

Abstract
Methamphetamine abuse escalates, but no approved therapeutics are available to treat addicted individuals. Methamphetamine increases extracellular dopamine in reward-relevant pathways by interacting at vesicular monoamine transporter-2 (VMAT2) to inhibit dopamine uptake and promote dopamine release from synaptic vesicles, increasing cytosolic dopamine available for reverse transport by the dopamine transporter (DAT). VMAT2 is the target of our iterative drug discovery efforts to identify pharmacotherapeutics for methamphetamine addiction. Lobeline, the major alkaloid in Lobelia inflata, potently inhibited VMAT2, methamphetamine-evoked striatal dopamine release, and methamphetamine self-administration in rats but exhibited high affinity for nicotinic acetylcholine receptors (nAChRs). Defunctionalized, unsaturated lobeline analog, meso-transdiene (MTD), exhibited lobeline-like in vitro pharmacology, lacked nAChR affinity, but exhibited high affinity for DAT, suggesting potential abuse liability. The 2,4-dicholorophenyl MTD analog, UKMH-106, exhibited selectivity for VMAT2 over DAT, inhibited methamphetamine-evoked dopamine release, but required a difficult synthetic approach. Lobelane, a saturated, defunctionalized lobeline analog, inhibited the neurochemical and behavioral effects of methamphetamine; tolerance developed to the lobelane-induced decrease in methamphetamine self-administration. Improved drug-likeness was afforded by the incorporation of a chiral N-1,2-dihydroxypropyl moiety into lobelane to afford GZ-793A, which inhibited the neurochemical and behavioral effects of methamphetamine, without tolerance. From a series of 2,5-disubstituted pyrrolidine analogs, AV-2-192 emerged as a lead, exhibiting high affinity for VMAT2 and inhibiting methamphetamine-evoked dopamine release. Current results support the hypothesis that potent, selective VMAT2 inhibitors provide the requisite preclinical behavioral profile for evaluation as pharmacotherapeutics for methamphetamine abuse and emphasize selectivity for VMAT2 relative to DAT as a criterion for reducing abuse liability of the therapeutic.
1. METHAMPHETAMINE ADDICTION
Psychostimulant abuse is an escalating problem, with 100,000 new methamphetamine (METH) users in the United States each year (Drug and Alcohol Services Information System (DASIS, 2008)). Methamphetamine use poses significant health risks, including long-term neuronal damage and concomitant deleterious effects on cognitive processes, such as memory and attention (Nordahl, Salo, & Leamon, 2003). The problem is complicated by the fact that treatment centers lack an effective means to combat its abuse (DASIS, 2008). Despite the serious consequences of METH use, there are currently no approved therapeutics available for those individuals suffering from METH addiction. Increasing emphasis has been placed on identifying the underlying mechanisms of METH action and relevant pharmacological targets for the development of novel therapeutic agents to treat METH addiction.
2. METHAMPHETAMINE: MECHANISM OF ACTION
Methamphetamine (Fig. 2.1), a powerful central nervous system (CNS) stimulant, exerts its pharmacological and behavioral effects through alterations in the brain dopaminergic reward circuitry, which is generally accepted as responsible for the rewarding effects of drugs of abuse (Di Chiara et al., 2004; Koob, 1992; Wise & Bozarth, 1987; Wise & Hoffman, 1992). Methamphetamine self-administration and conditioned place preference (CPP) in rodents are gold-standard assays used to demonstrate the reinforcing and rewarding effects of this drug (Hart, Ward, Haney, Foltin, & Fischman, 2001; Xu, Mo, Yung, Yang, & Leung, 2008; Yokel & Pickens, 1973). Amphetamines (including METH) enter dopaminergic pre-synaptic terminals by acting as substrates for the plasmalemma dopamine transporter (DAT) and by diffusion through the plasmalemma (Fig. 2.2; Johnson, Eshleman, Meyers, Neve, & Janowsky, 1998; Sulzer et al., 1995). Once inside the presynaptic terminal, amphetamines elicit the release of vesicular dopamine (DA) stores into the cytosol through an interaction with reserpine sites on the vesicular monoamine transporter-2 (VMAT2) protein (Ary & Komiskey, 1980; Liang & Rutledge, 1982; Peter, Jimenez, Liu, Kim, & Edwards, 1994; Philippu & Beyer, 1973; Pifl, Drobny, Reither, Hornykiewicz, & Singer, 1995) and via disruption of the vesicular proton gradient as a consequence of its weak basicity and high lipophilicity (Barlow & Johnson, 1989). Amphetamines promote DA release from synaptic vesicles into the cytosol of the dopaminergic presynaptic terminal, redistributing DA stores and increasing cytosolic DA concentrations (Pifl et al., 1995; Sulzer et al., 1995), and inhibit DA uptake from the cytosol by VMAT2 (Brown, Hanson, & Fleckenstein, 2000, 2001; Fleckenstein, Volz, Riddle, Gibb, & Hanson, 2007). As amphetamines also inhibit the activity of the mitochondrial enzyme monoamine oxidase (MAO), the elevated concentrations of cytosolic DA are not subjected to metabolism (Mantle, Tipton, & Garrett, 1976). With increased cytosolic DA concentrations, DA is available for release into the synaptic cleft via reversal of DAT (Ary & Komiskey, 1980; Fischer & Cho, 1979; Liang & Rutledge, 1982; Sulzer et al., 1995). Enhanced DA release and increased stimulation of post-synaptic DA receptors that follows lead to the rewarding effects and high degree of abuse liability associated with these psychostimulant drugs (Carr & White, 1983; Hiroi & White, 1991; Hoebel et al., 1983; Lyness, Friedle, & Moore, 1979; Wise & Bozarth, 1987). Furthermore, the demonstration that heterologous VMAT2 knockout mice exhibit reduced amphetamine conditioned reward, enhanced amphetamine locomotion, and enhanced sensitivity to amphetamine also indicates that VMAT2 plays a critical role in mediating the behavioral effects of this drug of abuse (Takahashi et al., 1997; Wang et al., 1997). Although the effects of METH on VMAT2 are not the only mechanism responsible for its behavioral effects, we have considered VMAT2 as a key pharmacological target for developing pharmacotherapies to treat psychostimulant abuse (Dwoskin & Crooks, 2002), because this protein is an essential cellular component contributing to the increased extracellular DA concentrations produced by the actions of METH.
Figure 2.1. Chemical structures.

Tetrabenazine (TBZ) is a benzoquinolizine compound that reversibly inhibits VMAT2 function. Lobeline is a lipophilic, nonpyridino alkaloid present in Lobelia inflata. Lobelane is a defunctionalized, saturated meso-analog of lobe-line. MTD is a lobelane analog bearing unsaturated phenethyl side chain linkers. GZ-793A is a lobelane analog bearing a dihydroxypropyl moiety on the central nitrogen atom and methoxy substituents in the para-position on the phenyl rings. AV-1-229 bears a high degree of structural similarity to lobelane, with substitution of a pyrrolidine ring for the piperidine ring. UKCP-110, also known as AV-1-228, is the demethylated nor-analog of AV-1-229.
Figure 2.2.

Model of the DA presynaptic terminal in the presence of methamphetamine (left) and in the presence of lobeline analog plus methamphetamine.
Furthermore, studies evaluating VMAT2 expression levels and function in both rodents and humans, together with results from imaging studies, led to the conclusion that VMAT2 protein levels and function are not changed by acute or chronic psychostimulant administration (Eiden & Weihe, 2011; Kilbourn et al., 2010). However, other investigators provide evidence that VMAT2 function and expression are altered by psychostimulant drugs of abuse (Riddle, Fleckenstein, & Hanson, 2005). Methamphetamine administration to rats reduced VMAT2 expression and clearance rates of DA in purified striatal vesicle preparations (Brown et al., 2000; Hogan, Staal, & Sonsalla, 2000), while cocaine increased Vmax of DA uptake in these preparations (Brown et al., 2001). Corroborating evidence revealed that administration of monoamine releasers, such as amphetamine or METH, resulted in a redistribution of vesicular pools in the presynaptic terminal, leading to a reduction in VMAT2 available to clear DA from the cytosol (Fleckenstein et al., 2007). Taken together, these findings implicate VMAT2 as a dynamic presynaptic target for psychostimulant drugs of abuse, specifically amphetamines, and that VMAT2 may be a viable target for the discovery of pharmacotherapeutics to treat psychostimulant abuse.
