

Abstract
Chutes and Ladders is an exciting up-and-down-again game in which players race to be the first to the top of the board. Along the way, they will find ladders to help them advance, and chutes that will cause them to move backwards. The development of nucleoside analogs for clinical treatment of hepatitis C presents a similar scenario in which taking shortcuts may help quickly advance a program, but there is always a tremendous risk of being sent backwards as one competes for the finish line. In recent years the treatment options for chronic hepatitis C virus (HCV) infection have expand due to the development of a replicon based in vitro evaluation system, allowing for the identification of multiple drugable viral targets along with a concerted and substantial drug discovery effort. Three major drug targets have reached clinical study for chronic HCV infection: the NS3/4A serine protease, the large phosphoprotein NS5A, and the NS5B RNA-dependent RNA polymerase. Recently, two oral HCV protease inhibitors were approved by the FDA and were the first direct acting anti-HCV agents to result from the substantial research in this area. There are currently many new chemical entities from several different target classes that are being evaluated worldwide in clinical trials for their effectiveness at achieving a sustained virologic response (SVR) (Pham et al., 2004; Radkowski et al., 2005). Clearly the goal is to develop therapies leading to a cure that are safe, widely accessible and available, and effective against all HCV genotypes (GT), and all stages of the disease. Nucleoside analogs that target the HCV NS5B polymerase that have reached human clinical trials is the focus of this review as they have demonstrated significant advantages in the clinic with broader activity against the various HCV GT and a higher barrier to the development of resistant viruses when compared to all other classes of HCV inhibitors.
Keywords: Hepatitis C virus, HCV, nucleoside analog, nucleotide analog, antiviral, clinical evaluation, prodrug, clinical trials, nucleosides
1. Introduction
The hepatitis C virus (HCV) was identified in 1989 as a member of the Flaviviridae family of positive-stranded RNA virus (Choo et al., 1989). Infection of a human host by HCV results in a serious infection affecting about 3% of the worldwide population, according to the World Health Organization (WHO). The WHO also estimates that 4 million people contract HCV each year (WHO, 2012). Although the early stages of HCV infection are usually asymptomatic, and about 20% of infected individuals will naturally clear the virus, a majority of infections progress to chronic infection. A significant number of chronically infected individuals will eventually develop more serious liver problems such as cirrhosis, hepatocellular carcinoma (HCC), or liver failure requiring liver transplantation (Darby et al., 1997; Poynard et al., 2000; Tong et al., 1995). The global health burden of HCV-associated morbidity and mortality is expected to increase substantially during the next decades, as many chronically infected individuals progress to end stage disease with associated complications (Davis et al., 2003; Manns et al., 2007). In fact, in industrialized nations, HCV infection is already the leading cause for liver transplantations. Unfortunately, re-infection of the transplanted liver from an unknown reservoir often occurs post-transplantation (Hoofnagle, 1997). The virus is transmitted parenterally by contaminated blood most often from sharing contaminated needles, but also from improper sterilization of medical, dental, body piercing, or tattoo equipment. Heterosexual transmission and vertical transmission (infected mother to her child during the birthing process) of HCV can also occur, but are rare (Fishman et al., 2009; Ghosn et al., 2009). Sexual practices that involve higher levels of trauma to the anogenital mucosa or that occur when there is a concurrent sexually transmitted infection, such as HIV or genital ulceration, do present a higher risk of HCV transmission (Tohme and Holmberg, 2010).
Three major drug targets have reached evaluation in humans for chronic HCV infection (Farnik and Zeuzem, 2012; Schaefer and Chung, 2012): the NS3/4A serine protease, the large phosphoprotein NS5A and NS5B RNA-dependent RNA polymerase (RdRp) (Bobeck et al., 2010; De Francesco and Migliaccia, 2005; Huang et al., 2006). The majority of drugs that target the HCV NS3/4A serine protease are peptidomimetics and represent the only two FDA approved direct acting antiviral agents for treatment of HCV infection. Although the ultimate role of NS5A in the HCV replication cycle is not fully understood, the 49 kDa NS5A protein is required for HCV replication as it is part of the membrane bound replication complex (Lemon et al., 2010). The two main categories of HCV NS5B RdRp inhibitors are non-nucleoside and nucleoside analog inhibitors (Brown, 2009; Burton and Everson, 2009; Legrand-Abravanel et al., 2010), which differ in their chemical structure, barrier to resistance, pan-genotypic activity and mode of action.
The current FDA-approved treatments for chronic HCV are limited to pegylated interferon-α (IFN) alone or in combination with ribavirin (RBV) with or without protease inhibitors (PI) boceprevir (Victrelis®) and telaprevir (Incivek®) (Fig. 1) (Hughes and Shafran, 2006; Pan et al., 2012; Sheridan, 2011). Non-pegylated IFNs have also been approved including the recombinant IFN alfacon1 and natural IFN, purified from the leukocyte fraction of human blood, multiferon. Unfortunately, these treatments have limited efficacy for the GT1 population, which is the most prevalent GT in the US and China and emphasize the urgent unmet medical need for new improved therapeutic agents that are pangenotypic (Manns et al., 2001; McCown et al., 2008). These treatments also have major side effects including depression, low red and/or white blood cell counts, central nervous system toxicity, fatigue, headache, flu-like symptoms, mild alopecia, thyroid dysfunction, itching, swelling of the face, eyes, lips, tongue, or throat, skin rashes, hemorrhoids, diarrhea and teratogenic effects (Fried, 2002; Gaetano and Reau, 2013; Gordon and Keller, 2005; Hezode et al., 2012; Keicher et al., 2005; Koch and Narjes, 2007; McHutchison et al., 2002; Pawlotsky, 2003; Wong and Lee, 2006).
Fig. 1.
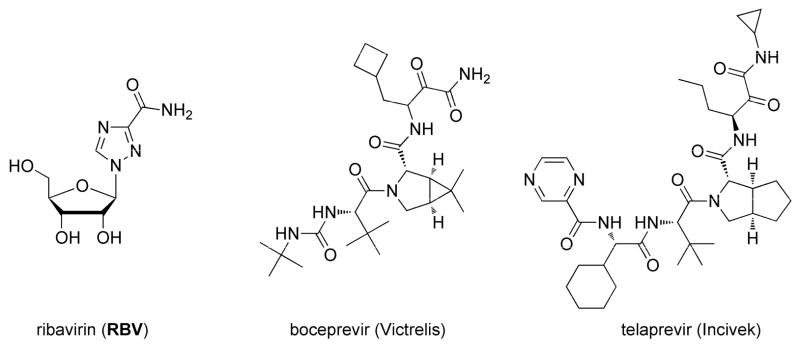
Structures for ribavirin (RBV), boceprevir (Victrelis) and telaprevir (Incivek).
In the clinic, the primary treatment goal for chronically infected HCV (CHC) persons is lifelong clearance of detectable HCV infection. When new anti-HCV chemical entities are evaluated in clinical trials, the prediction of lifelong clearance of HCV infection is often assessed by a surrogate endpoint termed sustained virologic response (SVR). SVR is determined at ≥ 12 weeks after discontinuation of antiviral therapy and defined as non-detectable HCV RNA in serum that is measured by sensitive assays such as a polymerase chain reaction (PCR) based determination. Two earlier predictors of SVR during clinical trials are rapid virologic response (RVR) and early virologic response (EVR) which are defined as reduction of serum HCV RNA to PCR-non-detectable levels by week 4 and achievement of ≥ 2 log10 reduction in serum HCV RNA level by week 12, respectively. Not all persons who eventually achieve an SVR will have met the EVR or RVR milestones and conversely reaching any or all of these three categories do not guarantee lifelong clearance of detectable HCV infection. However, these guidelines are predictive of ultimate therapy success and help clinicians determine, for example, if persons with significant side effects that fail to achieve EVR or RVR should discontinue treatment.
Nucleoside analogs that target the HCV NS5B polymerase that have reached human clinical trials will be the focus of this review as they have demonstrated significant advantages in the clinic with broader activity against the various HCV GT and a higher barrier to the development of resistant viruses when compared to all other classes of HCV inhibitors (McCown et al., 2008). The HCV NS5B polymerase catalytic site is highly conserved across the various genotypes resulting in little variation of potency of the nucleoside triphosphate inhibitors that target this protein (Asselah and Marcellin, 2012; Waheed et al., 013). Selection of HCV mutants in vitro has been quite difficult with most nucleoside inhibitors and selected mutants have been found markedly unfit with little to no clinical significance (Najera, 2013; Tong et al., 2013). There have been several nucleoside analogs discovered that exhibit selective anti-HCV activity, most with modifications at the 2′ and/or 4′ positions of the ribose sugar. However, nucleoside analogs as a class have potential problems such as poor passive cellular permeability and the modified nucleoside may not be a suitable substrate for the three successive cellular kinases that are necessary to transform it to the biologically active nucleoside analog triphosphate (NTP) (Fig. 2). Since HCV lacks nucleoside kinase expression, conversion to the biologically active nucleoside analog triphosphate relies on the human cellular kinases (Meppen et al., 2009). In addition, antiviral activity of nucleoside analogs is heavily influenced by the levels of natural dNTP or rNTP (Fig. 2) since they compete for incorporation by viral polymerases. Nucleoside analog inhibitors of viral RNA polymerization have the additional challenge of 10 to 100 times higher levels of rNTPs relative to the corresponding dNTP (Kennedy et al., 2010; Medivir, 2011a, b; Traut, 1994). Additionally, another significant obstacle in the development of nucleoside analogs for HCV therapy is their potential to be substrates and/or inhibitors of host polymerases. As an example, in the recent unfortunate events related to the 2′-methyl substituted guanosine analogs IDX184 and INX-189, the human mitochondrial RNA polymerase has become of significant concern (Arnold et al., 2012).
Fig. 2.

Nucleosides transform via three successive cellular kinases to the biologically active nucleoside analog triphosphates. Natural ribo- and 2′-deoxyribonucleoside bases.
Prodrugs are defined as a pharmacological substance administered in an inactive (or significantly less active) form that is metabolized into an active metabolite. Prodrugs are most often employed to improve absorption, distribution, metabolism, and/or elimination (ADME) properties of the parent pharmacological substance. One of the most common applications of prodrugs is for a drug that is poorly absorbed through the gastrointestinal tract to improve oral bioavailability. A prodrug may also be used to improve the selectively by which the drug interacts with the intended target cells or processes versus those that are not the drug’s intended target. In the HCV area, nucleoside prodrugs have demonstrated both improvement of oral bioavailability and the ability to selectively deliver nucleotide analogs to liver tissue by first pass metabolism. Nucleoside analogs themselves are prodrugs of the active triphosphate form, which is recognized and incorporated by viral polymerases or reverse transcriptases. Antiviral activity of a nucleoside analog can often be improved by conversion to a prodrug as this aids in absorption and/or cellular penetration. In the case of monophosphate prodrugs, the potential rate limiting initial phosphorylation step is bypassed leading to higher intracellular nucleoside analog monophosphate levels which in many cases leads to significantly higher triphosphate levels that can translate to a significant enhancement in antiviral activity (De Clercq and Field, 2006; Hecker and Erion, 2008; Li et al., 2008; Poijärvi-Virta and Lönnberg, 2006; Sofia, 2011; Sofia et al., 2010a).
Prodrugs have played a central role in the delivery of active agents to their target tissue with improved efficiency and fewer side effects for well over 100 years. One of the earliest and enduring prodrug applications was to salicylic acid which when converted to acylsalicylic acid (Gerhardt, 1853; Von Gilm, 1859), significantly reduced irritation to the mouth and gastrointestinal tract (Fig. 3). Salicylic acid has been used for thousands of years to ease pain and reduce fever, but it was not until the period from 1826 to 1838 that salicylic acid was isolated by a series of chemists from willow tree bark (Jeffreys, 2005).
Fig. 3.

Synthesis of acylsalicylic acid from salicylic acid.
In 1856 an industrial scale process to make salicylic acid was developed from phenol and carbon dioxide (Kolbe, 1860; Schmitt, 1885), but interestingly the correct chemical structure was not assigned until 1869 that the correct chemical structure was assigned (Schröder and Kraut, 1869). In 1897, approximately 44 years after the first synthesis, acylsalicylic acid was first prepared by Bayer and in less than two years aspirin was trademarked and was being sold worldwide. As of 2013 (160 years after the first synthesis) more than 41 kilotons (metric) of aspirin is consumed annually worldwide. Many modern prodrugs utilize the same simple ester masking of a free hydroxyl group as a prodrug strategy as is exemplified by 3′-L-valinyl ester prodrug NM283 (Fig. 4) or 2′,3′,5′-triisobutyrate ester prodrug R1626 (Fig. 5) and 3′,5′-diisobutyryl ester prodrug RG7128 (Scheme 4) which are among the first HCV clinical candidates.
Fig. 4.

Chemical structures of NM107 and NM283.
Fig. 5.

4′-Azidocytidine analogs used in the human clinical treatment of HCV.
Scheme 4.

Synthesis of RG7128. Reagents and conditions: (a) DMAP, THF/H2O, TEA, isobutyryl chloride, pH 6.4, 0 °C.
2. Valopicitabine (NM283): the first clinical HCV nucleoside
2.1. Preclinical development
2′-C-Methylcytidine (Harry-O’kuru et al., 1997) also known as NM107 (Sommadossi et al., 2008; Sommadossi and La Colla, 2001a, b; Sommadossi et al., 2004a; Sommadossi et al., 2004b, c; Storer et al., 2004), was discovered by Idenix Pharmaceuticals to be a potent inhibitor of RNA viruses replication such as bovine viral diarrhea virus (BVDV), West Nile virus (WNV), dengue-2 virus, yellow fever virus (YFV) and subsequently HCV (Kowdley et al., 2013; Sommadossi and La Colla, 2001b). Despite its broad spectrum in vitro activity, NM107 displayed low oral bioavailability. To overcome this issue, NM283 (Sommadossi et al., 2008; Sommadossi and La Colla, 2001a, b; Sommadossi et al., 2004a; Sommadossi et al., 2004b, c; Storer et al., 2004) also called valopicitabine, the 3′-O-L-valinyl ester prodrug of NM107, was developed (Pierra et al., 2005). In March 2006, Novartis announced to exercise its option from Idenix Pharmaceuticals to in-license the late stage development of valopicitabine.
2.2. Synthesis of valopicitabine (NM283)
Large scale synthesis of NM107 and NM283 were achieved from commercially available D-fructose by first reaction with calcium oxide and then treatment with oxalic acid to give the key 2′-C-Me-lactone 1. Subsequent benzoylation, reduction of the lactone with Red-Al and benzoylation of the lactol 3 afforded compound 4. At this stage, Vorbrüggen type coupling with cytosine using SnCl4 as Lewis acid followed by debenzoylation gives access to NM107. NM283 is finally obtained from NM107 after simple peptidic coupling with N-Boc-L-valine and Boc-removal under acidic condition (Scheme 1) (Pierra et al., 2005).
Scheme 1.

Synthesis of valopicitabine (NM283). Reagents and conditions: (a) i. CaO, water; ii. CO2, oxalic acid, 18%; (b) BzCl, TEA, DMAP, DME, 83%; (c) Red-Al/EtOH, toluene, 99%; (d) BzCl, TEA, DMAP, DME, 52%; (e) bis(trimethylsilyl)acetamide, cytosine, SnCl4, CAN; (f) MeONa, MeOH, r.t., 90% over two steps; (g) N-Boc-L-valine, CDI, DMAP, TEA, DMF/THF, 91%; (h) HCl, EtOH, 97%.
2.3. Clinical development
2.3.1. Phase I/II
In 2004, the first phase I/II study of valopicitabine was reported, wherein 48 subjects received valopicitabine at 50, 100, 200, 400 and 800 mg/day orally once daily for 15 days with 14 days of follow-up; a subset of subjects received 400 mg twice daily (Godofsky et al., 2004). During these escalation trials, a dose-related decrease of serum HCV RNA at Day 16 was observed ranging from 0.15 log10 at the lowest dose to 0.73 log10 at 400 mg/twice daily. At 400 mg/day, 50% of the subjects experienced transient mild nausea and 20% had vomiting, but all completed treatment without modifications.
2.3.2. Phase IIa
Following the discovery of a synergistic antiviral effect against BVDV, in vivo, between NM107, the parent compound of valopicitabine (NM283) and IFN, a combination study with treatment-naïve subjects involving valopicitabine and IFN was initiated (Afdahl et al., 2005). Valopicitabine was given for 24 weeks with escalation to 800 mg/day over the first week before IFN was administered (1.0 μg/kg weekly) on Day 8. Along with a satisfactory tolerance, HCV RNA reductions increased to 3.2 log10 IU/mL for the combination treatment group (compared to 1.0 log10 IU/mL for monotherapy group). Eleven of twelve subjects treated for at least 10 weeks had substantial HCV RNA reductions superior to 2 log10 and four subjects were PCR-negative (<10 IU/mL).
2.3.3. Phase IIb
In a Phase IIb clinical trial, a 48 weeks combination of valopicitabine and IFN was evaluated in treatment-naïve subjects with HCV GT1 (Dieterich et al., 2006; Lawitz et al., 2007). This study was divided into five treatment groups: (A) IFN alone (180 μg QW = once weekly) for 4 weeks, valopicitabine 800 mg/day starting at week 5; (B) valopicitabine 200 mg/day + IFN (180 μg QW); (C–E) valopicitabine 800 mg/day with various week 1 induction regimens + IFN (180 μg QW). However, due to high gastrointestinal (GI) side effects at 800 mg/day dose of valopicitabine, the protocol was amended to reduce the dose to 200 or 400 mg/day after 14–22 weeks of treatment. The rate of viral decline was shown to be valopicitabine dose-related through week 4. HCV RNA was below 600 IU/mL in most subjects by week 12 and below 20 IU/mL by week 24 and 36. Group B maintained suppression of HCV RNA through week 48 along with satisfactory tolerability.
2.3.4. Phase IIb
In another Phase IIb trial (Afdahl et al., 2005), a 72 week three arm combination study involving valopicitabine/IFN in non-responder subjects was designed. Subjects were randomized to: (A) valopicitabine monotherapy (800 mg/day) and 3 combinations arms: IFN (180 μg SQ/week) with RBV (1,000–1,200 mg/day) + (A) valopicitabine (400 mg/day); (C) dose escalation 400–800 mg/day; (D) 800 mg/day or IFN/RBV retreatment. Again, due to GI issues with the 800 mg/day dose, the maximum dose was reduced to 400 mg/day. At week 48, HCV RNA suppression of treated persons with doses of 800 mg/day or escalated-dose 400 to 800 mg/day of valopicitabine, was 6 fold greater (0.8 log10) than IFN/RBV treatment demonstrating the potency of valopicitabine for the treatment of non-responder persons.
2.4. Conclusions
Twelve days after Idenix announced on July 1st 2007, that a triple therapy with valopicitabine and IFN/RBV cleared HCV RNA in 72% of subjects after 12 weeks of treatment, the FDA requested that the clinical trials on valopicitabine be discontinued due “to the overall risk/benefit profile observed to date in clinical testing” (Idenix, 2007). From the beginning, this drug as plagued with GI issues and unfortunately was not potent enough to be used alone or even when combined with the standard of care (IFN/RBV).
3. R1479 and its prodrug R1626: balapiravir
3.1. Preclinical development
Discovered and developed by Hoffmann-La Roche, Inc., but aided by technology and compound libraries obtained by the 1994 purchase of Syntex Research, the 4′-azidocytidine derivative R1479 (Ahmed et al., 2007; Connolly et al., 2005; Devos et al., 2002; Harrington et al., 2008; Martin et al., 2004) was identified as a potent and selective inhibitor of HCV RNA polymerase. R1479 displayed an EC50 of 1.3 μM in an HCV replicon system and did not exhibit cytotoxic or cytostatic behavior in the Huh-7 replicon cell line (Smith et al., 2007). The triphosphate form of R1479 was found to be a substrate for HCV polymerase and it inhibited RNA chain extension after incorporation as a non-obligatory chain terminator with an IC50 of 320 nM (Klumpp et al., 2006). In vitro and in vivo assessment of R1479 revealed that like many other nucleoside analogs, R1479 was a polar molecule with a low aqueous solubility of 10 mg/mL that led to limited absorption after oral administration (Alfredson et al., 2006; Toniutto et al., 2008). Further, R1479 exhibited low Caco-2 permeability [(Artursson et al., 1996); Caco-2 cells are used as a model system to predict intestinal epithelial permeability] and oral bioavailability values ranging from 22% in monkey to 89% in rat. Oral absorption in human clinical studies showed 6–18% based on urine recovery.
Consequently, prodrug approaches were investigated (Alfredson et al., 2006) to improve the oral bioavailability of R1479. Prodrug approaches that increase lipophilicity of a compound increase the potential for passive transport across cellular membranes and thus potentially improve absorption and transport across cellular membranes. The first approach focused on using esters and amides at the 2′, 3′ or 5′ positions of sugar to increase the lipophilicity, the second approach focused on increased lipophilicity with liver targeted cleavage using carbamate prodrugs at N4 position of cytosine and the third approach focused on amino acid esters to potentially target the PepT1 transporter for increased active intracellular transport. A five-fold increase in plasma exposure was observed with tri-isobutyrate ester prodrug R1626 (Ahmed et al., 2007; Connolly et al., 2005; Devos et al., 2002; Harrington et al., 2008; Martin et al., 2004) in oral bioavailability in rats and monkeys. Further, results from Caco-2 experiments (Brandl et al., 2008) indicated that the hydrolysis of the prodrug is rapid and converted to R1479 by esterases in gastrointestinal epithelial cells. Exceptional enhancement of dose proportionality combined with a 3-fold enhancement of plasma exposure both in human studies versus oral administration of R1479 resulted in the selection of tri-isobutyrate ester prodrug R1626 for further clinical evaluation.
3.2. Synthesis of R1479 and its prodrug balapiravir (R1626)
The synthesis of 4′-azidocytidine (R1479) (Connolly et al., 2005) and its triisobutyrate ester prodrug R1626 (Sarma, 2007) is outlined in Scheme 2. Starting from uridine, 5′-iodo uridine was prepared then base eliminated to the 4′-exocyclic methylene nucleoside analog 7. Addition of iodoazide across the double bond and benzoylation gave the 5′-iodo,4′-azido bis-benzoylated nucleoside 8. Oxidative displacement of the 5′-iodo gave the 5′-m-chlorobenzyl ester 9 that was converted to the cytidine analog and debenzoylated to give R1479. R1626 was then formed in good yield by reaction with isobutyric anhydride in the presence of potassium hydroxide (Scheme 2).
Scheme 2.

