

Abstract
Oxidation of phenols by heterodinuclear CuIII(μ-O)2NiIII complexes containing nucleophilic oxo groups occurs by both proton coupled electron transfer (PCET) and hydrogen atom transfer (HAT) mechanisms; the exact mechanism depends on the nature of the phenol as well as the substitution pattern of the ligand bound to Cu.
The conversion of phenols to phenoxyl radicals plays a vital role in a number of biological systems. Photosystem II is the most prominent example,1-4 but evidence exists that similar processes are involved in the function of several other biochemical systems.5 The oxidative chemistry of phenols has also other potential biological application, notably their antioxidant properties.6-8 Additionally, oxidative dehydrodimerization of phenols is an important class of reactions, being involved in the first stages of natural processes such as lignin formation.9-10 Last but not least, phenol oxidation has important synthetic applications.11 In many cases,1-5 the phenoxyl radicals in biology are derived via oxidation of an active site tyrosine residue by a transition metal-oxo species. Thus, uncovering the mechanisms of metal-oxo mediated phenol oxidation is of interest from both fundamental viewpoints and the great relevance of these reactions to numerous natural and synthetic processes.
The formation of a phenoxyl radical from a neutral phenol can occur via direct hydrogen atom transfer (HAT) or via a proton-coupled electron transfer (PCET) (Scheme 1) process. For the HAT and PCET mechanisms, homolytic O-H bond cleavage constitutes the rate-determining step of the reaction. In the HAT mechanism, the proton and electron of the H· radical both come from the same orbital. Conversely, proton and electron transfers are both rate determining for the PCET process, but occur from different orbitals in a concerted mechanism.12 Alternatively the proton and electron transfers can be uncoupled (PT-ET) with either proton transfer (PT) or the electron transfer (ET) being the rate determining step (Scheme 1). A HAT mechanism has been established for the phenol oxidation mediated by the terminal MnV-oxo (TBP8Cz)MnVO (1) (TBP8Cz = octakis(para-tert-butylphenyl)corrolazinato3-)13 and CrIII-superoxo, [CrIII(TMC)(O2)(Cl)]+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) complexes.14 In contrast, the reactions of phenols with two distinct homodinuclear dicopper-dioxygen complexes having bis (μ-oxo)dicopper(III) and (μ-η2:η2-peroxo)dicopper(II) cores exhibit a PCET mechanism.15 Metal mediated oxidation of phenols by a PT-ET mechanism is unknown in the literature, although this mechanism has been previously invoked for the oxidation of phenols by organic radicals.16
Scheme 1.

Possible reaction pathways for the oxidation of phenols.
Very recently we reported the isolation and spectroscopic characterization of a novel mixed-metal NiIII-CuIII bis (μ-oxo) complex (Scheme 2), [(MeAN)CuIII(μ-O)2NiIIIL]+, (1, MeAN = N,N,N′,N′,N′–pentamethyl–dipropylenetriamine and L = [HC(CMeNC6H3(iPr)2)2]). Unlike typical homodinuclear bis(μ-oxo) complexes, this species contains nucleophilic oxo groups that can carry out deformylation of aldehydes.17 We initiated the present study to investigate whether the PCET mechanism established for phenol oxidation by homodinuclear Cu2O2 cores having electrophilic oxo groups also persists for the Cu(μ-O)2Ni core of 1, which has nucleophilic oxo groups. We report a systematic study of the oxidation of neutral phenols by 1 and provide deeper insights in the O-H bond activation mechanism towards substituted phenols by applying the Brønsted/Tafel analogy published by Ram and Hupp18 for electron transfer processes. A similar strategy has been previously utilized for studying the mechanism of electro-catalytic oxidation of guanidine19 and phenol oxidation by dicopper-dioxygen complexes.15
Scheme 2.

[(RAN)CuIII(μ-O)NiIII] (R = Me for 1 and R = H for 2) Complexes used in this study for the oxidation of phenols.