3. IDENTIFICATION AND CLASSIFICATION OF VESICULAR MONOAMINE TRANSPORTERS
The discovery of secretory vesicles (Hillarp, 1958) led to the subsequent discovery of the vesicular monoamine transporter (VMAT) and the characterization of the structure and function of this protein. Early research revealed several distinguishing features of this class of transporters, including a preference to transport monoamines (e.g., serotonin, DA, and norepinephrine), the dependence of substrate transport on an electrochemical gradient across the vesicular lipid bilayer membrane, and the inhibition of substrate transport by both reserpine and tetrabenazine (TBZ) (Fig. 2.1; Henry, Gasnier, Roisin, Isamber, & Scherman, 1987; Johnson, 1988; Kanner & Schuldiner, 1987; Njus, Kelley, & Harnadek, 1986). The molecular structure of VMAT was first elucidated using Chinese hamster ovary (CHO) cells via expression cloning from rat pheochromocytoma PC12 cells (Liu, Peter, et al., 1992). Successful expression of the transporter in CHO cells provided protection against the neurotoxic effects of 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of N-methyl-1,2,3,6-tetrahydropyridine (MPTP), demonstrating that intracellular sequestration of MPP+ via VMAT was responsible for the resistance to toxicity observed in PC12 cells (Liu, Peter, et al., 1992; Liu, Roghani, & Edwards, 1992). Gene transfer followed by plasmid rescue identified a cDNA clone for the VMAT isoform, designated VMAT1, which was predicted to be a 70-kDa protein having 12 transmembrane-spanning domains, internal carboxy and amino termini, and a large hydrophilic loop between transmembrane regions 1 and 2 (Liu, Peter, et al., 1992; Liu, Roghani, et al., 1992). Overall, the molecular structure for VMAT1 expressed in CHO cells was determined to be remarkably similar to the plasmalemma neurotransmitter transporters, yet its amino acid sequence showed homology to a multitude of bacterial resistance transporters (e.g., transporters encoded by tetracycline and methylenomycin resistance genes). An alternate isoform of VMAT was discovered contemporaneously by employing an expression cloning strategy using a mammalian cell line (CV-1), such that the expression of the protein by CV-1 cells resulted in transport of serotonin, DA, and norepinephrine (Erickson, Eiden, & Hoffman, 1992). The predicted molecular structure of this transporter, designated VMAT2, also suggested 12 transmembrane domains and internal carboxy and amino termini.
While having similar molecular structures, the development of polyclonal antibodies specific for each isoform revealed important differences between VMAT1 and VMAT2. Immunohistochemistry studies showed that VMAT1 is located primarily within the lipid bilayer membrane of large dense-core vesicles within chromaffin granule cells in the medulla of the adrenal, whereas scant expression of VMAT1 was found in the peripheral sympathetic neurons and blood platelets. In contrast, VMAT2 had a more ubiquitous expression pattern. VMAT2 is expressed widely by monoaminergic neurons of the CNS, histaminergic mast cells, peripheral sympathetic neurons, and β-cells of the pancreas (Erickson, Schafer, Bonner, Eiden, & Weihe, 1996; Peter et al., 1995; Weihe, Schafer, Erickson, & Eiden, 1995; Wimalasena, 2010). Isolation of VMAT1 to the peripheral nervous system and a likely minimal role in processes underlying psychostimulant abuse reduce the importance of this protein as a target for the discovery of pharmacotherapeutics for psychostimulant abuse.
4. VMAT2 FUNCTION
VMAT2 is critical as a functional component of monoaminergic neurotransmission. Within presynaptic terminals of monoaminergic neurons, VMAT2 translocates monoamines from the cytosol across the vesicle membrane into the vesicle lumen following neurotransmitter biosynthesis and/or clearance from the extracellular space. Intravesicular sequestration of monoamines by VMAT2 occurs despite the high concentrations (~0.5 M) of monoamines within the vesicle. Transport is driven by the transmembrane proton gradient, generated by a vesicular H+-ATPase, which shuttles protons into the vesicle by active transport. With respect to VMAT2, initial binding and movement of a hydrogen atom to the luminal face of VMAT2 induces a conformational change in the cytosolic face of the protein, exposing a high-affinity substrate binding site to which monoamines located in the cytosol bind. Following binding of substrate to the high-affinity site on VMAT2, a second proton binds to its luminal face, signaling a return to its original conformational state, which is accompanied by antiport of the hydrogen atom and monoamine molecule (Chaudhry, Edwards, & Fonnum, 2008; Wimalasena, 2010). Thus, intravesicular transport of a single monoamine molecule from the cytosol into the vesicle requires efflux of two protons from inside the vesicle to the cytosol.
5. VMAT2 BINDING SITES
Classically speaking, two distinct ligand binding sites have been identified on VMAT2: a reserpine binding site and a high-affinity TBZ binding site. Reserpine is obtained from Rauwolfia serpentina and was utilized originally to treat hypertension. Reserpine produces a long-lasting depletion of monoaminergic content from vesicles that express VMAT2. The use of reserpine as a first-line antihypertensive was discontinued due to its inhibition of VMAT2 on vesicles in monoaminergic neurons in the CNS and to the untoward side effect of depression resulting from vesicular monoamine depletion (Frize, 1954). Nonetheless, reserpine continues to be an important tool to evaluate the binding of novel compounds to and their subsequent functional inhibition of VMAT2. Reserpine binds in an irreversible manner to the substrate recognition site on VMAT2 following translocation of the initial proton from the vesicle lumen to the cytosol (Schuldiner, Liu, & Edwards, 1993). As such, reserpine is classified as a competitive monoamine uptake inhibitor at VMAT1 and VMAT2. In contrast, TBZ binding to VMAT2 is not affected by pH and is inhibited by high concentrations of monoamines (Darchen, Scherman, & Henry, 1989), suggesting a site of interaction different from that of reserpine. A single injection of reserpine in vivo inhibits TBZ binding to VMAT2 in rat striatal slices (Naudon et al., 1996). Furthermore, TBZ also inhibits reserpine binding to VMAT2 (Darchen et al., 1989; Schuldiner et al., 1993), suggesting either that reserpine and TBZ binding sites are overlapping or that each drug interacts with a functionally distinct conformation of the transporter (Liu & Edwards, 1997).
Several studies identified amino acid sequences in VMAT2 critical to protein function and that constitute the ligand binding sites. Photolabeling of purified VMAT2 revealed that the TBZ binding site is localized to the N-terminus of the protein (Sagne, Isambert, Vandekerckhove, Henry, & Gasnier, 1997; Sievert & Ruoho, 1997). Furthermore, mutagenesis of cysteine-439 resulted in >80% inhibition of dihydrotetrabenazine (DTBZ) binding, revealing a critical role of this amino acid in binding to the TBZ site (Thiriot & Ruoho, 2001). Mutation of aspartate-33 in the first transmembrane domain and of the serines 180–182 in the third transmembrane domain impaired substrate recognition by the transporter, suggesting that these amino acid residues are critical for interaction with the cationic amino groups and hydroxyl groups, respectively, of the monoamine substrate (Merickel, Rosandich, Peter, & Edwards, 1995). Additionally, mutation of transmembrane regions 9 through 12 revealed that tyrosine-434 and aspartate-461 are responsible for VMAT2 having greater affinity for TBZ, histamine, and serotonin compared with VMAT1 (Finn & Edwards, 1997).
6. ABERRANT VMAT2 EXPRESSION AND FUNCTION
Generation of VMAT2 knockout mice in 1997 allowed investigation of the role that VMAT2 plays in the physiological function of a number of different systems. Elimination of VMAT2 protein results in hypo-locomotion in comparison with wild-type and heterozygous littermates and, moreover, is lethal within 2 weeks following birth due to the failure to gain weight (Fon et al., 1997; Takahashi et al., 1997; Wang et al., 1997). Morphological abnormalities were not observed in VMAT2 knockout mice (Takahashi et al., 1997; Wang et al., 1997), and monoaminergic neurons exhibited normal dendritic processes and axonal projections, despite having decreased whole-brain levels of monoamines (Fon et al., 1997; Takahashi et al., 1997; Wang et al., 1997). VMAT2 heterozygotes exhibited physiological and behavioral similarities to wild-type littermates, including viability into adulthood, normal weight gain, expression of conditioned passive avoidance behavior and stress responses emitted upon exposure to a novel environment, and unchanged monoamine receptor densities and affinities (Takahashi et al., 1997). Presynaptic vesicles within monoaminergic neurons from heterozygote mice express VMAT2 protein at 50% of the expression levels found in wild-type mice, as evidenced by the amount of [3H]DTBZ binding to vesicle membranes (Takahashi et al., 1997). Cultures of midbrain neurons from heterozygote mice released ~50% less DA when depolarized compared to cultures from wild-type mice (Fon et al., 1997), revealing the necessity for VMAT2 expression and function for proper distribution and storage of monoamines for exocytotic release. VMAT2 heterozygotes administered amphetamine exhibited enhanced locomotor activity and a reduction in amphetamine reward in comparison with wild-type littermates in CPP assays (Takahashi et al., 1997). Following MPTP administration, the substantia nigra of heterozygotes exhibited twice the dopaminergic neuronal loss compared to wild-type mice (Takahashi et al., 1997), indicating enhanced sensitivity to its neurotoxic effects when VMAT2 protein levels were reduced. Collectively, these data indicate that appropriate VMAT2 expression is required for viability of the organism and that VMAT2 provides neuroprotection against some CNS toxins. Furthermore, VMAT2 appears to play a role in a variety of amphetamine-induced behaviors, which leads to the hypothesis that reversible inhibition of VMAT2 function may attenuate the behavioral effects of METH.