Synthesis of R1479 and its prodrug balapiravir (R1626). Reagents and conditions: (a) I2, PPh3, imidazole, THF, 83%; (b) NaOMe, MeOH, 75%; (c) BnN(Et)3Cl, NaN3, 4-NMM, I2, CH3CN; (d) BzCl, Pyr., 74% over two-steps; (e) K2HPO4, m-CPBA, CH2Cl2, 71%; (f) 1,2,4-triazole, POCl3, Et3N, MeCN, 88%; (g) NH4OH, MeOH, H2SO4-isopropanol, 95%; (h) isobutyric anhydride, KOH, DMAP, THF, 80%.
3.3. Clinical development
3.3.1. Phase I
Starting in 2006, balapiravir (R1626) was given to healthy subjects in single doses from 500 mg to 12,000 mg to evaluate the safety, tolerability and pharmacokinetics. High levels of R1479 was found in plasma accompanied by low levels of both mono and diester analogs. R1479 was found in plasma in a linear amount proportional to all doses studied. Forty healthy male subjects (13 to 34 years old) were randomized 3:1 for treatment with R1626 or placebo and were followed up for 7 days. The most common reported adverse effects were headache and irritation at the site of ECG electrode application. The phase I study concluded that balapiravir was efficiently absorbed, rapidly converted to R1479 and was tolerated up to a dose of 12,000 mg when orally administered in healthy subjects (Robson et al., 2007).
3.3.2. Phase Ib
This clinical trial was designed to study the effect of R1626 administered as monotherapy in HCV-infected persons. A total of fourty-seven GT1 subjects were randomized to oral treatment of R1626 with doses of 500, 1,500, 3,000, 4,500 mg and placebo for 14 days (Roberts et al., 2008). As seen in the phase I study, after oral administration, R1626 was efficiently converted to R1479 with dose proportional pharmacokinetics observed over the entire dose range. The mean viral concentration at baseline was 6.3 log10 IU/mL, after 2 weeks of dosing; the mean (and median) viral concentration decreased by 1.2 (0.8), 2.6 (2.7) and 3.7 (4.1) log10 at 1,500, 3,000 and 4,500 mg, respectively. R1626 was generally well tolerated up to 3,000 mg twice a day with mild side effects and no serious adverse events. However, there were increased adverse events at the highest dose of 4,500 mg BID. The main adverse effect seen was reversible, mild to moderate hematological changes. This study resulted in a maximum dose of 3,000 mg twice a day for R1626 in all future clinical trials. The study concluded that treatment with R1626 as monotherapy significantly reduced plasma HCV viral load.
3.3.3. Phase IIa
In this study drug resistance, efficacy and safety of the combination therapy of R1626 along with IFN with and without RBV was studied over four weeks in 107 GT1 treatment-naïve patents. Subjects were randomized to 1,500 mg or 3,000 mg BID of R1626 with IFN or 1,500 mg BID with RBV. Control group received standard of care (SOC) as IFN and RBV (Pockros et al., 2008). After the four-week treatment period all four arms continued on SOC out to 48 weeks. Results were promising at week 4 with undetectable HCV RNA (<15 IU/ml) in 29%, 69% and 74% of subjects receiving 1,500 mg BID dual therapy, 3,000 mg BID dual therapy, and 1,500 mg BID triple drug therapy, respectively, compared with 5% in the SOC arm. Sequence analysis of the entire NS5B coding region of the samples from these subjects revealed no known R1479 resistance mutations (S96T or S96T/N142T) when compared with the baseline sequence. The main adverse event noted was grade 4 neutropenia that occurred in 50% of the persons in the R1626 treatment groups and occurred with 78% of the persons in the 3,000 mg BID group compared to 10% in the SOC arm. Overall, this study had safety concerns related to the drug; however, most AEs were mild (80%) or moderate (16%), but 4% were rated as severe. Strong synergistic antiviral effect was seen with R1626, IFN and RBV, which suggested that the dose of one or more of these drugs could be lowered to improve the tolerability without significantly compromising efficacy.
3.3.4. Phase IIb
This double-blind phase II trial assessed the optimal treatment protocol for balapiravir plus IFN/RBV (Nelson et al., 2012). Seven arms of treatment-naïve GT1 infected subjects (N = 516) were randomized and received balapiravir 500, 1,000, or 1,500 mg twice daily, IFN 180 or 90 μg/week and RBV 1,000/1,200 mg/day or IFN/RBV. Safety concerns reduced the planned treatment duration with balapiravir from 24 to 12 weeks. From week 2 to week 12 undetectable HCV RNA was more common in all balapiravir groups. Ultimately, the efficacy assessment was compromised by high rates of dose modifications and discontinuations, which resulted in similar SVR rates in the balapiravir groups (32–50%) and IFN/RBV group (43%). Balapiravir showed dose related serious adverse events (especially hematological, infection, ocular events) and discontinued for safety reasons in 28–36% of patients (most often for lymphopenia). Balapiravir was considered possibly related to two deaths in the balapiravir/IFN/RBV combination groups.
3.4. Conclusions
In Oct-2008, following an unacceptable benefit to risk ratio revealed in later Phase IIb study and lack of superior efficacy, R1626 was withdrawn from further consideration of future drug development in the treatment of chronic hepatitis C (Klumpp and Smith, 2011; Roche, 2008). Interestingly, R1626 was evaluated in adult patients infected with dengue virus in a double-blind placebo controlled trial at 1,500 and 3,000 mg BID for five days. In August 2012, Roche reported R1626 to be well tolerated but it demonstrated no viremia improvement versus the placebo arm (Nguyen et al., 2013). In our hands the parent 4′-azidocytidine nucleoside was a weak inhibitor in the HCV replicon system (EC50 = 11 μM and EC90 = 32 μM) and it is unclear why this drug was selected to be developed in humans.
4. RG7348 (MB11362)
A collaboration and license agreement between Metabasis Therapeutics, Inc. and Roche began in 2008 for the development of new treatments for HCV infection utilizing pre-clinical candidate MB11362 and the proprietary HepDirect® liver-targeting technology. The lead HepDirect nucleoside, renamed RG7348, was declared a clinical candidate in the second quarter of 2009. In January 2010, Ligand acquired Metabasis and subsequently, Roche advanced RG7348 into Phase I clinical studies (Ligand, 2010).
4.1. Preclinical development
The structure of RG7348 has not yet been disclosed, but it is known that it is a double prodrug that acts as a potent and selective HCV NS5B polymerase inhibitor (Nieforth et al., 2012). RG7348 was designed to provide high intra-hepatic levels of 4′-azidouridine triphosphate (Fig. 6). Very little preclinical data has been disclosed for RG7348. After oral dosing, the double prodrug was subsequently transformed by esterases and CYP3A4 (cytochrome P450 3A4) to 4′-azidouridine monophosphate 10, which was converted by kinases to the active 4′-azidouridine triphosphate 11, a potent and selective inhibitor of HCV polymerase.
Fig. 6.

Metabolism and metabolites of RG7348: (a) esterases; (b) CYP3A4; (c) kinases.
4.2. Clinical development
4.2.1. Phase I
In 2012 Nieforth and co-workers reported a Phase I study on safety, pharmacokinetics (PK) and anti-HCV activity for GT1 subjects with RG7348. The study was a randomized, double-blind, placebo-controlled single ascending dose study in healthy subjects (part A: 60 subjects with 12/placebo and 8/cohort receiving 15 mg, 50 mg, 150 mg, 300 mg, 600 mg and 1,200 mg), and multiple dose study in chronic HCV GT1 subjects (part B: 12 male subjects: 4/placebo and 8/RG7348 150 mg BID). After RG7348 dosing, Cmax and AUCinf for (10) and (11) increased dose proportionally from 15 to 150 mg and less than dose proportionally at higher doses. RG7348 was well tolerated in chronic HCV persons in this proof-of-concept study. Although RG7348 was a novel liver-targeted HCV nucleotide polymerase inhibitor, which demonstrated promising safety and PK, there was limited observed antiviral effect (mean reduction in HCV RNA <0.8 log10 IU/mL at all time points) at the dose studied.
4.3. Conclusions
In November 2010, Roche terminated the HCV development with Ligand. There was no statement from either company that explained the basis of this decision. In January 2011, Ligand Pharmaceuticals Inc. announced a strategic relationship with Chiva Pharmaceuticals to develop multiple Ligand assets. Chiva was granted a non-exclusive HepDirect® technology license for the discovery, development and commercialization of new compounds for HCV treatment, including RG7348. No other advances for RG7348 have been reported to date. Although the HepDirect technology seemed interesting, it never delivered on the promise to be liver specific and to add value to the nucleoside analogs studied.
5. PSI-6130
5.1. Chemical synthesis
β-D-2′-Deoxy-2′-fluoro-2′-C-methyl-cytidine (PSI-6130) was conceived from a combination of 2′-methyl of the HCV active NM107 with the non-selective 2′-deoxy-2′-fluorocytidine (Cedilote et al., 2008; Clark, 2005). Also, at about the same time, Pharmasset scientists discovered the modest anti-HCV activity of gemcitabine which was another 2′-disubstitued nucleoside (Stuyver, 2003). PSI-6130 was first synthesized from commercial available cytidine in about 3% total yield (Clark et al., 2005), but a more efficient, diasteroselective and scalable process was developed using commercially available isopropylidene protected D-glyceraldehyde (Wang et al., 2009). Thus, Wittig type reaction was performed with aldehyde 12 to give the unsaturated derivative 13 (Scheme 3). After oxidative dihydroxylation, compound 15 was selectively fluorinated. Cyclisation of ester 16 under acidic conditions gave lactone 17, which after benzoylation, reduction of the lactone and further acetylation gave the key sugar 19. Finally, introduction of the cytosine base under Vorbrüggen type coupling condition and complete debenzoylation afforded PSI-6130 in a 6.4% overall yield.
Scheme 3.

Synthesis of PSI-6130. Reagents and conditions: (a) Ph3PC(Me)CO2Et, DCM, −40°C; (b) KMnO4, acetone, 0 °C, 53% over two steps; (c) i. SOCl2, TEA, DCM, 0 °C; ii. aq. NaOCl, TEMPO, NaHCO3, CH3CN, 0 °C; (d) i. TEAF, dioxane, 100 °C; ii. (MeO)2C(Me)2, conc. HCl, dioxane, r.t.; (e) EtOH, conc. HCl, r.t.; (f) BzCl, pyridine, r.t., 47% from (14); (g) i. Li(O-tBu)3AlH, THF, −20 °C; ii. Ac2O, DMAP, −20 °C; (h) i. silylated N4-benzoylcytosine, SnCl4, PhCl, 65 °C; ii. NH3, MeOH, r.t.. 29% from (18).
5.2. Preclinical development
PSI-6130 is a potent and specific HCV RNA-dependent RNA polymerase (RdRp) inhibitor with EC90 of 5.4 ± 2.6 μM in Huh-7 replicon cells (Clark et al., 2005; Stuyver et al., 2006). It is interesting to note that just a few years earlier the commonly used BVDV surrogate assay would not have identified the anti-HCV activity of PSI-6130 because PSI-6130 was found to be highly specific for HCV over other RNA or DNA viruses. The 5′-triphosphate of PSI-6130 was found to be equally active against the wild-type GT1b replicon clone and the S282T mutant replicon (Murakami et al., 2007). PSI-6130 showed no apparent cytotoxicity against various cell types, including human bone marrow and human peripheral blood mononuclear cells. No mitochondrial toxicity was observed with PSI-6130 (Stuyver et al., 2006). An in vitro triple combination study of PSI-6130 with IFN and RBV at 1:1:1 ratio indicated synergistic antiviral effect, with no apparent cytotoxicity effects with any of the combinations tested (Bassit et al., 2008). Cell-based metabolism studies with primary human hepatocytes indicated that PSI-6130 is a unique compound that gives rise to two nucleoside triphosphates that are both potent inhibitors of the HCV RdRp (Fig. 7) (Ma et al., 2007; Murakami et al., 2007; Murakami et al., 2008). PSI-6130 can be converted to an inactive uridine analog PSI-6206 (also known as RO2433) by human cytidine deaminase (CDA) and to PSI-6130-MP by human 2′-deoxycytidine kinase (dCK), respectively. The formed PSI-6130-MP produced two active nucleoside triphosphates through two separate pathways. Thus, phosphorylation of PSI-6130-MP to the 5′-di- and 5′-triphosphates was catalyzed by uridine-cytidine monophosphate kinase (YMPK) and nucleoside diphosphate kinase (NDPK), respectively. In addition, metabolism studies with PSI-6130 showed that the monophosphate of PSI-6130 was deaminated by 2′-deoxycytidylate deaminase (DCTD) to the uridine monophosphate derivative by and subsequently anabolized to the 5′-triphosphate RO2433-TP by YMPK and NDPK in a second pathway (Fig. 7). The inactive uridine analog PSI-6206 is not a substrate for dCK and cannot be converted to PSI-6206-MP directly. The first cellular activation step to monophosphate is the rate-limited step among the three phosphorylation steps. Interestingly, the monophosphate species of PSI-6130 and PSI-6206 are very rapidly transformed into their corresponding nucleoside triphosphates, since those compounds are found at the highest concentration among all phosphate species present. Finally, PSI-6130-TP was a more potent inhibitor of the wild-type and S282T mutant polymerase compared to PSI-6206-TP (Ali et al., 2008; Ma et al., 2007; Murakami et al., 2007). Notably, PSI-6206-TP had a significantly longer half-life than PSI-6130-TP (see below).
Fig. 7.

Metabolism of PSI-6130 leads to both the active triphosphate and the inactive nucleoside PSI-6206.
5.3. Clinical development
Early animal studies involving single-dose pharmacokinetics studies with PSI-6130 in rhesus monkeys showed low oral bioavailability for PSI-6130 (Asif et al., 2007). An early phase I study in humans found a significant conversion to the inactive uridine analog combined with a bioavailability of 25%.
5.4. Conclusions
Because of these factors, a prodrug strategy was explored to potentially enhance bioavailability and provide a more selective conversion to PSI-6130-TP. RG7128, a prodrug of PSI-6130 (refer to Section 6 of this manuscript), was ultimately selected for clinical evaluation based on its improvements in bioavailability and pharmacokinetics.
6. RG7128 (R7128, RO5024048, mericitabine)
6.1. Preclinical development and chemical synthesis
RG7128 (mericitabine) is a 3′,5′-diisobutyryl ester prodrug and was prepared directly from PSI-6130 with isobutyryl chloride in the presence of DMAP and TEA in THF/H2O at 0 °C (Scheme 4) (Chun et al., 2007).
Mericitabine has excellent oral bioavailability but has very little plasma exposure and is quickly converted to PSI-6130 in humans. Metabolism of PSI-6130 proceeds by a bifurcated pathway to more potent triphosphate species PSI-6130-TP and less active uridine analog PSI-6206-TP and both serve as non-obligate chain terminators (Ali et al., 2008; Ma et al., 2007; Murakami et al., 2007; Murakami et al., 2008). The prodrug mericitabine demonstrated potent inhibition of stable and transient replicons with EC50 values similar to those of PSI-6130. Both mericitabine and PSI-6130 inhibited HCV GT1a (H77 strain) and GT1b (Con1 strain) subgenomic RNA replication, with similar levels of potency (mean EC50 values, 0.31 μM and 0.51 μM, respectively) (Ali et al., 2008).
6.2. Clinical development
6.2.1. Phase I
In a two week phase I monotherapy clinical study with 32 HCV GT1 patients, mericitabine showed a mean 2.7 log10 reduction in viral RNA with a dose of 1,500 mg BID (Reddy et al., 2007). HCV RNA levels were unchanged at Day 15 in six subjects who were given oral placebo in this study. In a four week, randomized, placebo-controlled phase I trial, 88% and 85% of treatment-naïve subjects, who were given 1,000 mg or 1,500 mg BID of mericitabine in combination with SOC (IFN/RBV), achieved undetectable RNA levels, respectively (Rodriguez-Torres et al., 2008). By contrast, only 19% subjects in SOC control group achieved no virus detectable level.
6.2.2. Phase IIb
In one of the phase IIb clinical trials (PROPEL) (Jensen et al., 2010; Wedemeyer et al., 2013), 408 GT1 and GT4 treatment-naïve subjects were randomized into one of five treatment arms: arm A: mericitabine 500 mg BID for 12 weeks; arm B: mericitabine 1,000 mg BID for 8 weeks; arm C: mericitabine 1,000 mg BID for 12 weeks; arm D: mericitabine 1,000 mg BID for 12 weeks; and arm E: placebo BID for 12 weeks. All subjects received IFN/RBV at standard doses for 24 or 48 weeks during and after mericitabine/placebo treatment. Subjects in arm A, B and C who achieved extended rapid virologic response (eRVR) stopped all treatment at week 24. Subjects who did not have an eRVR, and persons in arm D and E continued SOC to complete the 48 weeks treatment. Overall, the virologic response rates were higher in arms A–D than in arm E at weeks 4 and 12. However, the arms D (50.6%) and E (placebo, 51.2%) had similar overall SVR24 rates (primary outcome) with lower rates in the response-guided therapy arms A, B and C (48.8%, 42.0% and 32.9%, respectively).
These data demonstrated that treatment with mericitabine plus IFN/RBV for 8 or 12 weeks provided potent suppression of HCV RNA, was well tolerated, did not result in selection of resistant variants; however, it did not increase SVR rates compared with placebo.
An analysis from the now completed phase IIb JUMP-C trial was reported by Pockros and colleagues (Pockros et al., 2013). A total of 166 treatment-naïve persons infected with HCV GT1 or GT4 were randomized to one of two treatment arms. Subjects randomized to arm A were given mericitabine BID plus SOC (IFN/RBV) with response guided therapy (RGT). Persons on arm A who achieved eRVR (extended rapid virologic response; means no virus detected at week 4 and week 12) stopped all treatment at week 24. Those persons who did not achieve an eRVR also stopped the mericitabine at week 24, but continued an additional 24 weeks of SOC treatment. The subjects in control group arm B received SOC treatment for 48 weeks. The results of JUMP-C phase IIb trials showed that mericitabine was safe, and associated with higher SVR rates when compared to SOC (56.8% vs. 36.5%). The virologic response obtained at weeks 4 and 12 in the JUMP-C trial was comparable to results observed with the PROPEL trials. However, the authors note that extending treatment period from 12 to 24 weeks in the PROPEL study was not as effective in increasing SVR rates as found in the JUMP-C study. The difference in SVR rates may have contributed to differences observed in viral breakthrough rates since the PROPEL study showed an almost two-fold increase in relapse as compared to JUMP-C results (Pockros et al., 2013).
It is worth noting that a combination of mericitabine plus SOC was also effective in treating HCV GT2/3 relapsers or non-responder subjects (Gane et al., 2010). Twenty persons with GT2/3 infections, who had not achieved SVR with previous treatments were given mericitabine 1,500 mg BID plus SOC for 4 weeks, followed by SOC alone for 22–24 weeks. Ninety percent (18/20) of persons achieved RVR and 65% (13/20) achieved SVR at 12 weeks. This trial demonstrated that mericitabine plus SOC provided an effective regimen for the treatment of HCV GT2 and GT3 infected persons who have failed previous treatments.
INFORM-SVR, an ongoing phase IIb clinical trial, aims to evaluate the safety and efficacy of an all-oral IFN-free treatment with mericitabine and ritonavir-boosted danoprevir (RG7227, protease inhibitor, Fig. 8), with or without RBV in GT1a/1b treatment-naïve subjects. Among the different arms of the study, a 15% virus breakthrough rate was reported for GT1a and 1b persons (Gane et al., 2012). Viral breakthrough was associated with virus harboring protease inhibitor resistance conferring mutations. Importantly, a transient S282T mutation, an NS5B polymerase mutation associated with mericitabine resistance, was selected in one person.
Fig. 8.

Structures of setrobuvir (ANA598, non-nucleoside polymerase inhibitor) and danoprevir (RG7227, protease inhibitor).
Finally, ANNAPURNA, an ongoing phase IIb clinical trial is evaluating the combination of setrobuvir (ANA598, RO5466731, non-nucleoside polymerase inhibitor, Fig. 8), danoprevir (RG7227, R05190591, protease inhibitor, Fig. 8) and ribavirin with or without mericitabine in patients with CHC who are either treatment-naïve or have previously experienced a null response to interferon-based treatment (Fig. 8). With an expected completion date of December 2013, this two-part five arm study will be looking at SVR12 and safety as end points. In part 2, based on results from part 1, further treatment-naïve subjects will receive a successful regimen from part 1, or a reduced intensity regimen, and interferon-based null-response persons will be added to the study (Roche, 2012).
6.3. Conclusions
The INFORM-SVR study is considered a milestone game changing combination since it demonstrated for the first time that an all-oral drug combination could suppress HCV replication to levels similar or exceeding SOC. It is also one of very few studies that show the selection of nucleoside analog associated resistance conferring mutations in humans, even though the apparently resistant virus was transient. The S282T mutation was not observed during other clinical triple combination trials with or without IFN, suggesting that the high barrier to resistance of mericitabine, and the low fitness of the mutant virus reduce the incidence of viral breakthrough caused by nucleoside analog resistance selection (Jensen et al., 2010; Le Pogam et al., 2010; Pockros et al., 2011; Pockros et al., 2013). The high rate of viral suppression with mericitabine combined with significant relapse rates without mutation selection indicates that longer treatments, higher doses, and/or other antiviral combinations may be necessary to make mericitabine a major player in HCV treatment. This may be due to the short half-life of PSI-6130-TP, PSI-6130-TP not reaching a viral reservoir(s), and/or overall lack of potency of PSI-6130-TP.
7. IDX184
7.1. Synthesis of IDX184
IDX184 (Cretton-Scott et al., 2010; Sommadossi et al., 2008; Surleraux and Gosselin, 2011, 2012) was designed by Idenix Pharmaceuticals as a 2′-C-methyl guanosine (2′-C-MeG) ribonucleoside that contains a S-acyl-2-thioethyl (SATE) phosphoramidate prodrug to selectively deliver the 5′-MP of 2′-C-methylguanosine ribonucleoside to human liver. The metabolism of IDX184 to 2′-MeG-MP takes place predominantly in liver cells and presumably involves cytochrome P450 (CYP450)-dependent and -independent processes (Zhou et al., 2011). By bypassing the rate-limiting monophosphorylation process, 2′-MeG-MP is then efficiently converted to its active 5′-TP within hepatocytes by cellular kinases. The complete metabolic pathway of IDX184 is yet to be fully elucidated. Liver targeting may result in a favorable therapeutic index by increasing the levels of the active 2′-MeG-TP in liver cells while lowering systemic exposure to the nucleoside, potentially minimizing the risk of systemic side effects (Cretton-Scott et al., 2008).
IDX184 was synthesized as shown in Scheme 5Error! Reference source not found. (Sommadossi et al., 2008). Preparation of the H-phosphonate monoester precursor was achieved in three steps from commercially available 2,2-dimethyl-3-hydroxypropanoic acid methyl ester 20, by protection of the alcohol followed by saponification, leading to compound 21 in 92% yield without purification. Installation of the side chain was performed by peptidic coupling between compound 21 and 2-mercaptoethanol, to generate alcohol 22. Finally, treatment of compound 22 with phosphorus acid and pivaloyl chloride, followed by quenching the reaction with triethylammonium bicarbonate (TEAB), generated H-phosphonate monoester precursor 23 in 90% over two steps.
Scheme 5.

