

Abstract
Mutations at solvent inaccessible core positions in proteins can impact function through many biophysical mechanisms including alterations to thermodynamic stability and protein dynamics. As these properties of proteins are difficult to investigate, the impacts of core mutations on protein function are poorly understood for most systems. Here, we determined the effects of alanine mutations at all 15 core positions in ubiquitin on function in yeast. The majority (13 of 15) of alanine substitutions supported yeast growth as the sole ubiquitin. The two null mutants (I30A and L43A) were both less stable to temperature-induced unfolding in vitro than wild-type, but were well folded at physiological temperatures. Heteronuclear NMR studies indicated that the L43A mutation reduces temperature stability while retaining a ground-state structure similar to wild-type. This structure enables L43A to bind to common ubiquitin receptors in vitro. Many of the core alanine ubiquitin mutants, including one of the null variants (I30A), exhibited an increased accumulation of high molecular weight species, suggesting that these mutants caused a defect in the processing of ubiquitin-substrate conjugates. In contrast, L43A exhibited a unique accumulation pattern with reduced levels of high molecular weight species and undetectable levels of free ubiquitin. When conjugation to other proteins was blocked, L43A ubiquitin accumulated as free ubiquitin in yeast. Based on these findings we speculate that ubiquitin's stability to unfolding may be required for efficient recycling during proteasome-mediated substrate degradation.
Keywords: thermodynamic stability, structural dynamics, protein function, proteasome, ubiquitin recycling
Introduction
Most proteins adopt structures where the relative positions of each atom are well defined in the ground state1; yet, each atom in proteins is in constant motion leading to structural dynamics that frequently have important functional consequences2. Compared to the ground state of proteins that can be interrogated through x-ray crystallography among other techniques, investigating protein dynamics and their connections to function often requires protein-specific approaches that can be difficult to develop and/or interpret3. Indeed, the challenges associated with studies of protein dynamics have limited our understanding of this important area of biochemistry. Many mutational studies, for example, focus on positions located on the solvent accessible surface of the native state in order to identify “hot spots” for binding4. This surface scanning approach, which provides efficient and valuable insights into protein function, often avoids positions located in the solvent-shielded protein core because of the challenges associated with investigating potential impacts on protein structure and dynamics. These practical factors have led to a relative dearth of understanding of the impacts of core mutations on protein function.
The effects of core mutations on protein thermodynamic stability have been rigorously investigated. For example, many studies have demonstrated that well-packed hydrophobic cores make dominant contributions to the thermodynamic stability of protein native states5, and that polar side chains in protein cores tend to form intramolecular hydrogen bonds6. While it is clear that core mutations can have a dominant impact on thermodynamic stability of the native state, how core mutations affect protein function has not been as extensively investigated. Simple models postulate that the functional impacts of mutations on protein stability are directly proportional to the ability of mutants to populate the native state7. However, recent experimental studies demonstrate that function in vivo is far more complex and cannot be accurately predicted by stability alone8; 9; 10; 11. In particular, the impact of mutations on protein dynamics and how these translate to function remains largely unknown for most proteins.
Our current understanding of protein dynamics and function are largely the fruits of NMR studies in vitro. NMR techniques have been developed to study protein motions that occur on timescales relevant to protein functions12. Protein dynamics have been found to play critical roles in enzyme efficiency and suggest that these motions are strongly temperature dependent, with catalytically optimum dynamics occurring near the growth temperature of the host organism13. Interestingly, protein engineering strategies have identified mutations that can increase enzyme efficiency across broad temperature ranges, indicating that the links between natural selection and protein dynamics can be complex14; 15.
In this work, we explored the interplay between protein stability, dynamics, and function in ubiquitin (UB). Ubiquitin is essential in all eukaryotes where it serves multiple functions via its ability to covalently attach to other proteins16. The covalent attachment of ubiquitin is mediated by a series of enzymes referred to as E1, E2, and E317. Multiple ubiquitin molecules can be linked to form poly-ubiquitin chains, with polymers of four ubiquitins being sufficient to target substrates for proteasome-mediated degradation18. The proteasome is a complex multi-protein assemblage that binds to poly-ubiquitylated substrates and then utilizes the energy of ATP to unfold and thread covalently attached substrate into an internal chamber for degradation19; 20. During the process of substrate degradation, ubiquitin is efficiently recycled by the de-ubiquitylase (DUB) activity of the intrinsic proteasomal subunit Rpn1121; 22 and the proteasome-associated DUBs Ubp6 (Usp14 in mammals)23; 24; 25 and Uch37 (in mammals)26. In vitro experiments with purified proteasomes demonstrate that ubiquitin recycling and substrate degradation are tightly coupled21. Non-covalent binding of ubiquitin to numerous receptors mediates many critical functions including delivery of substrates to the proteasome. Of note, binding of ubiquitin to important cellular receptors has recently been shown to depend on the structural dynamics of ubiquitin11; 27.
Here we investigated the effects of alanine mutations at all 15 core positions in ubiquitin. Of note, these core positions were not investigated in a previous alanine scan that focused on the surface of ubiquitin28. We find that yeast growth tolerates alanine substitutions at most (13 out of 15) core positions consistent with a previous high-throughput investigation of ubiquitin mutants in yeast29. The two mutants that failed to support growth (I30A and L43A) were both structurally stable at physiological temperature indicating that global unfolding was insufficient to explain the observed growth defects. In yeast cells, we observed that most of the core alanine ubiquitin mutants accumulated as both free ubiquitin and high molecular weight species, suggesting that they were compatible with enzymes involved in conjugation and recycling. The I30A mutant showed a strong accumulation of high molecular weight species suggesting that it may have a defect in targeting substrates to the proteasome as was previously observed for other core ubiquitin mutants11. L43A, the other growth defective mutant, exhibited a unique accumulation pattern: undetectable free ubiquitin and low levels of high molecular weight species. Importantly, the L43A mutant accumulated in yeast when substrate conjugation was prevented. NMR experiments with L43A demonstrate that it exhibits novel structural dynamics relative to wild-type (WT) ubiquitin. In particular, structure and backbone motions local to the L43A mutation are altered in the isolated protein. However, the mutant is capable of binding to partner molecules from the proteasome pathway, and upon binding assumes a structure that is virtually indistinguishable from wild type. Based on these observations, we propose that the stability, structure, and dynamics of ubiquitin are all critical for its function and that the L43A mutant perturbs these properties such that it exhibits a recycling defect.
