

Abstract
Polyglutamine (polyQ) amyloid fibrils are observed in disease tissue and have been implicated as toxic agents responsible for neurodegeneration in expanded CAG repeat diseases like Huntington’s disease (HD). Despite intensive efforts, the mechanism of amyloid toxicity remains unknown. As a novel approach to probing polyQ toxicity, we investigate here how some cellular and physical properties of polyQ amyloid vary with the chirality of the glutamine residues in the polyQ. We challenged PC12 cells with small amyloid fibrils composed of either L- or D-polyQ peptides and found that D-fibrils are as cytotoxic as L-fibrils. We also found using fluorescence microscopy that both aggregates effectively seed the aggregation of cell-produced L-polyQ proteins, suggesting a surprising lack of stereochemical restriction in seeded elongation of polyQ amyloid. To investigate this effect further, we studied chemically synthesized D- and L-polyQ in vitro. We found that, as expected, D-polyQ monomers are not recognized by proteins that recognize L-polyQ monomers. However, amyloid fibrils prepared from D-polyQ peptides can efficiently seed the aggregation of L-polyQ monomers in vitro, and vice versa. This result is consistent with our cell results on polyQ recruitment, but is inconsistent with previous literature reports on the chiral specificity of amyloid seeding. This chiral cross-seeding can be rationalized by a model for seeded elongation featuring a “rippled β-sheet” interface between seed fibril and docked monomers of opposite chirality. The lack of chiral discrimination in polyQ amyloid cytotoxicity is consistent with several toxicity mechanisms, including recruitment of cellular polyQ proteins.
Keywords: chiral cross-seeding, rippled β-sheet, toxicity, recruitment, Huntington’s disease
INTRODUCTION
The expanded polyglutamine (polyQ) diseases 1 form a family of genetic, progressive neurodegenerative diseases that includes Huntington’s disease (HD) 2, 3. There are currently nine well-characterized members of this family, each of which involves a CAG repeat expansion in the gene for a different protein 1, and this list itself will likely expand in the future 4–6. In these diseases, the expansion of the polyQ sequence beyond the normal range is correlated with both increased disease risk and decreased age-of-onset 1. A number of observations point to the possible key role of amyloid-like aggregates in the disease mechanism. Amyloid-like inclusions are observed in affected neurons in patient brains 7 and in tg mouse models 8, 9, and the repeat length dependence of spontaneous amyloid formation in vitro 10–12 and in animal models 13 parallels the repeat length dependence of incidence in most of the diseases 1.
A toxic role for the large, perinuclear inclusions that develop in many HD model cells has been questioned, due to inconsistent co-evolution of cytotoxicity with these large aggregates in some models 14. However, super-resolution fluorescence microscopy studies recently revealed that cells producing expanded polyQ also can possess smaller, isolated fibrils distributed to various cellular locations 15. In fact, even smaller amyloids might easily exist undetected in cells. Thus, while the massive HD inclusions observed in disease tissue can each contain up to nearly 109 polyQ disease protein molecules, and the fibrils visualized by Sahl et al. about 105 molecules, small, functional amyloid fibrils (such as those described in this paper) can be created in vitro that contain fewer than 103 protein molecules 16. Such observations suggest that previous studies may not have taken a full inventory of all aggregated forms of polyQ in the cell, and that relatively small, individual polyQ amyloid fibrils (in addition to non-amyloid aggregates 16) therefore remain viable candidates for the toxic species. Amyloid-like fibrils of polyQ 17 and polyQ-containing proteins 18 are well known to be cytotoxic to mammalian cells.
Based on extensive cell-free and cell biological experiments, a wide variety of mechanisms have been suggested to account for the toxicity of aggregates in neurodegenerative diseases. Some of these, such as interactions with membranes or other cell structures, might be expected to be guided by aggregate surface properties such as hydrophobicity, and therefore to be relatively structurally non-specific. Others, however, would appear to require potentially highly specific interactions with enzymes or other proteins, such as those tasked by the cell to recognize and destroy or divert protein aggregates. Another mechanism, the recruitment of cellular polyQ proteins into growing polyQ amyloid assemblies 11, 19, 20, is also expected to be a highly structurally specific mechanism, based on the well-characterized sensitivity of amyloid seeding and cross-seeding to fibril structure 21, amino acid sequence 22, 23 and amino acid chirality 24, 25. Thus, information on the dependence of polyQ cytotoxicity on polyQ chirality should be very useful in filtering various postulated molecular mechanisms of disease.
Previously it was shown that a dispersed suspension of small, L-polyQ amyloid fibrils can be taken up by cells in culture 17, and that these cytoplasmically localized fibrils are capable of recruiting ribosomally produced L-polyQ 26. If these synthetic aggregates are outfitted with a nuclear localization signal (NLS), the internalized aggregates are also extremely cytotoxic 17. Here, we exploit this model to carry out a direct comparison of L- and D-polyQ amyloid toxicity, and by so doing directly query the extent of stereochemical specificity in this obscure but critically relevant process.
In this study we prepared amyloid fibrils from D-polyQ peptides and determined their in vitro and cellular properties relative to L-polyQ fibrils. The study was based on an expectation that the gross surface properties of “mirror image” D- and L-polyQ amyloid would be quite similar, while their specific interactions with protein-based cellular machinery, as well as their efficiencies in seeding amyloid formation from other polyQ sequences, would be quite different. In the event, we found that D-polyQ amyloid is equipotent with L-polyQ amyloid in killing mammalian cells in culture. This lack of selectivity, however, does not rule out the recruitment mechanism, since we were surprised to find that cross-seeding between D-polyQ amyloid and L-polyQ monomers, both in vitro and in cells, is remarkably efficient. The data show an unanticipated promiscuity in chiral cross-seeding of amyloid fibrils. These data have implications for how polyQ fibrils are held together and propagated, and how their toxic effects are achieved.