Synthesis of IDX184. Reagents and conditions: (a) TrCl, Et3N, DMAP, CH2Cl2, reflux, overnight; (b) NaOH, dioxane, reflux, 16 h, 92% over two steps; (c) i. CDI, toluene/DMF (2/1, v/v), r.t., 30 min; ii. 2-mercaptoethanol, toluene/DMF (2/1, v/v), −10 °C, 3 h; (d) i. H3PO3, PivCl, pyridine, 0 °C to r.t., 3 h; ii. TEAB, 90% over two steps; (e) (23), PivCl, pyridine, −15 °C, 2 h, 32%; (f) Benzylamine, CCl4, r.t., 1 h, quantitative yield; (g) TFA, CH2Cl2, r.t., 30 min, 39%.
2′-C-Methylguanosine 24 and precursor 23 were then both treated with pivaloyl chloride to furnish intermediate 25, which was further reacted with benzylamine to generate Tr-protected phosphoramidate diester 26 in quantitative yield. Classical deprotection with trifluoroacetic acid led to the isolation of IDX184 in 39% yield (Scheme 5).
7.2. Preclinical development
In preclinical studies using an HCV replicon assay, IDX184 exhibited 10 times greater potency than nucleosides that were in clinical development at the time. In a seven-day preclinical toxicology study of IDX184, no toxicities, including gastrointestinal or hematological, were observed in monkeys at doses greater than or equal to 600 mg/kg/day. In HCV GT1 infected chimpanzees, once daily oral administration of 10 mg/kg of IDX184 produced a rapid and potent inhibition of HCV replication with mean viral load reductions of 2.5–4.0 log10 after four days of dosing (Standring et al., 2008, 2009). Studies were also performed to characterize resistance mutations profiles of IDX184. The S282T mutation was identified in all tested cell lines and emerged around 25–77 days. Although the S282T mutant gave IDX184 a 23-fold increase in EC50, low (5%) replicon capacity of S282T replicons was also observed as previously reported by Ludmerer et al. (Ludmerer et al., 2005). The signature S282T mutant and other drug-related resistance mutants were only established in vitro for cell-based administration as ultimately there were no mutant resistance found in clinical trials and no cross-resistance between IDX184 and other classes of HCV antiviral agents (McCarville et al., 2011; McCarville et al., 2010). IDX184 demonstrated additive antiviral activity in the HCV replicon in combination with HCV protease inhibitors and IFN and synergistic activity in combination with RBV. IDX184 also remained fully active in vitro against HCV containing known protease and non-nucleoside inhibitor drug resistance mutations (Zhou et al., 2011; Zhou et al., 2009). As a result of its favorable preclinical profile, IDX184 was selected as Idenix’s lead candidate in January 2008 and Phase I clinical trials were started in July of the same year.
7.3. Clinical development
7.3.1. Phase Ia
In July 2008, a randomized, placebo-controlled, dose-escalating study was designed to evaluate the safety and tolerability of IDX184 after oral administration in healthy subjects. Doses escalating from 5 mg to 100 mg were administered to fourty-eight healthy subjects for three days, and PK data were collected (Idenix, 2008b). The most common adverse event was dizziness, 5.6% of occurrences for the IDX184 treated group compared to 25% for the placebo cohort. Mild to moderate adverse events occurred and resolved by the end of the study. No severe adverse effects (SAE) or dose-limiting toxicity were observed. The level of IDX184 and 2′-C-MeG in plasma remained low and dose-dependent, consistent with a liver-targeting approach. The short half-life of IDX184 (around 1 h) in plasma suggested that IDX184 was unlikely going to accumulate over time. In comparison, 2′-MeG in plasma has a long half-life with t1/2 of approximate 18–42 h for doses above 25 mg, which reflected the long half-life of 2′-MeG-TP in the liver. The C24h of 2′-C-MeG is 0.58–2.88 ng/mL for dose higher than 25 mg of IDX184. Overall, IDX184 was safe and well tolerated at a single dose up to 600 mg/kg/day. The favorable PK supported once-daily dose administration therapy in future studies involving HCV infected individuals.
7.3.2. Phase Ib
In December 2008, a double-blind, dose-escalating study (Phase Ib) began to evaluate the safety and antiviral activity of IDX184 in fourty-one treatment-naïve subjects infected with GT1 chronic hepatitis C (Idenix, 2008a). These persons received 25 mg QD, 50 mg QD, 75 mg QD and 100 mg QD or placebo QD for 3 days. IDX184 was safe and well tolerated: the occurrences of adverse events were following similar pattern as the placebo-treated group with the most frequently adverse events reported being headache and dizziness. The PK analysis data in the study was consistent with those of the previous healthy volunteers study. The mean HCV RNA reduction (log10) ranged from 0.47 to 0.74 after treatment with 25 mg to 100 mg cohorts at day four. Significant HCV viral load decline occurred in persons receiving doses from 75 mg to 100 mg (Lalezari et al., 2012; Lalezari et al., 2009; Lalezari et al., 2010). This study helped to determine optimal doses that could be chosen for testing in later studies.
7.3.3. Phase IIa
In November 2009, a total of eighty subjects were enrolled to evaluate the short-term safety, the antiviral activity and PK profile of IDX184 in combination with IFN/RBV in subjects with GT1 chronic hepatitis C infection. Over 14 days, individuals enrolled in this phase IIa, randomized, double-blind study (Idenix, 2009) were given sequential doses of 50 mg QD, 50 mg BID, 100 mg QD, 100 mg BID, 150 mg QD, 200 mg QD of IDX184 in combination with IFN/RBV and IFN/RBV as the placebo cohort. Most adverse events were mild, and no serious adverse events were associated with IDX184 plus IFN/RBV treatment. The pattern of side effects was similar between treatment of IDX184 plus IFN/RBV and treatment of IFN/RBV alone. At day 15 of IDX184 combination treatment, mean viral load reduction (log10) was: 50 mg QD (−2.7), 50 mg BID (−4.0), 100 mg QD (−4.2), 100 mg BID (−4.1), 150 mg QD (−4.3), 200 mg QD (−3.7) of IDX184 in combination with IFN/RBV and −1.5 for IFN/RBV. For cohorts given IDX184 50 mg QD, 100 mg BID and 200 mg QD, undetectable viral level (< 15 IU/mL) were found in 13–29% of the persons, whereas for 50 mg BID, 100 mg QD and 150 mg QD, 40–50% of the individuals achieved undetectable viral levels. Moreover, all IDX184 treated cohorts led to a low level of mean alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels compared to IFN/RBV cohort. In addition, the mean plasma concentration of 2′-MeG, which was identified as predictor of viral load started to decline at Day 14 (Lalezari et al., 2013; Lalezari et al., 2010; Membreno and Lawitz, 2011).
7.3.4. Phase I combination of IDX184 and IDX320
In June 2010, a clinical drug-drug interaction study was initiated to evaluate the combination of IDX320 (a macrocyclic HCV protease inhibitor, Fig. 9) and IDX184 in healthy subjects (Idenix, 2010). This study was designed as a double blind, multiple-dose study of twenty subjects who were administered 100 mg/day of IDX184 in combination with 400 mg of IDX320. The first cohort (8:2) was either treated with IDX320/IDX184 matching placebo for seven days followed by seven additional days with IDX320/IDX184, or IDX320 matching placebo/IDX184 matching placebo. The second cohort (8:2) was given IDX184/IDX320 matching placebo for seven days followed by seven additional days IDX184/IDX320 or IDX184 matching placebo/IDX320 matching placebo. This study was designed to evaluate safety and tolerability and to assess the potential for a pharmacokinetic drug-drug interaction between IDX320 and IDX184 when the two drugs were administered in combination in healthy subjects. In September 2010, the FDA placed a clinical hold on IDX184 and IDX320 programs based on three serious adverse events (elevated liver function tests) that occurred during this combination study in healthy volunteers. In February 2011, the FDA removed the full clinical hold on IDX184 and placed it on partial clinical hold allowing Idenix to advance in the clinic under the partial clinical hold (Idenix, 2011a).
Fig. 9.

Chemical structure of HCV protease inhibitor IDX320.
7.3.5. Phase I
In April 2011, an open-label study was initiated to evaluate the relative bioavailability of IDX184 and any potential food effect in healthy male subjects (Idenix, 2011b). The purpose of this study was to compare the amount of drug in the blood after taking either a capsule or a tablet form of the drug while fasting. Additionally, the amount of drug present in the blood after eating a meal was investigated and the safety of the tablet form of IDX184 was evaluated in healthy volunteers. Twelve subjects received 50 mg BID IDX184 capsules without food on Day 1, 50 mg BID IDX184 tablets without food on Day 8, and 50 mg BID IDX184 tablets with food on Day 15. Blood levels of IDX184 were measured in each study participant to determine pharmacokinetic parameters including bioavailability of IDX184, AUC and Cmax of IDX184 and 2′-C-MeG. Results of this study have not been disclosed at the time of this publication.
7.3.6. Phase IIb
In July 2011, Idenix initiated a double-blind study of sixty-seven randomized subjects to evaluate the safety and the antiviral activity of IDX184 in combination with IFN/RBV at doses of 50 mg or 100 mg QD for 12 weeks. The common adverse events were fatigue, nausea and headache, which were generally mild to moderate. No treatment-related cardiac or renal serious adverse events were observed during the study and the occurrences of all adverse events were similar between IDX184-combination therapy and IFN/RBV-therapy. Echocardiograms and N-terminal pro B-type natriuretic peptide (NT-proBNP), a biomarker that aids in the detection of congestive heart failure and cardiac dysfunction, were added in this study to address potential cardiac concerns. Heart rate, AV conduction and cardiac depolarization remained normal. Several subjects in each group developed new T-wave inversions that were not judged to be clinically significant. The increases seen in NT-proBNP levels were greater than 200% of baseline, but reversible. In the 50 mg and 100 mg of IDX184 in combination with IFN/RBV cohorts, 94% and 87% of subjects, respectively, achieved undetectable virus level at a median of 8 weeks of treatment. At week 12, a 76% and 82% rate of complete EVR was observed in the 50 mg and 100 mg treatment arms, respectively (Lawitz et al., 2012a). In January 2012, Idenix submitted interim phase IIb data to the FDA requesting for the removal of the partial clinical hold on IDX184. In February 2012, the partial clinical hold on IDX184 was removed by the FDA allowing the initiation of dosing in the phase IIb study.
7.4. Conclusions
In August 2012, the FDA placed IDX184 and IDX19368 (another HCV nucleotide polymerase inhibitor for which Idenix had previously filed an IND, but had not initiated clinical studies) on partial clinical hold due to cardiac adverse events seen in the phase II clinical trial of BMS-986094 (refer to section 15 of this manuscript). All three drugs are 2′-C-methyl guanosine nucleotide prodrugs. In December 2012, Idenix completed the submission of requested cardiac safety data for IDX184 to the FDA. Idenix had found no evidence for severe cardiac effects in all of their clinical studies to date. In the meantime, in October 2012, Idenix started a three-year observational study to evaluate the durability of sustained viral response and the kinetics of antiviral-resistant HCV in subjects who participated in studies of Idenix anti-HCV direct acting antivirals, including IDX184. In February 2013, the FDA communicated that IDX184 (and IDX19368) programs would remain on clinical hold, and, as a result, Idenix decided to terminate the development of these programs. A reason for FDA concern over IDX184 and related compounds is the resultant NTP derived from IDX184 is identical to the NTP derived from BMS-986094. Cardiac toxicity for BMS-986094 was demonstrated to be dose-related and the postulated reason for the cardiac toxicity was mitochondrial toxicity. Of note, 2′-C-MeG-TP has been shown to be a substrate for incorporation by human mitochondrial RNA polymerase (Arnold et al., 2012). However, cell free assays can be misleading since the nucleoside may never be phosphorylated in mitochondria. Nevertheless, Idenix decided not to pursue this drug further since by then they had identified nucleoside analogs with superior virological and toxicological profiles.
8. PSI-6206 to PSI-7851 to GS-7977 (PSI-7977; sofosbuvir)
8.1. Discovery and preclinical profiling
The presence of 2′-deoxy-2′-fluoro-2′-C-methyluridine PSI-6206 metabolite along with its triphosphate PSI-6206-TP was recognized during metabolism studies on PSI-6130 in cell culture and in monkeys (see discussion Section 5.2). These compounds were evaluated for their anti-HCV activity in replicon assay (Ma et al., 2007). While the uridine nucleoside PSI-6206 was devoid of anti-HCV activity in culture, PSI-6206-TP demonstrated a potent and selective inhibition of HCV NS5B polymerase with mean IC50 = 1.19 μM. Subsequent stability studies using primary human hepatocytes showed that PSI-6206-TP had significantly longer half life (t1/2 = 38 h) than its analogous cytidine triphosphate PSI-6310-TP (t1/2 = 6 h) (Murakami et al., 2008).
Cellular pharmacology with PSI-6206 demonstrated that it was very poorly phosphorylated to the monophosphate nucleotide (PSI-6206-MP), which could explain the lack of anti-HCV activity. The phosphoramidate prodrug approach was taken to bypass the initial unproductive phosphorylation step (Murakami et al., 2010). After in vitro and in vivo profiling of multiple potential prodrugs, phosphoramidate prodrug PSI-7851 was selected as the lead compound. PSI-7851, an equal mixture of two phosphorous diastereoisomers PSI-7976 and PSI-7977 (Berrey et al., 2013; Chun et al., 2010; Clark, 2005; Cleary et al., 2013; Ross et al., 2010; Sofia et al., 2008) was shown to be a potent and selective inhibitor of HCV replication (EC50 = 0.52 μM) with no associated bone marrow, mitochondrial or other cellular toxicity. Its potential efficiency was further demonstrated by high levels of triphosphate (PSI-6206-TP) formation in primary human hepatocytes and in the livers of rats, dogs and monkeys when administered in vivo (Sofia et al., 2010a). In vitro studies, showed PSI-7851 had a broad range of antiviral activity across different HCV GTs (Lam et al., 2012). Based on impressive preclinical data, PSI-7851 was selected for future development and progressed into phase I human clinical trials.
8.2. Synthesis of PSI-7851 and PSI-7977 (GS-7977)
Subsequently, a method was developed to separate the two PSI-7977 and PSI-7976 diastereoisomers. The Sp diastereoisomer PSI-7977 (EC90 = 0.42 μM) was shown to be more potent inhibitor than the Rp diastereoisomer PSI-7976 (EC90 = 7.5 μM) in replicon assays (Sofia et al., 2010a). However, when both diastereoisomers were tested in primary human hepatocytes almost equal amounts of active triphosphate (PSI-6206-TP) was formed, suggesting both isomers may be equally active in the liver cells (Murakami et al., 2010). Enzymatic studies showed Cathepsin A (CatA) and carboxylesterase 1 (CES) could both catalyze the first step hydrolysis (of the carboxyl ester linkage between the carboxylic acid from amino acid moiety and the isopropyl alcohol) of phosphoramidate prodrugs PSI-7977 and PSI-7976. PSI-7977 was found to be a better substrate for CatA while PSI-7976 was preferentially hydrolyzed by CES1 (Murakami et al., 2010). The apparent discrepancy between replicon data and primary human hepatocytes was related to the different expression of these enzymes in replicon cell lines and primary human hepatocytes (Murakami et al., 2010; Sofia et al., 2010a). Both isomers were devoid of cytotoxicity when tested against a broad range of cell lines. Mitochondrial toxicity was evaluated up to 100 μM in both CEM and HepG2 cells and no toxicity was observed for PSI-7977 while PSI-7976 showed CC90 of 72 and 68.6 μM for inhibition of mtDNA and rDNA, respectively in HepG2 cells (Sofia et al., 2010a). Pure Sp isomer PSI-7977 was selected for future clinical studies due to its higher potency in replicon assay and better safety profile. To date, PSI-7977 (now called sofosbuvir or GS-7977) is the only nucleotide analog HCV polymerase inhibitor that has completed phase III clinical trials and this drug is expected to receive FDA approval by the end of 2013.
8.3. Synthesis of single isomer PSI-7977 (sofosbuvir, GS-7977)
The first generation synthesis of PSI-7977 involved direct reaction of phosphoryl chloride reagent 28a with 2′-deoxy-2′-fluoro-2′-C-methyluridine (PSI-6206) and separation of the two diastereoisomers by crystallization (Sofia et al., 2010a) (Scheme 6). Ultimately, with some modifications to the chemistry and workups during process development, the yield from (28a) to the mix of PSI-7976 and PSI-7977 (48:51) was improved to 45.7% from which the single isomer PSI-7977 was isolated in 15% yield after crystallization (Ross et al., 2010).
Scheme 6.