Results and Discussion
Effects of ubiquitin core alanine substitutions on yeast growth
The solvent inaccessible interior of ubiquitin (Figure 1a) is composed of 13 aliphatic amino acids that form a well packed hydrophobic core as well as two polar amino acids (Thr7 and Gln41). Both core polar amino acids form hydrogen bonds to solvent inaccessible polar main-chain atoms. To examine how each core position contributes to function, we generated individual alanine substitutions and measured their effects on yeast growth (Figure 1b,c). These experiments utilized the previously developed SUB328 ubiquitin shutoff strain30. Expression of ubiquitin in this strain is strictly dependent on galactose, and when switched to dextrose media ubiquitin levels rapidly decrease and growth stalls. The introduction of a plasmid that constitutively expresses ubiquitin rescues growth under shutoff conditions compared to a control plasmid lacking ubiquitin (Figure 1b). Under shutoff conditions, 13 out of the 15 alanine core substitutions support observable yeast growth as the sole ubiquitin protein in yeast (Figure 1b). In control experiments, all 15 of the alanine core substitutions permitted growth in the presence of galactose (Figure S1), indicating that none of the mutants are toxic when co-expressed with WT ubiquitin.
Figure 1. Effects of alanine mutations in the core of ubiquitin on yeast growth.

(a) Illustrations of the structure of ubiquitin highlighting the locations of core positions that are shielded from solvent. (b&c) Capability of mutants to function as the sole ubiquitin. Constitutively expressed mutants were introduced into the SUB328 shutoff strain, and growth under selective conditions was examined on plates (b) and in liquid culture (c). As a negative control, cells harboring a plasmid lacking the ubiquitin open reading frame (labeled as “no UB”) were analyzed.
To further quantify the impacts of the core alanine mutants on yeast growth, we measured growth rates in liquid culture under shutoff conditions (Figures 1c and S2). Observed growth rates in liquid culture paralleled those observed on plates and confirmed that 13 out of the 15 mutants were able to support yeast growth as the sole ubiquitin. Except for I30A and L43A, alanine substitutions in N-terminal regions up to position 50 exhibited growth rates indistinguishable from WT. Partial growth defects were observed for positions within and proximal to the C-terminus or residues 50-61 that connect the last two β-strands, suggesting that these regions may be sensitive to minor alterations to structure and/or dynamics. The C-terminus of ubiquitin, especially the last β-strand, is a known hot-spot of critical binding interactions28; 29; 31, and we have previously observed that the L67S and L69S core mutations in this β-strand disrupt binding to proteasomal receptors11. In both growth assays (Figure 1b,c), the I30A and L43A mutants failed to support yeast growth. The yeast system30 we used expresses ubiquitin mutants from a strong promoter (GPD) on a high copy plasmid in order to mimic the endogenous expression level of ubiquitin, which is ordinarily encoded at multiple genomic loci. Because of this experimental setup, increasing the expression level of ubiquitin mutants further is technically challenging.
Biophysical investigations of I30A and L43A
We sought to understand the biophysical basis for the null growth phenotypes of I30A and L43A, initially considering the possibility that these amino acid substitutions caused global unfolding of ubiquitin. To push the limits of ubiquitin stability that are compatible with yeast growth we generated double mutants of T7A with either V26A or L50A. As individual mutations, T7A, V26A, and L50A support yeast growth (Figure 1b,c), and both double mutants also support growth albeit with partial defects (Figure 2a). Thus, these double mutants have sufficient thermodynamic folding stability to support at least partial yeast growth. We compared the biophysical properties of these double mutants in purified form with I30A and L43A. All four variants exhibit circular dichroism (CD) spectra consistent with mixed α/β secondary structure (Figure 2b); though all four variants differ from WT in secondary structure content, with the I30A and L43A variants exhibiting the greatest differences. All four mutant variants exhibit cooperative temperature-induced unfolding transitions with midpoints near 60 °C (Figure 2c). Of note, urea denaturation experiments were performed to probe thermodynamic stability, but similarities in CD signal above 218 nm between the native and chemical denatured states prevented quantitative analyses of the free energy of unfolding. Based on temperature denaturation, all four variants reduce thermal stability compared to WT whose melting temperature is close to 100 °C32. Despite this reduction in stability relative to WT, all of the variants predominantly populate native conformations at physiological temperature (30 °C in our yeast experiments), suggesting that they are all capable of folding in cells. Furthermore, the I30A and L43A mutants that are the most severely growth deficient have slightly higher thermal stability than the T7A/V26A and T7A/L50A variants, indicating that thermal stability alone does not explain the growth defects of I30A and L43A ubiquitins.
Figure 2. Protein properties of ubiquitin mutants with severe growth defects.
