

Introduction
The Mayer–Rokitansky–Kuster–Hauser (MRKH) syndrome affects 1 out of 4,500 women [1]. It is a malformation of the female genitals due to interrupted embryonic development of the mullerian (paramesonephric) ducts in otherwise chromosomally, phenotypically, and endocrinologically normal female. It is second to Turner’s syndrome as a cause of primary amenorrhea and was described by Mayer (1829), Rokitansky (1838), Kuster (1910), and Hauser and Schreiner (1961) in various literature studies, which was later designated as MRKH syndrome. MRKH syndrome is broadly subdivided into type A (typical) having symmetric uterine remnants and normal fallopian tubes and type B (atypical) with asymmetric uterine buds and abnormally developed fallopian tubes and other organ system anomalies.
Gonadal agenesis or dysgenesis is a chromosomal aberration with a separate spectrum of anomalies having an overall incidence of about 1:2,500 live birth females, half of which have a mosaic pattern. These patients have multiple somatic abnormalities like short stature, broad chest, webbed neck, low hair line, sometimes low I.Q., cardiac abnormalities (1/3 cases), and renal abnormalities (35–70 %). They usually have pre-pubertal female genitalia, bilateral streak gonads, but usually normal uterus and vagina (Figs 1, 2).
Fig. 1.
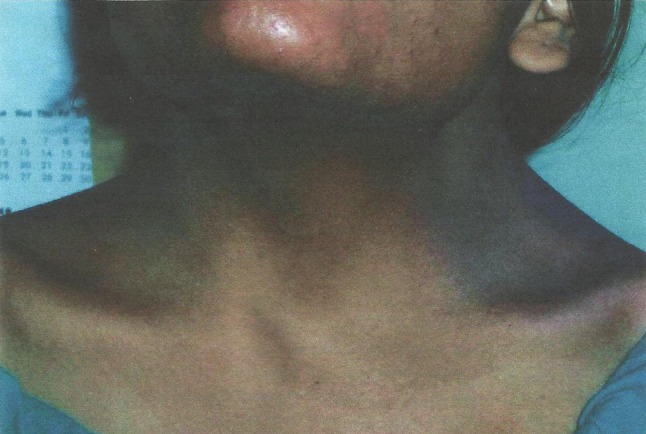
Photograph showing webbing of neck
Fig. 2.
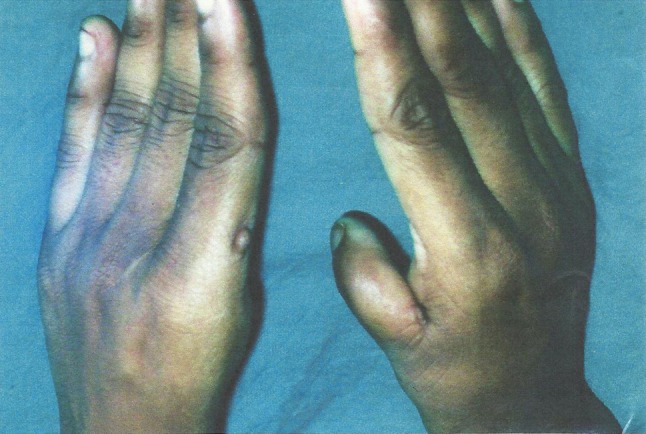
Photograph showing the absence of left thumb and rudimentary right thumb
Case Report
An 18-year-old girl with primary amenorrhea attended the O.P.D. with normal I.Q. (1st year college student) and without such family history. On examination, her height is 154 cm, weight—42 kg, with webbing of neck, telecanthus; and her left thumb was absent and right thumb rudimentary. Systemic examination was normal. Breasts were well developed and axillary and pubic hairs were present. On perineal examination, there was a hymenal fringe and blind vagina. Uterus was absent on per rectal examination.
Routine examination of blood, ECG, and chest X-ray reported normal. X-ray cervical spine showed fusion of the cervical vertebrae (Klippel Feil Syndrome). Serum FSH, LH, TSH, and testosterone were normal. USG [02/10/07] showed right-sided crossed fused ectopic kidney with normal corticomedullary differentiation, uterus absent, and normal gonads. Diagnostic laparoscopic [04/10/07] finding was asymmetric mullerian nodule [rt > lt] with rudimentary fallopian tubes, and both ovaries were normal with signs of ovulation. Karyotyping was Turner’s mosaics pattern (46XX/45XO). Diagnosis of MRKH type-B with gonadal dysgenesis was made. Vaginoplasty was done on 12/10/07 after proper counseling as parents wanted to arrange marriage. Post-operative period was uneventful.
Discussion
Several cases of MRKH syndrome have been reported both type A and B. Associated congenital abnormality of the upper urinary tract are seen in 40 % of cases, varying from renal agenesis to crossed ectopic kidney as seen in this case. Majority of the cases are sporadic; however, familial cases have also been described. Mode of inheritance is being autosomal dominant with incomplete penetrance and variable expressivity [1]. MRKH syndrome may be associated with MURCS, and the terms were first described by Duncan (1977). Renal agenesis or ectopia together with the MRKH and KF syndromes, known as the MURCS association (MU; Mullerian duct aplasia, R: renal agenesis/ectopia; CS: cervical somite dysplasia) [2]. Only atypical form of MRKH (type-B) is associated with renal, skeletal, and ovarian abnormalities [3]. The point of utmost importance is the association of Turner’s Mosaics with MURCS and the presence of scoliosis, hand, and finger abnormalities in this case. Presently ovarian dysgenesis or agenesis is not considered to be a part of MRKH or MURCS clinical spectrum. No single group shows random association between these two abnormalities.
Conclusion
We report this case as it is a very rare association of a chromosomal disorder and developmental mesodermal disorder. It is very difficult to say whether it is a sporadic association or genetic or any other influence plays any role for the occurrence. For such a conclusion, studies are needed on a large number of cases with MRKH. Till date to the best of our knowledge, after Medline search, no author has concluded or confirmed this association as a single clinical syndrome to be mentioned separately.
References
- 1.Guerrier D, Mouchel T, Pasquier L, et al. The Mayer–Rokitansky–Kuster–Hauser syndrome (congenital absence of uterus and vagina)—phenotypic manifestation and genetic approaches. J Negat Results Biomed. 2006;5:1. doi: 10.1186/1477-5751-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strubbe EH, Lemmens JA, Thijn CJ, et al. Spinal abnormalities and the atypical form of the Mayer–Rokitansky–Kuster–Hauser syndrome. Skeletal Radiol. 1992;21:459–462. doi: 10.1007/BF00190992. [DOI] [PubMed] [Google Scholar]
- 3.Kumar A, Mishra S, Dogra PN. Management of an unusual case of atypical Mayer–Rokitansky–Kuster–Hauser syndrome, with unilateral gonadal agenesis, solitary ectopic pelvic kidney, and pelviureteric junction obstruction. Int Urogynecol J. 2007;18:823–825. doi: 10.1007/s00192-006-0238-z. [DOI] [PubMed] [Google Scholar]