Synthesis of PSI-7976 and PSI-7977 (sofosbuvir, GS-7977) via column purification. Reaction conditions: (a) 70% aqueous acetic acid, 100 °C; (b) 25% methanolic ammonia, 0–15 °C, 78% overall yield; (c) (28a) (Rp:Sp = 1:1) (6.5 equiv), NMI (8 equiv), CH2Cl2, r.t., overnight, 6%, 1:1 mix of PSI-7976 and PSI-7977; (d) (28b) (Rp:Sp ≈ 1:1) (2 equiv), t-BuMgCl (2.1 equiv), THF, r.t., 48 h, 47%, 3:1 mix PSI-7977:PSI-7976.
This approach was further optimized by using p-nitrophenol as the leaving group of the phosphorylating agent as shown in (28b) (Scheme 6). In this case a 47% of a 3:1 mix of PSI-7977 and PSI-7976 were obtained. Separation of (28b) into its single diastereomers gave the desired chiral phosphorylating reagent 28c, which upon reaction under basic conditions gave clean inversion at the phosphorous centers to give PSI-7977 as the single desired Sp diastereomer. Pentafluorophenyl was used in a similar manner with the phosphorylating reagent 28d which allowed for a multigram synthesis of single Sp isomer PSI-7977 without the need for a chiral catalyst, chiral auxiliary or column purification (Ross et al., 2011; Ross et al., 2010).
8.4. Clinical trials for PSI-7977 (GS-7977; sofosbuvir)
8.4.1. Phase I
In March 2009, clinical trials for PSI-7851 (also known as GS-9851; mixture of diastereoisomers PSI-7976 and sofosbuvir) were completed in 42 healthy volunteers. The administration of single dosage of 25 mg up to 800 mg QD was shown to be generally safe and well tolerated with no adverse events when compared with the placebo arm and with a pharmacokinetic profile consistent with once-daily dosing (Denning et al., 2012). Phase Ia trials with PSI-7851 also evaluated safety and tolerability for three day monotherapy at different doses (50 mg, 100 mg, 200 mg, 400 mg) in treatment-naïve subjects. Reductions in viral load were observed without any serious adverse events when compared to the placebo arm (Lawitz et al., 2013c). In fact, the 400 mg QD dose led to a decrease of 1.76 log10 IU/mL after three days of treatment compared to the related mericitabine, which after 14 days at 1,500 mg with QD dosing, gave a 1.48 log10 IU/mL drop.
8.4.2. Phase IIa
The safety, antiviral activity and tolerability of three different doses of sofosbuvir in combination with IFN/RBV were assessed in treatment-naïve persons with GT1 HCV infection. Sixty-four subjects were randomized (1:1:1:1) to receive one of three once-daily doses of sofosbuvir (100, 200 and 400 mg) or placebo plus IFN/RBV for 28 days, which was followed by 44 weeks treatment with IFN/RBV alone therapy. All who received sofosbuvir experienced rapid virologic response (RVR) of 88%, 94% and 93% for 100, 200, and 400 mg dose, respectively. In comparison, only 21% of the subjects achieved RVR in the control arm. No viral breakthrough was observed during 28 days treatment; however, eight individuals had relapsed during IFN/RBV treatment (four on sofosbuvir 100 mg; two on sofosbuvir 400 mg and two on placebo arm). SVR24 rates were higher for subjects in all arms with sofosbuvir treatment (56% for 100 mg dose, 83% for 200 mg dose, and 84% for 400 mg dose) than those in the IFN/RBV arm (43% achieved SVR after 24 weeks). The most frequent adverse effects during 28 days treatment were fatigue, nausea, chills, headache and joint pain. No serious adverse events or discontinuation was reported in first 28 days. Sofosbuvir 200 and 400 mg doses were selected for future studies (Rodriguez-Torres et al., 2013).
8.4.3. Phase IIb
Several phase IIb clinical trials have addressed the safety and efficacy of sofosbuvir treatment in the treatment of HCV GTs. Results from four studies (PROTON, ELECTRON, QUANTUM and ATOMIC) are summarized below.
8.4.3.1 PROTON
In this double-blind, randomized study the efficacy of sofosbuvir in combination with IFN and RBV in treatment-naïve persons with HCV GT1, 2 and 3 was evaluated. A total of 121 treatment-naïve subjects with GT1 infection were randomly assigned to three different groups (2:2:1 ratio) to receive IFN/RBV with 200 mg sofosbuvir, 400 mg sofosbuvir, or placebo for 12 weeks followed by additional IFN/RBV treatment for 12 weeks or 36 weeks depending on the subject’s response. Among GT1 persons, 98% of subjects on triple therapy achieved RVR compared to 19% in the placebo group. 88% of those on 200 mg dose of sofosbuvir and 91% of those on 400 mg sofosbuvir achieved SVR12 while less than 50% achieved SVR in placebo cohort. All IL28-BT/T GT persons included in the study (13/13) achieved SVR12 following triple therapy. Finally, combination therapy with sofosbuvir was even more effective for treatment-naïve persons with GT2/3 HCV infections. In this cohort, all 24 subjects who stayed on therapy achieved SVR12 (Gane et al., 2013b; Lawitz et al., 2013b; Pham et al., 2004; Radkowski et al., 2005).
8.4.3.2. ELECTRON
This study was designed to evaluate the efficacy of sofosbuvir with RBV, with or without IFN in treatment-naïve or treatment-experienced persons with GT1, 2, or 3 HCV infections. Sofosbuvir monotherapy, reductions in RBV dose, and changes in duration of therapy were also evaluated. One hundred and fifty five subjects with all three HCV GTs were divided into eleven treatment groups (Radkowski et al., 2005). In a randomized cohort, fourty treatment-naïve subjects with HCV GT2 and GT3 infections were divided into four arms. All groups received sofosbuvir (400 mg QD) + RBV BID for 12 weeks. Three of the arms also received IFN for 4, 8 and 12 weeks, respectively. SVR24 was achieved for all subjects enrolled in the four arms (cohorts 1 to 4) (Gane et al., 2013b; Gane et al., 2012). In an additional arm (cohort 5), wherein ten treatment-naïve persons with HCV GT2 or GT3 received sofosbuvir monotherapy for 12 weeks, SVR24 rates were 60%. A shortened 8-week triple therapy arm (cohort 6) was also evaluated in ten GT2 or GT3 subjects and SVR24 was achieved for all participating subjects. In cohort 7, twenty-five treatment-naïve subjects with GT2 or GT3 were treated with sofosbuvir plus RBV for 8 weeks. Full (100%) SVR was achieved in this cohort. Cohort 8 evaluated treatment of ten GT2/3 subjects with sofosbuvir + reduced dose RBV (800 mg) and resulted in a reduced SVR rate of ~60%. Twenty-five treatment-experienced subjects with GT2 and GT3 HCV infections were treated with sofosbuvir and RBV (cohort 9). All subjects in this arm achieved RVR while 68% attained SVR at 12 weeks. Finally, a 12-week IFN-free sofosbuvir plus RBV treatment was evaluated in ten GT1 null-responders (cohort 10) and in twenty-five GT1 treatment-naïve subjects (cohort 11). Treatment-naïve subjects with GT1 achieved SVR12 of 84% while only one out of ten null-responder subjects achieved SVR12 (Table 1).
Table 1.
Participants with HCV infection and results of the ELECTRON study.
| Cohort # | # of Patients | Genotype | sofosbuvir (weeks) | RBV (weeks) | IFN (weeks) | SVR (%) |
|---|---|---|---|---|---|---|
| 1 | 10a | GT2 and 3 | 400 mg QD (12) | 1,000 mg BID (12) | (4) | SVR24 (100) |
| 2 | 10a | GT2 and 3 | 400 mg (12) | 1,000 mg BID (12) | (8) | SVR24 (100) |
| 3 | 10a | GT2 and 3 | 400 mg (12) | 1,000 mg BID (12) | (12) | SVR24 (100) |
| 4 | 10a | GT2 and 3 | 400 mg (12) | 1,000 mg BID (12) | none | SVR24 (100) |
| 5 | 10a | GT2 and 3 | 400 mg (12) | none | none | SVR24 (60) |
| 6 | 10a | GT2 and 3 | 400 mg (8) | 1,000 mg BID (8) | (8) | SVR24 (100) |
| 7 | 25a | GT2 and 3 | 400 mg (8) | 1,000 mg BID (8) | none | SVR24 (100) |
| 8 | 10a | GT2 and 3 | 400 mg (8) | 800 mg BID (8) | none | SVR24 (60) |
| 9 | 25b | GT2 and 3 | 400 mg (8) | 1,000 mg BID (8) | none | RVR (100) SVR12 (68) |
| 10 | 10c | GT1 | 400 mg (12) | 1,000 mg BID (12) | none | SVR12 (10) |
| 11 | 25a | GT1 | 400 mg (12) | 1,000 mg BID (12) | none | SVR12 (84) |
Treatment-naïve patients;
Treatment-experimented patients;
Null-responders
In summary, 12-week treatment with sofosbuvir and RBV resulted in 100% SVR24 in treatment-naïve, GT2/3 subjects. This regimen was less optimal for treatment-experienced subjects with GT2/3 or null-responders with GT1 infections. It was concluded that duration of less than 12 weeks or reduced RBV dose might adversely impact treatment efficacy. Reduced dosage of RBV in treatment-naïve difficult GT1 subjects in 24 weeks treatment was less efficient (SVR4 56%) than full dose RBV (77%). Addition of sofosbuvir to IFN + RBV regimen considerably improved the response rate among all GTs and shortened the duration of therapy. IFN-free combination showed high percent of cure rate in untreated individuals, but was less efficient in treatment-experienced patients or null-responders.
8.4.3.3. ATOMIC
This phase IIb clinical trial evaluated the combination of sofosbuvir with IFN/RBV for 12- or 24-weeks in treatment-naïve GT1, GT4, and GT6 persons (a total of 332 subjects were enrolled). Additionally, treatment-naïve persons with GT1 were treated with sofosbuvir in combination with IFN/RBV followed by 12 week treatment with sofosbuvir alone or in combination with RBV. The results from this study suggest that 12 week triple combination therapy was as effective as 24 week treatment for this group of subjects. Furthermore, it was found that sofosbuvir, with or without RBV after initial treatment, can be effective in achieving high SVR rate (Kowdley et al., 2013).
8.4.3.4. Phase II sponsored by the National Institute of Allergy and Infectious Diseases (NIAID)
Initiated in 2011, NIH investigators recruited 60 GT1 HCV treatment-naïve subjects, into a two-part open label study (Gilead, 2011). As a proof of concept, in the first part, ten patients with mild to moderate liver fibrosis were treated with 400 mg/day of sofosbuvir and weight-based RBV for 24 weeks. All nine subjects who completed the 24-week course had an undetectable level of the virus 24-weeks post treatment and were deemed cured. Subsequently, in the second group of fifty subjects where thirteen suffered from serious liver damage, all took 400 mg/day sofosbuvir with the subjects being split between weight-based and low-dose (600 mg/day) RBV. SVR-24 rates of 68% were observed in the weight-based group and 48% in the low-dose group (Osinusi et al., 2013). Seven grade 3 adverse events were reported (anemia, neutropenia, nausea, hypophosphatemia, and cholelithiasis or pancreatitis), but none resulted in discontinuation of treatment. This NIH-sponsored study of Gilead’s sofosbuvir combined with RBV demonstrated a safe and effective interferon-free approach for even some of the toughest HCV cases.
8.4.4. Phase III
Multiple phase III clinical trials have been initiated to evaluate the safety and efficacy of HCV treatments that contain sofosbuvir. Results from four trials (FUSION, FISSION, POSITRON and NEUTRINO) are summarized below.
8.4.4.1. FUSION
In June 2012, the randomized, double-blind trial was initiated in 200 treatment-experienced individuals with GT2 and GT3 who were assigned to two treatment groups to receive sofosbuvir (400 mg daily) with RBV (1,000 mg or 1,200 mg) for duration of 12 or 16 weeks. The study also included persons with compensated cirrhosis at baseline. In the 12-week treatment group the SVR12 rate was 86% for GT2 and 30% for GT3 infected subjects. In the 16-week treatment group SVR12 rate was 94% for GT2 and 62% for GT3 subjects. No adverse side effects were observed that were attributed to sofosbuvir treatment (Gilead, 2013d).
8.4.4.2. FISSION
This randomized, active-controlled study, initiated in December 2012, aimed to compare 12-week course with sofosbuvir 400 mg/RBV with 24-week standard of care treatment IFN/RBV treatment in genotype 2 and 3 treatment-naïve persons. The study met its primary efficacy end point with 67% of subjects achieving SVR12 in both groups. All common adverse effects (≥ 10%) occurred more frequently in IFN/RBV arm. Most frequently observed adverse events (≥ 10%) in the sofosbuvir/RBV group were fatigue, headache, nausea, insomnia and dizziness (Gilead, 2013a).
8.4.4.3. POSITRON
This randomized, double-blind, placebo controlled study was initiated in March 2012 and was designed to evaluate sofosbuvir plus RBV for persons who were IFN-intolerant or ineligible/unwilling to take IFN. Two hundred and seven subjects with GT2 and GT3 HCV infections were enrolled. Of those, 15% had compensated cirrhosis and 53% were GT2. Among individuals with GT2, 93% achieved SVR12, among those with GT3 and the small percentage that had cirrhosis, 61% also achieved SVR-12 (Gilead, 2012); however the overall SVR12 rate was 78% in GT3 and GT4 combined.
8.4.4.4. NEUTRINO
This study was initiated in June 2012 in 327 treatment-naïve subjects with GT1, GT4, GT5 and GT6 to evaluate treatment outcomes of a 12-week course of sofosbuvir with IFN and RBV treatment. Overall, triple combination was found to be superior with 90% of subjects achieving SVR12 compared to a predefined historic control SVR rate of 60 percent. The most common adverse events (reported in >20%) were fatigue, headache, nausea, insomnia and anemia (Gilead, 2013a).
8.5. Conclusions
Sofosbuvir, PSI-7977 (now GS-7977) is the most promising pan-genotypic antiviral drugs against HCV infection identified to date. In combination with SOC or other DAAs such as protease and NS5A inhibitors (see below), it demonstrated high percentage of cure rate among all GTs. Like earlier, anti-HCV nucleoside analogs sofosbuvir showed a clear synergistic effect when dosed in combination with RBV. Two small and separate early clinical studies showed that RBV could lower HCV viral load, but did not cure any subjects tested (Di Bisceglie et al., 1992; Reichard et al., 1991). In our hands, RBV has no anti-HCV activity in the Huh7 replicon system suggesting that the antiviral effect may be due to a host effect (Bassit et al., 2008).
SVR rates were consistently lower among GT3 patents in phase III studies reporting preliminary data. Early in-vitro genotype studies with sofosbuvir found similar EC90 values for inhibition of GT1b (Con1)-, GT1a (H77)-, and GT2a (JFH-1)-derived replicons and for GT1b chimeric replicons containing the NS5B region from the J6 GT2a isolate and from GT2b and GT3a isolates (Lam et al., 2012). Thus, there was no published preclinical indication of lower efficacy versus GT3 and it remains to be seen if this pattern holds true as more data are disclosed.
On April 8, 2013, based on these clinical studies, Gilead submitted a new drug application to U.S. FDA for the use of sofosbuvir and RBV as an all-oral regimen in GT2 and three individuals and in combination with IFN/RBV for the treatment-naïve persons with GT1, 4, 5 and 6 HCV infection (Gilead, 2013c). On October 25, 2013, an FDA advisory committee voted unanimously (15 to 0) in favor of approval of the use of sofosbuvir in persons infected with GT2 and GT3 in combination with RBV. The committee also recommended (15-0) sofosbuvir in combination with IFN and RBV for treatment of chronic HCV in treatment-naïve adults with GT1 and GT4 infections.
9. Combinations of sofosbuvir with other DAAs
9.1. Sofosbuvir (PSI-7977; GS-7977) plus daclatasvir (BMS-790052)
The all-oral, IFN/RBV-free, combination of direct acting antivirals (DAA) involving sofosbuvir an NS5B polymerase inhibitor, and daclatasvir (BMS-790052) (Fig. 12), an NS5A inhibitor, has been explored as an attractive approach toward HCV treatment (Sulkowski et al., 2012a; Sulkowski et al., 2012b).
Fig. 12.

Structures of daclatasvir, ledipasvir and simeprevir.
9.1.1. Phase IIa
The first combination study of sofosbuvir and daclatasvir with or without RBV was reported in a phase IIa trial (AI444-040 study) in treatment-naïve subjects with GT1a/1b, GT2, or GT3 HCV infection (Sulkowski et al., 2012a). Eighty-eight subjects were enrolled to receive sofosbuvir (400 mg QD) and daclatasvir (60 mg QD) in combinations with or without RBV for 24 weeks. HCV RNA levels were monitored up to 48 weeks after discontinuation of antiviral therapy. Based on the interim results, overall, more than 95% of all treatment-naïve subjects with any GT tested achieved SVR4, but in GT1a/1b subjects the SVR4 was 100% (HCV RNA < 10 IU/mL). The combination of sofosbuvir and daclatasvir was generally well tolerated, and, of special significance, RBV only contributed to adverse effects and had no effect on virologic response.
An additional eighty-two GT1a/1b subjects were randomized to the AI444-040 study to receive sofosbuvir (400 mg QD) and daclatasvir (60 mg QD) in combination with or without RBV for 12 weeks. In a recent update of the study (Sulkowski et al., 2012b), with a total of 170 treatment-naïve individuals with GT1a/1b, GT2, or GT3 HCV infection enrolled to receive sofosbuvir (400 mg QD) and daclatasvir (60 mg QD) in combinations with or without RBV for 24 weeks, in GT1a/1b, GT2, or GT3 and 12 weeks in GT1a/1b more than 93% of all treatment-naïve subjects achieved SVR. Importantly, 43 out of 44 (98%) GT1 subjects achieved SVR24 and 41 out of 44 (93%) of GT2 and GT3 subjects achieved SVR24. IL28B GT, viral subtype, and the administration of RBV had no effect on virologic response. The combination of sofosbuvir and daclatasvir were generally safe and well tolerated. The development of anemia in ≤ 21% subjects was only observed with RBV treatment.
9.1.2. Phase II
The safety and efficacy of sofosbuvir (400 mg QD) and daclatasvir (60 mg QD) in combination with or without RBV for 24 weeks was evaluated in fourty-one GT1 subjects who experienced virologic failure with previous telaprevir- or boceprevir-based triple therapy. Sofosbuvir plus daclatasvir arm and sofosbuvir plus daclatasvir plus RBV arm demonstrated high SVR12 rate of 100% and 95%, respectively (Sulkowski et al., 2013).
9.2. Sofosbuvir plus ledipasvir (GS-5885)
The multipart phase II ELECTRON study aimed to evaluate the safety and efficacy of sofosbuvir-containing regimens with or without IFN and/or RBV in GT1, GT2 and GT3 individuals (Gane et al., 2013b) (See section 8.3.3.2 for additional details). One arm of this study focused on an all-oral regimen in GT1 treatment-naïve and null-responders in which subjects were treated with sofosbuvir and RBV over 12 weeks (Gane et al., 2013a). Other arms of the study were similar in design, but also included ledipasvir (GS-5885, Fig. 12), a direct acting antiviral NS5A inhibitor. Among treatment-naïve subjects (N = 25) and null-responders (N = 10) who received sofosbuvir plus RBV without ledipasvir, SVR12 rates were 84% and 10%, respectively. However, all individuals [treatment-naïve (N = 25) and null-responders (N = 10)] receiving ledipasvir in combination with sofosbuvir and RBV showed 100% SVR12 regardless of treatment history (Gane et al., 2013a). Common adverse events included anemia, depression and headache.
There are currently several ongoing phase II and phase III clinical studies for evaluating the safety, tolerability and antiviral efficacy of a once daily fixed-dose combination tablet containing ledipasvir (90 mg), and sofosbuvir (400 mg) with or without RBV (1,000 or 1,200 mg/day divided BID) in GT1 HCV infected subjects. The latest updates from these trials are summarized in the sections below (Gilead, 2013b).
9.2.1. ION-1
In October 2012, Gilead launched a phase III trial for evaluating safety and efficacy of sofosbuvir and ledipasvir with and without RBV for 12 or 24 weeks among treatment-naïve GT1 subjects. Each arm enrolled fifty individuals (total 200 subjects). Based on the initial results of the 12-week arms, the trial will expand to include 800 subjects (Gilead, 2013b).
9.2.2. ION-2
A second phase III study for fixed-dose sofosbuvir/ledipasvir began in January 2013. The study plans to enroll 400 treatment-experienced GT1 subjects who have failed previous IFN-containing regimens (with or without protease inhibitors). This study will evaluate treatment response in a 12-week regimen with RBV and a 24-week regimen with or without RBV (Gilead, 2013b).
9.2.3. LONESTAR
A new Phase II clinical trial is now underway to study sofosbuvir/ledipasvir for 12 weeks and of sofosbuvir/ledipasvir with and without RBV for 8 weeks. This is the first trial to enroll hundred GT1 treatment-naïve persons for the shorter 8-week treatment duration. Treatment with sofosbuvir plus ledipasvir (with or without RBV) for 12 weeks will also be evaluated for treatment-experienced GT1 persons who have failed the protease inhibitor-containing regimen (Gilead, 2013b).
9.2.4. ION-3
In May 2013, Gilead started a Phase III, multicenter, randomized, open-label study to evaluate the safety, tolerability and antiviral efficacy of sofosbuvir/ledipasvir (400/90 mg) with or without RBV (1,000 or 1,200 mg/day divided BID) administered once daily for eight weeks versus sofosbuvir/ledipasvir (SOF/LDV) administered once daily for 12 weeks in treatment-naïve non-cirrhotic subjects with chronic GT1 HCV infection (Gilead, 2013e). The primary outcomes of this study are to determine the sustained virologic response twelve weeks after discontinuation of therapy (SVR12), as well as monitoring the number/frequency of adverse events and potential laboratory abnormalities.
9.3. PSI-7977 (GS-7977; sofosbuvir) plus simeprevir (TMC435)
Simeprevir (TMC435) (Fig. 12) is an HCV protease inhibitor currently in phase III clinical trials for HCV infection. In November 2011, a phase II clinical trial (the COSMOS study) evaluating the combination of sofosbuvir and simeprevir with or without RBV in individuals who are HCV GT1 null-responders to prior IFN/RBV with METAVIR stage either F0-F2 (cohort 1) or F3-F4 (cohort 2) was initiated. The study aims to randomize ninety subjects per cohort in a 2:1:2:1 ratio to the following arms: simeprevir 150 mg once daily + sofosbuvir 400 mg once daily for 24 weeks with or without RBV, or simeprevir 150 mg once daily + sofosbuvir 400 mg once daily for 12 weeks with or without RBV. For cohort 1, a safety and efficacy interim analysis was conducted when approximately 20% of the planned randomized patients reached the end of treatment or permanently discontinued treatment early. Interim data demonstrated RVR in 13/15, 8/11, 20/23, 5/10 in simeprevir with sofosbuvir and RBV (24 weeks), simeprevir and sofosbuvir (24 weeks), simeprevir with sofosbuvir and RBV (12 weeks), and simeprevir plus sofosbuvir (12 weeks), respectively (Lawitz et al., 2013a). Similar RVR rates were observed with simeprevir plus sofosbuvir + RBV treated for 12 or 24 weeks. However, lower RVR rates were observed without RBV for 12 (50%) and 24 week (72%) treatments. Interim SVR4 results from the 12 week arms of cohort 2 in the ongoing COSMOS study, including treatment-naïve or previous null-responder HCV patients all with METAVIR score F3-F4 were recently reported. Treatment for 12 weeks with simeprevir and sofosbuvir, with or without ribavirin, led to SVR4 rates of 96% and 100%, respectively (Medivir, 2013). All arms of this study at 12 weeks were generally considered safe and well tolerated. However, anemia and increased bilirubin was observed in 13.0% and 9.3% of the patients in the RBV containing arms, respectively. These events were not observed in the non-RBV containing arms.
9.4. Sofosbuvir (GS-7977; PSI-7977) a pyrimidine plus PSI-938 a purine nucleotide analog
For information on the phase II NUCLEAR and QUANTUM trials refer to sections 11.3.2 and 11.3.3 of this manuscript.
10. R7432
In 2010, Roche initiated a phase I clinical trial of R7432 in a dose-escalation study with sixty persons enrolled (Roche, 2010). This study was discontinued by February of 2012 (Roche, 2011a, b). Unfortunately, the chemical structure of R7432, the clinical trial results and an explanation for the discontinuation has not been disclosed.
11. PSI-938 (PSI-352938)
Purine nucleoside derivatives of mericitabine were designed to be second-generation agents with improved potency, pharmacokinetics, and resistance profiles. The guanosine derivative PSI-938 (also known as PSI-352938) was a result of these studies (Chun et al., 2010; Clark, 2005; Du et al., 2009; Reddy et al., 2010; Sofia et al., 2010b). This derivative is a cyclic phosphate prodrug of a 2′-α-F-2′-β-C-methyl class of nucleosides. The choice for this particular prodrug was based on observations from previous studies which showed that nucleotide cyclic phosphate prodrugs were precursors of the nucleotide monophosphate with enhanced cellular permeability in vitro (Gunic et al., 2007).
11.1. Synthesis of PSI-938
Pharmasset reported a multi-kilogram process synthesis of PSI-938 requiring no chromatography (Reddy et al., 2011; Wang et al., 2009). 2′-α-F-2′-β-C-methyl-2′-deoxy lactone 29 was prepared as described in Scheme 8. Benzoylation was originally used to complete the synthesis of a key intermediate similar in structure to compound 29 (Scheme 8). However, a switch to 4-chlorobenzyl protection afforded more crystalline intermediates that allowed for a multi-step synthesis that did not require any column chromatography.
Scheme 8.

Synthesis of PSI-938 intermediate lactone 29. Reagents and conditions: (a) p-Cl-BzCl, pyr., r.t., 70%.
The conclusion of the PSI-938 synthesis is shown in Scheme 9. The p-chlorobenzoyl protected lactone 29 was reduced to lactol 30, and it is interesting to note that upon standing the β/α anomeric mixture isomerized to the thermodynamically more stable β-lactol. An Appel reaction at low temperature then furnished bromo derivative 31. Glycosylation of compound 31 with 2-amino-6-chloro purine followed by treatment with sodium ethoxide in ethanol provided the unprotected 6-ethoxy purine nucleoside 32. After extensive investigation, conditions were found for the formation of the 3′,5′-cyclic phosphate PSI-938 (cis-RP configuration) in high diastereoselectivity (>95%): it was finally isolated in 52% yield and greater than 99.5% diastereoselectivity after direct crystallization.
Scheme 9.

Synthesis of PSI-938. Reagents and conditions: (a) LiAl(t-BuO)3H, THF, −20 °C, 61%; (b) CBr4, PPh3, CH2Cl2, −20 °C, 1 h, 79%; (c) 2-amino-6-chloropurine, t-BuOK, t-BuOH, CH3CN, 50 °C, 16 h, 65%; (d) NaOEt, EtOH, reflux, 1 h, 92%; (e) isopropyldichlorophosphate, NMI, Et3N, CH2Cl2, 52%.
11.2. Preclinical development
In an HCV replicon cell assay, PSI-938 had an EC90 of 1.37 μM with no cytotoxicity up to 100 μM (Reddy et al., 2010). It demonstrated prolonged stability in simulated gastrointestinal fluid, simulated intestinal fluid, human plasma, and human liver S9 (in vitro model for liver stability). Expanded cytotoxicity data showed no cytotoxicity in a variety of cell lines and no mitochondrial toxicity. Rats treated with the compound had high levels of the triphosphate in the liver. Interestingly, PSI-938 was equally active against the NS5B S282T mutant replicon. It also showed a high barrier of resistance and a different resistance profile than that of RG-7128 (Lam et al., 2011). PSI-938 was found to be metabolized by a phosphorylation pathway distinct from RG-7128. These promising results lead Pharmasset to nominate PSI-938 as a preclinical candidate in June 2009 (Pharmasset, 2009).
11.3. Clinical development
11.3.1. Phase I
A phase I single ascending dose trial began in early 2010 (Pharmasset, 2010). Healthy male and female volunteers received a single dose of PSI-938 ranging from 100 mg to 1,600 mg. No dose-limiting toxicity was identified. In August 2010, a multidose ascending dose trial was conducted in fourty subjects chronically infected with HCV GT1. Subjects received 100 mg QD, 200 mg QD, 300 mg QD, and 100 mg BID for seven days. PSI-938 demonstrated potent antiviral activity with an average drop of 4 log10 IU/mL in mean HCV RNA from baseline. A consistent decline of HCV RNA levels was observed with no viral breakthrough. PSI-938 was shown to be safe and well tolerated. No adverse events were reported as well as any subject discontinuations (Pharmasset, 2011c).
11.3.2. Phase IIa, NUCLEAR
The combination of sofosbuvir and PSI-938 represented the first once daily dual-nucleotide IFN/RBV-free study for HCV treatment (Lawitz et al., 2011). Sofosbuvir, a pyrimidine, and PSI-938, a purine, are both NS5B polymerase inhibitors. In March 2011, Pharmasset published the results of the Phase IIa NUCLEAR trial in HCV GT1 treatment-naïve subjects. The study evaluated the safety, PK and antiviral activity of PSI-938 and sofosbuvir administered as monotherapy or in combination for seven to fourteen days. Forty subjects were enrolled in cohorts 1–4 receiving PSI-938 (300 mg QD) as monotherapy and/or sofosbuvir (400 mg QD) in combinations, and the treatment regimens resulted in median drops of HCV RNA that ranged from 5.0 to 5.2 log10 IU/mL. PSI-938 was generally safe and well tolerated over seven to fourteen days, with no adverse events or subject discontinuations. HCV RNA rapidly declined in the first three days followed by continued decrease until end of treatment or assay limit of detection (LOD) was reached. Of the 24 subjects who received the combination treatment, 22 (92%) achieved HCV RNA levels of <15 IU/mL after fourteen days of treatment. Of note, sofosbuvir and PSI-938 had similar HCV RNA reductions over seven days. The NUCLEAR study suggested that the combination of sofosbuvir and PSI-938 was generally safe and well tolerated with no viral breakthrough and no significant pharmacokinetic interaction during the seven to fourteen day therapy. These promising results were followed by FDA Fast Track designation in August 2011 (Pharmasset, 2011d).
11.3.3. Phase IIb, QUANTUM
In September 2011, Pharmasset initiated the Phase IIb QUANTUM trial in approximately 450 adults of all viral genotypes to assess safety and SVR rates at 12 or 24 weeks for PSI-938, PSI-7977 (sofosbuvir), and various combinations of the drugs with and without RBV (Pharmasset, 2011c). The trial evaluated interferon-free regimens of sofosbuvir 400 mg QD and PSI-938 300 mg QD with and without RBV over 12 or 24 weeks in treatment-naïve patients. The trial also evaluated PSI-938 monotherapy. SVR24 was the primary endpoint of the trial with patients equally randomized across the following arms: 1) PSI-938 only; 2) PSI-938 and sofosbuvir; 3) sofosbuvir and RBV; 4) PSI-938, sofosbuvir, and RBV and 5) a placebo control of 24 weeks.
Unfortunately, routine safety monitoring detected “laboratory abnormalities associated with liver function” among the 235 individuals in the study who were receiving treatment with PSI-938 alone or in combination with sofosbuvir or sofosbuvir and RBV (Pharmasset, 2011a). The remaining 12 and 24-week sofosbuvir and RBV combination arm continued unchanged to generate data in support of the NEUTRINO trial (see section 8.4.4.4).
11.4. Conclusions
In December 2011, PSI-938 was dropped from the follow-up phase IIb QUANTUM trial and further clinical evaluation due to abnormalities in liver function (Pharmasset, 2011b). No future plans for PSI-938 have been reported. It is unfortunate that a relatively high dose of this drug was used for the main studies, since it is likely that a lower dose would have also been effective with no apparent untoward effects especially when used with sofosbuvir.
12. TMC649128
12.1. Preclinical development
TMC649128 (Johansson et al., 2008; Jonckers et al., 2011) is a nucleoside NS5B polymerase inhibitor and has demonstrated an attractive pre-clinical profile: it displayed in vitro activity across multiple HCV GTs with a high genetic barrier to resistance. It was disclosed in May 2008, when Medivir entered into an R&D agreement with Ortho Biotech Products LP, an affiliate of Tibotec, to develop medications to treat HCV infection targeting NS5B virus polymerase. Under the agreement, Medivir and Tibotec jointly performed preclinical studies to identify potential compounds. As an effort to explore 4′-azido-2′-modified cytidines as potential HCV polymerase inhibitors known for their antiviral HCV activities, Medivir’s scientists designed and synthesized 4′-azido-2′-deoxy-2′-methyl-cytidine 33 (Nilsson et al., 2012) (Fig. 13), which exhibited a moderate anti-HCV activity (Johansson et al., 2008). Although all the ester prodrug derivatives only exhibited similar or slightly lower activity compared with (33), the isobutyryl ester (TMC649128) demonstrated greatly improved mean maximum plasma concentration (Cmax = 4.65 μM) and hence a larger area under the curve (AUC0-t = 12.7 3 μM × h) and oral bioavailability (F = 65%).
Fig. 13.