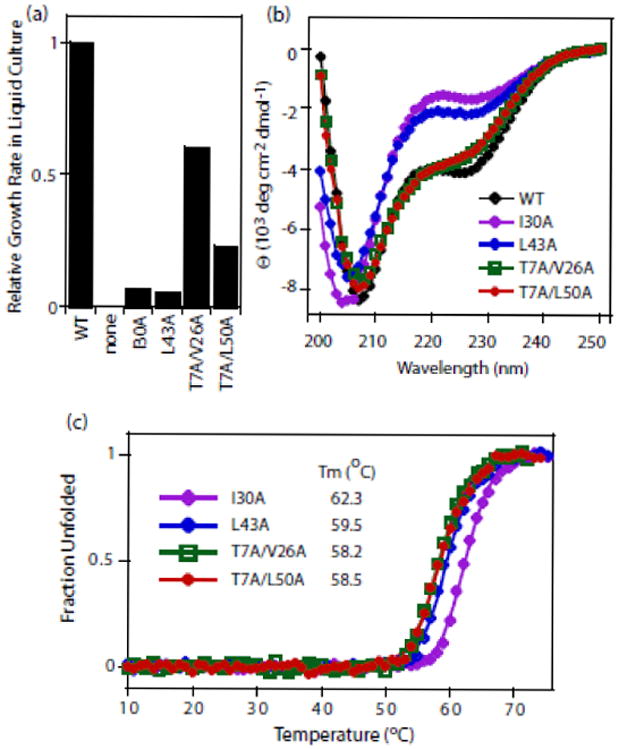
(a) Relative growth rates of yeast with I30A or L43A ubiquitin are slower than yeast with T7A/V26A or T7A/L50A ubiquitin. (b) Circular dichroism spectra of purified ubiquitin proteins at 23 °C. (c) Stability of purified ubiquitin mutants to temperature denaturation.
Effects of mutants on accumulation of free and conjugated ubiquitin in yeast
In seeking alternative explanations for the growth defects of the I30A and L43A mutants, we surveyed our entire panel of variants for the accumulation of both free ubiquitin and high molecular weight covalent conjugates in yeast (Figure 3). As further described in the Methods, these experiments were performed using yeast grown under shutoff conditions for sufficient time to deplete the pool of WT ubiquitin in cells lacking a rescue mutant (Figure 3a, second lane). Many of the alanine mutants accumulated higher levels of free and/or high molecular weight ubiquitin species (Figure 3a,b). While there are many potential explanations for these observations, the increased expression level of some mutants may indicate selective compensation for a biochemical defect as has been previously observed to occur in yeast9. Similarly, the increased accumulation of high molecular weight species may indicate a deficiency in targeting substrates to the proteasome as we previously observed for the L67S and L69S mutants11. Interestingly, we observed a unique accumulation pattern for L43A ubiquitin, one of the two mutants null for yeast growth.
Figure 3. Accumulation of ubiquitin mutants in yeast.
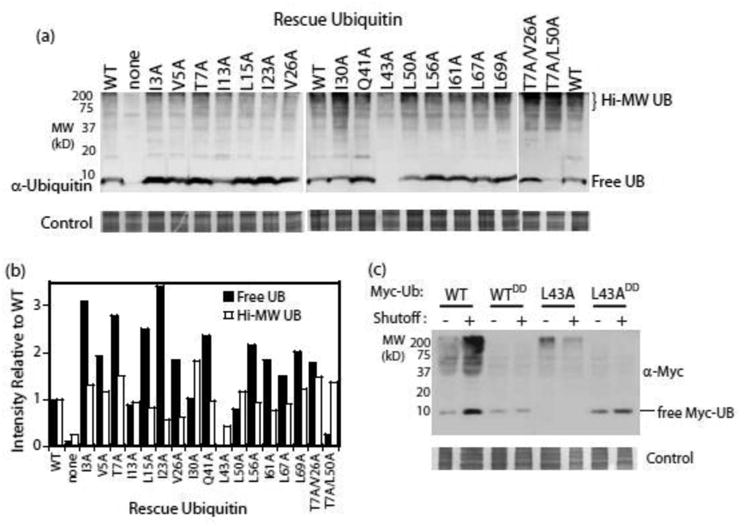
(a) Western blot analyses of yeast whole cell lysates. Cells were grown in dextrose for 24 hours in order to shut off expression of WT ubiquitin. (b) The intensity of the free ubiquitin band as well as of high molecular weight species in panel (a) were quantified and compared to a WT control present on each blot. (c) Western blot analyses of lysates from yeast expressing epitope-tagged ubiquitin variants. The G75D/G76D mutations (annotated as DD) were used to block activation by E1 and hence eliminate all covalent attachment reactions. Under shutoff conditions, the epitope-tagged version is the only ubiquitin protein in the cells.
In yeast, L43A ubiquitin accumulated a reduced amount of high molecular weight species and no appreciable free ubiquitin (Figures 3a,b). To confirm this unusual accumulation pattern, we re-cloned the L43A mutant with an N-terminal epitope tag previously shown to be compatible with yeast growth33. We examined the accumulation pattern of epitope-tagged WT and L43A ubiquitins (Figure 3c) with and without a double aspartic acid (DD) mutation at the C-terminus that prevents conjugation to substrates. Epitope-tagged WT ubiquitin accumulates in both conjugated and free forms. Of note, when co-expression of untagged ubiquitin is shut off, the overall levels of epitope-tagged ubiquitin increase. This may indicate the selection for increased expression as has been previously observed in yeast on these timescales9. In contrast, epitope-tagged WTDD ubiquitin accumulates as free ubiquitin only, and its expression level is independent of co-expression of untagged ubiquitin, consistent with the DD mutation preventing conjugation and an absence of any selective advantage of increased WTDD expression. By contrast, epitope-tagged L43A ubiquitin accumulates in reduced amounts of high molecular weight species and no observable free species. When conjugation is prevented, the L43ADD variant accumulates as free ubiquitin in yeast. These observations strongly indicate that the reduced accumulation of L43A ubiquitin is dependent on conjugation to substrate, and are consistent with an increased propensity for co-degradation of substrate-conjugated L43A by the proteasome.