RESULTS
Preparation and characterization of aggregates
We obtained chemically synthesized samples of peptides of the sequence PKKKRKVGGQ25KK (Methods) in which the polyQ segment following the NLS is in either the L or D configuration. We also obtained analogous peptides of sequence PKKKRKVGGQ25CKK in which the fluorophore Cy5 was attached to the Cys residue (Methods). Previously we found that the large amyloid-like structures normally obtained when polyQ is incubated at 37 °C 27 are not capable of efficiently entering mammalian cells 17. We therefore used these peptides to prepare uniform dispersions of small amyloid fibrils that we previously showed to be required for cell uptake 17. First, solutions of polyQ monomers in PBS were snap-frozen in liquid N2, then placed at −20 °C and incubated until most of the polyQ had formed amyloid fibrils. By freezing small aliquots and withdrawing these at different times for analysis, we confirmed that aggregation occurs in a time-dependent fashion in the frozen state (Fig. 1a), presumably facilitated by the well-known phenomenon of freeze-concentration 27, 28. Thus, t = 0 time points, which were snap-frozen in liquid N2 and immediately thawed, without incubation at −20 °C, showed no aggregation, while frozen aliquots stored at −20 °C for one day before thawing exhibited about 90% aggregate formation (Fig. 1a). The resulting fibrils (Fig. 1b, left panel) were subjected to a combination of sonication and membrane filtration to generate a suspension of uniform particle size in the 40 – 65 nm size range as assessed by EM (Fig. 1b) and DLS (Fig. 1c). At the level of resolution of our instrumentation, these fibrils of L- and D-polyglutamine are identical in appearance in EM (Fig. 1b). In particular, both preparations feature the same small aggregates that we know to be required for cellular uptake exhibiting relatively short (25–100 nm) curvilinear filaments with diameters in the 6–8 nm range, sizes in excellent agreement with the DLS measurements. As expected for ordered structures of opposite chirality, the L- and D-poyQ fibrils exhibit mirror image CD spectra (Fig. 1d). These fibrils are indistinguishable from each other, and from L-polyQ fibrils grown at 37 °C, by both FTIR (Fig. 1e) and thioflavin T fluorescence (ThT) (Fig. 1f). The second derivative FTIR spectra show the characteristic triplet of peaks for polyQ amyloid consisting of a central band assigned to β-sheet flanked by bands associated with ordered side chain amide groups 29. Ability to alter the fluorescence of ThT is a characteristic feature of amyloid structures 30. These data suggest that the D- and L-polyQ fibrils formed at −20 °C have very similar molecular structures to each other and to fibrils grown at 37 °C.
Figure 1.

Preparation and physical characterization of amyloids. (a) Time-dependent aggregation of an L-K2Q25K2 peptide incubated in PBS in the frozen state at −20 °C; (b) negative stained EM images of poly-Q amyloids grown at −20 °C, either without (left panel) or with (center and right panels) sonication; (c) size analysis of aggregates by dynamic light scattering (DLS); (d) circular dichroism of polyQ amyloid fibrils grown at −20 °C; (e) Second derivative Fourier transform infrared spectra of polyQ amyloid fibrils; (f) ThT fluorescence shift in the presence of equal weights of various polyQ amyloid fibrils.
Cell experiments
Using our previously described methods 17 we challenged WT PC12 cells with the L- and D-polyQ versions of PKKKRKVGGQ25KK fibrils. The results (Fig. 2a, blue bars) confirm our previous report 17 of concentration dependent cytotoxicity of similarly prepared L-polyQ amyloid fibrils. Importantly, the new results also show that, within error, D-polyQ amyloid (Fig. 2a, red bars) is just as toxic to cells as is L-polyQ amyloid. These data therefore suggest that whatever the polyQ amyloid cytotoxicity mechanism might be, it is chirally indiscriminate. This would be consistent, for example, with a cellular target – such as a membrane - that can interact with the amyloid via generalized surface chemistry and not via exquisite stereo-chemical surface complementarity. Given the chiral specificity of protein folding and binding 31, as well as previous reports that amyloid seeding is chirally specific 24, 25, we expected that these cytotoxicity results would also effectively rule out amyloid-linked recruitment mechanisms of polyQ amyloid toxicity. To confirm this expected chiral specificity, we conducted our own polyQ recruitment experiment in cells.
Figure 2.

Comparative cellular activities of L- and D-polyQ versions of PKKKRKVGGQ25KK amyloid fibrils. (a) Percent of WT PC12 cells undergoing lytic death (LDH release) after 24 hrs exposure to different concentrations of L- or D-polyQ versions of PKKKRKVGGQ25KK amyloid; (b) percent of Htt-exon1-Q25-EGFP cells treated with 2.5 μM (monomer equivalent) of PKKKRKVGGQ25Ccy5KK amyloid that develop green puncta (see Figure 3) at different growth times. Values in a and b are from three independent experiments and are expressed as mean ± SEM (**, p < 0.01, ***, p < 0.001).
In these experiments we utilized a PC12 cell line stably transfected with a Q25 version of htt exon1 fused to EGFP under the control of an ecdysone promoter 32. When these cells are induced with 1 μM ponasterone, diffusely distributed EGFP is clearly visible by fluorescence microscopy after a few hours, but no green fluorescent inclusions or puncta are visible even after 24 hrs (Fig. 3a). However, when the cells are treated with aggregated Cy5-labelled PKKKRKVGGQ25CCy5KK (at 2.5 μM monomer-equivalent concentration) simultaneously with ponasterone induction of htt exon1 synthesis, EGFP puncta are observed by confocal microscopy as early as 7 hrs after the start of the experiment (Fig. 2b; Fig. S1). In addition, many of these EGFP-tagged puncta co-localize with internalized aggregates. Consistent with a previous report 26, after 24 hrs 95% of Q25-htt-exon1-EGFP-producing cells treated with L-polyQ amyloid develop green fluorescent puncta (Fig. 2b; Fig. 3b), while untreated cells develop no puncta (Fig. 2b; Fig. 3a). Surprisingly, we found that 28% of cells treated with D-polyQ amyloid also develop puncta after 24 hrs (Fig. 2b; Fig. 3b). This is unlikely to be a non-specific effect, since previously Ren et al. found that Aβ amyloid taken up by similar htt exon1 producing cells does not induce htt exon1 puncta formation 26. Thus, while the ability of D-polyQ amyloid to recruit an L-polyQ protein in cells is diminished compared to that of L-polyQ amyloid, seeded elongation is by no means chirally specific. If this cell effect is genuine, it is capable of explaining the observed lack of chiral specificity in cytotoxicity (Fig. 2a). We therefore set out to further investigate the chiral selectivity or specificity of polyQ amyloid seeding in vitro.
Figure 3.

Fluorescence microscopy of Htt-exon1-Q25-EGFP cells with and without added amyloid. (a) Htt-exon1-Q25-EGFP cells induced with 1 μM ponesterone and grown for the times shown. (b) Htt-exon1-Q25-EGFP expressing PC12 cells induced with 1 μM ponesterone and simultaneously treated with cy5-labelled D- or L- polyQ amyloid after 24 hrs. Nuclear staining (blue); in vitro prepared amyloid added to cell culture (red puncta); cell-produced exon1 (diffuse and punctate green); aligned chemically- and cell-produced aggregates (orange-yellow puncta) in merged images. Images were taken with an Olympus Fluoview 1000 confocal microscope (x100 oil lens, 5–6X digital zoom).