The Cu(μ-O)2Ni complex 1 was generated by the reaction of equimolar amounts of [(MeAN)CuI]BF4 and [L(Ni)O2]20 complexes at -90 °C in CH2Cl2 as previously reported.17 Treatment of a preformed solution of 1 with excess 2,4-di-tert-butylphenol led to pseudo-first order decay of the characteristic absorption feature of 1 at 895 nm (Figure 1A). The rate constant increases proportionally with the substrate concentration (Figure 1B), affording a second-order rate constant, k2, of 2.55 M−1s−1 at -90 °C. These kinetic behaviors indicate that the reaction between 2,4-di-tert-butylphenol and 1 is a simple bimolecular process. Analysis of the reaction mixture shows the formation of the C-C coupling dimer 2,2′,4,4′-tetra-tert-butyl-6,6′-biphenol in 45% yield based on 2,4-di-tert-butylphenol. Thus 1 acts as a two-electron oxidant in its reaction with 2,4-di-tert-butylphenol to produce two equivalents of the corresponding phenoxyl radical at -90 °C that spontaneously dimerizes to give the C-C coupling product (Scheme 2).21 In support of this mechanism, the X-band EPR spectrum of a reaction mixture of 1 and 2,4,6-tri-tert-butylphenol shows a characteristic S=1/2 signal for a phenoxyl radical. Spin quantification studies demonstrate the formation of the radical in near quantitative yield based on 2,4,6-tri-tert-butylphenol. The second-order rate constants (k2) for the oxidation of several substituted phenols by 1 (Figure S1) were determined in a similar fashion at temperatures ranging from -50 to -90 °C, and are listed in Table 1 together with the ArOH/ArOH+ potentials (Eox) of the phenols and the yields of the oxidation products.
Figure 1.

A) Changes in the absorption spectra associated with the reaction of 1 (0.2 mM in CH2Cl2) with 2,4-di-tert-butylphenol (100 equiv) at -90 °C. The inset shows the time trace of the decay of the 895 nm band upon addition of 2,4-di-tert-butylphenol, which can be fitted to a pseudo first-order kinetic model to obtain a first-order rate constant (kobs). Figures 1B and 1C depict the linear dependence of kobs on the concentrations of 2,4-di-tert-butylphenol and the mono-deuterated analogue for the reactions with 1 and 2, respectively.
Table 1.
Oxidation potential (Eox) of ArOH and the second order rate constants (k2) for the oxidation of ArOH by 1 and 2.
| Complex 1 | Eox / V | k2 / M-1s-1 | T / K | (RT/F) lnk2 |
|---|---|---|---|---|
| 2,4-DTBP | 1.46 | 2.55 | 183.16 | 0.015 |
| 2,6-DTBP | 1.62 | 0.03 | 223.16 | -0.067 |
| 2,4,6-TTBP | 1.58 | 0.15 | 223.16 | -0.037 |
| 4-phenylphenol | 1.52 | 4.23 | 183.16 | 0.023 |
| 4-phenoxylphenol | 1.49 | 10.92 | 183.16 | 0.038 |
|
| ||||
| Complex 2 | Eox / V | k2 / M-1s-1 | T / K | (RT/F) lnk2 |
|
| ||||
| 2,4-DTBP | 1.46 | 24.08 | 183.16 | 0.050 |
| 2,6-DTBP | 1.62 | 0.05 | 193.16 | -0.050 |
| 2,4,6-TTBP | 1.58 | 0.18 | 213.16 | -0.031 |
| 4-Br-2,6-DTBP | 1.64 | 0.02 | 183.16 | -0.062 |
| 4-phenylphenol | 1.52 | 4.48 | 183.16 | 0.023 |
| 4-phenoxylphenol | 1.49 | 3.24 | 183.16 | 0.019 |
DTBP = di-tert-butylphenol, TTBP = tri-tert-butylphenol, Eox / V vs SCE values are taken from ref 15 except for 4-Br-2,6-DTBP, which has been determined by cyclic voltammetry (see Electronic Supplementary Information for details). Reaction of complex 1 with 4-Br-2,6-DTBP is very slow, and hence, no kinetic measurements could be performed.