The role of VMAT2 in the protection of monoaminergic neurons from toxic insult gained attention in the clinical arena. VMAT2 sequesters MPP+ from the cytosol in cell lines expressing VMAT2 (Liu, Roghani, & Edwards, 1992), thus limiting MPP+ exposure and damage to the mitochondria and cellular respiration processes and the development of Parkinson’s-like behavioral symptoms. Furthermore, elevated cytosolic DA concentrations are of particular concern, as DA inhibits mitochondrial respiration and DA auto-oxidation generates harmful free radicals (Ben-Shachar, Zuk, & Glinka, 1995; Hastings, Lewis, & Zigmond, 1996). Parkinson’s disease is characterized by reduced motor coordination with an underlying etiology of loss of dopaminergic neurons in the substantia nigra pars compacta, which innervates the striatum. Given that VMAT2 provides neuroprotection from MPP+-induced Parkinson’s-like symptoms, clinicians employed a variety of neuroimaging and genetic screens to evaluate VMAT2 expression levels in Parkinson’s patients to determine the role of this transporter in the disease. Results indicated that polymorphisms in the coding sequence of VMAT2 are extremely rare and, thus, likely did not account for the incidence of Parkinson’s disease (Glatt et al., 2001). However, investigators found numerous gain-of-function haplotypes present in the genetic sequence responsible for the transcription of VMAT2 that seemed to confer a neuro-protective effect, specifically in women (Glatt, Wahner, White, Ruiz-Linares, & Ritz, 2006). Results from clinical studies employing positron emission tomography and photon emission computed tomography imaging techniques led to the speculation that the ratio of DAT to VMAT2 expression may be a critical parameter in predicting vulnerability of monoaminergic neurons to neurotoxic insult (Frey et al., 1996; Kazumata et al., 1998; Madras et al., 1998; Miller, Gainetdinov, Levey, & Caron, 1999; Uhl, 1998). Toxins gain rapid access to the presynaptic terminal through DAT and are removed from the cytosol by VMAT2 at a slower rate. Thus, toxins have increased access to vital intracellular organelles in individuals with a high DAT to VMAT2 expression ratio. Taken together, results from VMAT2 heterozygous mouse studies and from clinical studies suggest that inhibition of VMAT2 may attenuate the behavioral effects of METH. Further, a criterion for VMAT2 inhibitors developed as therapeutics to treat METH abuse is selectivity at VMAT2 relative to DAT in order to minimize abuse liability of the therapeutic.
7. CONCLUSION
7.1. Lobeline
This review focuses on the discovery of novel compounds that interact with VMAT2 and inhibit the pharmacological effect of METH. Lobeline, the major lipophilic, nonpyridino alkaloidal constituent of Lobelia inflata (also known as Indian tobacco), inhibits DA uptake by VMAT2 (Teng, Crooks, & Dwoskin, 1998; Teng, Crooks, Sonsalla, & Dwoskin, 1997). Lobeline also has high affinity for the [3H]DTBZ binding site on VMAT2 protein (Fig. 2.2; Kilbourn, Lee, Vander Borght, Jewett, & Frey, 1995; Miller et al., 2004). Importantly, lobeline decreases amphetamine-evoked DA release from superfused rat striatal slices, but did not inhibit field stimulation-evoked DA release, indicating a selective inhibition of the effect of amphetamine (Miller et al., 2001).
Lobeline also decreases METH-induced hyperactivity, behavioral sensitization, and self-administration in rats (Harrod, Dwoskin, Crooks, Klebaur, & Bardo, 2001; Miller, Green, et al., 2000; Miller et al., 2001). The development of locomotor sensitization has been suggested to result from neuroadaptations in response to repeated METH administration and may play a role in the development of dependence (Kalivas & Stewart, 1991; Vezina, 1996). In Miller, Green, et al. (2000), adult male rats were pretreated with lobeline (3 mg/kg) 10 min prior to METH (1 mg/kg) for 10 consecutive days. Lobeline significantly reduced locomotor activity in response to METH relative to saline-pretreated rats on each day. Thus, lobeline attenuates the induction of METH behavioral sensitization, leading to the prediction that lobeline may attenuate METH dependence. Lobeline decreased responding for food reinforcers; however, tolerance developed to this effect of lobeline (Harrod et al., 2001). Moreover, the lobeline-induced decrease in responding for METH persisted, revealing specificity upon repeated administration. Furthermore, this effect of lobeline was not surmountable upon increasing the unit dose of METH. Importantly, lobeline is not self-administered and does not produce CPP (Harrod, Dwoskin, Green, Gehrke, & Bardo, 2003). Lobeline did not substitute for METH and did not reinstate responding for METH following extinction, suggesting that the alkaloid does not act as a substitute reinforcer and that it lacks abuse liability (Harrod et al., 2001, 2003). Based on these preclinical findings, lobeline has been evaluated as a treatment for METH abuse and has completed phase 1b clinical trials, which determined that lobeline was safe in METH addicts (http://www.clinicaltrials.gov/ct2/show/NCT00439504?term=NCT00439504&rank=1).
The interaction of lobeline with VMAT2 was hypothesized to be the mechanism underlying the ability of lobeline to decrease the neurochemical and behavioral effects of METH (Dwoskin & Crooks, 2002; Miller et al., 2001; Teng et al., 1997). Lobeline, in a manner comparable to METH, inhibits the transport of cytosolic DA into synaptic vesicles via potent inhibition of VMAT2 function; however, lobeline does not inhibit MAO. Moreover, lobeline is more potent in inhibiting DA uptake at VMAT2 than it is in releasing DA from the vesicles, whereas amphetamine is more potent in releasing DA from the vesicles than it is in inhibiting DA uptake into the vesicles. Importantly, lobeline reduces METH-evoked DA release in vitro while concurrently increasing extracellular dihydroxyphenylacetic acid (DOPAC) concentrations (Miller et al., 2001; Wilhelm, Johnson, Eshleman, & Janowsky, 2008). Collectively, these results suggest that the decreased reinforcing effects of METH are the result of lobeline decreasing the cytosolic pool of DA available for METH-induced reverse transport by DAT (Dwoskin & Crooks, 2002). However, lobeline is not selective for VMAT2, acting as an inhibitor of DAT and the serotonin transporter (SERT; Miller et al., 2004) and as a potent inhibitor of [3H]nicotine (NIC) and [3H]methyllycaconitine (MLA) binding to rat brain membranes. Additionally, lobeline decreases NIC-evoked 86Rb+ efflux from rat thalamic synaptosomes, suggesting that it acts as an alpha4beta2* and alpha7* nicotinic acetylcholine receptor (nAChR) antagonist (Damaj, Patrick, Creasy, & Martin, 1997; Flammia, Dukat, Damaj, Martin, & Glennon, 1999; Miller, Crooks, & Dwoskin, 2000; Miller et al., 2004).
7.2. meso-Transdiene
To enhance selectivity, a number of analogs similar in structure to lobeline were synthesized and evaluated for selectivity for VMAT2 and affinity for the DA uptake site on VMAT2. Lobeline consists of a central piperidine ring with phenyl rings attached at C-2 and C-6 of the piperidine ring by ethylene linkers containing hydroxyl and keto functionalities at the C8 and C10 positions on the linkers, respectively (Fig. 2.1). Both affinity at and selectivity for VMAT2 were improved as a result of alterations in structure, with the emergence of two new lead compounds, N-methyl-2,6-bis(cis-phenylethenyl) piperidine (meso-transdiene, MTD; Fig. 2.1) and N-methyl-2,6-bis(cis-phenylethyl)piperidine (lobelane; Fig. 2.1; Miller et al., 2004; Nickell et al., 2010; Zheng, Dwoskin, Deaciuc, Norrholm, & Crooks, 2005). MTD is a lobeline analog with defunctionalized and unsaturated (double bonds) linker units. Compared with lobeline, MTD exhibited similar affinity for the [3H]DTBZ binding site on VMAT2 and dramatically decreased affinity for alpha4beta2* and alpha7* nAChRs, thus revealing increased selectivity for VMAT2 (Miller et al., 2004; Zheng, Dwoskin, Deaciuc, Norrholm, et al., 2005). Importantly, MTD inhibited METH-evoked DA release from superfused rat striatal slices (Nickell et al., 2010). Furthermore, MTD specifically decreased METH self-administration in rats, but only at the highest dose (17 mg/kg) evaluated (Horton, Siripurapu, Norrholm, et al., 2011). However, MTD exhibited a 100-fold higher affinity for DAT and a 6.5-fold lower affinity for SERT, compared with lobeline (Miller et al., 2004). The affinity of MTD at DAT was similar to that for cocaine and methylphenidate (Ki =300 and 100 nM, respectively; Han & Gu, 2006), suggesting the potential for abuse liability. Due to the observation that MTD inhibited DAT more potently than VMAT2, suggesting the potential for abuse liability, and given that MTD had limited solubility, its potential was diminished for development as a pharmacotherapy for METH abuse. As such, mechanistic studies evaluating the effect of MTD on depolarization-stimulated DA release were not pursued and other more promising analogs were evaluated.