In vitro experiments
We first conducted experiments to ensure that polyQ monomers exhibit the expected high degree of stereochemical discrimination in standard biochemical reactions involving protein-protein interactions. Such stereochemical discrimination is a central tenant of protein biology and evolution: protein molecules, themselves composed of L-amino acids, evolve specificity in their structures and functions in such a way that these cannot easily be replicated by the same sequence of D-amino acids 31. Indeed, in line with this expectation, we found that while the L-polyQ monomer is recognized by the anti-polyQ monoclonal antibody MW1 33, this antibody does not detectibly bind to the D-polyQ peptide (Fig. 4a). The structural basis of this particular chiral discrimination is apparent from examination of the co-crystal structure of the all-L MW1 antibody and an L-polyQ antigen 34 (Fig. 5). The X-ray crystal structure suggests that a D-polyQ peptide inserted into the MW1 binding site would suffer steric clashes and/or exposure of buried surface area at most residue positions (Fig. 5). In another example of stereochemical discrimination by proteins for polypeptide ligands, we found that proteinase K effectively digests L-polyQ monomers (Fig. 4b, top panel), but has no effect of D-polyQ (Fig. 4b, bottom panel). Together, these confirmations of L-protein discrimination against our D-polyQ peptides unequivocally eliminate any uncertainty about the chirality of the material. The exquisite chiral sensitivity of these protein-protein interactions also validates our a priori expectation that a structurally intimate protein-protein interaction like the assembly of monomers onto an L-polyQ amyloid fibril would equally strongly discriminate against D-polyQ monomers. The cell results described above, however, gave a contradictory result. This underscores the importance of further in vitro studies to explore this possible chiral cross-seeding in a more defined system.
Figure 4.

Biological activities of D- and L-polyQ monomers. (a) binding of anti-polyQ MAb MW1 to dot-blotted polyQ monomers (LC = loading control stained with Ponceau S); (b) proteinase K digestion of polyQ monomers (top panel, L-polyQ; bottom panel, D-polyQ), with RP-HPLC profiles of injected samples of polyQ before (black traces) and after (red traces) digestion. Arrow indicates migration position of intact peptide.
Figure 5.
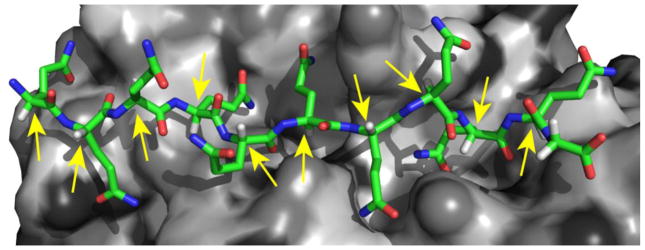
The role of D,L chirality in a typical protein-protein interactions. A strand of L-polyQ (colored stick model) nestled into its binding site in a cleft in the MW1 antibody (gray surface model) in the X-ray crystal structure (PDB file 2OTU) of the complex 34. Yellow arrows indicate the α-carbons that are the enantiomeric centers for D/L chirality. The short, white / light gray rods emerging from these α-carbons are the Cα hydrogens that would switch with the (green) Cα side chains in a global L to D epimerization.
We therefore set about to examine in vitro the seeding specificities of the L- and D-polyQ fibrils toward K2Q25K2 monomers of matched and opposite chiralities. When aggregation reactions are initiated with 12% by weight of fibril seeds to monomer, we found that, as reported previously 35, L-fibrils greatly stimulate aggregation of L-monomers (Fig. 6a,
 ) compared with the unseeded reaction (Fig. 6a, ●). Importantly, we found that under these conditions the reaction of L-monomers with D-fibrils (Fig. 6a,
) compared with the unseeded reaction (Fig. 6a, ●). Importantly, we found that under these conditions the reaction of L-monomers with D-fibrils (Fig. 6a,
 ) also proceeds at a rate much faster than that of the unseeded reaction, but slightly more slowly than the reaction seeded by L-aggregates. The morphologies of the fibrils produced by seeding of L-polyQ monomers with either L- or D-amyloid fibrils are indistinguishable by EM analysis (Fig. 7c,d) and are similar in appearance to amyloid fibrils generated by spontaneous aggregation from similar, simple polyQ monomers 35. Identical rates were observed for the reactions with the opposite chirality combinations, i.e. the aggregation of D-K2Q25K2 monomers either unseeded (Fig. 6b, ●) or seeded with either D- (Fig. 6b,
) or L- (Fig. 6b,
) polyQ fibrils. An even smaller difference in kinetics between L- and D- amyloid seeded aggregation was observed when reactions were conducted at higher monomer concentration and a higher (25%) weight ratio of seeds to monomer (Fig. 6c,d). Thus, these in vitro seeding reactions are entirely consistent with the cellular recruitment data described above in indicating a lack of chiral specificity in polyQ amyloid cross-seeding reactions.
) also proceeds at a rate much faster than that of the unseeded reaction, but slightly more slowly than the reaction seeded by L-aggregates. The morphologies of the fibrils produced by seeding of L-polyQ monomers with either L- or D-amyloid fibrils are indistinguishable by EM analysis (Fig. 7c,d) and are similar in appearance to amyloid fibrils generated by spontaneous aggregation from similar, simple polyQ monomers 35. Identical rates were observed for the reactions with the opposite chirality combinations, i.e. the aggregation of D-K2Q25K2 monomers either unseeded (Fig. 6b, ●) or seeded with either D- (Fig. 6b,
) or L- (Fig. 6b,
) polyQ fibrils. An even smaller difference in kinetics between L- and D- amyloid seeded aggregation was observed when reactions were conducted at higher monomer concentration and a higher (25%) weight ratio of seeds to monomer (Fig. 6c,d). Thus, these in vitro seeding reactions are entirely consistent with the cellular recruitment data described above in indicating a lack of chiral specificity in polyQ amyloid cross-seeding reactions.
Figure 6.

Interactions of polyQ amyloid (PKKKRKVGGQ25KK) and monomers (K2Q25K2). Panels in left column (a,c,e) show studies with L-polyQ monomers; panels in right column (b,d,f) show studies with D-polyQ monomers. (a–d) seeded elongation reactions at 37 °C in PBS; (e,f) binding of biotin-tagged polyQ monomers to polyQ amyloids in PBS at 25 °C; (a,b) 30–40 μM monomer and 12% (w/w) amyloid; (c,d) 90 μM monomer and 25% (w/w) amyloid; (e,f) varied concentrations of biotinyl-K2Q30K2 monomers incubated with 245 ng polyQ amyloid. In a–f, amyloid seed: none (●); L-polyQ (
 ); D-polyQ (
); D-polyQ (
 ).