The k2 values (Table 1) obtained for the reaction of 1 with different 4-substituted phenols (ArOH) were found to be dependent on the Eox values of the phenols; in general, k2 increased with decreasing Eox for all investigated phenols with the exception of 2,4-di-tert-butyl phenol. Plots of (RT/F)lnk2 versus Eox afford a good linear correlation with a slope of -0.81±0.05 (Figure 2A); the corresponding value for 2,4-di-tert-butyl phenol is again significantly below the trend line, which may be suggestive of a change in mechanism for this substrate. If electron transfer from phenols to 1 is rate-determining, and is followed by fast proton transfer, then according to Ram and Hupp18 the slope of the (RT/F)lnk2 versus Eox plot should be 0.5, as one would expect from Marcus theory22 for a pure electron transfer reaction. On the other hand, if proton transfer is rate determining and the electron transfer is in equilibrium then the slope should be -1.0. If the rates of electron transfer and proton transfer are comparable and thereby coupled to each other (PCET mechanism), a value between -0.5 and -1.0 would be obtained.15,18,19 In contrast, the k2 values for a HAT mechanism are expected to be constant irrespective of the Eox values, as has been reported previously for the HAT reactions with N,N-dimethylanilines23 and for the oxidation of phenols by cumylperoxyl radical.15 The slope of -0.81 in Figure 2A therefore establishes a PCET mechanism for the oxidation of all investigated phenols by 1 except 2,4-di-tert-butylphenol, for which a different mechanism may be involved.
Figure 2.
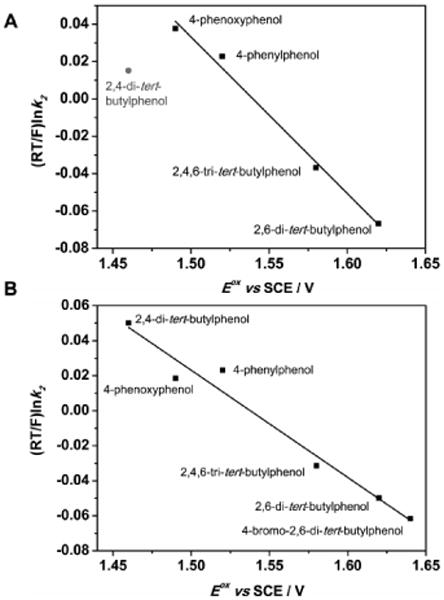
Plots of (RT/F)ln(k2) against the oxidation potential (Eox) of ArOH for the reactions of ArOH with 1 (Figure 2A) and 2 (Figure 2B) in CH2Cl2.
In order to obtain additional mechanistic insights, deuterium kinetic isotope effects (KIE) on the second-order rate constant k2(O-H)/k2(O-D) were measured for 4-phenoxyphenol and 2,4-di-tert-butylphenol. A deuterium KIE of 1.66 (Figure S4) at -90 °C was determined for 4-phenoxyphenol, indicating that proton transfer is involved in the rate determining step. This value is smaller than the KIE values obtained for most metal-oxo mediated HAT reactions (3 – 25). 13,14,24 It is nearly the same as the KIE reported for the PCET reaction of guanidine with Ru(bpy)33+,19 as well as those of 4-substituted phenols and dicopper-dioxygen complexes.15 Interestingly, 2,4-di-tert-butylphenol exhibited a significantly larger KIE of 4.72 (Figure 1B), which is in the typical range for a HAT process. The KIE values determined for the oxidations of 4-phenoxyphenol and 2,4-di-tert-butylphenol by 1 may, therefore, point to the involvement of a PCET mechanism for 4-phenoxyphenol and a HAT mechanism for 2,4-di-tert-butylphenol, which would be also consistent with the (RT/F)lnk2 versus Eox plot shown in Figure 2A.