Nevertheless, taking into account these encouraging findings, but tempered by the limitations associated with the potential for abuse liability, modifications to the MTD molecule were evaluated in the search for preclinical candidates for the treatment of METH abuse (Horton, Siripurapu, Norrholm, et al., 2011). Structural modifications of MTD were made, including lengthening the linkers and either introducing substituents (4-methoxy,4-methyl, or2,4-dichloro)into the phenyl rings or replacing the phenyl rings with furan or thiophene rings. The effects of altering the geometry of the C5 double bond of the piperidine ring were evaluated in analogs with longer linkers or an aromatic 2,4-dicholor-substituted phenyl ring. In this series of analogs, affinity for VMAT2 was retained and affinity for DAT was reduced by 50- to 1000-fold, importantly increasing selectivity for VMAT2 over DAT, compared with MTD. UKMH-105 and UKMH-106, 2,4-dicholorophenyl analogs (Fig. 2.1), exhibiting 20- to 30-fold higher potency at VMAT2 over DAT, were evaluated for inhibition of METH-evoked DA release. Interestingly, the effect of METH was inhibited by UKMH-106, but not by UKMH-105, revealing an important geometric specificity of inhibition and suggesting that these isomers may interact with different sitesonVMAT2toinhibitDAuptakeandMETH-evokedDArelease(Horton, Siripurapu, Norrholm, et al., 2011). Although UKMH-106 and UKMH-105 exhibit no differences in selectivity for the substrate uptake site on VMAT2, differences in affinity for the DTBZ binding site on VMAT2 have been noted. Thus, incorporation of the phenylethylene moiety of MTD into the piperidine ring and the addition of the dichloro groups resulted in a novel compound, UKMH-106, which had improved water solubility, reduced affinity for DAT and nAChRs, increased selectivity for VMAT2, and, importantly, inhibited the effect of METH to increase the extracellular concentration of DA.
7.3. Lobelane
Defunctionalization of lobeline afforded lobelane, with the hydroxyl and keto groups of lobeline eliminated from the saturated linkers. Lobelane is also a minor alkaloid of L. inflata. Preclinical evaluation of lobelane, as well as analogs based on the lobelane structural scaffold, revealed that this structural modification was sufficient to eliminate affinity for nAChRs, to provide a 10-fold increase in affinity for the DA uptake site on VMAT2 and a two-fold enhancement in the affinity for the DTBZ binding site on VMAT2, compared with lobeline (Beckmann et al., 2010; Nickell et al., 2010; Zheng, Dwoskin, Deaciuc, Zhu, et al., 2005). Kinetic analysis revealed that lobelane inhibited DA uptake at VMAT2 via a competitive mechanism. Furthermore, lobelane exhibited 35-fold greater potency for VMAT2 than for DAT (i.e., reduced affinity for DAT). Moreover, lobelane potently and nearly completely inhibited METH-evoked DA release from superfused rat striatal slices (Nickell et al., 2010). Taking into account these in vitro results, considerable enthusiasm was generated for the potential for lobelane as a lead compound.
Preclinical assessment of the ability of lobelane to decrease the behavioral effects of METH revealed that lobelane dose-dependently decreased METH self-administration while having no effect on sucrose-maintained responding (Neugebauer et al., 2007). However, lobelane appeared to have a shorter duration of action than lobeline in the self-administration assay. Furthermore, only the highest dose (10 mg/kg) of lobelane decreased locomotor activity. Unfortunately, tolerance developed to the effect of lobelane to decrease METH self-administration, revealing a transient effect of this alkaloid across seven consecutive sessions. The development of tolerance with repeated lobelane administration was in contrast with results obtained with lobeline using similar behavioral procedures. Lobelane appears to be a better substrate for hepatic metabolic oxidation compared to lobeline, which is consistent with the shorter duration of action of lobelane and metabolic issues being a factor in the development of tolerance to the effects of lobelane. Furthermore, lobelane exhibited decreased water solubility and as such reduced drug-likeness due to its decreased polarity as a result of the elimination of the keto and hydroxyl functionalities in lobeline.
Further structural modification of lobelane revealed that the most potent (Ki =13–16 nM) and selective analogs included para-methoxyphenyl nor-lobelane, para-methoxyphenyl lobelane, and 2,4-dichlorphentyl lobelane (Nickell, Zheng, Deaciuc, Crooks, & Dwoskin, 2011; Zheng, Dwoskin, Deaciuc, Zhu, et al., 2005). Kinetic analyses revealed a competitive mechanism of inhibition of VMAT2 function. Also, these analogs had >100-fold higher affinity for VMAT2 than DAT, predicting low abuse liability.
7.4. GZ-793A
To improve water solubility and enhance drug-likeness properties of the lobelane scaffold, the N-methyl moiety of the central piperidine ring of lobelane was replaced with a chiral N-1,2-dihydroxypropyl (N-1,2-diol) moiety, which was predicted to enhance water solubility based on computational modeling. As such, a new series of lobelane analogs was synthesized by incorporating the chiral N-1,2-diol, altering the configuration of this moiety, and incorporating phenyl ring substituents (2-methoxy, 3-methoxy, 4-methoxy, 3-flouro, 2,4-flouro, and 3,4-methylenedioxy) into both phenyl rings or replacing the phenyl rings with naphthalene or biphenyl rings. Emerging from the in vitro pharmacological evaluation of this series of analogs was (R)-3-[2,6-cis-di(4-methoxyphenethyl)piperidin-1-yl]propane-1,2-diol (GZ-793A; Fig. 2.1), a potent, VMAT2-selective, drug-like lead (Horton, Siripurapu, Zheng, Crooks, & Dwoskin, 2011). GZ-793A was a potent competitive inhibitor of DA uptake at VMAT2, exhibiting a Ki value of 29 nM and an Imax of 86%. Thus, the pharmacophore for inhibiting VMAT2 function accommodates the N-1,2-diol moiety, which not only improved water solubility and drug-likeness but also enhanced the pharmacological characteristics of the molecule. For example, GZ-793A was 50-fold selective for inhibiting VMAT2 function over DAT or SERT, suggesting that GZ-793A would have low abuse liability at relevant doses. Importantly, GZ-793A inhibited METH-evoked DA release from superfused striatal slices (Horton, Siripurapu, Zheng, et al., 2011), and GZ-793A reduced the duration of the METH-induced increase in extracellular DA in nucleus accumbens in microdialysis studies (Meyer et al., 2013). Taking a more molecular approach, GZ-793A was found recently to inhibit METH-evoked DA release from isolated striatal synaptic vesicles via a surmountable allosteric inhibition and to interact with VMAT2 at several distinct sites on the protein, both extravesicular and intravesicular (Horton, Nickell, Zheng, Crooks, & Dwoskin, 2013). These results from the neurochemical assays support VMAT2 as an important pharmacological target and GZ-793A as a lead compound for reducing the neurochemical effects of METH.
Moreover, GZ-793A, administered peripherally across a reasonable dose range (3–30 mg/kg), decreased METH self-administration in rats, without altering responding for food reinforcers (Beckmann et al., 2012). Similar doses of GZ-793A blocked METH CPP, and GZ-793A itself did not induce place preference. Importantly, tolerance did not develop to the GZ-793A-induced decrease in responding for METH in the self-administration assay (Beckmann et al., 2012). Furthermore, the results of this study showed that increasing the unit dose of self-administered METH did not surmount the GZ-793A-induced decrease in responding for METH. GZ-793A was evaluated also for its ability to attenuate cue- and METH-induced reinstatement of METH-seeking after a period of extinction (Alvers et al., 2012). GZ-793A (15 mg/kg) was found to decrease reinstatement following exposure to conditioned cue or METH administration. GZ-793A did not produce response suppressive effects in the absence of the conditioned cue stimuli; however, this lead compound did produce response suppressive effects in the METH reinstatement experiments. Thus, GZ-793A specifically blocks the primary and conditioned rewarding effects of METH and also decreases reinstatement of METH-seeking following presentation of conditioned cues and is predicted to have low abuse liability. In a subsequent investigation, oral dosing of GZ-793A (240 mg/kg) was shown to attenuate METH self-administration 85% relative to the control baseline responding rate while producing no alteration in responding for food (Wilmouth, Zheng, Crooks, Dwoskin, & Bardo, 2013). Thus, GZ-793A is orally bioavailable. Together, these results support the hypothesis that analogs that exhibit potent and selective inhibition of VMAT2 function translate to the requisite behavioral profile in preclinical studies, supporting their clinical evaluation as a pharmacotherapies for METH abuse.