Structure of prodrug TMC649128 and related nucleosides.
12.2. TMC649128 synthesis
TMC649128 was synthesized as shown in Scheme 10: Glycosylation of 4 with silylated uracil in the presence of tin chloride allowed for the formation of compound 33. Treatment of compound 33 with ammonia in methanol led to the clean deprotection of the benzoyl group, leading to compound 34 in quantitative yield. Protection of the 3′- and 5′-hydroxyl groups with a TIPDS protective group, followed by deoxygenation via free-radical chemistry gave compound 36 in 97% yield with selectivity of 93:7 (Li and Piccirilli, 2003). Deprotection of the TIPDS group with TBAF generated compound 37 in high yield. Nucleoside 37 was then converted to the olefin compound 38 (Verheyden and Moffatt, 1975). Treating exocyclic olefin with iodine and benzyltriethylammonium azide, followed by benzoylation, provided 4′-azido nucleoside 40 in 81% yield over two steps. Oxidative nucleophilic substitution of compound 40 with m-chloroperbenzoic acid/m-chlorobenzoic acid in DCM afforded protected azido derivative 41, which was readily converted to 4′-azido-2′-deoxy-2′-methyl cytidine (42) by treatment with 1-H-tetrazole and 4-chlorophenyldichlorophosphate followed by ammonia deprotection. Final acylation of both 3′- and 5′-hydroxy groups led to the formation of TMC649128.
Scheme 10.

Synthesis of 4′-azido-2′-deoxy-2′-methyl cytidine (TMC649128). Reagents and conditions: (a) bis(trimethylsilyl)uracil, SnCl4, CH3CN, r.t., 20 h, 96%; (b) NH3, CH3OH, 0–4 °C, 2 days, >99%; (c) TIPDSCl2, imidazole, DMF, r.t., 2 h, 65%; (d) ClCOCO2Me, DMAP, CH3CN, r.t., 1 h; (e) n-Bu3SnH, AIBN, toluene, reflux, 2 h, 97% (β/α 93:7, de 86%) over two steps; (f) TBAF/THF, r.t., 1 h, >99% (β/α 93:7); (g) I2, PPh3, imidazole, THF, 76%; (h) NaOMe, MeOH, 75%; (i) i. I2, NMO, Bn(Et)3NN3; ii. BzCl, NMP, DMAP, THF, 81% over 2 steps; (j) m-CPBA, (NH4)2SO4, DCM, 68%; (k) i. pyridine, 2-chlorophenyldichlorophosphate, 1-H-tetrazole; ii. dioxane, NH3; iii. NH3, MeOH, 61% over 3 steps; (l) i-PrCOCl, TEA, DCM, r.t., >99%.
12.3. Clinical development
12.3.1. Phase Ia
In January 2011, Medivir announced the start of phase Ia trial of the hepatitis C polymerase inhibitor TMC649128. This first in human, double-blind, randomized, placebo-controlled trial, was successfully completed in June 2011 to assess the safety, tolerability, and pharmacokinetics of increasing single oral doses of TMC649128 in nine healthy volunteers. After each administration of the drug and up to four weeks after the last administration, safety assessments (blood and urine tests, blood pressure, pulse, electrocardiogram, and physical examination) were performed (Medivir, 2011b; Tibotec, 2011b).
12.3.2. Phase Ib
In June 2011, Tibotec initiated a phase Ib, double-blind, randomized and placebo-controlled study in GT1 HCV-infected individuals to evaluate the safety, tolerability, pharmacokinetics and antiviral activity of multiple ascending doses of TMC649128 given as monotherapy and in combination with IFN and RBV. The pharmacokinetic/pharmacodynamic relationship for antiviral activity, active metabolite and safety of TMC649128 and its metabolite were assessed. The short-term safety and tolerability of the co-administration of TMC649128 and IFN plus RBV during multiple dosing for 14 days in treatment-naïve GT1 HCV-infected participants was explored. The short-term antiviral effect of the combination of TMC649128 with IFN and RBV was also preliminary assessed (Medivir, 2011c; Tibotec, 2011a).
12.3. Conclusions
In November 2011, this study was terminated, since the antiviral activity was considered insufficient to warrant further clinical development of TMC649128 (Medivir, 2011a).
13. GS-6620
13.1. Preclinical development
GS-6620 (Butler et al., 2011; Butler et al., 2009; Cho et al., 2011a; Cho et al., 2011b; Cho and Wolckenhauer, 2011) is a pangenotypic, liver-targeted nucleotide prodrug developed by Gilead Sciences for the treatment of HCV. GS-6620 was shown to have a favorable inhibitory profile in vitro whereby the incorporation of corresponding triphosphate resulted in chain-termination. Cell culture resistance selection assays reported the selection of the S282T mutation in the NS5B viral polymerase. This mutation conferred >200-fold resistance in in vitro experiments with purified recombination S282T mutant enzyme (Fenaux et al., 2011). Oral administration of GS-6620 to animals resulted in efficient delivery of the nucleoside triphosphate metabolite into the liver (Lawitz et al., 2012b).
13.2. GS-6620 synthesis
The synthesis of GS-6620 is outlined in Scheme 11 (Cho et al., 2013). The tribenzylated 2′-C-Me sugar 43 was efficiently connected to the heterocyclic base 44 under lithium-halogen exchange conditions. The 1′-cyano group was introduced under Lewis acid conditions to give 46 which was followed by a boron trichloride promoted debenzylation to give C-nucleoside analog 47. Formation of the 5′-phosphoramidate and 3′-isobutyl ester are preformed utilizing standard nucleoside protocols to give GS-6620.
Scheme 11.

Synthesis of GS-6620. Reagents and conditions: (a) (44), 1,1,4,4-tetramethyl-1,4-dichlorodisilylethylene (1.0 equiv) and n-BuLi (3.3 equiv), THF, −78 °C followed by addition of (43), 1 h, 65–80%; (b) TMSCN (6 equiv), TMSOTf (4 equiv), CH2Cl2, −15 to 0 °C, 2 h, 85–95%; (c) BCl3 (3 equiv), CH2Cl2, 0 °C, 1 h, 90%; (d) N-methylimidazole, trimethylphosphate and THF, 0 °C to r.t., 2 h, overall yield 59%; (e) i. N,N-dimethylformamide dimethyl acetal, CH3CN, r.t., 1 h; ii. isobutyric acid, DCC, CH3CN, r.t., 48 h; iii. water-TFA, 0 °C to r.t., 64 h, overall yield 73%.
13.3. Clinical development
The favorable preclinical characteristics of this compound made it a candidate for phase I clinical trials wherein safety, tolerability, pharmacokinetics and antiviral activity were evaluated in a multi-center, randomized, placebo controlled, multiple-dose escalation first-in-man study in 2010. Out of the ninety treatment-naïve subjects enrolled in the study, twelve showed signs of mild adverse events related to treatment. Two subjects who received the highest dose of treatment (900 mg BID) had virus levels below the limit of detection at the end of the five-day dosing period.
13.4. Conclusions
Although GS-6620 was safe and well tolerated over five day of administration period, it was found that exposure to the drug was markedly lower in humans as compared to pre-clinical animal studies with substantial intra- and inter-subject variability seen in all cohorts. Gilead Sciences indicated that without further optimization of drug delivery to improve PK and antiviral activity, future clinical development would not occur for GS-6620 (Lawitz et al., 2012b).
14. ALS-2200 (VX-135) and ALS-2158
In June 2011, Vertex licensed both a purine and pyrimidine nucleoside prodrug inhibitors of HCV NS5B polymerase from Alios Biopharma (Vertex, 2011). Both uridine nucleoside analog ALS-2200 (VX-135) and adenosine nucleotide analog ALS-2158 (Beigelman et al., 2010; Beigelman et al., 2012a; Beigelman et al., 2012b; Smith et al., 2012) entered phase I clinical trials in December 2011. The exact structure of these nucleoside analogs has not yet been disclosed.
14.1. ALS-2258 clinical development
In an early clinical study, the purine ALS-2158 did not show sufficient antiviral activity and the study was discontinued in September 2012 (Vertex, 2012b).
14.2. ALS-2200 clinical development
On the other hand, preliminary seven-day viral kinetic data with the pyrimidine prodrug ALS-2200(VX-135) administered at 200 mg QD (Marcellin et al., 2012) demonstrated a median drop of 4.18 log10 (range −3.6, −5.2) in HCV RNA in combination with RBV in treatment-naïve chronic HCV subjects with GT1. Five of eight subjects achieved HCV RNA levels below the limit of quantification, and two of these five achieved HCV RNA levels below the limit of detection after seven days of dosing. Similar results were obtained with monotherapy of ALS-2200 (200 mg, once-daily, seven days). Adverse events during the study were mild to moderate with no subject discontinuations.
14.2.1. Phase II
As of November 2012 (Vertex, 2012a, d) future multiple phase II developments of ALS-2200 (VX-135) were envisaged: the first one was initiated with GlaxoSmithKline’s NS5A inhibitor GSK2336805 (Vertex, 2012d) (Fig. 14) but was terminated in June 2013 when Vertex and GSK mutually decided to cease the collaboration for a phase II study and prioritized other projects. A second study is planned for the second half of 2013 with VX-135 in combination with Janssen’s protease inhibitor simeprevir (TMC435, Fig. 12) in patients with GT1 HCV (Vertex, 2012a). Vertex also planned to begin a study of VX-135 in combination with protease inhibitor Incivek® (telaprevir), the company’s existing drug for persons with chronic GT1 HCV. Two additional studies include evaluating the safety and efficacy of VX-135 plus RBV in treatment-naïve subjects with chronic Hepatitis C and planned study to evaluate the safety, efficacy, pharmacokinetics and pharmacodynamics of VX-135 and daclatasvir in GT1 chronic hepatitis C subjects. All of these Phase II studies will evaluate safety, tolerability and SVR12 using 12 week all oral regimens in subjects with GT1 HCV. So far, Vertex completed dosing of 100 mg and 200 mg of VX-135 in combination with RBV in Europe and both doses were well tolerated with no discontinuations. No serious adverse events have been reported and no liver or cardiac safety issues have been identified. Most recently, in April 2013, Vertex started a study of 100 and 200 mg doses of VX-135 in combination with daclatasvir (BMS-790052), an NS5A inhibitor being developed by BMS (Vertex, 2012c), with results expected in early 2014. However, in July 2013, the FDA notified Vertex that a partial clinical hold had been placed on Vertex’s ongoing phase II US study of VX-135 (Vertex, 2013). This partial hold prevented further evaluation of the 200 mg dose of VX-135 due to reversible elevated liver enzymes in patients receiving 400 mg of VX-135 in combination with RBV in a European phase II study. Evaluation of the 100 mg dose of VX-135 in combination with RBV in the U.S. continued. In addition, the recently initiated dosing of 100 and 200 mg of VX-135 in combination with daclatasvir as part of a phase II study in New Zealand also continued.
Fig. 14.
Structure of GSK2336805.
15. BMS-986094 (INX-189/INX-08189)
15.1. Synthesis of BMS-986094
BMS-986094 (Chamberlain et al., 2010; McGuigan and Perrone, 2008) is a double prodrug of 2′-C-methylguanosine triphosphate developed by Inhibitex and later acquired by Bristol-Myers Squibb (Fig. 15). The synthetic route started with trimethylsilyl (TMS) triflate mediated glycosylation of tetrabenzoyl 2′-C-methyl sugar 4 and 6-chloroguanine. The resulting benzoylated 6-chloro nucleoside 49 was then converted to unprotected 6-O-methyl analog 50 by substitution of the chlorine with sodium methoxide in methanol at room temperature. For the prodrug portion, the phosphorochloridate 53 was prepared in two steps by treatment of 1-naphthol with phosphorus oxychloride in presence of triethylamine followed by reaction with t-BuCH2-L-valine ester. Finally, reaction of (53) with (50) was conducted in THF in presence of N-methylimidazole (NMI) to generate the target compound, BMS-986094, in 18% yield as a mixture of diastereoisomers (Scheme 12) (McGuigan et al., 2010a; McGuigan et al., 2010b). As the anti-HCV activity and estimated half-life values were very similar between the two phosphorous diastereomers, BMS-986094 was tested as a mixture in the latter studies.
Fig. 15.

Structure of BMS-986094.
Scheme 12.

Synthesis of BMS-986094. Reagents and conditions: (a) 2-amino-6-chloropurine, DBU, MeCN, TMSOTf, 65 °C, 4 h, 79%; (b) NaOMe, MeOH, r.t., 24 h, 76%; (c) POCl3, Et3N, Et2O, −78 °C, 1 h then r.t., overnight, 95%; (d) tBuCH2-L-valine ester, Et3N, DCM, −78 °C, 1 h then r.t., 2 h; (e) NMI, THF, r.t., overnight, 18%.
This 1-naphthyl neopentyl alanine phosphoramidate derivative of 6-O-methoxy-2′-C-methylguanosine showed potent inhibition in an HCV replicon 1b assay (EC50 = 0.01 μM, EC90 = 0.04 μM) and demonstrated a 500 fold greater potency than the parent 6-O-methoxy nucleoside (Kolykhalov et al., 2009; McGuigan et al., 2010a; McGuigan et al., 2010b). However, in comparison with other clinical anti-HCV agents, BMS-986094 was relatively toxic in multiple cell lines, with CC50 values of 0.32, 2.23, 2.81, and 15.0 μM in Huh7 (human hepatoma), HepG2 (human hepatoma), BxPC3 (human pancreatic), and CEM (human T lymphocyte) cells, respectively (Furman et al., 2011). Other studies showed that BMS-986094 caused bone marrow toxicity in the low micromolar range along with toxicity in kidney, blood, skeletal muscle, CNS and intestinal cells. Additional studies suggested that BMS-986094 could cause unique tissue specific toxicity based on in vitro assay results, with the lowest CC50 values in bone marrow cells (Hutchins et al., 2009; Standring, 2012). Furthermore, in a 14-day study, no appreciable nucleoside analog-associated mitochondrial toxicity was observed in CEM and HepG2 cells treated with BMS-986094 up to 1 μM (Vernachio et al., 2011). It is not clear why higher concentrations were not reported.
15.2. Clinical development
15.2.1. Phase Ia
In a first-in-human, single-ascending oral dose (3 mg to 100 mg or a matching placebo) phase Ia study, BMS-986094 was well tolerated with pharmacokinetics that supported once daily dosing (Patti et al., 2011). In a separate double-blind, placebo-controlled, dose-escalation study, 70 treatment-naïve HCV GT1 subjects dosed with BMS-986094 at 9, 25, 50, 100, 200 mg once-daily for seven days resulted in a mean decrease in viral load of −0.62, −1.02, −1.53, −2.54 and −4.25 log10 IU/mL, respectively. Doses of 9, 25 and 100 mg once-daily were also investigated in combination with RBV over seven days, producing −0.75, −1.56 and −3.79 log10 IU/mL reduction in viral load, respectively. The drug was well tolerated at all doses (Rodriguez-Torres et al., 2011).
15.2.2. Phase II
After BMS-986094 was granted Fast-Track Status by the FDA in March 2011, various Phase II trials were scheduled to commence in 2011 to continue investigating drug combinations for longer treatment periods. After Bristol-Myers Squibb bought Inhibitex in January 2012, INX-189 was renamed BMS-986094. A 12 week Phase II clinical study in GT2/3 treatment-naïve individuals, in which BMS-986094 was dosed at either 0, 25, 50 or 100 mg in combination with RBV and IFN resulted in RVR rates of 58%, 82%, 78% and 75%, cEVR rates of 92%, 93%, 100% and 88%, and eRVR rates of 58%, 75%, 78% and 71%, respectively. No virologic breakthroughs were observed over the course of therapy (Lawitz et al., 2012c).
15.3. Conclusions
The development of BMS-986094 was discontinued as a result of a series of significant adverse events: in August 2012, a subject receiving 200 mg once daily dosing died of heart failure and another eight subjects were diagnosed with severe kidney and heart damage (Sheridan, 2012). The study was suspended and further development of BMS-986094 was discontinued; however, a follow-up safety evaluation in subjects who participated in prior clinical studies is ongoing (BMS, 2012). The cause(s) of these serious side effects are still under investigation at this time. It is not clear if the observed toxicity was due to the nucleoside triphosphate itself or due to potential toxic metabolites, such as neopentyl alcohol, 1-naphthanol and methanol, released during the enzymatic cleavage of the prodrug moiety or because of the extremely high levels of nucleoside triphosphates produced. Cardiac toxicity for BMS-986094 was demonstrated to be dose-related and the postulated reason for the cardiac toxicity was mitochondrial toxicity. As mentioned above, 2′-C-MeG-TP has been shown to be a substrate for incorporation by human mitochondrial RNA polymerase (Arnold et al., 2012). Toxicities observed during preclinical in vitro studies should have resulted in closer monitoring of clinical toxicities that would have led to an earlier discontinuation of the clinical study of BMS-986094.
16. Lessons Learned
To date, nucleoside inhibitors appear to be the most promising of the three classes of direct-acting antiviral agents currently in phase III clinical studies. Recent progress in the development of nucleoside prodrugs, particularly sofosbuvir, solidifies our belief that this agent could become the best-in-class and backbone of future HCV combination chemotherapy. As such, sofosbuvir is an important milestone in the fight against HCV. The unforeseen surprises noted with nucleoside inhibitors in development are drug specific and not a class effect. The de-risking events based on carefully conducted preclinical developments and clinical trials suggest that we are on the verge of a huge game changer in the field of HCV with proof-of-concept already established for an all-oral cure for HCV. It is clear that early preclinical toxicology and pharmacology can make or break a newly drug preferably before clinical trials are begun. The tragic events related to BMS-986094 only re-emphasizes the need for researchers to fully preclinically evaluate compounds using robust predictive toxicological tests and thorough toxicity screening in various cell-based systems and particularly mitochondrial toxicity which is paramount for the success of a new chemical entity, especially for nucleoside analogs. To assess mitochondrial toxicity, the use of the newly developed mitovir (mitochondrial dysfunction caused by antiviral ribonucleoside) score was recently proposed (Arnold et al., 2012). The mitovir score can be readily obtained during preclinical studies and may be useful to quantify the mitochondrial toxicity potential of compounds. The possibility exists that some infected persons might be more susceptible to mitochondrial toxicity when treated with nucleoside analogs due to their inherent mitochondrial or nuclear transcription dysfunction. The bar has been raised with the FDA when it comes to toxicity associated with nucleoside analogs as is reflected by their actions with IDX184 and VX-135, although exact details and guidance has not yet been forthcoming.
It is also clear that the prodrug selected may enhance oral bioavailability, but may also concurrently increase toxicity by delivering the active metabolite in high levels to undesirable compartments causing significant off-target side effects and even death. Thus, for treatment of HCV infection it is important that the drug is primarily liver targeted due to first pass effect with extensive hepatic extraction and that nucleoside triphosphate production and metabolism remain largely localized in the liver, the site of HCV replication with low systemic exposure. In the context of a viral liver infection, it is ideal for the nucleoside prodrug to be quickly metabolized and preferentially activated in the liver such that it does not go into systemic circulation and result in off-target exposure of the nucleoside or its triphosphate. For example, the side effects that were noted with both IDX184 and INX-189 would indicate that the drug was escaping first pass metabolism and reaching some degree of systemic exposure. Additionally, the intracellular half-life of the nucleoside triphosphate might be more important than the amount of NTP formed. After all, the active metabolite has to persist in the liver long enough to suppress and eliminate viral load. Furthermore, an oral prodrug that is unstable in gastric fluid/GI tract or the plasma may never reach its target to deliver the nucleoside monophosphate to the liver. It is also clearer now that the most desirable nucleoside analogs are those that are pangenotypic and will be effective in all HCV infected persons, especially the difficult-to-treat persons with advanced liver disease including those with decompensated cirrhosis. It is also becoming apparent that one pill may not fit all especially for underdeveloped countries where access and cost of the best combinations may become a severe obstacle to cures (Hagan and Schinazi, 2013).
The HCV drug development tsunami will continue and be broadly felt across many areas of healthcare in the next decade. It is anticipated that the prospects of a cure for millions of infected persons will in time become possible despite the cost of treatment since we have a moral obligation to provide life saving medicines that cure globally. However, it will take courageous leadership at various levels of society and government to succeed in eradicating HCV globally (Hagan and Schinazi, 2013).
Fig. 10.

Structure of 2′-F-2′-C-methyluridine, PSI-6206, and its triphosphate PSI-6206-TP.
Fig. 11.