).
Figure 7.

Results of seeded elongation experiments. (a,b) Pseudo-first order seeded elongation kinetics plots. Loss of monomer over time was determined by the HPLC sedimentation assay for solutions of ~ 80 μM starting concentration of K2Q25K2 monomers seeded at 25% (w/w) with amyloid-like aggregates of PKKKRKVGGQ25KK peptides. (a) L-polyQ monomers seeded by L-polyQ (◆) or D-polyQ (◇) amyloid; (b) D-polyQ monomers seeded by L-polyQ (◆) or D-polyQ (◇) amyloid. (c,d) Negative-stain EM images of the amyloid fibrils produced in PBS at 37 °C by seeding of L-K2Q25K2 monomers with amyloid fibrils of L- (c) or D- (d) PKKKRKVGGQ25KK grown at −20 °C (Fig. 1b) as described.
In analogy to protein crystallization, seeding of polymerization can occur with different levels of structural specificity. Highly specific seeding occurs when a growth edge of a fibril or crystal serves as a template for the incoming monomer. Some protein aggregates can also seed elongation via processes of less well-understood specificity, such as the “secondary nucleation” that can sometimes occur when monomers interact with the lateral surface of a filament 36. Polymerization can also be nucleated by non-specific processes involving extraneous agents with fortuitous complementarity in a process termed “heterogeneous nucleation” 37. This can even include enhancement of amyloid formation by generalized hydrophobic interfaces 38. To investigate the nature of the templating surface of the polyQ seed fibrils in these surprisingly robust chiral cross-seeding reactions, we studied the binding of biotin-tagged D- and L-K2Q30K2 monomers to the growth sites on the D- and L-fibril seeds 39, under conditions limiting binding to one measureable monomer per site (Methods). As expected for a seeded elongation reaction involving an initial monomer binding event 40, we found that the binding of L-polyQ monomers to L-polyQ amyloid (Fig. 6e,
) exhibits a cooperative, saturable binding curve indicative of a limited number of similar sites. We observed a similar binding curve of L-polyQ monomer to D-polyQ amyloid (Fig. 6e,
), but with a higher midpoint of the binding curve suggesting ~3-fold weaker binding. Entirely complementary data were obtained for binding of biotinylated D-polyQ monomers to D- (Fig. 6f,
) and L- (Fig. 6f,
) polyQ amyloid. These data together suggest that the seeding of L-monomers by D-aggregates, and vice versa, involves the same kind of specific interactions with a limiting, structured fibril growth face as those expected in homologous seeding reactions.
From the amplitudes of the titration curves in Figure 6e we calculated the molar concentrations of fibril growth points in the D- and L-aggregates. These values, in combination with pseudo-first order rate constants of seeded elongation conducted under conditions where the fibril growth faces are saturated (Fig. 7a,b), were used to determine k+, the second order rate constant, for elongation under these conditions. We found a value of k+ = 1.92 × 104 M−1sec−1 for the seeding of L-monomers by L-amyloid, and 0.75 × 104 M−1sec−1 for seeding of L-monomers by D-amyloid. Thus, at saturating concentrations of monomer, seeding of L-monomers by D-fibrils is only about 2.5-fold less efficient than seeding of L-monomers by L-fibrils. Similarly, seeded elongation of D-monomers by D-amyloid or L-amyloid exhibited k+ values of 2.48 × 104 and 0.58 × 104 M−1sec−1, respectively, for a difference in seeding efficiency of 4.3. These data strongly support the observation that D-poly amyloid can recruit L-polyQ proteins in cells (Fig. 2b; Fig. 3c). Having confirmed the cellular result, however, we were now confronted with the need to provide a structural rationale for this highly efficient, and unprecedented 24, 25, cross-seeding effect.
A rippled β-sheet interface in chiral cross-seeding?
To better understand the molecular basis of chiral cross-seeding in polyQ amyloid elongation, we employed the anti-parallel β-sheet structure based on the analysis of Perutz’s X-ray fiber diffraction data 41 by Sikorski and Atkins 42 as a model for the L-fibril end. Next, we constructed a D-polyQ molecule in a β-extended configuration by making a mirror image of an L-polyQ β-strand excised from the fibril structure. This conformation is consistent with basic principles of protein folding and chirality, and is also consistent with the data in Figure 1, which shows that D-polyQ fibrils exhibit an FTIR spectrum similar to L-polyQ amyloid but a mirror image CD spectrum. We then alligned this D-polyQ strand with the edge of the L-polyQ β-sheet in such a way as to maximize backbone amide H-bonds (Fig. 8a). The mixed D/ L structure so obtained forms, at the edge of the L-fibril, a two-stranded “rippled” β-sheet 43 in which main chain H-bonds are all satisfied and there are no side chain steric clashes (Fig. 8b). At the same time, the lineup of Gln side chains in the all-L anti-parallel β-sheet structure, that allows for strand-to-strand H-bonding between side chains across the surface of the β-sheet, is broken at the D/ L interface, as the side-chains are now staggered across the interface between the L- and D-β-strands (Fig. 8b). It also is structurally unlikely that new H-bonds could form with Gln side chains in adjacent β-sheets within the fibrils (Fig. S2). This lack of side-chain hydrogen bonding is noteworthy, as it might provide a rationale for at least some of the diminished signal in the binding (Fig. 6f), the in vitro seeded elongation (Fig. 6b,d), and the intracellular polyQ recruitment (Fig. 2b; Fig. 3b) described above. It is worth reiterating that the construction and visualization of this structure in Figure 8 is primarily meant as a model to rationalize the unexpected, but experimentally clearly observed, lack of chiral specificity in the cross-seeding of polyQ amyloid.
Figure 8.

Visualization of modeled compatibility between an all-D β-strand and an all-L polyQ amyloid-like assembly. (a) Schematic D-polyQ strand (top strand, light blue) aligned in an anti-parallel fashion with an internally anti-parallel L-polyQ amyloid assembly model (gray) as proposed by Sikorski & Atkins 42. Thick dashed lines (blue) show inter-backbone hydrogen bonding between the incoming D-strand and a pre-existing L-fibril, in analogy to the bonding patterns (thick dashed black lines) within the L-fibril. (b) Side-view of the hypothesized model showing the staggered side chains of the L-(gray) and D-strands (light blue), as well as the rippled profile of the β-sheet, viewing down the plane of a single β-sheet. (c) In a cross-seeding experiment, a single “rippled β-sheet” interface would join the pre-aggregated parent fibril segment to the newly propagated fibril segment of opposite chirality.