7.5. 2,5-Disubstituted pyrrolidine analogs
To further generate the pharmacophore, a recent approach with respect to structural modification of the lobelane molecule was the introduction of a smaller, more conformationally constrained pyrrolidine ring in place of the central piperidine ring in the lobelane scaffold, which afforded a novel series of 2,5-disubstituted pyrrolidine analogs. Changes in the chemical structure also incorporated various linker lengths of the phenylethyl side chain and the addition of various substituents to the phenyl rings (Fig. 2.3). The pyrrolidine analogs were synthesized in the Department of Pharmaceutical Sciences in the College of Pharmacy at the University of Kentucky. The chemical structures were verified by 1H and 13C NMR spectroscopy, mass spectrometry, and X-ray crystallography.
Figure 2.3. Chemical structures of the novel 2,5-disubstituted pyrrolidine analogs.

Analogs are grouped according to structural similarity: Group 1: analogs with either lengthened or shortened side chain linkers in comparison with AV-1-229; Group 2: analogs incorporating methoxy and hydroxyl moieties on the phenyl rings; and Group 3: analogs incorporating phenylethanol and phenylmethanol substituents on the nitrogen atom on the central pyrrolidine ring.
These 2,5-disubstituted pyrrolidine analogs were evaluated first for inhibition of [3H]DTBZ binding to VMAT2 using modifications of a previously published method (Teng et al., 1998). Briefly, vesicular suspension (15 μg protein/100 μl) from whole rat brain was added to each well of a 96-well plate, containing 5 nM [3H]DTBZ, 50 μl of analog (1 nM–1 mM), and 50 μl of assay buffer. Samples were incubated at room temperature for 30 min. Non-specific binding was determined in the presence of (2R,3S,11bS)-2-ethyl-3-isobutyl-9,10-dimethoxy-2,2,4,6,7,11b-hexahydro-1H-pyrido[2,1a] isoquinolin-2-ol (Ro4-1284; 10 μM). Following incubation, plates were filtered, filters washed with 350 μl of ice-cold buffer (25 mM HEPES, 100 mM potassium tartrate, 5 mM MgSO4, and 10 mM NaCl, pH 7.5), dried, sealed, and 40 μl of scintillation cocktail added to each well, and radioactivity determined by liquid scintillation spectrometry.
All of the pyrrolidine analogs inhibited [3H]DTBZ binding (Table 2.1), although only AV-2-190 (Ki =0.27 μM) exhibited greater affinity for VMAT2 than lobelane (Ki = 0.97 μM). Thus, AV-2-190, a nor-analog incorporating an additional methylene in each of its methylene linker units, exhibited the highest affinity at the DTBZ binding site in this series of analogs. The majority of analogs inhibited [3H]DTBZ binding at concentrations of 1–10 μM, although several exceptions with low affinity were noted, including AV-3-156B (Ki =96.6 μM), AV-3-158 (Ki =74.7 μM), AV-3-159 (Ki =89.9 μM), and AV-3-162 (Ki =82.6 μM). Low-affinity analogs had methoxy groups in the ortho-position on the phenyl rings. Interestingly, analogs bearing methoxy groups in the para-position on the phenyl rings, including AV-1-252B and AV-2-256B, exhibited relatively high affinity (Ki =1.23 and 1.39 μM, respectively) for the DTBZ binding site on VMAT2. Generally, nor-pyrrolidine analogs inhibited [3H]DTBZ binding with higher affinity than their respective pyrrolidine N-methylated analogs.
Table 2.1.
Ki and Imax values for 2,5-disubstituted lobelane analog inhibition of [3H]DTBZ binding and [3H]DA uptake at VMAT2
| Compound | [3H]DTBZ binding | VMAT2 [3H]DA uptake | ||
|---|---|---|---|---|
| Ki (μM) | Imax (%) | Ki (μM) | Imax (%) | |
| Standards | ||||
| TBZ | 0.013±0.001a | 100 | 0.054±0.015 | 92±3 |
| Lobelane | 0.97±0.19 | 90±3 | 0.045±0.002 | 100 |
| AV-1-229A | 8.80±2.30 | 97±2 | 0.27±0.035 | 100 |
| AV-1-228 | 2.66±0.37 | 100 | 0.028±0.0010 | 100 |
| Group 1 | ||||
| AV-2-192 | 1.64±0.12 | 100 | 0.0093±0.00075 | 100 |
| AV-2-197 | 2.46±0.42 | 96±1 | 0.019±0.00091 | 100 |
| AV-2-190 | 0.27±0.0067 | 100 | 0.014±0.0011 | 100 |
| AV-2-195 | 2.78±0.63 | 100 | 0.12±0.011 | 100 |
| AV-3-165 | 5.18±0.47 | 100 | 0.042±0.0035 | 100 |
| AV-2-241 | 11.2±1.17 | 97±2 | 0.21±0.019 | 98±1 |
| Group 2 | ||||
| AV-2-256B | 1.39±0.062 | 100 | 0.16±0.0071 | 100 |
| AV-3-161 | 25.6±7.31 | 64±1 | 2.23±0.29 | 100 |
| AV-3-158 | 74.7±14.4 | 47±2 | 3.38±0.27 | 100 |
| AV-3-159 | 89.9±5.14 | 50±2 | 2.39±0.53 | 100 |
| AV-3-162 | 82.6±11.9 | 57±6 | 1.78±0.47 | 100 |
| Group 3 | ||||
| AV-3-155A | 1.49±0.77 | 58±1 | 0.38±0.066 | 99±1 |
| AV-3-156A | 7.55±1.45 | 60±2 | 0.69±0.19 | 100 |
| AV-3-156B | 96.6±21.9 | 56±2 | 2.42±0.20 | 98±1 |
| AV-3-155B | 3.94±2.29 | 55±2 | 0.43±0.052 | 100 |
| AV-2-252A | 1.06±0.047 | 100 | 0.0053±0.00045 | 100 |
| AV-2-199 | 2.23±0.58 | 85±2 | 0.40±0.087 | 99±1 |
| AV-2-194 | 1.84±0.35 | 91±1 | 0.51±0.024 | 100 |
| AV-1-227A | 4.34±0.71 | 85±2 | 1.29±0.24 | 100 |
| AV-1-227B | 5.05±1.05 | 97±2 | 1.94±0.47 | 99±1 |
| AV-1-258A | 2.75±0.63 | 74±8 | 0.14±0.049 | 97±3 |
Data represent mean±SEM; n=three to four rats per compound.
The ability of the pyrrolidine analogs to inhibit vesicular [3H]DA uptake was determined using our reported method employing synaptic vesicle preparations (Teng et al., 1997). Briefly, rat striatal vesicular suspension (100 μl) was added to assay tubes containing buffer, various concentrations of analog (0.1 nM–10 mM), and 0.1 μM [3H]DA in a final volume of 500 μl. Samples were incubated at 37 °C for 8 min and filtered, and radioactivity remaining on the filter determined. Pyrrolidine analogs exhibited >90% inhibition of VMAT2 function (Table 2.1). Many of these analogs exhibited greater affinity for the DA uptake site on VMAT2 compared to lobelane (Ki =0.045 μM). The nor-pyrrolidine analog AV-2-192, with a reduced number of methylene linker units having cis geometry, had an affinity of <10 nM. The nor-pyrrolidine analog AV-1-228 (Fig. 2.1; also known as UKCP-110) with the greatest structural resemblance to lobelane also inhibited VMAT2 function with high affinity (Ki =28 nM). AV-2-252A and AV-2-252B, incorporating methoxy moieties in the para-position on the phenyl rings, exhibited high affinity (Ki =5.3 and 50 nM, respectively) for the DA uptake site on VMAT2. In all cases, the nor-pyrrolidine analogs exhibited higher affinity for the DA uptake site than the respective pyrrolidine N-methylated analogs.