Structures, anti-HCV replication activity and cytotoxicity of phosphoramidate prodrug PSI-7851 and its diastereoisomers PSI-7976 and PSI-7977.
Scheme 7.
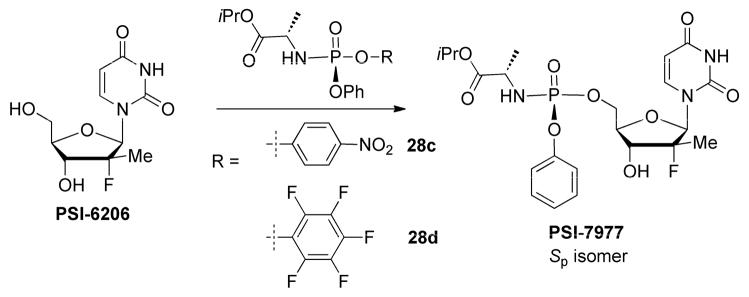
Synthesis of single isomer PSI-7977 (sofosbuvir, GS-7977). Reaction conditions: (a) THF, t-BuMgCl (2.1 equiv) added over 30 min at r.t., stirred 30 min then (28c), r.t., 60 h, 40 %; (b) THF, t-BuMgCl (2.1 equiv) added over 30 min at −5 °C, stirred 30 min, warmed up to 20 °C and stirred for 30 min and then cooled to 5 °C then (28d) (1.2 equiv) was added over 30 min, stirred for 18 h at 5 °C, 68%.
Nucleoside analogs target the hepatitis C (HCV) NS5B RNA-dependent RNA polymerase.
At least twelve nucleoside analogs have reached clinical study for HCV infection.
Nucleoside analogs have activity against the various HCV genotypes.
Nucleoside analogs have a high barrier to resistant virus compared to other classes of inhibitors.
Nucleoside analogs that have reached human clinical trials are the focus of this review.
Acknowledgments
Acknowledgements & Disclosures
This work was supported in part by CFAR NIH grant 2P30AI-050409 (to RFS) and by the Department of Veterans Affairs (to RFS). Dr. Schinazi is the founder and a major shareholder of RFS Pharma, LLC. Emory received no funding from RFS Pharma, LLC to perform this work and vice versa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afdahl N, Rodriguez-Torres M, Lawitz E, Godofsky E, Chao G, Fielman B, Knox S, Brown N. Enhanced antiviral efficacy for valopicitabine (NM283) plus Peg-Interferon in hepatitis C patients with HCV genotype-1 infection: Results of a Phase IIa multicenter trial. J Hepatol. 2005;42:39–40. [Google Scholar]
- Ahmed HA, Alfredson TV, Birudaraj K, Brandl MT, Phuapradit W, Shah NH, Stefanidis D. HCV prodrug formulation. WO 2007068615 PCT Int Appl. 2007
- Alfredson T, Sarma K, Brandl M, Larrabee S, Wu X, Tu Y, Liu H, Smith D, Birudaraj R, Li F, Tran T, Hadig X, Hill G, Daley R, Blue D, Berger N, Symons J, Cammack N, Ipe D, Berns H, Lakatos I, Swallow S, Klumpp K. Design and evaluation of novel HCV polymerase inhibitor prodrugs with enhanced oral bioavailability. AAPS Annual Meeting; San Antonio, TX. 2006. [Google Scholar]
- Ali S, Leveque V, Le Pogam S, Ma H, Philipp F, Inocencio N, Smith M, Alker A, Kang H, Najera I, Klumpp K, Symons J, Cammack N, Jiang WL. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob Agents Chemother. 2008;52:4356–4369. doi: 10.1128/AAC.00444-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Sharma SD, Feng JY, Ray AS, Smidansky ED, Kireeva ML, Cho A, Perry J, Vela JE, Park Y, Xu Y, Tian Y, Babusis D, Barauskus O, Peterson BR, Gnatt A, Kashlev M, Zhong W, Cameron CE. Sensitivity of Mitochondrial Transcription and Resistance of RNA Polymerase II Dependant Nuclear Transcription to Antiviral Ribonucleosides. PLoS Pathog. 2012;8:e1003030. doi: 10.1371/journal.ppat.1003030. doi:1003010.1001371/journal.ppat.1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 1996;22:67–84. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- Asif G, Hurwitz SJ, Shi J, Hernandez-Santiago BI, Schinazi RF. Pharmacokinetics of the antiviral agent β-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine in rhesus monkeys. Antimicrob Agents Chemother. 2007;51:2877–2882. doi: 10.1128/AAC.00193-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselah T, Marcellin P. Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver Int. 2012;32 (Suppl 1):88–102. doi: 10.1111/j.1478-3231.2011.02699.x. [DOI] [PubMed] [Google Scholar]
- Bassit L, Grier J, Bennett M. Combinations of 2′-C-methylcytidine analogues with interferon-alpha2b and triple combination with ribavirin in the hepatitis C virus replicon system. Antivir Chem Chemother. 2008;19:25–31. doi: 10.1177/095632020801900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigelman L, Blatt L, Wang G. Substituted nucleoside and nucleotide analogs. WO 2010108140 PCT Int Appl. 2010
- Beigelman L, Blatt L, Wang G, Rajwanshi VK, Dyatkina NB, Smith BD. Substituted nucleotide analogs. WO 2012040126 PCT Int Appl. 2012a
- Beigelman L, Smith BD, Deval J, Rajwanshi VK. Cyclic nucleotide analogs. WO 2012088155 PCT Int Appl. 2012b
- Berrey MM, Hindes R, Symonds WT, Ray AS, Mo H, Hebner CM. Methods and compositions for treating hepatitis C virus. WO 2013066748 PCT Int Appl. 2013
- BMS. A Follow-up Evaluation for Safety in Subjects Who Participated in a Phase 1 Study with BMS-986094 (INX-08189) Clinical trial NCT017322848. 2012 retrieved from: http://clinicaltrials.gov/show/NCT01732848.
- Bobeck DR, Coats SJ, Schinazi RF. Advances in Nucleoside Monophosphate Prodrugs as Anti-hepatitis C Virus. Agents Antivir Ther. 2010;15:935–950. doi: 10.3851/IMP1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl M, Wu X, Holper M, Hong L, Jia Z, Birudaraj R, Reddy M, Alfredson T, Tran T, Larrabee S, Hadig X, Sarma K, Washington C, Hill G, Smith DB. Physicochemical Properties of the Nucleoside Prodrug R1626 Leading to High Oral Bioavailability. Drug Dev Ind Pharm. 2008;34:683–691. doi: 10.1080/03639040701836636. [DOI] [PubMed] [Google Scholar]
- Brown NA. Progress towards improving antiviral therapy for hepatitis C with hepatitis C virus polymerase inhibitors. Part I: Nucleoside analogues. Expert Opin Investig Drugs. 2009;18:709–725. doi: 10.1517/13543780902854194. [DOI] [PubMed] [Google Scholar]
- Burton JRJ, Everson GT. HCV NS5B Polymerase Inhibitors. Clin Liver Dis. 2009;13:453–465. doi: 10.1016/j.cld.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Butler T, Cho A, Graetz BR, Kim CU, Metobo SE, Saunders OL, Waltman AW, Xu J, Zhang L. Processes and intermediates for the preparation of 1′-substituted carba-nucleosides. WO 2011035250 PCT Int Appl. 2011
- Butler T, Cho A, Kim CU, Saunders OL, Zhang L. 1′-Substituted carba-nucleoside analogs for antiviral treatment. WO 2009132135 PCT Int Appl. 2009
- Cedilote M, Cleary TP, Zhang P. Alternate process for preparing 3,5-O-micron-acyl-2-fluoro-2-C-methyl-D-ribono-gamma-lactone. WO 2008090046 PCT Int Appl. 2008
- Chamberlain S, Hutchins J, Medela K, McGuigan C, Vernachio J, Aljarah M, Gilles A. Phosphoramidate derivatives of guanosine nucleoside compounds for treatment of viral infections. WO 2010081082 PCT Int Appl. 2010
- Cho A, Kim CU, Metobo S, Ray AS, Xu J. 2′-Fluoro substituted carba-nucleoside analogs for antiviral treatment. WO 2011035231 PCT Int Appl. 2011a
- Cho A, Kim CU, Ray AS, Zhang L. 1′-Substituted-carba-nucleoside prodrugs for antiviral treatment. WO 2011150288 PCT Int Appl. 2011b
- Cho A, Wolckenhauer SA. Methods for the preparation of diastereomerically pure phosphoramidate prodrugs. WO 2012012465 PCT Int Appl. 2011
- Cho A, Zhang L, Xu J, Lee R, Butler T, Metobo S, Aktoudianakis V, Lew W, Ye H, Clarke M, Doerffler E, Byun D, Wang T, Babusis D, Carey AC, German P, Sauer D, Zhong W, Rossi S, Fenaux M, McHutchison JG, Perry J, Feng J, Ray AS, Kim CU. Discovery of the First C-Nucleoside HCV Polymerase Inhibitor (GS-6620) with Demonstrated Antiviral Response in HCV Infected Patients. J Med Chem. 2013 doi: 10.1021/jm400201a. ASAP. [DOI] [PubMed] [Google Scholar]
- Choo QL, Kuo G, Weiner A, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derivied from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- Chun B-K, Clark J, Sarma K, Wang P. Antiviral nucleosides. WO 2007065829 PCT Int Appl. 2007
- Chun B-K, Du J, Rachakonda S, Ross BS, Sofia MJ, Pamulapati Reddy G, Chang W, Zhang HR, Nagarathnam D. Synthesis of purine nucleosides. WO 2010075554 PCT Int Appl. 2010
- Clark J. Modified fluorinated nucleoside analogues. WO 2005003147 PCT Int Appl. 2005
- Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman FA, Stec WJ, Patterson SE, Pankiewicz KW. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J Med Chem. 2005;48:5504–5508. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- Cleary D, Reynolds CJ, Berrey MM, Hindes RG, Symonds WT, Ray AS, Mo H, Hebner CM, Oliyai R, Zia V, Stefanidis D, Pakdaman R, Casteel MJ. Compositions and methods for treating hepatitis C virus. WO 2013082003 PCT Int Appl. 2013
- Connolly TJ, Durkin K, Sarma K, Zhu J. Processes for preparing 4′azido nucleoside derivatives. WO 05000864 PCT Int Appl. 2005
- Cretton-Scott E, Gupta K, Hernandez-Santiago B, Larsson M. Compounds and pharmaceutical compositions for the treatment of viral infections. WO 2010014134 PCT Int Appl. 2010
- Cretton-Scott E, Perigaud C, Peyrottes S, Licklider L, Camire M, Larsson M, La Colla P, Hildebrand E, Lallos L, Bilello J, McCarville J, Seifer M, Liuzzi M, Pierra C, Badaroux E, Gosselin G, Surleraux D, Standring DN. In vitro antiviral activity and pharmacology of IDX184, a novel and potent inhibitor of HCV replication. J Hepatol. 2008;48:S220. [Google Scholar]
- Darby SC, Ewart DW, Giangrande PLF, Spooner RJ, Rizza CR, Dusheiko GM, Lee CA, Ludlam CA, Preston FE. Mortality from liver cancer and liver disease in haemophilic men and boys in UK given blood products contaminated with hepatitis C. Lancet. 1997;350:1425–1431. doi: 10.1016/s0140-6736(97)05413-5. [DOI] [PubMed] [Google Scholar]
- Davis GL, Albright JE, Cook SF, Rosenberg DM. Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 2003;9:331–338. doi: 10.1053/jlts.2003.50073. [DOI] [PubMed] [Google Scholar]
- De Clercq E, Field HJ. Antiviral prodrugs - the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. 2006;146:1–11. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco R, Migliaccia G. Challenges and successes in developing new therapies for hepatitis C. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- Denning J, Cornpropst MT, Flach SD, Berrey MM, Symonds WT. Pharmacokinetics, safety, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor for hepatitis C virus, following single ascending doses in healthy subjects. Antimicrob Agents Chemother. 2012;57:1201–1208. doi: 10.1128/AAC.01262-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos RR, Hobbs CJ, Jiang W-R, Martin JA, Merrett JH, Najera I, Shimma N, Tsukuda T. 4′-Substituted nucleosides. WO 02100415 PCT Int Appl. 2002
- Di Bisceglie AM, Shindo M, Fong TL, Fried MW, Swain MG, Bergasa NV, Axiotis CA, Waggoner JG, Park Y, Hoofnagle JH. A pilot study of ribavirin therapy for chronic hepatitis C. Hepatol. 1992;16:649–654. doi: 10.1002/hep.1840160307. [DOI] [PubMed] [Google Scholar]
- Dieterich D, Lawitz E, Nguyen CT, Santoro J, Gitlin N, McEniry D, Chasen R, Goff J, Knox S, Kleber K, Belanger B, Brown N. Ealy clearance of HCV RNA with valopicitabine (NM283) plus Peg-interferon in treatment-naïve patients with HCV-1 infection: First results from a phase IIb trial. J Hepatol. 2006;44:S271–272. [Google Scholar]
- Du J, Nagarathnam D, Pamulapati Reddy G, Ross BS, Sofia MJ. Nucleoside cyclicphosphates. WO 2009152095 PCT Int Appl. 2009
- Farnik H, Zeuzem S. New antiviral therapies in the management of HCV infection. Antivir Ther. 2012;17:771–783. doi: 10.3851/IMP2127. [DOI] [PubMed] [Google Scholar]
- Fenaux M, Cheng G, Mabery E, Ku K, Eng S, Vela J, Feng J, Delaney W, Mo H, Ray A, Zhong W. GS-6620, A Novel Anti-hepatitis C Virus Nucleotide Prodrug, Has a High in vitro Barrier to Resistance. 46th Annual Meeting of the European Association for the Study of the Liver (EALS); Berlin, Germany. 2011. [Google Scholar]
- Fishman S, Childs K, Dieterich D, Carriero D, Uriel A, Mullen M, Factor S, Fierer D, Branch A. Age and risky behaviors of HIV-infected men with acute HCV infection in New York City are similar, but not identical, to those in a European outbreak. 16th Conference on Retroviruses and Opportunistic Infections (CROI); Montreal, Canada. 2009. [Google Scholar]
- Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology. 2002;36:S237–S244. doi: 10.1053/jhep.2002.36810. [DOI] [PubMed] [Google Scholar]
- Furman PA, Murakami E, Niu C, Lam AM, Espiritu C, Bansal S, Bao H, Tolstykh T, Micolochick SH, Keilman M, Zennou V, Bourne N, Veselenak RL, Chang W, Ross BS, Du J, Otto MJ, Sofia MJ. Activity and the metabolic activation pathway of the potent and selective hepatitis C virus pronucleotide inhibitor PSI-353661. Antivir Res. 2011;91:120–132. doi: 10.1016/j.antiviral.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetano JN, Reau N. Hepatitis C: management of side effects in the era of direct-acting antivirals. Curr Gastroenterol Rep. 2013;15:305. doi: 10.1007/s11894-012-0305-1. [DOI] [PubMed] [Google Scholar]
- Gane EJ, Rodriguez-Torres M, Nelson DE, Jacobson IM, McHutchison JG, Duca A, Hyland R, Hill G, Albanis EM, Symonds WT, Berrey MM. Sustained virologic response (SVR) following RG7128 1500 mg BID/Peg-IFN/RBV for 28 days in HCV genotype 2/3 prior nonresponders. J Hepatol. 2010;52:S16. [Google Scholar]
- Gane EJ, Stedman CA, Hyland RH, Ding X, Pang PS, Symonds WT. 20th Conference on Retroviruses and Opportunistic Infections (CROI); March 3–6 2013; Atlanta, GA. 2013a. [Google Scholar]
- Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013b;368:34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- Gane EJ, Stedman CA, Hyland RH, Pang PS, Sorensen RD, Symonds WT, Hindes RD, Berrey MM. Once daily sofosbuvir (GS-7977) regimens in HCV genotype 1–3: The ELECTRON trial. 63rd Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); Boston, MA. 2012. [Google Scholar]
- Gerhardt C. Untersuchungen über die wasserfreien organischen Säuren. Ann Chem Pharma. 1853;87:149–179. [Google Scholar]
- Ghosn J, Larsen C, Piroth L, Duval X, Auperin I, Delarocque-Astagneau E, Gervais A, Alric L, Pol S, Chaix M-L. Evidence for ongoing epidemic sexual transmission of HCV (2006 to 2007) among HIV-1-infected men who have sex with men: France. 16th Conference on Retroviruses and Opportunistic Infections (CROI); Montreal, Canada. 2009. [Google Scholar]
- Gilead. GS-7977 Alone or with Ribavirin for Hepatitis C. Clinical trial NCT01441180. 2011 retrieved from: http://clinicaltrials.gov/ct2/show/NCT01441180?term=01441180&rank=01441181.
- Gilead. Gilead announces sustained virologic response rate of 78% from phase 3 study of sofosbuvir for genotype 2/3 hepatitis C infected patients. Press release. 2012 retrieved from: http://www.gilead.com/news/press-releases/2012/11/gilead-announces-sustained-virologic-response-rate-of-78-from-phase-3-study-of-sofosbuvir-for-genotype-23-hepatitis-c-infected-patients.
- Gilead. Gilead announces sustained virologic response rates from two phase 3 studies of sofosbuvir for hepatitis C. Press release. 2013a retrieved from http://investors.gilead.com/phoenix.zhtml?c=69964&p=irol-newsArticle&id=1780873.
- Gilead. Gilead Provides Update on Hepatitis C Development Programs. Press release. 2013b retrieved from: http://investors.gilead.com/phoenix.zhtml?c=69964&p=irol-newsArticle&id=1771657.
- Gilead. Gilead submits new drug application to U.S. FDA for Sofosbuvir for the treatment of Hepatitis C. Press release. 2013c retrieved from: http://investors.gilead.com/phoenix.zhtml?c=69964&p=irol-newsArticle&id=1804362.
- Gilead. Gilead’s Sofosbuvir for hepatitis C meets primary endpoint in fourth pivotal phase 3 study. Press release. 2013d retrieved from: http://www.gilead.com/news/press-releases/2013/2/gileads-sofosbuvir-for-hepatitis-c-meets-primary-endpoint-in-fourth-pivotal-phase-3-study.
- Gilead. Safety and Efficacy of Sobosbuvir/Ledipasvir Fixed-Dose Combination +/− Ribavirin for the Treatment of HCV (ION-3) Clinical trial NCT01851330. 2013e retrieved from: http://clinicaltrials.gov/ct2/show/NCT01851330?term=ion-01851333&rank=01851331.
- Godofsky E, Afdahl N, Rustgi V, Shick L, Duncan L, Zhou XJ, Chao G, Fang C, Fielman B, Myers M, Brown NA. First clinical results for a novel antiviral treatment for Hepatitis C: a phase I/II dose escalation trial assessing tolerance, pharmacokinetics, and antiviral activity of NM283. J Hepatol. 2004;S1:35. [Google Scholar]
- Gordon CP, Keller PA. Control of hepatitis C: a medicinal chemistry perspective. J Med Chem. 2005;48:1–20. doi: 10.1021/jm0400101. [DOI] [PubMed] [Google Scholar]
- Gunic E, Girardet JL, Ramasamy K, Stoisavljevic-Petkov V, Chow S, Yeh LT, Hamatake RK, Raney A, Hong Z. Cyclic monophosphate prodrugs of base-modified 2′-C-methyl ribonucleosides as potent inhibitors of hepatitis C virus RNA replication. Bioorg Med Chem Lett. 2007;17:2452–2455. doi: 10.1016/j.bmcl.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Hagan LM, Schinazi RF. Best strategies for global HCV eradication. Liver Int. 2013;33(Suppl 1):68–79. doi: 10.1111/liv.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington PJ, Hilbrand S, Sarma K. Process for preparation of 4′-azido cytidine derivatives. WO 2008071571 PCT Int Appl. 2008
- Harry-O’kuru RE, Smith JM, Wolfe MS. A Short, Flexible Route toward 2′-C-Branched Ribonucleosides. J Org Chem. 1997;62:1754–1759. [Google Scholar]
- Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- Hezode C, Dorival C, Zoulim F, Poynard T, Mathurin P, Pol S, Larrey D, Cacoub P, De Ledinghen V, Bourlière M, Bernard PH, Riachi G, Alric L, Samuel D, Barthe Y, Fontaine H, Carrat F, Bronowicki J-P. Safety of telaprevir or boceprevir in combination with peginterferon alfa/ribavirin, in cirrhotic non responders. First results of the french early access program (ANRS CO20-CUPIC); International Liver Congress; 2012; 2012. retrieved from: http://mobile.ilcapp.eu/EASL_161/poster_23756/program.aspx. [Google Scholar]
- Hoofnagle JH. Hepatitis C: The clinical spectrum of disease. Hepatology. 1997;26:15S–20S. doi: 10.1002/hep.510260703. [DOI] [PubMed] [Google Scholar]
- Huang Z, Murray MG, Secrist IJA. Recent development of therapeutics for chronic HCV infection. Antivir Res. 2006;71:351–362. doi: 10.1016/j.antiviral.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Hughes CA, Shafran SD. Chronic hepatitis C virus management: 2000–2005 Update. Ann Pharmacother. 2006;40:74–82. doi: 10.1345/aph.1G263. [DOI] [PubMed] [Google Scholar]
- Hutchins J, Chamberlain S, Chang C, Ganguly B, Gorovits E, Hall A, Henson G, Kolykhalov A, Liu Y, Muhammad J, Perrone P, Gilles A, Holl S, Madela K, McGuigan C, Patti J. In vitro activity and in vivo pharmacokinetics of highly potent phosphoramidate nucleoside analogue inhibitors of hepatitis C NS5B. Antivir Res. 2009;82:A28. [Google Scholar]
- Idenix. Valopicitabine development program placed on clinical hold in the United States. Press release. 2007 retrieved from: http://www.thefreelibrary.com/Valopicitabine+Development+Program+Placed+on+Clinical+Hold+in+the...-a0166344307.
- Idenix. Double-blind, Dose-Escalating Study of IDX184 in Chronic Hepatitis C Treatment-Naïve Subjects. Clinical trial NCT00807001. 2008a retrieved from: http://clinicaltrials.gov/ct2/show/NCT00807001?term=idx00807184&rank=00807005.
- Idenix. Single-dose escalation study of IDX184 in healthy volunteers. Clinical trial NCT00730431. 2008b retrieved from: http://clinicaltrials.gov/ct2/show/NCT00730431?term=idx00730184&rank=00730436.
- Idenix. Safety and antiviral activity of IDX184 in combination with pegylated interferon and ribavirin. Clinical trial NCT01011166. 2009 retrieved from: http://clinicaltrials.gov/ct2/show/NCT01011166?term=idx01011184&rank=01011161.
- Idenix. A drug-drug interaction study evaluating the combination of IDX320 and IDX184 in healthy subjects. Clinical trial NCT01157104. 2010 retrieved from: http://clinicaltrials.gov/ct2/show/NCT01157104?term=idx01157184&rank=01157102.
- Idenix. Idenix Pharmaceuticals Provides Updates on Three Clinical Development Programs. Press release. 2011a retrieved from: http://ir.idenix.com/releasedetail.cfm?ReleaseID=611432.
- Idenix. A open-label study to evaluate the relative bioavailability of IDX184 and food effect in healthy male subjects. Clinical trial NCT01335607. 2011b retrieved from: http://clinicaltrials.gov/ct2/show/NCT01335607?term=idx01335184&rank=01335603.
- Jeffreys D. Aspirin: the remarkable story of a wonder drug. New York: Bloomsbury; 2005. pp. 38–40. [Google Scholar]