Karlin et al.25 have previously reported that the electronic structure of the dicopper-dioxygen cores supported by the RAN (MeAN for R = CH3; and AN for R = H) ligands is strongly dependent on the substitution pattern of the ligand. While the (AN)CuI complex (AN = 3,3′-iminobis-(N,N-dimethylpropylamine)) forms a CuIII2(μ-O)2 core in the presence of dioxygen, the corresponding (MeAN)CuI complex affords a CuII2(μ-peroxo) structure. In an effort to understand the role of the ligand structure of the copper precursor on the structure and reactivity of heterodinuclear bis-μ-oxo intermediates, we treated (AN)CuI with the [L(Ni)O2] complex at -90 °C in CH2Cl2 to generate 2 in near-quantitative yields (from EPR quantification studies). Complex 2 possesses a half-life of 9000 seconds at -50 °C, an order of magnitude higher than 1 (t1/2 = 900 s at -50 °C).17 However, the accumulated spectroscopic (absorption, X-ray absorption near-edge, resonance Raman, and electron paramagnetic resonance) properties (Figures S5-S6) of 2 are almost indistinguishable from those of 1, indicating that 2 also possesses a heterodinuclear CuIII(μ-O)2NiIII core similar to 1 (Scheme 2). Thus, unlike homodinuclear dicopper-dioxygen chemistry,25 where MeAN stabilized a μ-peroxo core, and AN stabilized a bis (μ-oxo) core, both of the MeAN and AN ligands lead to formation of a heterodinuclear Cu-Ni bis (μ-oxo) complex.
The reactivity of 2 was also investigated with various 4-substituted phenols and the second-order rate constants (Table 1, Figure S7) were compared with those obtained for 1. A plot of (RT/F)ln k2 versus Eox for the reaction of 2 affords a good linear correlation with a slope of -0.61±0.03 (Figure 2B), which supports a PCET mechanism for the oxidation of phenols. In particular, reaction of 2,4-di-tert-butylphenol with 2 follows the linear correlation, in contrast to 1, where it appeared outside the linear trend. The KIEs for the second-order rate constant k2(O-H)/k2(O-D) was also determined, with values of 1.71 (Figure 1C) and 1.77 (Figure S8) for the reactions of 2 with 2,4-di-tert-butylphenol and 4-phenoxyphenol, respectively. These KIEs are similar to the KIE value of 1.66 obtained for the reaction with 1 with 4-phenoxyphenol, but significantly lower than the value of 4.72 obtained for the corresponding reaction of 1 with 2,4-di-tert-butylphenol.
In summary, the oxidation of phenols mediated by the heterodinuclear CuIII(μ-O)2NiIII complex 2 is shown to proceed via a proton coupled electron transfer mechanism based on the rate dependence of the reaction on the one-electron oxidation potentials of the phenol substrates, as well as deuterium kinetic isotope effects of magnitude less than 2. Thus, while the oxygen atoms of the CuIII(μ-O)2NiIII core in 2 are nucleophilic, they prefer to oxidize phenol by a concerted PCET mechanism similar to what has been observed before for the corresponding CuIII(μ-O)2CuIII species involving electrophilic oxygen atoms. In contrast, for complex 1, which differs from 2 with respect to the substitution pattern of the ligand attached to the Cu center, but possesses identical spectroscopic properties, both HAT and PCET mechanisms may be feasible for the oxidation of phenols. Specifically, the oxidation of 2,4-di-tert-butylphenol proceeds by a HAT mechanism, while oxidations of 2,6 di-tert-butylphenol, 2,4,6-tri-tert-butylphenol, 4-phenylphenol and 4-phenoxyphenol proceed by a PCET mechanism. The different mechanisms observed for the oxidation of 2,4-di-tert-butylphenol by 1 and 2, therefore, highlights the importance of subtle electronic changes in modulating the reactivity of biologically relevant metaldioxygen intermediates.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work from the Cluster of Excellence “Unifying Concepts in Catalysis” (EXC 314/1), Berlin. XAS data were obtained on beamline X3B of the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY, USA), which is operated by the Case Western Reserve University Center for Synchrotron Biosciences, supported by NIH Grant P30–EB–009998. NSLS is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE–AC02–98CH10886.We also thank Prof. Dr. Peter Hildebrandt and Dr. Uwe Kuhlmann for the resonance Raman measurement of 2 and Prof. Dr. Matthias Driess and Dr. Shenglai Yao for the supply of the nickel superoxide precursor.