Structural and Dynamic Analyses of L43A Ubiquitin by NMR
The 1H-15N HSQC spectrum of L43A ubiquitin shows a well-spread set of backbone amide resonances (Figure 4a), clearly indicating that the protein is well folded. However, a large number of amides show significant spectral differences (up to 1.2 ppm) from WT ubiquitin. The largest chemical shift perturbations (CSPs) are observed in the α which is packed directly against the side chain of L43A (Figure 4b,c). The surrounding regions, including strands β2 and β5, as well as the β3 strand (containing the mutation site) also exhibit signal shifts. These perturbations reflect changes in the electronic environment of the nuclei under observation, and are likely caused by small rearrangements of the hydrophobic core of the protein because all of the main secondary structure elements in ubiquitin (including the α-helix) remain intact in L43A (Figure S3a). In order to examine possible global conformational changes34 caused by the mutation, we measured residual dipolar couplings (RDCs) for L43A in a weakly aligned liquid crystalline medium, and compared these data against the structure of WT ubiquitin. The excellent agreement between the experimental RDCs and those back-calculated using WT ubiquitin structure (Figure 4d) strongly indicates that the overall three-dimensional structure of L43A is very similar to that of WT ubiquitin.
Figure 4. Structure and backbone dynamics of L43A ubiquitin by NMR.

(a) Overlay of 1H-15N NMR spectra of L43A (red) and WT (black) ubiquitin and (b) quantification of the spectral differences between the two proteins in terms of amide chemical shift perturbations (CSPs) plotted as a function of the residue number. (c) Mapping the CSPs due to the L43A mutation on ubiquitin structure. L43 is shown in spheres, and the coloring represents CSPs in decreasing order: Red> Orange > Yellow > Green. (d) The 1H-15N residual dipolar couplings (RDCs) measured for L43A and the back-calculated RDCs using solution structure of WT ubiquitin (1D3Z.PDB) are in close agreement, indicating that the 3D structure of ubiquitin is not grossly affected by the mutation. The Pearson's correlation coefficient is r=0.976, the quality factor61 is 0.146 (62 residues included). (e) Conformational exchange (Rex) contributions detected in L43A (red) and WT ubiquitin (blue). (f) Backbone order parameters for L43A (red) and WT ubiquitin (blue) indicate similarity in the backbone motions on a sub-nanosecond time scale.
To examine the effect of the L43A mutation on backbone dynamics, we measured 15N relaxation rates (R1, R2, and heteronuclear NOEs), extracted backbone microdynamic parameters, and compared them with the corresponding data for WT ubiquitin. Our results (Figures 4e,f and S3b-f) demonstrate that fast, sub-nanosecond motions in the backbone of the protein are not affected by the mutation. However, a large number of residues in L43A showed the presence of strong conformational exchange contributions (Rex) to R2 indicative of the presence of slower, ms-μs motions in the mutant (Figure 4e). The distribution of residues exhibiting conformational exchange mirrors that of residues with large chemical shift perturbations (CSPs, Figure 4c). This suggests that the CSPs observed in L43A versus WT ubiquitin reflect conformational averaging between the ground state (which is structurally similar to that of WT ubiquitin, based on the RDC data) and one or more excited states. These findings are in line with our previous observations for another ubiquitin core mutant (L69S) that also had little effect on its 3D structure, but experienced increase in ms-μs and slower motions11.
These high-resolution NMR measurements of the structural changes caused by the L43A substitution are consistent with the changes observed at lower resolution by CD (Figure 2b). Both methods indicate that the L43A substitution caused structural changes to the α-helix. Specifically, the significant chemical shift perturbations and large conformational exchange contributions throughout the helix suggest the presence of structural rearrangements on a μs-ms time scale. While on first glance the change in CD spectra appears to indicate a more dramatic structural change than the NMR observations, there are two methodological factors that are important to consider. First, because the signature of a helical residue has many fold greater CD intensity (at 208 and 220 nm) than residues in other secondary structures, and because ubiquitin has a single α-helix, the CD spectra of ubiquitin may be ultra-sensitive to changes in the helix. Second, CD spectra are not a pure measurement of secondary structure35; 36, and accurate secondary structure predictions from CD require training on spectra of samples from proteins of known structure. The CD spectra we observed for WT and L43A ubiquitins were not sufficiently similar to those in the CD database to provide accurate estimates of their secondary structure.
L43A Ubiquitin Binds to Common Effector Domains
Having confirmed that the structure of L43A ubiquitin is intact, we examined whether the mutation affected its ability to bind to receptor proteins. To do this, we screened L43A binding to all known ubiquitin-receptors from the ubiquitin-proteasome pathway: the UBA domains of shuttle proteins ubiqulin-1 (UQ1) (aka hPLIC-1, human homologue of Dsk2) and of hHR23a (human homologue of Rad23), the UIM domain of proteasome subunit Rpn10, and the pleckstrin-like receptor (Pru) domain of proteasome subunit hRpn13. These ubiquitin-binding domains cover a range of architectures, ranging from a single helix (UIM) to three-helix bundle (UBA) to an α/β protein fold (Pru)37; 38; 39.