DISCUSSION
The ability of amyloid fibrils to seed the elongation of protein monomers, a process that underpins the biological propagation of prions 44–47 and possibly other neurotoxic protein aggregates 48, has been shown to be efficient only in cases of appreciable shared amino acid sequence homology 22, 23. Given this, and the strong chiral specificity of most protein activities in biology 31, previous results 24, 25 showing stringent chiral specificity in amyloid seeding reactions have not been considered surprising. We show here, however, that amyloid fibrils of D-polyQ can efficiently seed the elongation of L-polyQ monomers, and vice versa. This remarkable chiral cross-seeding of amyloid growth is consistent with peptide binding data suggesting that the template for both seeding and chiral cross-seeding is a limiting, ordered structure that is most likely the fibril terminus. We also observe similar cross-seeding when D-polyQ amyloid prepared in vitro is introduced into cells expressing an L-polyQ sequence.
The fundamental stereochemical compatibility of β-extended chains of L- and D-polypeptides in an anti-parallel arrangement, that apparently underlies the chiral cross-seeding observed here, was anticipated in a theoretical structural analysis by Pauling and Corey that showed that mixed β-sheets of alternating L- and D-polypeptides are stereochemically feasible, producing a motif they termed the “rippled β-sheet” 43. In fact, several groups have reported examples of stable, mixed β-sheets formed from L- and D-polypeptides of various sequences 49–51, although this does not appear to be a universal phenomenon 52. In contrast to such alternating D/L β-sheet models, however, the cross-seeding reported here would only require a single “rippled” strand-strand interface between the seed fibril and daughter fibril (Fig. 8c). That is, once amyloid elongation is initiated, the new fibril element whose growth is supported by the monomer pool (which in our seeding reactions is chirally pure) is expected to be essentially chirally homogeneous, with only the interface between seed segment and daughter segment of the fibril being “rippled” (Fig. 8c). Based on both our hypothetical model and on various experimental observations, it seems that this interface is relatively fragile and may therefore not always survive in the final seeded elongation reaction product. Indeed, this kind of fracturing of the D/L interface may well rationalize the incomplete superposition of red and green puncta in our cellular cross-seeding experiments (Fig. 3b). Fracturing of even chirally pure fibrils is recognized as a common and important aspect of fibril growth reactions, playing a central role in seeding and prion propagation 53.
If the suggested model for how a rippled β-sheet interface might mediate D/L polyQ cross-seeding is correct, it might be expected that one could demonstrate the formation of mixed L plus D fibrils in an in vitro experiment. Preliminary data on such a reaction has proved inconclusive, however (Supplemental Discussion, Figs. S3 – S6, and Table S1). In fact, providing convincing evidence for a single amyloid fibril composed of a mixture of two polyQ peptides is expected to be far from trivial. Even the convincing demonstration of the existence of a mixed fibril may not directly elucidate the structural basis for cross-seeding. Further, the suggestion from the data provided here, and of the model we constructed to explain the data, is that a co-aggregated fibril composed of regularly alternating L and D polyQ might not be as stable as all-D or all-L fibrils, and hence would not be thermodynamically favored. In fact, in a simple peptide system accessible to solution NMR analysis Chung and Nowick showed that, while L plus D co-assembly does occur spontaneously, it is thermodynamically less favorable than L plus L 49. This thermodynamically disfavored mode would not rule out the cross-seeding described in this paper.
Why did previous studies 24, 25 fail to report D/L cross-seeding? This is possibly due to different geometrical constraints in those particular amyloid structures, or, alternatively, observation of cross-seeding may require particular experimental conditions, such as relatively high concentrations of monomer and/or seed. The robust cross-seeding observed here, that appears to be primarily dependent on the alignment of H-bond donors and acceptors in the extended polypeptide backbone (Fig. 8a), suggests a possible molecular rationale for the low, but measureable, levels of cross-seeding detected between fibrils and monomers of different amino acid sequences 22.
The mechanism(s) by which neurons lose normal functionality in HD and a number of other neurodegenerative diseases has been of great interest for decades. In spite of enormous efforts, the HD field has not reached a consensus on this central question, which has remained “stubbornly unclear” 54. A variety of mechanisms, many involving the mediation of protein aggregates, have been suggested 3, 55–62, and work continues to sort out this experimentally complex issue. Hundreds of expanded polyQ huntingtin protein interaction partners, at least some of which were likely identified due to their binding to the aggregated state 63, have been described 64, 65, any one of which could be a key cellular target of polyQ toxicity. In the cell model we describe here, in which exogenous aggregates of chemically synthesized peptides are applied to cells, essentially equivalent toxicity was previously observed 17 in three distinct assays: (a) LDH release, which senses the integrity of the outer membrane, (b) propidium iodide exclusion, which senses the integrity of the nuclear membrane, and (c) MTT reduction, which monitors the activity of NAD(P)H-dependent cellular oxidoreductase enzymes. Since all three of these functions are expected to degrade entirely with cell death, their parallel declines suggest that in this case they are all essentially reporting on a similar cell death endpoint. Previous studies also explored the basis of the cell death mechanism in this cell model 66. While aggregate-treated cells exhibit some time-dependent changes normally associated with apoptosis, such as caspase activation and sensitivity to caspase inhibition and protein synthesis inhibitors, other apoptosis-associated phenomena, such as DNA fragmentation, were not observed 66. It is precisely because of the experimental challenge of determining the cellular mechanism of aggregate toxicity that we devised the approach described in this paper. Specifically, we endeavored here to set limits on the structural specificity of the toxicity mechanism, by asking whether that mechanism exhibits the kind of chiral specificity common to many cellular biochemical phenomena. The result, that polyQ aggregate cytotoxicity in this model is independent of chirality, is unequivocal. The interpretation, as discussed in this paper, is, not surprisingly, less clear. There remain a number of viable cytotoxicity mechanisms that are expected to be insensitive to chirality.