In an effort to further generate the pharmacophore via a thorough analysis of structure–activity relationship on this series of compounds, analogs were divided into three groups (Fig. 2.3; Table 2.1) based upon similarities in chemical structure. The pyrrolidine analog most structurally similar to lobelane was AV-1-229A (Fig. 2.1), with one less methylene group in the central heterocyclic ring. AV-1-229A exhibited a Ki of 270 nM at the DA uptake site on VMAT2, which had six-fold lower affinity compared with lobelane. The N-methylated analog AV-2-195, with an increased number of methylene linker units (Fig. 2.3, Group 1), exhibited a two-fold higher affinity for the DA uptake site on VMAT2, relative to AV-1-229A. Interestingly, the N-methylated analog AV-2-197, with only one methylene linker unit, exhibited 14 orders-of-magnitude increased affinity for the DA uptake site on VMAT2 compared with AV-1-229A. Demethylation of the central pyrrolidine N-atom to afford nor-pyrrolidines AV-2-192 and AV-2-190 resulted in increased affinity at both the DTBZ binding and DA uptake sites on VMAT2. The greatest impact of N-demethylation occurred with analogs incorporating three methylene linker units, for example, AV-2-190, which exhibited an order-of-magnitude higher affinity than AV-2-195. Thus, the loss of one methylene moiety from each linker unit in AV-1-229A produced the greatest increase in affinity for the DA uptake site.
Pyrrolidine analogs incorporating either a methoxy or hydroxyl moiety at various positions of the phenyl rings (Fig. 2.3, Group 2) had reduced affinity at both the [3H]DTBZ binding and DA uptake sites on VMAT2 in comparison with AV-1-229A. Furthermore, a final group of pyrrolidine analogs consisting of various structural modifications was evaluated. These analogs (Fig. 2.3, Group 3) had altered linker lengths, methoxy substituents in the phenyl rings or N-phenethanol or N-phenmethanol groups in place of the N-methyl of AV-1-229A. Generally, these analogs had reduced affinity at both uptake and binding sites compared to AV-1-229A.
Most analogs exhibited affinities for the DA uptake site at least 10-fold higher than their respective affinities for the [3H]DTBZ binding site on VMAT2, and a positive correlation between uptake and binding affinity was found (Spearman r=0.72; p<0.001; Fig. 2.4). These results are in contrast with previous results evaluating this relationship for a different series of lobelane analogs with phenyl ring substituents (Nickell et al., 2011). At least for the pyrrolidine analog series, the high-throughput binding assay serves as a predictive screen for inhibition of DA uptake by VMAT2. While a positive correlation was found, affinity for the majority of analogs at the DTBZ binding site was within a relatively restricted range of 1–10 μM, whereas a greater range of 0.005–0.7 μM was obtained for affinity at the DA uptake site. Thus, structural modification did not alter interactions with the binding site to as great a degree as interactions with the DA uptake site.
Figure 2.4. Positive correlation between 2,5-disubstituted pyrrolidine analog affinity for the DTBZ binding site and the DA uptake site.
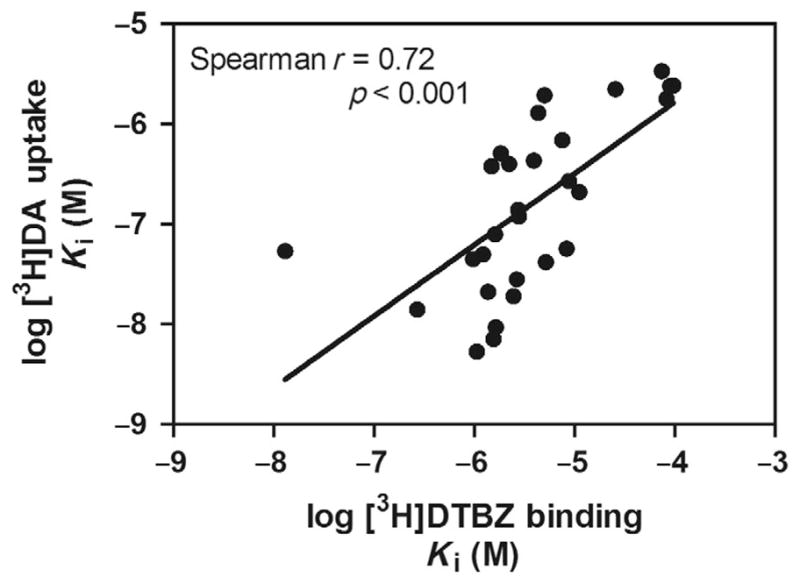
Data are Ki values obtained from concentration–response curves for analog-induced inhibition of [3H]DTBZ binding and [3H]DA uptake (data not shown). Spearman analysis revealed a positive correlation (Spearman r =0.72; p <0.001) between pyrrolidine analog affinity for the DTBZ binding site and the DA uptake site on VMAT2.
Furthermore, the current results support previous findings of alternative recognition sites on VMAT2, including a ketanserine binding site near the N-terminal of the VMAT2 protein and a TBZ site located near the C-terminus of the VMAT2 protein (Sievert & Ruoho, 1997; Weaver & Deupree, 1982). Interestingly, studies investigating interactions of novel compounds at VMAT2 and DAT reveal that compounds exhibiting large differences in affinity between binding and uptake sites act as transporter substrates, while compounds equipotent inhibiting binding and uptake act purely as uptake inhibitors (Andersen, 1987; Matecka et al., 1997; Partilla et al., 2006; Rothman, Ayestas, Dersch, & Baumann, 1999). If allowed to extrapolate from these previous studies, all of the pyrrolidine analogs evaluated herein, with the exception of AV-1-227A and AV-1-227B, would be predicted to act as substrates for VMAT2 and to inhibit DA uptake in a competitive manner.
To determine the mechanism of inhibition of [3H]DA uptake (i.e., competitive vs. noncompetitive) for the lead pyrrolidine analogs, kinetic analyses were performed. The methylated pyrrolidine AV-2-192 and its nor-counterpart AV-2-197 (Fig. 2.3, Group 1) were selected for evaluation of mechanism of inhibition of DA uptake at VMAT2, as both compounds displayed high affinity for the DA uptake site and moderate affinity for the [3H]DTBZ binding site. Concentrations of these lead compounds, AV-2-192 and AV-2-197 (9.3 and 19 nM, respectively), were chosen based on Ki values obtained from the [3H]DA uptake assays. Incubations were initiated by the addition of 50 μl of vesicular suspension to 150 μl assay buffer, 25 μl of inhibitor or assay buffer (control), and 25 μl of a range of concentrations of [3H]DA (0.001–5.0 μM). Nonspecific uptake was determined in the presence of Ro4-1284 (10 μM). Following an 8 min incubation, [3H]DA uptake was terminated by filtration, and radioactivity retained by the filters determined as previously described. Both AV-2-192 and AV-2-197 increased ( p<0.05) the Km value relative to control, without altering Vmax (Fig. 2.5; Table 2.2), indicating that the analogs competitively inhibit [3H]DA uptake by VMAT2, in agreement with the earlier-mentioned prediction and indicating that methylation of the pyrrolidine N-atom was not an important factor in the interaction of these analogs with VMAT2.
Figure 2.5. Kinetic analysis of the inhibition of [3H]DA uptake at VMAT2 by AV-2-192 and AV-2-197.
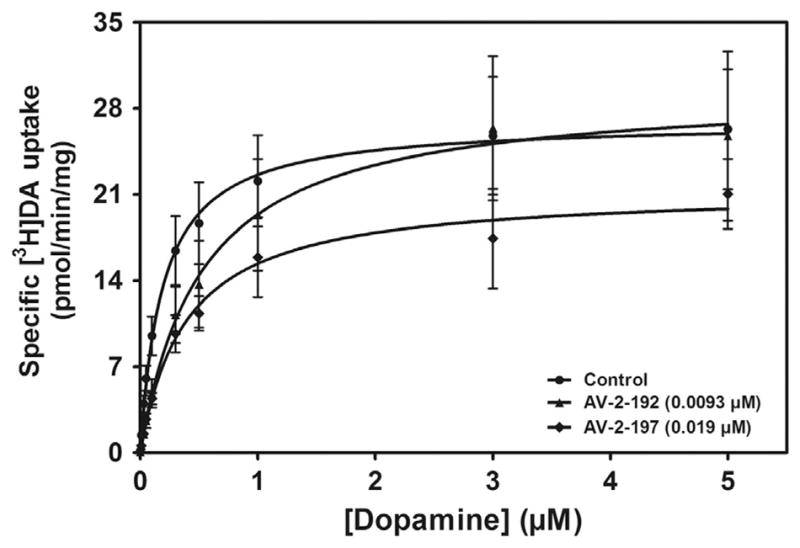
The nor-pyrrolidine analog (AV-2-192) containing single methylene unit side chain linkers in the cis conformation and its N-methyl counterpart (AV-2-197) were evaluated to determine mechanism of inhibition of [3H]DA uptake. Concentrations of analog utilized (in parentheses) for the kinetic analysis were the respective Ki concentrations from the inhibition curves (data not shown). Vmax and Km values (±SEM) are provided in Table 2.2 (n =4–6 rats/analog).