NMR titration experiments (Figure 5) demonstrate that L43A ubiquitin does bind UQ1 UBA. Several amides in L43A showed strong signal attenuations in the course of titration, which is typical for slow exchange (slow off-rates) on the NMR time scale that indicates strong binding. Based on the observed spectral perturbations, we mapped the UBA-binding surface on L43A. The binding surface is very similar to that for WT ubiquitin and includes the surface hydrophobic patch comprising residues L8, I44, and V7031. Interestingly, we observed strong similarity between the NMR spectra of L43A and WT ubiquitin in the UQ1 UBA-bound state (Figure 5d), in contrast with the spectra of free L43A and WT ubiquitins that show significant differences (Figure 4a). This suggests that although the conformational ensembles of the two unbound proteins are different, the UQ1 UBA binding selects a similar conformation for both WT and L43A ubiquitins. To determine the stoichiometry of L43A/UBA, we measured longitudinal 15N relaxation time (T1=1/R1) for L43A in the UBA-bound state. 15N T1 directly reflects the overall tumbling rate of a protein and therefore can be used as a “molecular weight ruler”40. The result (T1 = 677 ± 32 ms) is slightly below that for WT ubiquitin in complex with UQ1 UBA (731 ± 37 ms)41 and in excellent agreement with the expected molecular weight (14.5 kDa) of a 1:1 ubiquitin/UBA complex. These NMR analyses demonstrate that L43A binds UQ1 UBA in the same stoichiometry and manner as WT ubiquitin. Fitting of NMR titration curves (Figure 5e,f) revealed that L43A binds to UQ1 UBA slightly weaker (Kd = 26 ± 13 μM) than WT ubiquitin (4.2 ± 4.9 μM).
Figure 5. Recognition of L43A and WT ubiquitins by the ubiquitin-associated domain (UBA) of the proteasomal shuttle protein ubiquilin-1 (UQ1).

Comparison of spectral perturbations induced by UQ1 UBA binding to (a) L43A ubiquitin and (b) WT ubiquitin. Light-gray bars mark residues that show significant attenuation (disappearance) of the NMR signals in the course of titration with UQ1 UBA. Horizontal bars at the top of the plots indicate elements of ubiquitin's secondary structure. The spectral perturbation profiles indicate that a hydrophobic surface patch including L8, I44, and V70 mediates binding of both WT and L43A ubiquitin with this UBA domain. (c) Map of the UBA-induced spectral perturbations on the surface of ubiquitin. Shown is the structure of WT- ubiquitin/UQ1 UBA complex (2JY6.PDB) facing the hydrophobic patch surface, with ubiquitin residues showing perturbations (CSPs>0.15 ppm and/or signal attenuations) colored red for L43A (left) and WT ubiquitin (right). (d) Overlay of 1H-15N HSQC spectra of L43A (red) and WT (black) at the endpoint of titrations reveals a striking similarity of the UQ1 UBA-bound states of the two ubiquitin variants. Titration curves for L43A (e) and WT ubiquitin (f) plotted as CSPs versus the UBA: ubiquitin molar ratio. Titration data were fit with a 1:1 binding model. The average Kd values (over ∼10 residues) are 28±11 μM (L43A) and 4.2±4.9 μM (WT).
Similar results were obtained from the analyses of L43A binding to the other ubiquitin-receptors (Figure S4, Table S1). Specifically, our NMR data indicate that the L43A mutation did not impair ubiquitin's ability to bind to the UBA2 domain of hHR23A, the UIM domain of Rpn10, or the Pru domain of Rpn13. All these ubiquitin-receptors bind to L43A through similar contacts as to WT ubiquitin and in the same (1:1) stoichiometry. By contrast, previous studies of the core mutations L69S and L67S indicated that this type of destabilizing mutation can abolish binding to the Rpn10 UIM11. Overall, we conclude that the L43A mutation is compatible with ubiquitin recognition by effector proteins. Furthermore, L43A can be efficiently polymerized in vitro (Figure S5) by E2-25K that generates K48-linked chains and by Ubc13/MMS2 that makes K63-linked chains40; 42; 43. Thus, L43A ubiquitin is compatible with both binding to effector domains and enzyme-mediated polymerization. Futhermore, K48-linked chains formed of L43A ubiquitin are recognized and disassembled by both general (Usp2) and K48-linkage selective (OTUB1) DUBs (Figure S6).
Mechanistic Model
Our results demonstrate that L43A ubiquitin is stably folded at physiological temperature, capable of binding to common effector domains, but unable to accumulate in yeast unless attachment to substrates is prevented. These results suggest that the L43A protein in yeast is turned over rapidly when it is capable of covalent attachment. While we have not measured the half life of L43A, nor directly assessed the recycling efficiency of this variant, our results are consistent with a model where the folding stability of ubiquitin is coupled to its recycling efficiency (Figure 6). As previously demonstrated, ubiquitin polymers direct binding of substrates to the proteasome, and an accessible region on the substrate then serves to initiate degradation and is threaded into the internal proteasome cavity where degradation occurs16; 44; 45. We speculate that substrate threading into the proteasome is obstructed when the substrate-conjugated (proximal) ubiquitin is encountered. This obstruction can be dealt with by multiple competing pathways including hydrolysis of the ubiquitin-substrate isopeptide bond by the inherent proteasomal de-ubiquitylase, Rpn1121; 22; or unfolding of the proximal ubiquitin and threading it into the proteasome degradation cavity (lower pathway in Figure 6). Of note, the proteasome is capable of simultaneously threading two polypeptide chains based on its ability to initiate degradation at internal loops46; 47. Based on our results, we propose that the high stability of WT ubiquitin32 results in a strong obstruction to processive substrate degradation by the proteasome. This mechanistic model is consistent with the observations that tightly-folded domains in two substrates have been shown to cause partial proteasome degradation48, and that unfolding is frequently rate-limiting in the degradation of substrates by similar bacterial AAA+ proteases49. In future work, it will be interesting to directly investigate the recycling efficiency and half-life of ubiquitin variants including L43A.
Figure 6. Proposed mechanistic model of ubiquitin recycling by the proteasome.

In this speculative model, the stability of wild-type ubiquitin forms a strong obstruction to processing and degradation, increasing the probability that the Rpn11 subunit will remove ubiquitin before it can be unfolded and translocated into the core particle for degradation.