Footnotes
Electronic Supplementary Information (ESI) available: [Synthesis and characterization of 2; additional kinetic data]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Stubbe J, van der Donk WA. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 2.Tommos C, Babcock GT. Biochim Biophys Acta. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 3.Renger G. Biochim Biophys Acta. 2004;1655:195–204. doi: 10.1016/j.bbabio.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Meyer TJ, Huynh MHV, Thorp HH. Angew Chem Int Ed. 2007;46:5284–5304. doi: 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]
- 5.Miller AF. Acc Chem Res. 2008;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 6.Williams LL, Webster RD. J Am Chem Soc. 2004;126:12441–12450. doi: 10.1021/ja046648j. [DOI] [PubMed] [Google Scholar]
- 7.Cotelle N, Hapiot P, Pinson J, Rolando C, Vézin H. J Phys Chem B. 2005;109:23720–23729. doi: 10.1021/jp0550661. [DOI] [PubMed] [Google Scholar]
- 8.Webster RD. Acc Chem Res. 2007;40:251–257. doi: 10.1021/ar068182a. [DOI] [PubMed] [Google Scholar]
- 9.Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph Sa, Christensen JH, Boerjan W. Phytochem Rev. 2004;3:29–60. [Google Scholar]
- 10.Vanholme R, Morreel K, Ralph J, Boerjan W. Curr Opin Plant Biol. 2008;11:278–285. doi: 10.1016/j.pbi.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Morrow GW. Anodic oxidation of oxygen-containing compounds. In: Lund H, Hammerich O, editors. Organic Electrochemistry. 4th. Marcel Dekker; New York: 2001. pp. 589–620. [Google Scholar]
- 12.Huynh MHV, Meyer TJ. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lansky DE, Goldberg DP. Inorg Chem. 2006;45:5119–5125. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]
- 14.Cho J, Woo J, Eun Han J, Kubo M, Ogura T, Nam W. Chem Sci. 2011;2:2057–2062. [Google Scholar]
- 15.Osako T, Ohkubo K, Taki M, Tachi Y, Fukuzumi S, Itoh S. J Am Chem Soc. 2003;125:11027–11033. doi: 10.1021/ja029380+. [DOI] [PubMed] [Google Scholar]
- 16.(a) Litwinienko G, Ingold KU. J Org Chem. 2003;68:3433–3438. doi: 10.1021/jo026917t. [DOI] [PubMed] [Google Scholar]; (b) Litwinienko G, Ingold KU. J Org Chem. 2004;69:5888–5896. doi: 10.1021/jo049254j. [DOI] [PubMed] [Google Scholar]; (c) Foti MC, Daquino C, Geraci C. J Org Chem. 2004;69:2309–2314. doi: 10.1021/jo035758q. [DOI] [PubMed] [Google Scholar]
- 17.Kundu S, Pfaff FF, Miceli E, Zaharieva I, Herwig C, Yao S, Farquhar ER, Kuhlmann U, Bill E, Hildebrandt P, Dau H, Driess M, Limberg C, Ray K. Angew Chem Int Ed. 2013;52:5622–5626. doi: 10.1002/anie.201300861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ram MS, Hupp JT. J Phys Chem. 1990;94:2378–2380. [Google Scholar]
- 19.Weatherly SC, Yang IV, Thorp HH. J Am Chem Soc. 2001;123:1236–1237. doi: 10.1021/ja003788u. [DOI] [PubMed] [Google Scholar]
- 20.Yao S, Bill E, Milsmann C, Wieghardt K, Driess M. Angew Chem Int Ed. 2008;47:7110–7114. doi: 10.1002/anie.200802234. [DOI] [PubMed] [Google Scholar]
- 21.The formation of the bis(hydroxo)CuIINiII product in scheme 2 is suggested on the basis of the analysis of the reaction mixture by ESI-MS (Figure S2) and EPR (Figure S3) spectroscopic methods. We emphasize, however, that the formation of other metal-containing products in addition to the bis(hydroxo) species cannot be excluded at this point.
- 22.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 23.Fukuzumi S, Shimoosako K, Suenobu T, Watanabe Y. J Am Chem Soc. 2003;125:9074–9082. doi: 10.1021/ja035156o. [DOI] [PubMed] [Google Scholar]
- 24.(a) Que L., Jr Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]; (b) Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (c) Hohenberger J, Ray K, Meyer K. Nat Commun. 2012;3:720–733. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]
- 25.Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbühler AD, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2002;124:4170–4171. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.