Previously, a number of groups provided data supporting a recruitment-sequestration model of polyQ toxicity 19, 20, 67–70 in which polyQ aggregates are posited to recruit critically important cellular molecules whose removal from solution leads to loss of activity and a subsequent toxic response. While such recruitment phenomena might be mediated by other protein elements 68, a convenient version of this general model is the well-understood ability of one polyQ amyloid to act as a seed for the elongation or recruitment of other polyQ proteins 11. Many transcription factors contain polyQ segments 71. While in most such proteins these are of repeat lengths too short to efficiently nucleate amyloid formation in vitro 35 or cause spontaneous polyQ disease in vivo 1, they are sufficiently long to support good elongation when a polyQ amyloid seed is provided 11. This recruitment mechanism is supported by several pieces of data from cell models featuring cell-internalized, in vitro-produced polyQ amyloid 17, 26, 72. First, dispersed microaggregates of polyQ amyloid are clearly capable of recruiting a different polyQ protein produced within the cell 26 (Fig. 2 a,b). Second, the cytotoxicity of such aggregates can be blocked by a structure-based inhibitor of polyQ amyloid elongation 72. The recent report that cultured mammalian cells producing expanded polyQ htt exon1 develop relatively small, individual polyQ amyloid fibrils 15 not unlike those used in the experiments reported here is also consistent with such a recruitment mechanism.
In this paper we show that the cytotoxicity of polyQ amyloid fibrils delivered into mammalian cells is insensitive to the chirality of the polyQ chains in the fibrils. One plausible interpretation of this lack of stereochemical specificity is that the molecular steps triggering toxicity involve interactions of relatively low order, such as hypothesized disruptive interactions with lipid membranes. However, we also show that the ability of D-polyQ amyloid to drive the aggregation of L-polyQ monomers is surprisingly robust and nearly as efficient as the elongation of L-polyQ fibrils with L-polyQ monomers. This lack of stereochemical discrimination means that toxicity mechanisms involving recruitment of polyQ-containing cellular proteins by polyQ aggregates are also consistent with the cytotoxicity of D-polyQ amyloid.
MATERIALS AND METHODS
Materials and general methods
Crude synthetic peptides were obtained (Keck Biotechnology Resource Laboratory of Yale University) and purified to homogeneous repeat lengths by reverse-phase chromatography using a ZORBAX SB C3 (Agilent Technologies) column. Structures and purities were confirmed by LC-MS. Different variations of Q25 peptides were obtained. For in vitro amyloid elongation reactions, monomers were of the sequence K2Q25K2. For cellular cytotoxicity and recruitment studies, sequences included the N-terminal nuclear localization sequence (NLS), PKKKRKV, with a GG spacer as well as a C-terminal Cys residue for attaching fluorescent tags (see below). For in vitro seeding experiments and to optimize preparation of fluorescently tagged amyloid, the same peptide was also obtained without the Cys residue. In all D-polyQ versions of the above peptides, only the Gln residues – and not the NLS amino acids or Lys or Cys residues - are in D-configuration.
Lyophilized, purified peptides were disaggregated and prepared in PBS as previously described 73. Most starting peptide solutions were filtered through a 20-nm filter unit (Anotop 10, Whatman) 35. For studies of aggregation kinetics at 37 °C only, 0.05% (w/v) sodium azide was added. To clearly distinguish the mechanism of fibril formation at −20 °C, and in particular to determine whether fibrils grow in a time-dependent manner at −20 °C or are generated by freeze-thaw cycles, we designed a kinetics experiment in which multiple aliquots of the peptide solution were frozen by immersion in liquid N2, then incubated at −20 °C and removed individually at different times for analysis. Aggregation kinetics data were obtained using a previously described sedimentation assay 73. Thioflavin T fluorescence data for isolated polyQ aggregates were obtained as described 29. Titration of growing ends on polyQ amyloid aggregates was conducted at 25 °C as previously described using L- or D- versions of biotinyl-K2Q30K2 73. Electron microscopy was conducted as described 35 in the Structural Biology Department’s EM facility using a Tecnai T12 microscope (FEI) operating at 120 kV and 30,000× magnification and equipped with an UltraScan 1000 CCD camera (Gatan).
Proteinase K digestion and MW1 binding were conducted on freshly disaggregated monomer solutions in PBS. For the protease digestion, polyQ monomer solutions (~15 μM) in PBS were incubated for 45 mins at 37 °C with a ratio of 41 units of proteinase K (CalBiochem) per mg peptide substrate, then reaction mixtures were analyzed by analytical reverse phase HPLC. For the antibody binding, aliquots of L- and D-polyQ solutions containing 50 ng to10 μg monomer were transferred to a polyvinylidene fluoride membrane (Millipore) using a dot blot apparatus (Bio-Rad), and matched loadings were confirmed using Ponceau S protein stain. Membranes were incubated overnight with MW1 antibody in blocking buffer (LiCor), and antibody binding was detected using Odyssey compatible IR-680 goat-anti-mouse (Invitrogen) and visualized using a LiCor Odyssey system.
Fluorescent labeling of peptides
A solution of ~ 24 mM Cy5 maleimide Mono-Reactive dye (GE Healthcare, Life Sciences) in ~50 μl DMSO was added to a solution of ~ 200 μM peptide in 6 M GdnHCl, 20 mM Tris.HCl, pH 7.5 and 2 mM Tris-(2-Carboxethyl) Phosphine (TCEP), and the mixture stirred at 25 °C and monitored by analytical HPLC. When the reaction was 90% complete (~ 2 hrs) the labeled peptide was purified by HPLC.
Preparing and quantifying aggregates
Stocks of polyQ amyloid fibrils of defined size and known concentration were prepared by a modification of published protocols 17. Monomer solutions of approximately 150 μM in PBS were snap frozen in liquid N2, then incubated at −20 °C until aggregation was at least 90% complete. Thawed, aggregated samples contained long, single filament fibrils and loose clusters of fibrils. These were subjected to multiple rounds of sonication with the reduction in average particle size monitored by DLS. Typically, 10 rounds, each consisting of 30 secs sonication followed by 30 secs rest, were performed using a micro-probe sonicator (Sonic Dismembrator Model 500-Fisher Scientific (20 kHz)) with amplitude at 40%, with the sample cooled on ice. The resulting aggregate suspension was then filtered using a 0.2 μm filter (VWR), yielding as filtrate a final stock suspension with a 40–65 nm size range by DLS. The concentration of the aggregate suspensions were determined as described 73. Aggregates labeled with Cy5 were obtained using the above procedure, except that we started with a mixture of one part Cy5-labelled peptide to 15 parts unlabelled peptide. We found that using higher percentages of labeled peptide led to formation of other aggregate morphologies.
Spectroscopy
Fourier transform infrared spectroscopy on aggregates was performed as described 35 using an MB3000 series spectrophotometer (ABB) and PROTA software (BioTools, Inc.). DLS measurements were conducted at 37 °C on a DynaPro plate reader (Wyatt Technology) on samples aliquoted into wells of a 384-well microtiter plate. Data were analyzed using Dynamics V7.1.0 software (Wyatt Technology) 35. CD spectroscopy was conducted on freshly sonicated samples using a JASCO J-810 spectropolarimeter using a 0.1 cm cuvette 29.