In our proposed obstruction model the efficiency of ubiquitin recycling is determined by competition between DUB-mediated cleavage of the link between substrate and the proximal ubiquitin and unfolding/degradation of the proximal ubiquitin. In AAA+ bacterial proteases related to the proteasome, unfolding of substrates is frequently rate limiting for degradation, and correlates strongly with the mechanical stability of the substrate (and not global thermodynamic stability)49; 50. Similarly, the degradation propensity of ubiquitin variants engaged with the proteasome may primarily be determined by mechanical stability, which does not necessarily correspond directly with thermodynamic stability. This provides a potential rationale for the greater accumulation of the T7A/V26A and T7A/L50A ubiquitin mutants in yeast compared to L43A despite reduced temperature stability. Previous studies have observed that during the degradation of natively conjugated substrates by proteasomes in vitro essentially all of the ubiquitin molecules avoid degradation and are instead efficiently recycled18; 21. In these studies, de-ubiquitylated substrates are not readily observable, indicating that substrates were committed to degradation before ubiquitin was recycled. Our obstruction model may provide a simple mechanism where ubiquitin recycling follows substrate commitment. Consistent with previous observations21 and our results and model, recent electron-microscopy analyses indicate that the Rpn11 is located near the pore that leads into the proteasome degradation cavity20. Together these findings suggest that de-ubiquitylation is only efficient when the proximal ubiquitin is in a position where it obstructs degradation. This speculative hypothesis is consistent with our results, though future experiments beyond the scope of this work will clearly be required to further address this issue.
Materials and Methods
Growth Rate of Yeast with Ubiquitin Mutants
Ubiquitin mutants were generated in p427GPD and transformed into the Sub328 shutoff strain30 as previously described11. Yeast cells harboring these mutants were grown in synthetic complete (SC) media with G418 and containing 1% raffinose and 1% galactose (RGal) unless otherwise noted. To analyze growth on plates, yeast were pre-grown for 6 hours in SC+G418 media with 2% dextrose (Dex), then serially diluted onto SC+G418 plates with either RGal or Dex and grown for 2 days at 30 °C. To measure growth rates in liquid culture, strains were grown to mid-log phase in SC+G418 RGal media, diluted into SC+G418 Dex media and grown in exponential phase for 16 hours at 30 °C to shutoff and deplete WT ubiquitin. Cells were then diluted to early log phase (OD600=0.1) in SC+G418 Dex media, grown at 30 °C and OD600 monitored over time.
Accumulation of ubiquitin in cells
For the accumulation of un-tagged ubiquitin expressed from p427GPD, cells were grown in SC+G418 with either RGal or Dex for 24 hours at 30 °C. Approximately 50 mL of cells at an OD600 of 0.8 were collected by centrifugation, resuspended in 1 mL of water, transferred to a microfuge tube, pelleted by centrifugation, aspirated and frozen at -80 °C. After thawing, pellets were lysed by bead beating and boiling in SDS. After pelleting insoluble debris, the protein concentration of these whole cell lysates was quantified using the BCA assay (Pierce). 30 μg of total protein was loaded onto an SDS PAGE gel, which was then transferred to a PVDF membrane and probed with α-ubiquitin antibody (Upstate Biotech 07-375). Myc-tagged plasmids for wt, wtDD (G75D/G76D), L43A and L43ADD were transformed into Sub328 and processed as above except that α-Myc antibody (Roche 9E10) was used. For the detection of epitope-tagged ubiquitin mutants, a His6-Myc sequence that is compatible with ubiquitin function in cells33 was cloned upstream of ubiquitin in p427GPD.
Preparation of purified proteins
Ubiquitin with a C-terminal His6 tag was utilized for temperature melts. This protein was expressed from pET21 in BLR(DE3) cells and purified on a Nickel-NTA column and an ion-exchange column. For NMR studies, L43A ubiquitin was expressed as a fusion construct with the chitin-binding domain of intein in E. coli BL21 (DE3) using unlabeled or 15N-labeled ZYP auto-inducing minimal media 51 and purified as described52. Wild-type ubiquitin was prepared as detailed elsewhere42. Final protein samples for NMR studies contained 20mM sodium phosphate (pH 6.8), 0.02% NaN3, and 7% D2O. The ubiquilin-1 UBA domain (UQ1-UBA) construct used in this study was the same as in41, except for a five-residue deletion (Q541-V545) in the flexible N-terminal extension and the F540Y mutation introduced to improve accuracy of concentration measurements by UV-absorbance. The protein was expressed and purified as previously described41. Rpn10 UIM construct was expressed and purified as previously described53. UBA2 domain of hHR23a was expressed and purified as detailed elsewhere40. Rpn13 Pru was expressed and purified as previously described54. The proteins were placed into 20 mM sodium phosphate buffer, pH 6.8. The buffer for hHR23a UBA2 and Rpn13 Pru samples also contained 1 mM TCEP. For the Rpn13 Pru binding studies the buffer also contained 150 mM NaCl, and the pH was 6.5.
Stability to Temperature Denaturation
Temperature stability of purified ubiquitin protein was analyzed using a circular dichroism (CD) readout on folding and a peltier system to control temperature. Purified ubiquitin at 50 μM concentration was prepared in 50 mM potassium phosphate (pH 6.8) and analyzed in a 1 mm path length cuvette. The temperature was increased from 10 °C in 1 °C increments, equilibrated for 1 minute, and the CD signal at 222 nm recorded. The CD readings were plotted versus temperature and fit to a six-parameter model including linear pre- and post-transition baselines as well as the transition temperature as previously described55. The pre- and post-transition baselines were utilized to convert from CD signal to fraction unfolded.