- Jensen DM, Wedemeyer H, Herring RW, Ferenci P, Ma MM, Zeuzem S, Rodriguez-Torres M, Bzowej NH, Pockros P, Vierling JM, Ipe D, Hill GZ. High rates of early viral response, promising safety profile and lack of resistance-related breakthrough in HCV GT 1/4 patients treated with RG7128 plus Peg-IFN alfa-2a (40KD)/RBV: Planned week 12 interim analysis from the PROPEL study. Hepatology. 2010;52:360A. [Google Scholar]
- Johansson N-G, Kalayanov G, Martin JA, Smith BD, Winqvist A. HCV Nucleoside inhibitor. WO 2008043704 PCT Int Appl. 2008
- Jonckers TH, Raboisson P, Vandyck K, Pelcman M, Sund BC, Wahling HJ, Passos Pinho PM, Winqvist A, Nilsson KM. Phosphoramidate derivatives of nucleosides. WO 2011039221 PCT Int Appl. 2011
- Keicher JD, Roberts CD, Dyatkina NB. Nucleoside compounds for treating viral infections. US 0107312 PCT Int Appl. 2005
- Kennedy EM, Gavegnano C, Nguyen L, Slater R, Lucas A, Fromentin E, Schinazi RF, Kim B. Ribonucleoside triphosphates as substrate of human immunodefficency virus type 1 reverse transcriptase in human macrophages. J Biol Chem. 2010;285:39980–39991. doi: 10.1074/jbc.M110.178582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K, Leveque V, Le Pogam S, Ma H, Jiang WR, Kang H, Granycome C, Singer M, Laxton C, Hang JQ, Sarma K, Smith DB, Heindl D, Hobbs CJ, Merrett JH, Symons J, Cammack N. The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J Biol Chem. 2006;281:3793–3799. doi: 10.1074/jbc.M510195200. [DOI] [PubMed] [Google Scholar]
- Klumpp K, Smith DB. Discovery and Clinical Evaluation of the Nucleoside Analog Balapiravir (R1626) for the Treatment of HCV Infection. In: Kazmierski WM, editor. Antiviral Drugs: From Basic Discovery through Clinical Trials. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2011. [DOI] [Google Scholar]
- Koch U, Narjes F. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr Top Med Chem. 2007;7:1302–1329. doi: 10.2174/156802607781212211. [DOI] [PubMed] [Google Scholar]
- Kolbe H. Uber Synthese der Salicylsäure. Ann Chem Pharma. 1860;113:125–127. [Google Scholar]
- Kolykhalov A, Chamberlain S, Gorovits E, Hutchins J, Liu Y, Muhammad J, Raja N, Vernachio J, Henson G, Madela K, Gilles A, Mcguigan C, Patti J. HEP DART: Frontiers in Drug Development for Viral Hepatitis. Hawaii, USA: 2009. In vitro characterization of INX-189, a highly potent phosphoramidate nucleoside analogue inhibitor of HCV. [Google Scholar]
- Kowdley KV, Lawitz E, Crespo I, Hassanein T, Davis MN, DeMicco M, Bernstein DE, Afdahl N, Vierling JM, Gordon SC, Anderson JK, Hyland RH, Dvory-Sobol H, An D, Hindes RG, Albanis E, Symonds WT, Berrey MM, Nelson DR, Jacobson IM. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naïve patients with hepatitis C geotype-1 infection (ATOMIC): an open-label, randomised, multicenter phase 2 trial. Lancet. 2013;381:2100–2107. doi: 10.1016/S0140-6736(13)60247-0. [DOI] [PubMed] [Google Scholar]
- Lalezari J, Asmuth D, Casiro A, Vargas H, Lawrence S, Dubuc-Patrick G, Chen J, McCarville J, Pietropaolo K, Zhou XJ, Sullivan-Bolyai J, Mayers D. Short-term monotherapy with IDX184, a liver-targeted nucleotide polymerase inhibitor, in patients with chronic hepatitis C virus infection. Antimicrob Agents Chemother. 2012;56:6372–6378. doi: 10.1128/AAC.01521-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalezari J, Asmuth D, Casiro A, Vargas H, Patrick GD, Liu W, Pietropaolo K, Zhou XJ, Sullivan-Bolyai J, Mayers D. Antiviral activity, safety and pharmacokinetics of IDX184, a liver-targeted nucleotide HCV polymerase inhibitor, in patients with chronic hepatitis C. 60th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); Boston, MA. 2009. [Google Scholar]
- Lalezari J, Box T, O’Riordan W, Mehra P, Nguyen T, Poordad F, Dejesus E, Kwo P, Godofsky E, Lawrence S, Dubuc-Patrick G, Chen J, McCarville J, Pietropaolo K, Zhou X-J, Sullivan-Bolyai J, Mayers D. IDX184 in combination with peginterferon alfa-2a and ribavirin for two weeks in treatment-naïve patients with chronic hepatitis C. Antivir Ther. 2013 doi: 10.3851/IMP2552. EPub ahead of print. [DOI] [PubMed] [Google Scholar]
- Lalezari J, Poordad F, Mehra P, Nguyen T, Dejesus E, Godofsky E, Dubuc-Patrick G, Chen J, Pietropaolo K, Zhou XJ, Sullivan-Bolyai JZ, Mayers D, Group ICI. Antiviral activity, pharmacokinetics and safety of IDX184 in combination with pegylated interferon (pegIFN) and ribavirin (RBV) in treatment-naïve HCV genotype 1-infected subjects. J Hepatol. 2010;52:S469. [Google Scholar]
- Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Niu C, Zennou V, Keilman M, Zhu Y, Lan S, Otto MJ, Furman PA. Genotype and Subtype Profiling of PSI-7977 as a Nucleotide Inhibitor of Hepatitis C Virus. Antimicrob Agents Chemother. 2012;56:3359–3368. doi: 10.1128/AAC.00054-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Zennou V, Otto MJ, Furman PA. In vitro Selection of HCV Replicons with Reduced Sensitivity to PSI-352938, A Cyclicphosphate Prodrug of β-D-2′-α-F-2′-β-C-Methylguanosine. Antivir Res. 2011;90:A22. [Google Scholar]
- Lawitz E, Box T, Pruitt M, Rodriguez-Torres M, Nguyen T, Lalezari J, Vargas H, Goff J, Hinestrosa F, Luketic V, Mehra P, Dubuc-Patrick G, Donovan E, Chen J, Pietropaolo K, Zhou XJ, Sullivan-Bolyai JZ, Mayers D. High rates of rapid virologic response (RVR) and complete early virologic response (cEVR) with IDX184, pegylated interferon and ribavirin in genotype 1 HCV-infected subjects: interim results. Annual Meeting of the American Association for the Study of Liver Diseases (AALSD); Nov. 9–13 2012; Boston, MA. 2012a. [Google Scholar]
- Lawitz E, Ghalib R, Rodriguez-Torres M, Nguyen T, Lalezari J, Vargas H, Goff J, Hinestrosa F, Luketic V, Mehra P, Dubuc-Patrick G, Donovan E, Chen J, Pietropaolo K, Zhou XJ, Sullivan-Bolyai JZ, Mayers D. Suppression of viral load through 4 weeks post-treatment results of a once-daily regimen of Simeprevir + Sofosbuvir with or without ribavirin in heptitis C virus GT1 null responders. 20th Conference on Retroviruses and Opportunistic Infections (CROI); March 3–6 2013; Atlanta, GA. 2013a. Paper #155LB. [Google Scholar]
- Lawitz E, Hill J, Marbury T, Hazan L, Gruener D, Webster L, Majauskas R, Morrison R, DeMicco M, German P, Stefanidis D, Svaroskaia E, Arterburm S, Ray A, Rossi S, McHutchison J, Rodriguez-Torres M. GS-6620, a liver-targeted nucleotide prodrug, exhibits antiviral activity and favorable safety profile over 5 days in treatment-naive chronic HCV genotype 1 subjects. 47th Annual Meeting of the European Association for the study of the Liver; Barcelona, Spain. 2012b. [Google Scholar]
- Lawitz E, Lalezari J, Freilich B, Ghalib R, Gambarin-Gelwan M, Tarum HA, Shiffman ML, DeMicco MP, Gitlin N, Marbury TC, Nguyen TT, Beavers KL, Wright D, Kowdley KV, Younossi ZM, Rustgi VK, Patti J, Campaneria R, Wenzel ED, Zamparo J, Boparai N, Yin P, Barry AN, Hughes EA. BMS-986094 (INX-08189) plus peginterferon alfa-2a and ribavirin results in treatment-naïve HCV-genotype 2/3 patients compared to peginterferon alfa-2a and ribavirin: week 12 results. Hepatology. 2012c;56:566A. [Google Scholar]
- Lawitz E, Lalezari J, Hassanein T, Kowdley KV, Poordad F, Sheikh AM, Afdahl NH, Bernstein DE, DeJesus E, Freilich B, Nelson DR, Dieterich DT, Jacobson IM, Jensen D, Abrams GA, Darling JM, Rodriguez-Torres M, Reddy KR, Sulkowski MS, Bzowej NH, Hyland RH, Mo H, Lin M, Mader M, Hindes R, Albanis E, Symonds WT, Berrey MM, Muir A. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naïve patients with genotype 1, 2, and 3 hepatitis C infecton: a randomised, double-blind, phase 2 trial. Lancet Infect Dis. 2013b;13:401–408. doi: 10.1016/S1473-3099(13)70033-1. [DOI] [PubMed] [Google Scholar]
- Lawitz E, Nguyen T, Younes Z, Santoro J, Gitlin N, McEniry D, Chasen R, Goff J, Knox S, Kleber K, Belanger B, Brown N. Clearance of HCV RNA with valopicitabine (NM283) plus peg-interferon in treatment-naïve patients with HCV-1 infection: Results at 24 and 28 weeks. J Hepatol. 2007;46:S9. [Google Scholar]
- Lawitz E, Rodriguez-Torres M, Denning J, Cornpropst MT, Clemons D, McNair L, Berrey MM, Symonds WT. Once Daily Dual-Nucleotide Comtination of PSI-938 and PSI-7977 Provides 94% HCV RNA < LOD at day 14: First Purine/Pyrimidine Clinical Combination Data (The NUCLEAR Study) J Hepatol. 2011;54:S543. [Google Scholar]
- Lawitz E, Rodriguez-Torres M, Denning JM, Albanis E, Cornpropst MT, Berrey MM, Symonds WT. Pharmacokinetics, pharmacodynamics, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor, follwoing multiple ascending doses in patients with chronic hepatitis C infection. Antimicrob Agents Chemother. 2013c;57:1209–1217. doi: 10.1128/AAC.01263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pogam S, Yan JM, Kosaka A, Ji Y, Gonzaludo N, Ewing A, Klumpp K, Najera I. No evidence of drug resistance or baseline S282T resistance mutation among GT1 and GT4 HCV infected patients on nucleoside polymerase inhibitor RG7128 and Peg-IFN/RBV combination treatment for up to 12 weeks: Interim analysis from the PROPEL study. Hepatology. 2010;52:701A. [Google Scholar]
- Legrand-Abravanel F, Nicot F, Izopet J. New NS5B polymerase inhibitors for hepatitis C. Expert Opin Investig Drugs. 2010;19:963–975. doi: 10.1517/13543784.2010.500285. [DOI] [PubMed] [Google Scholar]
- Lemon SM, McKeating JA, Pietschmann T, Frick DN, Glenn JS, Tellinghuisen TL, Symons J, Furman PA. Development of novel therapies for hepatitis C. Antivir Res. 2010;86:79–92. doi: 10.1016/j.antiviral.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci. 2008;97:1109–1134. doi: 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- Li N, Piccirilli JA. Synthesis of the phosphoramidite derivatives of 2′-deoxy-2′-C-methyl cytidine. J Org Chem. 2003;68:6799–6802. doi: 10.1021/jo034263y. [DOI] [PubMed] [Google Scholar]
- Ligand. Roche’s progress of RG7348 into phase I clinical trial triggers $6.5M milestone payment to Ligand. Press release. 2010 retrieved from: http://investor.ligand.com/Investors/News-and-Events/Press-Releases/Press-Release-Details/2010/Ligand-Completes-Acquisition-of-Metabasis/default.aspx.
- Ludmerer SW, Graham DJ, Boots E, Murray EM, Simcoe A, Markel EJ, Grobler JA, Flores OA, Olsen DB, Hazuda DJ, LaFemina RL. Replication fitness and NS5B drug sensitivity of diverse hepatitis C virus isolates characterized by using a transient replication assay. Antimicrob Agents Chemother. 2005;49:2059–2069. doi: 10.1128/AAC.49.5.2059-2069.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Jiang WR, Robledo N, Leveque V, Ali S, Lara-Jaime T, Masjedizadeh M, Smith DB, Cammack N, Klumpp K, Symons J. Characterization of the Metabolic Activation of Hepatitis C Virus Nucleoside Inhibitor β-D-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (PSI-6130) and Identification of a Novel Active 5′-Triphosphate Species. J Biol Chem. 2007;282:29812–29820. doi: 10.1074/jbc.M705274200. [DOI] [PubMed] [Google Scholar]
- Manns MP, Foster GR, Rockstroh JK, Zeuzem S, Zoulim F, Houghton M. The way forward in HCV treatment—finding the right path. Nat Rev Drug Discov. 2007;6:991–1000. doi: 10.1038/nrd2411. [DOI] [PubMed] [Google Scholar]
- Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- Marcellin P, Popa S, Berliba A, Moscalu I, Boyer N, Kauffman RS, Koziel MJ, Garg V, Tong M, Chanda SM, Zhang Q, Westland C, Beigelman L, Blatt LM, Fry J. ALS-2200, a novel once-daily nucleotide HCV polymerase inhibitor, demonstrates potent antiviral activity over 7 days in treatment-naïve genotype 1 (GT1) patients. Hepatology. 2012;56:235A. [Google Scholar]
- Martin JA, Sarma K, Smith BD, Smith M. Antiviral nucleoside derivatives. WO 2004046159 PCT Int Appl. 2004
- McCarville JF, Dubuc G, Donovan E, Mayers D, Seifer M, Standring D. No resistance to IDX184 was detected in 3-day and 14-day clinical studies of IDX184 in genotype 1-infected HCV subjects. J Hepatol. 2011;54:S488–S489. [Google Scholar]
- McCarville JF, Li B, Panzo RJ, Gillum JM, Soubasakos MA, Bilello JP, Lallos LB, Seifer M, Standring DN. In vitro and in vivo resistance profile of IDX184, a novel nucleotide analog for the treatment of HCV infection. 5th International Workshop on Hepatitis C - Resistance and New Compounds; Boston, MA. 2010. [Google Scholar]
- McCown MF, Rajyaguru S, Le Pogam Sl, Ali S, Jiang WR, Kang H, Symons J, Cammack N, Najera I. The hepatitis C virus replicon presents a higher barier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob Agents Chemother. 2008;52:1604–1612. doi: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C, Gilles A, Madela K, Aljarah M, Holl S, Jones S, Vernachio J, Hutchins J, Bryant D, Gorovits E, Ganguly B, Hall A, Kolykhalov A, Muhammad J, Chamberlain S, Henson G. Phosphoramidate ProTides of 2′-C-methylguanosine as highly potent inhibitors of hepatitis C virus. Study of their in vitro and in vivo properties. J Med Chem. 2010a;53:4949–4957. doi: 10.1021/jm1003792. [DOI] [PubMed] [Google Scholar]
- McGuigan C, Madela K, Aljarah M, Gilles A, Brancale A, Zonta N, Chamberlain S, Vernachio J, Hutchins J, Hall A, Ames B, Gorovits E, Ganguly B, Kolykhalov A, Wang J, Muhammad J, Patti JM, Henson G. Design, synthesis and evaluation of a novel double pro-drug: INX-08189 A new clinical candidate for hepatitis C virus. Bioorg Med Chem Lett. 2010b;20:4850–4854. doi: 10.1016/j.bmcl.2010.06.094. [DOI] [PubMed] [Google Scholar]
- McGuigan C, Perrone P. Chemical compounds. WO 2008062206 PCT Int Appl. 2008
- McHutchison JG, Manns M, Patel K, Poynard T, Lindsay KL, Trepo C, Dienstag J, Lee WM, Mak C, Garaud JJ, Albrecht JK. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterol. 2002;123:1061–1069. doi: 10.1053/gast.2002.35950. [DOI] [PubMed] [Google Scholar]
- Medivir. Annual report. 2011a retrieved from: http://medivir.se/v4/images/pdf/2012/Medivir_annualreport.pdf.
- Medivir. Medivir announces start of phase 1a trial of the hepatitis C polymerase inhibitor TMC64128. Press release. 2011b retrieved from: http://www.medivir.se/v5/en/uptodate/pressrelease.cfm?releaseid=2D62DF545CEECD11&year=2011.
- Medivir. TMC649128 enters phase 1b trial in patients chronically infected with genotype 1 hepatitis C virus. Press release. 2011c retrieved from: http://www.medivir.se/v5/en/uptodate/pressrelease.cfm?releaseid=1639EA2C5103B43D&year=2011.
- Medivir. Medivir announces interim results from Cohort 2 of the COSMOS study evaluating Simeprevir and Sofosbuvir in HCV patients with METAVIR scores F3–F4. Press release. 2013 retrieved from: http://www.medivir.se/v5/en/uptodate/pressrelease.cfm?releaseid=FEBC5055C8C2C046&year=2013.
- Membreno FE, Lawitz EJ. The HCV NS5B Nucleoside and Non-Nucleoside Inhibitors. Clin Liver Dis. 2011;15:611–626. doi: 10.1016/j.cld.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Meppen M, Pacini B, Bazzo R, Koch U, Leone JF, Koeplinger KA, Rowley M, Altamura S, Di Marco A, Fiore F, Giuliano C, Gonzalez-Paz O, Laufer R, Pucci V, Narjes F, Gardelli C. Cyclic phosphoramidates as prodrugs of 2′-methylcytidine. Eur J Med Chem. 2009;44:3765–3770. doi: 10.1016/j.ejmech.2009.04.043. [DOI] [PubMed] [Google Scholar]
- Murakami E, Bao H, Ramesh M, McGrayer TR, Whitaker T, Micolochick Steuer HM, Schinazi RF, Stuyver LJ, Obikhod A, Otto MJ, Furman PA. Mechanism of activation of beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob Agents Chemother. 2007;51:503–509. doi: 10.1128/AAC.00400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami E, Niu C, Bao H, Micolochick Steuer HM, Whitaker T, Nachman T, Sofia MJ, Wang P, Otto MJ, Furman PA. The mechanism of action of beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to beta-D-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother. 2008;52:458–464. doi: 10.1128/AAC.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami E, Tolstykh T, Bao T, Niu C, Micolochick Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem. 2010;285:34337–34347. doi: 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najera I. Resistance to HCV nucleoside analogue inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Current Opin Virol. 2013;3:508–513. doi: 10.1016/j.coviro.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Nelson DR, Zeuzem S, Andreone P, Ferenci P, Herring R, Jensen DM, Marcellin P, Pockros P, Rodriguez-Torres M, Rossaro L, Rustgi VK, Sepe T, Sulkowski M, Thomason IR, Yoshida EM, Chan A, Hill G. Balapiravir plus peginterferon alfa-2a (4-KD)/ribavirin in a randomized trial of heptitis C genotype 1 patients. Ann Hepatol. 2012;11:15–31. [PMC free article] [PubMed] [Google Scholar]
- Nguyen NM, Tran CN, Phung LK, Duong KT, Huynh Hle A, Farrar J, Nguyen QT, Tran HT, Nguyen CV, Merson L, Hoang LT, Hibberd ML, Aw PP, Wilm A, Nagarajan N, Nguyen DT, Pham MP, Nguyen TT, Javanbakht H, Klumpp K, Hammond J, Petric R, Wolbers M, Nguyen CT, Simmons CP. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis. 2013;207:1442–1450. doi: 10.1093/infdis/jis470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieforth KA, Morcos PN, Chang L, Davies B, Li R, Smith PF. Study, pharmacokinetics (PK), and antiviral activity of RG7348, a novel Hepdirect™ liver-targeted double prodrug hepatitis C virus (HCV) nucleoside polymerase inhibitor. Clin Pharmacol Ther. 2012;91:S96–S135. (poster session III (PIII 131–110)) [Google Scholar]
- Nilsson M, Kalayanov G, Winqvist A, Pinho P, Sund C, Zhou XX, Wähling H, Belfrage AK, Pelcman M, Agback T, Benckestock K, Wikström K, Boothee M, Lindqvist A, Rydegård C, Jonckers TH, Vandyck K, Raboisson P, Lin TI, Lachau-Durand S, De Kock H, Smith DB, Martin JA, Klumpp K, Simmen K, Vrang L, Terelius Y, Samuelsson B, Rosenquist S, Johansson NG. Discovery of 4′-azido-2′-deoxy-2′-C-methyl cytidine and prodrugs thereof: A potent inhibitor of Hepatitis C virus replication. Bioorg Med Chem Lett. 2012;22:3265–3268. doi: 10.1016/j.bmcl.2012.03.021. [DOI] [PubMed] [Google Scholar]
- Osinusi A, Meissner EG, Lee YJ, Bon D, Heytens L, Nelson A, Sneller M, Kohli A, Barrett L, Proschan M, Herrmann E, Shivakumar B, Gu W, Kwan R, Teferi G, Talwani R, Silk R, Kotb C, Wroblewski S, Fishbein D, Dewar R, Highbarger H, Zhang X, Kleiner D, Wood BJ, Chavez J, Symonds WT, Subramanian M, McHutchison J, Polis MA, Fauci AS, Masur H, Kottilil S. Sofosbuvir and Ribavirin for Hepatitis C Genotype 1 in Patients with Unfavorable Treatment Characteristics. JAMA. 2013;310:804–811. doi: 10.1001/jama.2013.109309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Peppelenbosch MP, Janssen HL, De Knegt RJ. Telaprevir/boceprevir era: from bench to bed and back. World J Gastroenterol. 2012;18:6183–6188. doi: 10.3748/wjg.v18.i43.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti J, Matson M, Boehlecke B, Barry A, Wenzel E, Pentikis H, Alam J, Henson G. A study of the safety and pharmacokinetics of single ascending oral doses of INX-08189, a nucleotide polymerase inhibitor, in healthy subjects. Hepatology. 2011;54:S187. [Google Scholar]
- Pawlotsky JM. Mechanisms of antiviral treatment efficacy and failure in chronic hepatitis C. Antivir Res. 2003;59:1–11. doi: 10.1016/s0166-3542(03)00088-3. [DOI] [PubMed] [Google Scholar]
- Pham TNQ, MacParland SA, Mulrooney PM, Cooksley H, Naoumov NV, Michalak TI. Hepatitis C Virus Persistence after Spontaneous or Treatment-Induced Resolution of Hepatitis C. J Virol. 2004;78:5867–5874. doi: 10.1128/JVI.78.11.5867-5874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharmasset. Pharmasset nominates PSI-938 as a new nucleotide analog inhibitor of hepatitis C for Preclinical development. Press release. 2009 retrieved from: http://www.prnewswire.com/news-releases/pharmasset-nominates-psi-938-as-a-new-nucleotide-analog-inhibitor-of-hepatitis-c-for-preclinical-development-62126747.html.
- Pharmasset. Pharmasset Reports Positive Preliminary Antiviral Data with PSI-938 for the Treatment of Hepatitis. Press release. 2010 retrieved from: http://www.virtualpressoffice.com/publicsiteContentFileAccess?fileContentId=396775&fromOtherPageToDisableHistory=Y&menuName=News&sId=&sInfo.