Cell experiments
Cells (PC12 “Schweitzer morph A cells” 74, either WT or transfected 32) were maintained in Dulbecco’s modified Eagle’s media (DMEM) containing 25mM HEPES (Cellgro), 5% supplemented calf serum (Hyclone), 5% horse serum (Hyclone), 2 mM l-glutamine, penicillin and streptomycin on collagen IV coated plates (Trevigen) at 37°C in 9.5% CO2. Cell media was changed every 3 days. Medium for transfected (Htt-exon1-Q25-EGFP) cells also included 0.5 mg/ml G418 (Mediatech) and 1 μM ponasterone. For aggregate internalization 17, freshly sonicated aggregates, prepared as described above, were diluted into OptiMEM (Gibco) medium supplemented with antibiotics. Cell toxicity was assessed by LDH release using the CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega).
For confocal microscopy analysis of cellular aggregates, cells were plated in collagen coated glass chamber slides (Nunc). Htt-exon1-Q25-EGFP PC12 cells were incubated with L- or D-polyQ-cy5 aggregates (2.5 μM) and simultaneously induced for exon1 expression with 1μM ponasterone. At specific times, cells were fixed with 4% paraformaldehyde (Cytofix, EB) and the nuclei stained with Hoechst 33342 (Invitrogen). Confocal images were collected using an Olympus Fluoview 1000 confocal microscope (×100 oil immersion lens) at room temperature. Random fields were scored (≥ 200 cells per condition over 3 experiments) for the percentage of cells presenting EGFP and Cy5 puncta using ImageJ software (NIH).
Data analysis and statistics
For the in vitro aggregation assays, error bars are standard deviations from analyses in duplicate. Data sets were fit in Origin 7.5 software (OriginLab). Most reaction profiles were fit to B-spline curves. Semi-log plots for heavily seeded elongation reactions used to determine elongation rate constants were fit by linear regression. Growing end titration data were fit to one site saturation ligand binding curves using Sigma Plot 10.0. Cellular punta counts and toxicity data were analyzed by GraphPad PRISM or Microsoft Excel. Significance was determined using one-way ANOVA, followed by post-hoc analysis (Student’s t-tests with Bonferroni correction) using P < 0.05.
Supplementary Material
HIGHLIGHTS.
D-polyQ amyloid fibrils are equipotent with L-fibrils in killing cells in culture
D-polyQ fibrils seed aggregation of L-polyQ monomers in cells and in vitro
Achiral cross-seeding can be rationalized by a D,L rippled β-sheet interface
The results have implications for molecular mechanisms of Huntington’s disease
Relaxed stringency in amyloid propagation also has implications for prion biology
Acknowledgments
We acknowledge funding support from NIH grants R01 AG019322 (RW and PvdW) and R21 AG033757 (RW). We thank Pawel Sikorski for providing coordinates for the polyQ amyloid fibril model, Erik Schweitzer for providing the PC12 cell lines, and Paul Patterson and Jan Ko for the sample of the MW1 antibody. We acknowledge James Conway and Alexander Makhov for access to the Structural Biology Department’s cryo-EM facility, and Ravindra Kodali for help in obtaining the EM images and FTIR spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bates GP, Benn C. The polyglutamine diseases. In: Bates GP, Harper PS, Jones L, editors. Huntington’s Disease. Oxford University Press; Oxford, U.K: 2002. pp. 429–472. [Google Scholar]
- 2.Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–81. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- 3.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 4.Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–69. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 5.Paulson HL. If it’s not one thing, it’s another. Nat Genet. 2006;38:743–4. doi: 10.1038/ng0706-743. [DOI] [PubMed] [Google Scholar]
- 6.Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu XF, Greiner E, Park CS, Wang N, Sopher BL, La Spada AR, Osmand A, Margolis RL, Sun YE, Yang XW. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron. 2011;70:427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 8.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 9.Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci U S A. 1999;96:4604–9. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–82. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 12.Wetzel R. Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J Mol Biol. 2012;421:466–90. doi: 10.1016/j.jmb.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–22. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 15.Sahl SJ, Weiss LE, Duim WC, Frydman J, Moerner WE. Cellular inclusion bodies of mutant huntingtin exon 1 obscure small fibrillar aggregate species. Sci Rep. 2012;2:895. doi: 10.1038/srep00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wetzel R. Order, disorder, and conformational flux in the structural biology of Huntington’s disease. In: Bates GP, Tabrizi SJ, Jones L, editors. Huntington’s Disease. Oxford University Press; Oxford, U.K: 2014. In press. [Google Scholar]
- 17.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 18.Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:9679–84. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–70. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME, Gusella JF. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat Cell Mol Genet. 1998;24:217–33. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- 21.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s b-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 22.O’Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 23.Krebs MR, Morozova-Roche LA, Daniel K, Robinson CV, Dobson CM. Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci. 2004;13:1933–8. doi: 10.1110/ps.04707004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esler WP, Stimson ER, Fishman JB, Ghilardi JR, Vinters HV, Mantyh PW, Maggio JE. Stereochemical specificity of Alzheimer’s disease beta-peptide assembly. Biopolymers. 1999;49:505–14. doi: 10.1002/(SICI)1097-0282(199905)49:6<505::AID-BIP8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 25.Wadai H, Yamaguchi K, Takahashi S, Kanno T, Kawai T, Naiki H, Goto Y. Stereospecific amyloid-like fibril formation by a peptide fragment of beta(2)-microglobulin. Biochemistry. 2005;44:157–164. doi: 10.1021/bi0485880. [DOI] [PubMed] [Google Scholar]
- 26.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nature Cell Biology. 2009;11:219–U232. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–9. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 28.Franks F. Storage stabilization of proteins. In: Franks F, editor. Protein Biotechnology: Isolation, Characterization and Stabilization. Humana Press; New York: 1993. pp. 489–531. [Google Scholar]
- 29.Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R. Slow amyloid nucleation via alpha-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J Mol Biol. 2012;415:881–99. doi: 10.1016/j.jmb.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorscent dye, thioflavine T. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 31.Milton RC, Milton SC, Kent SB. Total chemical synthesis of a D-enzyme: the enantiomers of HIV-1 protease show reciprocal chiral substrate specificity [corrected] Science. 1992;256:1445–8. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 32.Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16:546–55. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Ko J, Ou S, Patterson PH. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull. 2001;56:319–29. doi: 10.1016/s0361-9230(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 34.Li P, Huey-Tubman KE, Gao T, Li X, West AP, Jr, Bennett MJ, Bjorkman PJ. The structure of a polyQ-anti-polyQ complex reveals binding according to a linear lattice model. Nat Struct Mol Biol. 2007;14:381–7. doi: 10.1038/nsmb1234. [DOI] [PubMed] [Google Scholar]