NMR Studies
All NMR experiments were performed using standard or in-house pulse sequences on a Bruker Avance III 600 MHz NMR spectrometer equipped with cryoprobe. The temperature was set to 23°C. All NMR resonance signals were assigned based on 1H-15N HSQC and 15N-edited TOCSY and NOESY. 15N relaxation rates (R1 and R2) and steady-state heteronuclear 15N{1H} NOEs were measured as described56. In-house software was used to analyze 15N relaxation data, and the programs ROTDIF 57 and DYNAMICS 58; 59 were used to characterize the overall and backbone dynamics. Amide chemical shift perturbations (CSPs) were calculated using the following equation: Δδ = [ΔδH2 +(ΔδN/5)2]1/2, where ΔδH and ΔδN are the perturbations for 1H and 15N, respectively. NMR titration experiments were performed by adding increasing amounts of (unlabeled) protein (ligand) to 15N-labeled ubiquitin (L43A or WT), and monitoring binding through changes in 1H-15N NMR spectra recorded at each titration step. Dissociation constants for individual residues in each titration were obtained by fitting titration data to a 1:1 stoichiometry model using program KD_FIT as detailed in43, the reported dissociation constants (Kd) were averaged over a selection of residues, mainly centered around the binding site. To measure residual dipolar couplings (RDCs), the protein was placed in a weakly aligned liquid crystalline medium containing a mixture of PEG/n-hexanol60. The RDCs were quantified as the difference in the apparent 1H-15N couplings observed in the IPAP spectra recorded in the presence and absence of the aligning medium. The analysis of RDCs was performed using computer program ALTENS42; 43.
Supplementary Material
Highlights.
Diverse core mutant effects indicate complex coupling between function and dynamics
L43A mutant only accumulated in yeast if conjugation was prevented
L43A can adopt wild type structure and binds to proteasomal ubiquitin-receptors
We speculate that L43A may be deficient for recycling during substrate degradation
Acknowledgments
We are appreciative to O. Rando, R. Gilmore, E. Baehrecke, A. Ballesteros, and M. Moore for useful comments. The SUB328 yeast strain was kindly provided by D. Finley; and the Rpn13 Pru clone was kindly provided by K. Walters. This work was supported in part by the following grants from the National Institutes of Health: R01-GM083038 to D.N.A.B., and R01-GM065334 to D.F.
Abbreviations
- UB
ubiquitin
- UBD
ubiquitin binding domain
- WT
wild-type
- DUB
deubiquitinase
- CD
circular dichroism
- RDC
residual dipolar coupling
- CSP
chemical shift perturbation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci U S A. 2005;102:6679–85. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mittermaier AK, Kay LE. Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci. 2009;34:601–11. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–5. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 5.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–55. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 6.Bolon DN, Marcus JS, Ross SA, Mayo SL. Prudent modeling of core polar residues in computational protein design. J Mol Biol. 2003;329:611–22. doi: 10.1016/s0022-2836(03)00423-6. [DOI] [PubMed] [Google Scholar]
- 7.Zeldovich KB, Chen P, Shakhnovich EI. Protein stability imposes limits on organism complexity and speed of molecular evolution. Proc Natl Acad Sci U S A. 2007;104:16152–7. doi: 10.1073/pnas.0705366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kacser H, Fell DA. The control of flux: 21 years on. Biochem Soc Trans. 1995;23:341–366. doi: 10.1042/bst0230341. [DOI] [PubMed] [Google Scholar]
- 9.Jiang L, Mishra P, Hietpas RT, Zeldovich KB, Bolon DN. Latent effects of Hsp90 mutants revealed at reduced expression levels. PLoS Genet. 2013;9:e1003600. doi: 10.1371/journal.pgen.1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bershtein S, Mu W, Serohijos AW, Zhou J, Shakhnovich EI. Protein quality control acts on folding intermediates to shape the effects of mutations on organismal fitness. Mol Cell. 2013;49:133–44. doi: 10.1016/j.molcel.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haririnia A, Verma R, Purohit N, Twarog MZ, Deshaies RJ, Bolon D, Fushman D. Mutations in the hydrophobic core of ubiquitin differentially affect its recognition by receptor proteins. J Mol Biol. 2008;375:979–96. doi: 10.1016/j.jmb.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kern D, Eisenmesser EZ, Wolf-Watz M. Enzyme dynamics during catalysis measured by NMR spectroscopy. Methods Enzymol. 2005;394:507–24. doi: 10.1016/S0076-6879(05)94021-4. [DOI] [PubMed] [Google Scholar]
- 13.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature. 2007;450:913–6. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 14.Giver L, Gershenson A, Freskgard PO, Arnold FH. Directed evolution of a thermostable esterase. Proc Natl Acad Sci U S A. 1998;95:12809–13. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloom JD, Arnold FH. In the light of directed evolution: pathways of adaptive protein evolution. Proc Natl Acad Sci U S A. 2009;106:9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 18.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith DM, Benaroudj N, Goldberg A. Proteasomes and their associated ATPases: a destructive combination. J Struct Biol. 2006;156:72–83. doi: 10.1016/j.jsb.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–91. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, 3rd, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–5. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 22.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–7. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 23.Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. Embo J. 2001;20:5187–96. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- 25.Guterman A, Glickman MH. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem. 2004;279:1729–38. doi: 10.1074/jbc.M307050200. [DOI] [PubMed] [Google Scholar]
- 26.Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, Cohen RE. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol. 2006;8:994–1002. doi: 10.1038/ncb1460. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Zhou L, Rouge L, Phillips AH, Lam C, Liu P, Sandoval W, Helgason E, Murray JM, Wertz IE, Corn JE. Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nat Chem Biol. 2013;9:51–8. doi: 10.1038/nchembio.1134. [DOI] [PubMed] [Google Scholar]