Table 2.2.
Km and Vmax values for AV-2-192 and AV-2-197 from kinetic analysis of [3H]DA uptake at VMAT2
| Compounda | Km (μM) | Vmax (pmol/min/mg) |
|---|---|---|
| Controlb | 0.19±0.021c | 29.9±4.30 |
| AV-2-192 | 0.51±0.025d | 29.6±7.28 |
| AV-2-197 | 0.43±0.13d | 22.0±4.36 |
Concentrations of analogs utilized for kinetic analyses were the Ki concentrations from the inhibition curves (AV-2-192, 0.0093 μM; AV-2-197, 0.019 μM).
Control represents the absence of analog.
Data are mean±SEM for Km and Vmax values.
p<0.05 different from control, two-tailed t test (n=4–6 rats/analog).
The ability of the leads, AV-2-192 and AV-2-197, to inhibit [3H]DA uptake at DAT in synaptosomal preparations was determined according to previous methods (Teng et al., 1997). Synaptosomal suspension (25 μl) was added to assay tubes containing assay buffer and various concentrations of analog (100 μM–1 nM) and incubated at 34 °C for 5 min. Nonspecific uptake was determined in the presence of nomifensine (10 μM). Samples were placed on ice, and 50 μl of 0.1 μM [3H]DA was added, and then, incubation proceeded for 10 min at 34 °C. Samples were filtered and radioactivity retained by the filters determined. No difference was found between AV-2-192 and AV-2-197 affinities for DAT (Ki =2.63±0.35 and 5.40±0.53 μM, respectively). Both analogs were 1- to 2-orders-of-magnitude more potent inhibiting DA uptake at VMAT2 relative to inhibiting DA uptake at DAT; thus, these leads are not likely to possess abuse liability.
The ability of AV-2-192 and AV-2-197 to inhibit METH-evoked DA release from striatal slices was determined as previously reported (Teng et al., 1997). Coronal striatal slices were incubated in Krebs buffer at 34 °C in a metabolic shaker for 60 min. Slices were superfused (1 ml/min) for 60 min with Krebs buffer. Then, two 5 min basal samples (1 ml into 100 μl of 0.1 M perchloric acid) were collected. Superfusion continued for 30 min in the absence or presence of a single concentration (0.3–30 μM) of AV-2-192 or AV-2-197. Then, METH (5 μM) was added to the buffer and superfusion continued for 15 min. Slices were superfused for another 20 min with analog in the absence of METH. In each experiment, a striatal slice was superfused for 90 min in the absence of analog or METH, serving as the buffer control. Duplicate slices were superfused with METH in the absence of analog, serving as the METH control. Samples (500 μl) were processed immediately by adding 20 μl of ascorbate oxidase (81 U/1 ml), vortexing for 30 s, followed by injection of 100 μl onto the HPLC with electrochemical detection (Table 2.3).
Table 2.3.
Summary of data for lead compounds: Affinity for nAChRs, DTBZ site on VMAT2 affinity, substrate site on VMAT2 and DAT, mechanism of VMAT2 inhibition, potency for inhibition of METH-evoked DA release, and inhibition of the behavioral effects of METH
| Compound | nAChR Ki value (μM) | DTBZ site Ki value (μM) | VMAT2 substrate site Ki value (μM) | Mechanism of VMAT2 inhibition | DAT Ki value (μM) | IC50 and Imax values for analog inhibition of METH-evoked DA release (μM) | Inhibition of METH behavioral effects |
|---|---|---|---|---|---|---|---|
| Lobeline | [3H] NIC=0.016 [3H] MLA=11.6 |
2.04 | 0.47 | Competitive | 31.6 | IC50 =0.42 Imax =56% |
Hyperactivity Self-administration CPP Discrimination Reinstatement |
| Lobelane | [3H] NIC=77.3 [3H] MLA=43.1 |
0.97 | 0.045 | Competitive | 1.57 | IC50 =0.65 Imax =73% |
Hyperactivity Self-administration |
| MTD | [3H] NIC=>100 [3H] MLA=>100 |
9.88 | 0.46 | Competitive | 0.039 | IC50 =0.44 Imax =76% |
Self-administration |
| UKMH-105 | [3H] NIC=>100 [3H] MLA=>100 |
4.60 | 0.22 | Competitive | 6.27 | No inhibition | Not evaluated |
| UKMH-106 | [3H] NIC=>100 [3H] MLA=>100 |
41.3 | 0.32 | Competitive | 6.90 | IC50 =0.38 Imax =50% |
Not evaluated |
| GZ-793A | [3H] NIC=>100 [3H] MLA=>100 |
8.29 | 0.029 | Competitive | 1.44 | IC50 =10.6 Imax =85% |
Self-administration CPP Reinstatement |
| AV-2-192 | [3H] NIC=Not evaluated [3H] MLA=Not evaluated |
1.64 | 0.0093 | Competitive | 2.63 | IC50 =5.2 Imax =55% |
Not evaluated |
| AV-2-197 | [3H] NIC=Not evaluated [3H] MLA=Not evaluated |
2.46 | 0.019 | Competitive | 5.40 | IC50 =4.2 Imax =63% |
Not evaluated |
Analysis of the effect of AV-2-192 and AV-2-197 on fractional DOPAC release during the 30 min period prior to addition of METH to the superfusion buffer revealed a concentration-dependent increase in DOPAC (Fig. 2.6). The increase in extracellular DOPAC levels was observed consistent with findings using lobeline and lobelane (Nickell et al., 2010). The lowest concentration of AV-2-192 and AV-2-197 to increase extracellular DOPAC was 3 and 10 μM, respectively. Across the concentration range, addition of either AV-2-192 or AV-2-197 to the superfusion buffer (15–45 min) alone did not alter fractional DA release (Fig. 2.7), indicating that the lead compounds lacked intrinsic activity with respect to releasing DA. METH (5 μM) increased fractional DA release compared to the buffer control condition. Both AV-2-192 and AV-2-197 attenuated METH-evoked DA release. When the data were expressed as total DA overflow (Fig. 2.8), the IC50 value for AV-2-192 to inhibit this effect of METH was 5.2±0.7 μM and the Imax was 55.0±3.5%, and similarly, the IC50 value for AV-2-197 was 4.2±0.8 μM and the Imax was 63.0±4.2%. Thus, AV-2-192 and AV-2-197 were equipotent but incompletely inhibited METH-evoked DA release.
Figure 2.6. AV-2-192 and AV-2-197 stimulate fractional DOPAC release from rat striatal slice preparations.

Top and bottom panels illustrate the time course for AV-2-192 and AV-2-197, respectively, to stimulate fractional DOPAC release. Each striatal slice was superfused with either a single concentration (0.3–30 μM) of AV-2-192 or AV-2-197 alone during the 30 min time period prior to the inclusion of 5 μM methamphetamine (METH) in the buffer. Arrow denotes time point at which analog was added to the buffer. METH was added to the buffer after 30 min and remained in the buffer for 15 min. Buffer control represents slices superfused in the absence of analog and METH. Data are mean (±SEM) pg/min/ml slice weight. *p <0.05 different from buffer control condition.
Figure 2.7. Time course of AV-2-192 and AV-2-197 inhibition of methamphetamine (METH)-evoked fractional DA release.

Top and bottom panels illustrate the time course for AV-2-192 and AV-2-197, respectively, to inhibit METH-evoked fractional DA release. Each striatal slice was superfused with either a single concentration (0.3–30 μM) of AV-2-192 or AV-2-197 alone during the 30-min time period prior to the inclusion of 5 μM METH in the buffer. Arrow denotes time point at which analog was added to the buffer. METH was added to the buffer after 30 min and remained in the buffer for 15 min. METH control represents slices superfused with METH in the absence of analog. Data are mean (±SEM) pg/min/ml slice weight. *p <0.05 different from the METH control condition.
Figure 2.8. AV-2-192 and AV-2-197 inhibit methamphetamine-evoked total DA overflow from rat striatal slices.

Data are total DA overflow following methamphetamine administration as a function of concentration of AV-2-192 (top panel) and AV-2-197 (bottom panel). CON denotes slice superfused with 5 μM methamphetamine in absence of analog. Data are mean (±SEM) pg/ml/mg slice weight. *p <0.05 and **p <0.01 different from the methamphetamine control condition.