- Pharmasset. Pharmasset Announces Intent to Amend QUANTUM Trial. Press release. 2011a retrieved from: http://www.hivandhepatitis.com/hepatitis-c/hepatitis-c-topics/hcv-treatment/3412-pharmasset-halts-testing-of-hcv-polymerase-inhibitor-psi-3938-due-to-liver-problems.
- Pharmasset. Pharmasset Halts Hepatitis C Drug in Test. The Wall Street Journal. 2011b retrieved from: http://online.wsj.com/article/SB10001424052970204553904577102383041237416.html.
- Pharmasset. Pharmasset Initiates QUANTUM, a Phase 2b Interferon-Free Trial of PSI-7977 and PSI-938 for All HCV Genotypes. Press release. 2011c retrieved from: http://www.hivandhepatitis.com/hepatitis-c/hepatitis-c-topics/hcv-treatment/3251-pharmasset-starts-trial-of-psi-7977-and-psi-3938-oral-regimens-for-all-hcv-genotypes.
- Pharmasset. PSI-938 Receives Fast Track Designation from the FDA for the Treatment of Chronic Hepatitis C Infection. Press release. 2011d retrieved from: http://www.hivandhepatitis.com/hepatitis-c/hepatitis-c-topics/hcv-treatment/3199-hcv-polymerase-inhibitor-psi-3938-gets-fast-track-status.
- Pierra C, Benzaria S, Amador A, Moussa A, Mathieu S, Storer R, Gosselin G. NM283, an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. Nucleos Nucleot Nucl. 2005;24:767–770. doi: 10.1081/ncn-200060112. [DOI] [PubMed] [Google Scholar]
- Pockros P, Jensen D, Tsai N, Taylor RM, Ramji A, Cooper C, Dickson R, Tice A, Stancic S, Ipe D, Thommes JA, Vierling JM. First SVR data with the nucleoside analogue polymerase inhibitor mericitabine (RG7128) combined with peginterferon/ribavirin in treatment-naïve HCV G1/4 patients: interim analysis from the JUMP-C trial. J Hepatol. 2011;54:S538. [Google Scholar]
- Pockros PJ, Jensen D, Tsai N, Taylor R, Ramji A, Cooper C, Dickson R, Tice A, Kulkarni R, Vierling JM, Munson ML, Chen YC, Najera I, Thommes J. JUMP-C: A randomized trial of mericitabine plus peginterferon alfa-2a/ribavirin for 24 weeks in treatment-naïve HCV genotype 1/4 patients. Hepatology. 2013;58:514–523. doi: 10.1002/hep.26275. [DOI] [PubMed] [Google Scholar]
- Pockros PJ, Nelson D, Godofsky E, Rodriguez-Torres M, Everson GT, Fried MW, Ghalib R, Harrison S, Nyberg L, Shiffman ML, Najera I, Chan A, Hill G. R1626 plus peginterferon α-2a provides potent suppression of hepatitis C virus RNA and significant antiviral synergy in combination with ribavirin. Hepatology. 2008;48:285–297. doi: 10.1002/hep.22357. [DOI] [PubMed] [Google Scholar]
- Poijärvi-Virta P, Lönnberg H. Prodrug approaches of nucleotides and oligonucleotides. Curr Med Chem. 2006;13:3441–3465. doi: 10.2174/092986706779010270. [DOI] [PubMed] [Google Scholar]
- Poynard T, Ratziu V, Benhamou Y, Opolon P, Cacoub P, Bedossa P. Natural history of HCV infection. Best Pract Res Clin Gastroenterol. 2000;14:211–228. doi: 10.1053/bega.1999.0071. [DOI] [PubMed] [Google Scholar]
- Radkowski M, Gallegos-Orozco JF, Jablonska J, Colby TV, Walewska-Zielecka B, Kubicka J, Wilkinson J, Adair D, Rakela J, Laskus T. Persitence of Hepatitis C Virus in Patients Successfully Treated for Chronic Hepatitis C. Hepatol. 2005;41:106–114. doi: 10.1002/hep.20518. [DOI] [PubMed] [Google Scholar]
- Reddy PG, Bao D, Chang W, Chun BK, Du J, Nagarathnam D, Rachakonda S, Ross BS, Zhang HR, Bansal S, Espiritu CL, Keilman M, Lam AM, Niu C, Steuer HM, Furman PA, Otto MJ, Sofia MJ. 2′-Deoxy-2′-α-fluoro-2′-β-C-methyl 3′,5′-cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: Discovery of PSI-352938. Bioorg Med Chem Lett. 2010;20:7376–7380. doi: 10.1016/j.bmcl.2010.10.035. [DOI] [PubMed] [Google Scholar]
- Reddy PG, Chun BK, Zhang HR, Rachakonda S, Ross BS, Sofia MJ. Stereoselective Synthesis of PSI-352938: A β-D-2′-Deoxy-2′-α-fluoro-2′-β-C-methyl-3′,5′-cyclic Phosphate Nucleotide Prodrug for the Treatment of HCV. J Org Chem. 2011;76:3782–3790. doi: 10.1021/jo200060f. [DOI] [PubMed] [Google Scholar]
- Reddy R, Rodriguez-Torres M, Gane E, Robson R, Lalezari J, Everson GT, DeJesus E, McHutchison JG, Vargas HE, Beard A, Rodriguez CA, Hill GZ, Symonds W, Berrey M. Antiviral activity, pharmacokinetics, safety, and tolerability of R7128, a novel nucleoside HCV RNA polymerase inhibitor, following multiple, ascending, oral doses in patients with HCV genotype 1 infection who have failed prior interferon therapy. Hepatology. 2007;46:862A–863A. [Google Scholar]
- Reichard O, Andersson J, Schvarcz R, Weiland O. Ribavirin treatment for chronic hepatitis C. Lancet. 1991;337:1058–1061. doi: 10.1016/0140-6736(91)91707-2. [DOI] [PubMed] [Google Scholar]
- Roberts SK, Cooksley G, Dore GJ, Robson R, Shaw D, Berns H, Hill G, Klumpp K, Najera I, Washington C. Robust antiviral activity of R1626, a novel nucleoside analog: a randomized, placebo-controlled study in patients with chronic hepatitis C. Hepatology. 2008;48:398–406. doi: 10.1002/hep.22321. [DOI] [PubMed] [Google Scholar]
- Robson R, Berns H, Brandl M, Fettner S, Haznedar J, Hill G, Ipe D, Love J, Mannino M, O’Mara E, Tu Y, Washington C. Safety, tolerability and pharmacokinetics of R1626, a novel nucleoside analog targeting HCV polymerase: results from a phase 1 single dose escalation trial in healthy subjects. Clin Pharmacol Ther. 2007;81:S98. (Poster PIII-41 Abstract) [Google Scholar]
- Roche. A study of HCV polymerase inhibitor prodrug in combination with PEGASYS plus COPEGUS compared with PEGASYS plus COPEGUS in patients with chronic hepatitis C genotype 1 infection. Clinical trial NCT00517439. 2008 retrieved from: http://clinicaltrials.gov/ct2/show/NCT00517439?term=00517439&rank=00517431.
- Roche. Annual report. 2010 retrieved from: http://www.roche.com/gb10e01.pdf.
- Roche. Annual report. 2011a retrieved from: http://www.roche.com/static/app/annual_report_2011/_data/pipeline/pipeline.html.
- Roche. Committed to innovation and profitable growth. Nine months YTD 2011 sales. 2011b retrieved from: http://www.roche.com/irp3q11e.pdf.
- Roche. ANNAPURNA: A Study of The Combination of RO5466731, RO5190591, Ritonavir and Copegus (Ribavirin) With or Without RO5024048 in Patients With Chronic Hepatitis C Who Are Either Treatment-Naïve or Have Previously Experienced a Null Response to Interferon-Based Treatment. Clinical trial NCT01628094. 2012 retrieved from: http://clinicaltrials.gov/ct2/show/NCT01628094?term=01628094&rank=01628091.
- Rodriguez-Torres M, Lalezari J, Gane E, DeJesus E, Nelson DR, Everson GT, Jacobson IM, Reddy KR, McHutchison JG, Beard A, Walker S, Symonds W, Berrey MM. Potent antiviral response to the HCV nucleoside polymerase inhibitor R7128 for 28 days with PEG-IFN and ribavirin: subanalysis by race/ethnicity, weight and HCV genotype. Hepatology. 2008;48:1160A. [Google Scholar]
- Rodriguez-Torres M, Lawitz E, Hazan L, Barry A, Wenzel E, Alam J, Henson G, Patti J. Antiviral activity and safety of INX-08189, a nucleotide polymerase inhibitor, following 7-days of oral therapy in naïve genotype-1 HCV patients. Hepatology. 2011;54:535A. [Google Scholar]
- Rodriguez-Torres M, Lawitz E, Kowdley KV, Nelson DR, DeJesus E, McHutchison JG, Cornpropst MT, Mader M, Albanis E, Jiang D, Hebner CM, Symonds WT, Berrey MM, Lalezari J. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: A randomized, 28-day, dose-ranging trial. J Hepatol. 2013;58:663–668. doi: 10.1016/j.jhep.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Ross BS, Ganapati Reddy P, Zhang HR, Rachakonda S, Sofia MJ. Synthesis of Diastereomerically Pure Nucleotide Phosphoramidates. J Org Chem. 2011;76:8311–8319. doi: 10.1021/jo201492m. [DOI] [PubMed] [Google Scholar]
- Ross BS, Sofia MJ, Pamulapati Reddy G, Rachakonda S, Zhang H-R, Chun B-K, Wang P. N-[(2′R)-2′-deoxy-2′-fluoro-2′-methyl-P-phenyl-5′-uridylyl]-L-alanine 1-methylethyl ester and process for its production. WO 2010135569 PCT Int Appl. 2010
- Sarma K. Selective O-acylation of nucleosides. US 2007066815 PCT Int Appl. 2007
- Schaefer EA, Chung RT. Anti-hepatitis C virus drugs in development. Gastroenterol. 2012;142:1340–1350. doi: 10.1053/j.gastro.2012.02.015. [DOI] [PubMed] [Google Scholar]
- Schmitt R. Beitrag zur Kenntniss der Kolbe’schen Salicylsäure Synthese. J Prakt Chem. 1885;31:397–411. [Google Scholar]
- Schröder P, Kraut K. Uber Salicylverbindungen. Ann Chem Pharma. 1869;150:1–20. [Google Scholar]
- Sheridan C. New Merck and Vertex drugs raise standard of care in hepatitis C. Nat Biotechnol. 2011;29:553–554. doi: 10.1038/nbt0711-553. [DOI] [PubMed] [Google Scholar]
- Sheridan C. Calamitous HCV trial casts shadow over nucleoside drugs. Nat Biotechnol. 2012;30:1015–1016. doi: 10.1038/nbt1112-1015. [DOI] [PubMed] [Google Scholar]
- Smith BD, Deval J, Dyatkina NB, Beigelman L, Wang G. Substituted nucleotide analogs. WO 2010040127 PCT Int Appl. 2012
- Smith DB, Martin JA, Klumpp K, Baker SJ, Blomgren PA, Devos R, Granycome C, Hang J, Hobbs CJ, Jiang W-R, Laxton C, Le PS, Leveque V, Ma H, Maile G, Merrett JH, Pichota A, Sarma K, Smith M, Swallow S, Symons J, Vesey D, Najera L, Cammack N. Design, Synthesis and Antiviral Properties of 4′-substituted Ribonucleosides as Inhibitors of Hepatitis C Virus Replication: The Discovery of R1479. Bioorg Med Chem Lett. 2007:17. doi: 10.1016/j.bmcl.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Sofia MJ. Nucleotide prodrugs for HCV therapy. Antivir Chem Chemother. 2011;22:23–49. doi: 10.3851/IMP1797. [DOI] [PubMed] [Google Scholar]
- Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, H-RZ, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HMM, Niu C, Otto MJ, Furman PA. Discovery of a β-D-2′-Deoxy-2′-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J Med Chem. 2010a;53:7202–7218. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- Sofia MJ, Du J, Wang P, Nagarathnam D. Nucleoside phosphoramidate prodrugs. WO 2008121634 PCT Int Appl. 2008
- Sofia MJ, Du J, Wang P, Nagarathnam D. Nucleoside phosphoramidates. WO 2010075549 PCT Int Appl. 2010b
- Sommadossi J-P, Gosselin G, Pierra C, Perigaud C, Peyrottes S. Compounds and pharmaceutical compositions for the treatment of viral infections. WO 2008082601 PCT Int Appl. 2008
- Sommadossi J-P, La Colla P. Methods and compositions for treating Flaviviruses and Pesiviruses. WO 0192282 PCT Int Appl. 2001a
- Sommadossi J-P, La Colla P. Methods and compositions for treating hepatitis C virus. WO 01090121 PCT Int Appl. 2001b
- Sommadossi J-P, La Colla P, Standring D, Bichko V, Qu L. 2′-Branched nucleosides and Flaviridae mutation. WO 2004046331 PCT Int Appl. 2004a
- Sommadossi J-P, La Colla P, Storer R, Gosselin G. 2′ and 3′-Nucleoside prodrugs for treating Flaviridae infections. WO 200400300 PCT Int Appl. 2004b
- Sommadossi J-P, La Colla P, Storer R, Gosselin G. Modified 2′ and 3′-nucleoside prodrugs for treating flaviridae infections. WO 2004002999 PCT Int Appl. 2004c
- Standring D. IDX184 and novel nucleotides for the treatment of HCV. 20th International Round Table on Nucleosides, Nucleotides and Nucleic Acids; Montreal, Canada. 2012. [Google Scholar]
- Standring DN, Lanford R, Li B, Panzo RJ, Seifer M, Larsson M, Good SS, Zhou XJ. Potent antiviral activity of second generation nucleoside inhibitors, IDX102 and IDX184, in HCV-infected chimpanzees. J Hepatol. 2008;48:S30. [Google Scholar]
- Standring DN, Lanford R, Li B, Panzo RJ, Seifer M, Larsson M, Good SS, Zhou XJ. Antiviral activity of the liver-targeted nucleotide HCV polymerase inhibitor IDX184 correlates with trough serum levels of the nucleoside metabolite in HCV-infected chimpanzees. J Hepatol. 2009;50:S37. [Google Scholar]
- Storer R, Moussa A, Chaudhuri N, Waligora F. Process for the production of 2-branched nucleosides. WO 2004052899 PCT Int Appl. 2004
- Stuyver LJ. Dosing regimen for Gemcitabine HCV therapy. WO 03068164 PCT Int Appl. 2003
- Stuyver LJ, McBrayer TR, Tharnish PM, Clark J, Hollecker L, Lostia L, Nachman T, Grier J, Bennett MA, Xie MY, Schinazi RF, Morrey JD, Julander JL, Furman PA, Otto MJ. Inhibition of hepatitis C replicon RNA synthesis by β-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine: a specific inhibitor of hepatitis C virus replication. Antivir Chem Chemother. 2006;17:79–87. doi: 10.1177/095632020601700203. [DOI] [PubMed] [Google Scholar]
- Sulkowski M, Gardiner D, Lawitz E, Hinestrosa F, Nelson D, Thuluvath P, Rodriguez-Torres M, Lok A, Schwartz H, Reddy K, Eley T, Wind-Rotolo M, Huang SP, Gao M, McPhee F, Hindes R, Symonds B, Pasquinelli C, Grasela D. Potent viral suppression with all-oral combination of daclatasvir (NS5A inhibitor) and GS-7977 (NS5B inhibitor), +/− ribavirin, in treatment-naïve patients with chronic HCV GT1, 2, or 3. Hepatology. 2012a;56:S560. [Google Scholar]
- Sulkowski M, Gardiner D, Rodriguez-Torres M, Reddy K, Hassanein T, Jacobson I, Lawitz E, Lok A, Hinestrosa F, Thuluvath P, Schwartz H, Nelson D, Eley T, Wind-Rotolo M, Huang S-P, Gao M, McPhee F, Sherman D, Hindes R, Symonds W, Pasquinelli C, Grasela D. High rate of sustained virologic response with the all-oral combination of daclatasvir (NS5A inhibitor) plus sofosbuvir (nucleotide NS5B inhibitor), with or without ribavirin, in treatment-naïve patients chronically infected with HCV genotype 1, 2, or 3. Hepatology; Annual Meeting of the American Association for the Study of Liver Diseases (AALSD); Nov. 09–13 2012; Boston, MA. 2012b. p. abstract LB-02. [Google Scholar]
- Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, Lawitz E, Lok AS, Hinestrosa F, Thuluvath PJ, Schwartz H, Nelson DR, Everson GT, Eley T, Wind-Rotolo M, Huang S-P, Gao M, Hernandez D, McPhee F, Sherman D, Hindes R, Symonds W, Pasquinelli C, Grasela DM. Sustained Virologic Response with daclatasvir plus sofosbuvir ± ribavirin (RBV) in chronic HCV genotype (GT) 1-infected patients who previously failed telaprevir (TVR) or boceprevir (BOC). 48th Annual Meeting of the European Association for the Study of the Liver (EASL); April 24–28, 2013; Amsterdam, The Netherlands: 2013. p. abstract 1417. AI4444-040 Study group. retrieved from file://localhost/%20http/:: http://www.natap.org:2013:EASL:EASL_1422.htm. [Google Scholar]
- Surleraux D, Gosselin G. Compounds and pharmaceutical compositions for the treatment of viral infections. WO 2011123586 PCT Int Appl. 2011
- Surleraux D, Gosselin G. Compounds and pharmaceutical compositions for the treatment of viral infections. WO 2012154321 PCT Int Appl. 2012
- Tibotec B. A Trial in Genotype 1 Hepatitis C Virus (HCV) - Infected Participants to Determine the Safety, Tolerability, Pharmacokinetics and Antiviral Activity of TMC649128, Alone and Combined With Pegylated Interferon + Ribavirin. Clinical trial NCT01391117. 2011a http://clinicaltrials.gov/ct2/show/NCT01391117?term=TMC01649128&rank=01391111.
- Tibotec P. First In-human Trial to Examine Safety, Tolerability, and Pharmacokinetics (How the Drug is Absorbed Into the Bloodstream) of Increasing Single Oral Doses of TMC649128 in Healthy Volunteers. Clinical trial NCT01288677. 2011b retrieved from: http://clinicaltrials.gov/ct2/show/NCT01288677?term=TMC01649128&rank=01288672.
- Tohme RA, Holmberg SD. Is sexual contact a major mode of hepatitis C virus transmission? Hepatology. 2010;52:1497–1505. doi: 10.1002/hep.23808. [DOI] [PubMed] [Google Scholar]
- Tong MJ, El-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusion-Associates hepatitis C. New Engl J Med. 1995;332:1463–1466. doi: 10.1056/NEJM199506013322202. [DOI] [PubMed] [Google Scholar]
- Tong X, Pogam SL, Li L, Haines K, Piso K, Baronas V, Yan J-M, So S-S, Klumpp K, Nájera I. In Vivo Emergence of a Novel Mutant L159F/L320F in the NS5B Polymerase Confers Low-Level Resistance to the HCV Polymerase Inhibitors Mericitabine and Sofosbuvir. 2013 doi: 10.1093/infdis/jit1562. Epub Oct 23. [DOI] [PubMed] [Google Scholar]
- Toniutto P, Fabris C, Bitetto D, Fumolo E, Fornasiere E, Pirisi M. R-1626, a specific oral NS5B Polymerase Inhibitor of Hepatitis C Virus. IDrugs. 2008;11:738–749. [PubMed] [Google Scholar]
- Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- Verheyden JPH, Moffatt JG. 4′-Substituted nucleosides. I Synthesis of 4′-methoxyuridine and related compounds. J Am Chem Soc. 1975;97:4386–4395. doi: 10.1021/ja00848a042. [DOI] [PubMed] [Google Scholar]
- Vernachio J, Bleiman B, Byrant KD, Chamberlain S, Hunley D, Hutchins J, Ames B, Gorovits E, Ganguly B, Hall A, Kolykhalov A, Liu Y, Muhammad J, Raja N, Walters R, Wang J, Williams K, Patti JM, Henson G, Madela K, Aljarah M, Gilles A, McGuigan C. INX-08189, a phosphoramidate prodrug of 6-O-methyl-2′-C-methyl guanosine, is a potent inhibitor of hepatitis C virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob Agents Chemother. 2011;55:1843–1851. doi: 10.1128/AAC.01335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertex. Vertex and Alios BioPharma Announce Exclusive Worldwide Licensing Agreement for Two Nucleotide Drug Candidates, Broadening Vertex’s Efforts to Develop New Combinations of Medicines for Hepatitis C. Press release. 2011 retrieved from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=584023.
- Vertex. Vertex Announces Collaboration with Janssen To Initiate Phase 2 All-Oral Study of VX-135 and Simeprevir (TMC435) for the Treatment of Hepatitis C. Press release. 2012a retrieved from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=717772.
- Vertex. Vertex Announces New Data on ALS-2200 in People With Hepatitis C, Supporting Advancement into Phase 2 All-Oral Studies in 2012; Ends Development of ALS-2158. Press release. 2012b retrieved from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=709250.
- Vertex. Vertex enters agreement with Bristol-Meyers Squibb for Phase 2 all-oral studies of VX-135 in combination with daclatasvir for the treatment of Hepatitis C. Press release. 2012c retrieved from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=754494.
- Vertex. Vertex Enters Agreement with GlaxoSmithKline for Phase 2 All-Oral Study of VX-135 and GSK2336805 for the Treatment of Hepatitis C. Press release. 2012d retrieved from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=717777.
- Vertex. Vertex Provides Update on Ongoing All-Oral Studies of VX-135 in Hepatitis C. Press release. 2013 retrieved from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=780456.
- Von Gilm H. Acetylderivate der Phloretin und Salicylsäure. Ann Chem Pharma. 1859;112:180–185. [Google Scholar]
- Waheed Y, Bhatti A, Ashraf M. RNA dependent RNA polymerase of HCV: A potential target for the development of antiviral drugs. Infect Genet Evol. 14:247–257. doi: 10.1016/j.meegid.2012.12.004. 013. [DOI] [PubMed] [Google Scholar]
- Wang P, Chun BK, Rachakonda S, Du J, Khan N, Shi J, Stec W, Cleary D, Ross BS, Sofia MJ. An Efficient and Diastereoselective Synthesis of PSI-6130: A Clinically Efficacious Inhibitor of HCV NS5B Polymerase. J Org Chem. 2009;74:6819–6824. doi: 10.1021/jo901345j. [DOI] [PubMed] [Google Scholar]
- Wedemeyer H, Jensen D, Herring RJ, Ferenci P, Ma MM, Zeuzem S, Rodriguez-Torres M, Bzowej N, Pockros P, Vierling J, Ipe D, Lou Munson M, Chen YC, Najera I, Thommes J. PROPEL: A randomized trial of mericitabine plus peginterferon alfa-2a/ribavirin therapy in treatment-naïve HCV Genotype 1/4 patients. Hepatology. 2013;58:524–537. doi: 10.1002/hep.26274. [DOI] [PubMed] [Google Scholar]
- WHO. Hepatitis C. Fact Sheet #164. 2012 retrieved from: http://www.who.int/mediacenter/factsheet/fs164/en/
- Wong T, Lee SS. Hepatitis C: a review for primary care physicians. Can Med Assoc J. 2006;174:649–659. doi: 10.1503/cmaj.1030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Pietropaolo K, Chen J, Khan S, Sullivan-Bolyai J, Mayers D. Safety and Pharmacokinetics of IDX184, a liver-targeted nucleotide polymerase inhibitor of hepatitis C virus, in healthy subjects. Antimicrob Agents Chemother. 2011;55:76–81. doi: 10.1128/AAC.01101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Pietropaolo K, Sullivan-Bolyai J, Kuca B, Liu W, Xu L, Belanger B, Khan S, Mayers D. IDX184, a liver-targeted nucleotide HCV polymerase inhibitor: results of a first-in-man safety and pharmacokinetic study. J Hepatol. 2009;50:S351. [Google Scholar]