- 28.Sloper-Mould KE, Jemc JC, Pickart CM, Hicke L. Distinct functional surface regions on ubiquitin. J Biol Chem. 2001;276:30483–9. doi: 10.1074/jbc.M103248200. [DOI] [PubMed] [Google Scholar]
- 29.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol. 2013;425:1363–77. doi: 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol. 1995;15:1265–73. doi: 10.1128/mcb.15.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C. Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci U S A. 1996;93:861–6. doi: 10.1073/pnas.93.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wintrode PL, Makhatadze GI, Privalov PL. Thermodynamics of ubiquitin unfolding. Proteins. 1994;18:246–53. doi: 10.1002/prot.340180305. [DOI] [PubMed] [Google Scholar]
- 33.Spence J, Gali RR, Dittmar G, Sherman F, Karin M, Finley D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell. 2000;102:67–76. doi: 10.1016/s0092-8674(00)00011-8. [DOI] [PubMed] [Google Scholar]
- 34.Tolman JR. Dipolar couplings as a probe of molecular dynamics and structure in solution. Curr Opin Struct Biol. 2001;11:532–9. doi: 10.1016/s0959-440x(00)00245-1. [DOI] [PubMed] [Google Scholar]
- 35.Pancoska P, Bitto E, Janota V, Urbanova M, Gupta VP, Keiderling TA. Comparison of and limits of accuracy for statistical analyses of vibrational and electronic circular dichroism spectra in terms of correlations to and predictions of protein secondary structure. Protein Sci. 1995;4:1384–401. doi: 10.1002/pro.5560040713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 2008;8:25. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J. 2006;399:361–72. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–21. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 39.Fushman D, Wilkinson KD. Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep. 2011;3:26. doi: 10.3410/B3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varadan R, Assfalg M, Raasi S, Pickart C, Fushman D. Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol Cell. 2005;18:687–98. doi: 10.1016/j.molcel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 41.Zhang D, Raasi S, Fushman D. Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J Mol Biol. 2008;377:162–80. doi: 10.1016/j.jmb.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varadan R, Walker O, Pickart C, Fushman D. Structural properties of polyubiquitin chains in solution. J Mol Biol. 2002;324:637–47. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 43.Fushman D, Varadan R, Assfalg M, Walker O. Determining domain orientation in macromolecules by using spin-relaxation and residual dipolar coupling measurements. Progress in NMR Spectroscopy. 2004;44:189–214. [Google Scholar]
- 44.Prakash S, Inobe T, Hatch AJ, Matouschek A. Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol. 2009;5:29–36. doi: 10.1038/nchembio.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 46.Lee C, Prakash S, Matouschek A. Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J Biol Chem. 2002;277:34760–5. doi: 10.1074/jbc.M204750200. [DOI] [PubMed] [Google Scholar]
- 47.Piwko W, Jentsch S. Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat Struct Mol Biol. 2006;13:691–7. doi: 10.1038/nsmb1122. [DOI] [PubMed] [Google Scholar]
- 48.Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–53. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- 49.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–20. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 50.Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci U S A. 2005;102:1390–5. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 52.Castaneda CA, Liu J, Kashyap TR, Singh RK, Fushman D, Cropp TA. Controlled enzymatic synthesis of natural-linkage, defined-length polyubiquitin chains using lysines with removable protecting groups. Chem Commun (Camb) 2011;47:2026–8. doi: 10.1039/c0cc04868b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia Q, Guo Y, Zhang Z, Li D, Xuan Z, Li Z, Dai F, Li Y, Cheng D, Li R, Cheng T, Jiang T, Becquet C, Xu X, Liu C, Zha X, Fan W, Lin Y, Shen Y, Jiang L, Jensen J, Hellmann I, Tang S, Zhao P, Xu H, Yu C, Zhang G, Li J, Cao J, Liu S, He N, Zhou Y, Liu H, Zhao J, Ye C, Du Z, Pan G, Zhao A, Shao H, Zeng W, Wu P, Li C, Pan M, Li J, Yin X, Li D, Wang J, Zheng H, Wang W, Zhang X, Li S, Yang H, Lu C, Nielsen R, Zhou Z, Wang J, Xiang Z, Wang J. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx) Science. 2009;326:433–6. doi: 10.1126/science.1176620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, Groll M. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature. 2008;453:548–52. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Minor DL, Jr, Kim PS. Measurement of the beta-sheet-forming propensities of amino acids. Nature. 1994;367:660–3. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 56.Hall JB, Fushman D. Characterization of the overall and local dynamics of a protein with intermediate rotational anisotropy: Differentiating between conformational exchange and anisotropic diffusion in the B3 domain of protein G. J Biomol NMR. 2003;27:261–75. doi: 10.1023/a:1025467918856. [DOI] [PubMed] [Google Scholar]
- 57.Walker O, Varadan R, Fushman D. Efficient and accurate determination of the overall rotational diffusion tensor of a molecule from (15)N relaxation data using computer program ROTDIF. J Magn Reson. 2004;168:336–45. doi: 10.1016/j.jmr.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 58.Hall JB, Fushman D. Variability of the 15N chemical shielding tensors in the B3 domain of protein G from 15N relaxation measurements at several fields. Implications for backbone order parameters. J Am Chem Soc. 2006;128:7855–70. doi: 10.1021/ja060406x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fushman D, Cahill S, Cowburn D. The main-chain dynamics of the dynamin pleckstrin homology (PH) domain in solution: analysis of 15N relaxation with monomer/dimer equilibration. J Mol Biol. 1997;266:173–94. doi: 10.1006/jmbi.1996.0771. [DOI] [PubMed] [Google Scholar]
- 60.Ruckert M, Otting G. Alignment of Biological Macromolecules in Novel Nonionic Liquid Crystalline Media for NMR Experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 61.Clore GM, Garrett DS. R-factor, Free R, and Complete Cross-Validation for Dipolar Coupling Refinement of NMR Structures. J Am Chem Soc. 1999;121:9008–9012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.