yin J, et al. I(2)(PP2A) regulates p53 and Akt correlatively and leads the neurons to abort apoptosis. Neurobiol Aging. 2012;33:254–264. doi: 10.1016/j.neurobiolaging.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFela FM. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2006;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magariños AM, Li CJ, Gal Toth J, Bath KG, Jing D, Lee FS, et al. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2011;21:253–264. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Kay K, Matsouka R, Rozkalne A, Betensky RA, Hyman BT. Calcineurin inhibition with systemic FK506 treatment increases dendritic branching and dendritic spine density in healthy adult mouse brain. Neurosci Lett. 2011;487:260–263. doi: 10.1016/j.neulet.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.