- 35.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328–36. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powers ET, Ferrone F. Kinetic models for protein misfolding and association. In: Dobson CM, Kelly JW, Ramirez-Alvarado M, editors. Protein Misfolding Diseases: Current and Emerging Principles and Therapies. Wiley; New York: 2009. pp. 73–92. [Google Scholar]
- 37.Elias H-G. Macromolecules: Physical Structures and Properties. Vol. 3. Wiley; Weinheim: 2008. [Google Scholar]
- 38.Brovchenko I, Singh G, Winter R. Aggregation of amyloidogenic peptides near hydrophobic and hydrophilic surfaces. Langmuir. 2009;25:8111–6. doi: 10.1021/la9006058. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharyya AM, Thakur AK, Wetzel R. polyglutamine aggregation nucleation: thermodynamics of a highly unfavorable protein folding reaction. Proc Natl Acad Sci U S A. 2005;102:15400–5. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannon MJ, Williams AD, Wetzel R, Myszka DG. Kinetic analysis of beta-amyloid fibril elongation. Anal Biochem. 2004;328:67–75. doi: 10.1016/j.ab.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 41.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sikorski P, Atkins E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecules. 2005;6:425–32. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- 43.Pauling L, Corey RB. Two rippled-sheet configurations of polypeptide chains, and a note about the pleated sheets. Proc Natl Acad Sci U S A. 1953;39:253–6. doi: 10.1073/pnas.39.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 45.Jarrett JT, Lansbury PJ. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie?. [Review] Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 46.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–3. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 47.Chien P, DePace AH, Collins SR, Weissman JS. Generation of prion transmission barriers by mutational control of amyloid conformations. Nature. 2003;424:948–51. doi: 10.1038/nature01894. [DOI] [PubMed] [Google Scholar]
- 48.Aguzzi A. Cell biology: Beyond the prion principle. Nature. 2009;459:924–5. doi: 10.1038/459924a. [DOI] [PubMed] [Google Scholar]
- 49.Chung DM, Nowick JS. Enantioselective molecular recognition between beta-sheets. Journal of the American Chemical Society. 2004;126:3062–3063. doi: 10.1021/ja031632z. [DOI] [PubMed] [Google Scholar]
- 50.Dzwolak W, Ravindra R, Nicolini C, Jansen R, Winter R. The diastereomeric assembly of polylysine is the low-volume pathway for preferential formation of beta-sheet aggregates. Journal of the American Chemical Society. 2004;126:3762–3768. doi: 10.1021/ja039138i. [DOI] [PubMed] [Google Scholar]
- 51.Swanekamp RJ, Dimaio JT, Bowerman CJ, Nilsson BL. Coassembly of Enantiomeric Amphipathic Peptides into Amyloid-Inspired Rippled beta-Sheet Fibrils. J Am Chem Soc. 2012 doi: 10.1021/ja301642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koga T, Matsuoka M, Higashi N. Structural control of self-assembled nanofibers by artificial beta-sheet peptides composed of D- or L-isomer. Journal of the American Chemical Society. 2005;127:17596–17597. doi: 10.1021/ja0558387. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–9. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 54.Blum ES, Schwendeman AR, Shaham S. PolyQ disease: misfiring of a developmental cell death program? Trends in Cell Biology. 2013;23:168–174. doi: 10.1016/j.tcb.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selkoe DJ. Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2011:3. doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Musiek ES, Holtzman DM. Origins of Alzheimer’s disease: reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-beta and tau involvement. Curr Opin Neurol. 2012;25:715–20. doi: 10.1097/WCO.0b013e32835a30f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stefani M. Structural features and cytotoxicity of amyloid oligomers: implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog Neurobiol. 2012;99:226–45. doi: 10.1016/j.pneurobio.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 59.Orr HT. Polyglutamine neurodegeneration: expanded glutamines enhance native functions. Curr Opin Genet Dev. 2012;22:251–5. doi: 10.1016/j.gde.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cisbani G, Cicchetti F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis. 2012;3:e382. doi: 10.1038/cddis.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci. 2013;55:26–36. doi: 10.1016/j.mcn.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 62.Hirsch EC, Jenner P, Przedborski S. Pathogenesis of Parkinson’s disease. Mov Disord. 2013;28:24–30. doi: 10.1002/mds.25032. [DOI] [PubMed] [Google Scholar]
- 63.Davranche A, Aviolat H, Zeder-Lutz G, Busso D, Altschuh D, Trottier Y, Klein FAC. Huntingtin affinity for partners is not changed by polyglutamine length: aggregation itself triggers aberrant interactions. Human Molecular Genetics. 2011;20:2795–2806. doi: 10.1093/hmg/ddr178. [DOI] [PubMed] [Google Scholar]
- 64.Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, Yang XW. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Culver BP, Savas JN, Park SK, Choi JH, Zheng S, Zeitlin SO, Yates JR, 3rd, Tanese N. Proteomic Analysis of Wild-type and Mutant Huntingtin-associated Proteins in Mouse Brains Identifies Unique Interactions and Involvement in Protein Synthesis. J Biol Chem. 2012;287:21599–614. doi: 10.1074/jbc.M112.359307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang W. PhD Dissertation. The University of Tennessee, ProQuest, UMI Dissertations Publishing; 2003. The role of polyglutamine aggregate cytotoxicity in Huntington’s disease. document number 3151870. [Google Scholar]
- 67.McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 68.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–8. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 70.McCampbell A, Fischbeck KH. Polyglutamine and CBP: fatal attraction? Nat Med. 2001;7:528–30. doi: 10.1038/87842. [DOI] [PubMed] [Google Scholar]
- 71.Gerber HP, Seipel K, Georgiev O, Hofferer M, Hug M, Rusconi S, Schaffner W. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science. 1994;263:808–11. doi: 10.1126/science.8303297. [DOI] [PubMed] [Google Scholar]
- 72.Thakur AK, Yang W, Wetzel R. Inhibition of polyglutamine aggregate cytotoxicity by a structure-based elongation inhibitor. FASEB J. 2004;18:923–5. doi: 10.1096/fj.03-1238fje. [DOI] [PubMed] [Google Scholar]
- 73.O’Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, Thiagarajan G, Wetzel R. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- 74.Schweitzer ES, Paddock S. Localization of human growth hormone to a subset of cytoplasmic vesicles in transfected PC12 cells. J Cell Sci. 1990;96(Pt 3):375–81. doi: 10.1242/jcs.96.3.375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.