The current results obtained with superfusion of striatal slices with AV-2-192 and AV-2-197 alone (Figs. 2.6 and 2.7) are consistent with our previous findings when employing the parent compounds lobeline and lobelane. This family of compounds inhibits DA uptake at VMAT2 and promotes DA release from the synaptic vesicles via an interaction with VMAT2, presumably increasing the cytosolic concentration of DA. The increase in cytosolic DA is then exposed to intracellular MAO, an avid enzyme that metabolizes the cytosolic DA to DOPAC, and the observed outcome is an increase in DOPAC, but not DA, in superfusate (the extracellular compartment). Thus, this family of compounds clearly does not inhibit MAO activity. The increased DOPAC concentrations in superfusate, in combination with the lack of change in DA concentration, are the result of MAO metabolizing the increased cyotosolic DA as a component of the analog effect. Furthermore, consistent with the interaction of these lead analogs at the DA uptake site on VMAT2, methylation of the pyrrolidine N-atom was not a factor in the inhibition of METH-evoked DA release from intact striatal slices.
The results presented herein show that 2,5-disubstituted pyrrolidine analogs constitute a promising series of analogs for further preclinical evaluation as potential pharmacotherapeutics for METH addiction. Specifically, the pyrrolidine analogs with a short linker length (AV-2-192 and AV-2-197) afforded modest affinity at the DTBZ binding site on VMAT2, high affinity at DA uptake site, and competitive inhibition at the DA uptake site on VMAT2; were not potent inhibitors of DAT function; and, moreover, inhibited the effect of METH to release DA from intact striatal slices. Future studies will determine the ability of these lead analogs in the 2,5-disubstituted pyrrolidine series to decrease reinforcement produced by METH in behavioral studies.
Acknowledgments
This research was supported by NIH grants U01 DA013519, UL1TR000117, and T32 DA016176.
ABBREVIATIONS
- AV-2-192
cis-2,5-di(2-benzyl)-pyrrolidine
- AV-2-197
1-methyl-cis-2,5-di(2-benzyl)-pyrrolidine
- CNS
central nervous system
- CHO
Chinese hamster ovary
- CPP
conditioned place preference
- DA
dopamine
- DAT
dopamine transporter
- DTBZ
dihydrotetrabenazine
- DOPAC
dihydroxyphenylacetic acid
- GZ-793A
(R)-3-[2,6-cis-di(4-methoxyphenethyl)piperidin-1-yl]propane-1,2-diol
- METH
methamphetamine
- MTD
meso-transdiene
- MLA
methyllycaconitine
- MAO
monoamine oxidase
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
N-methyl-1,2,3,6-tetrahydropyridine
- NIC
nicotine
- nAChR
nicotinic acetylcholine receptor
- SERT
serotonin transporter
- TBZ
tetrabenazine
- UKMH-105
(3Z,5E)-3,5-bis(2,4-dichlorobenzylidene)-1-methylpiperidine
- UKMH-106
(3Z,5Z)-3,5-bis(2,4-dichlorobenzylidene)-1-methylpiperidine
- VMAT
vesicular monoamine transporter
- VMAT2
vesicular monoamine transporter-2
Footnotes
CONFLICT OF INTEREST
The University of Kentucky holds patents on the compounds described in the current work, some of which have been licensed by Yaupon Therapeutics/Ceptaris Inc. A potential royalty stream to LPD and PAC may occur consistent with the University of Kentucky policy. Both LPD and PAC are founders of, and have financial interest in, Yaupon Therapeutics/Ceptaris Inc.
References
- Alvers KM, Beckmann JS, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. The effect of VMAT2 inhibitor GZ-793A on the reinstatement of methamphetamine-seeking in rats. Psychopharmacology. 2012;224:255–262. doi: 10.1007/s00213-012-2748-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PH. Biochemical and pharmacological characterization of [3H]GBR 12935 binding in vitro to rat striatal membranes: Labeling of the dopamine uptake complex. Journal of Neurochemistry. 1987;48:1887–1896. doi: 10.1111/j.1471-4159.1987.tb05752.x. [DOI] [PubMed] [Google Scholar]
- Ary TE, Komiskey HL. Phencyclidine: Effect on the accumulation of 3H-dopamine in synaptic vesicles. Life Sciences. 1980;26:575–578. doi: 10.1016/0024-3205(80)90322-7. [DOI] [PubMed] [Google Scholar]
- Barlow RB, Johnson O. Relations between structure and nicotine-like activity: X-ray crystal structure analysis of (−)-cytisine and (−)-lobeline hydrochloride and a comparison with (−)-nicotine and other nicotine-like compounds. British Journal of Pharmacology. 1989;98:799–808. doi: 10.1111/j.1476-5381.1989.tb14608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann JS, Denehy ED, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. The effect of a novel VMAT2 inhibitor, GZ-793A, on methamphetamine reward in rats. Psychopharmacology. 2012;220:295–403. doi: 10.1007/s00213-011-2488-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann JS, Siripurapu KB, Nickell JR, Horton DB, Denehy ED, Vartak A, et al. The novel pyrrolidine nor-lobelane analog UKCP-110 [cis-2,5-di-(2-phenethyl)-pyrrolidine hydrochloride] inhibits VMAT2 function, methamphetamine-evoked dopamine release, and methamphetamine self-administration in rats. The Journal of Pharmacology and Experimental Therapeutics. 2010;335:841–851. doi: 10.1124/jpet.110.172742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar D, Zuk R, Glinka Y. Dopamine neurotoxicity: Inhibition of mitochondrial respiration. Journal of Neurochemistry. 1995;64:718–723. doi: 10.1046/j.1471-4159.1995.64020718.x. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hanson GR, Fleckenstein AE. Methamphetamine rapidly decreases vesicular dopamine uptake. Journal of Neurochemistry. 2000;74:2221–2223. doi: 10.1046/j.1471-4159.2000.0742221.x. [DOI] [PubMed] [Google Scholar]
- Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter-2: A novel mechanism for cocaine and other psychostimulants. The Journal of Pharmacology and Experimental Therapeutics. 2001;296:762–767. [PubMed] [Google Scholar]
- Carr GD, White NM. Conditioned place preference from intra-accumbens but not intra-caudate amphetamine injections. Life Sciences. 1983;33:2551–2557. doi: 10.1016/0024-3205(83)90165-0. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Edwards RH, Fonnum F. Vesicular neurotransmitter transporters as targets for endogenous and exogenous toxic substances. Annual Review of Pharmacology and Toxicology. 2008;48:277–301. doi: 10.1146/annurev.pharmtox.46.120604.141146. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Patrick GS, Creasy KR, Martin BR. Pharmacology of lobeline, a nicotinic receptor ligand. The Journal of Pharmacology and Experimental Therapeutics. 1997;282:410–419. [PubMed] [Google Scholar]
- Darchen F, Scherman D, Henry JP. Reserpine binding to chromaffin granules suggests the existence of two conformations of the monoamine transporter. Biochemistry. 1989;28:1692–1697. doi: 10.1021/bi00430a040. [DOI] [PubMed] [Google Scholar]
- DASIS, Drug and Alcohol Services Information System (DASIS) The DASIS Report. Primary methamphetamine/amphetamine admissions to substance abuse treatment: 2005. 2008 http://www.oas.samhsa.gov/2k8/methamphetamineTx/meth.htm.
- Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, et al. Dopamine and drug addiction: The nucleus accumbens shell connection. Neuropharmacology. 2004;47:227–241. doi: 10.1016/j.neuropharm.2004.06.032. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Crooks PA. A novel mechanism and potential use for lobeline as a treatment for psychostimulant abuse. Biochemical Pharmacology. 2002;63:89–98. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- Eiden LE, Weihe E. VMAT2: A dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse. Annals of the New York Academy of Sciences. 2011;1216:86–98. doi: 10.1111/j.1749-6632.2010.05906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JD, Eiden LE, Hoffman BJ. Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10993–10997. doi: 10.1073/pnas.89.22.10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JD, Schafer MK, Bonner TI, Eiden LE, Weihe E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5166–5171. doi: 10.1073/pnas.93.10.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn JP, III, Edwards RH. Individual residues contribute to multiple differences in ligand recognition between vesicular monoamine transporters 1 and 2. The Journal of Biological Chemistry. 1997;272:16301–16307. doi: 10.1074/jbc.272.26.16301. [DOI] [PubMed] [Google Scholar]
- Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: Evidence for an exchange diffusion model. The Journal of Pharmacology and Experimental Therapeutics. 1979;208:203–209. [PubMed] [Google Scholar]
- Flammia D, Dukat M, Damaj MI, Martin B, Glennon RA. Lobeline: Structure-affinity investigation of nicotinic acetylcholinergic receptor binding. Journal of Medicinal Chemistry. 1999;42:3726–3731. doi: 10.1021/jm990286m. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annual Review of Pharmacology and Toxicology. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun B, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Frey KA, Koeppe RA, Kilbourn MR, Vander Borght TM, Albin RL, Gilman S, et al. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Annals of Neurology. 1996;40:873–884. doi: 10.1002/ana.410400609. [DOI] [PubMed] [Google Scholar]
- Frize ED. Mental depression in hypertensive patients treated for long periods with high doses of reserpine. The New England Journal of Medicine. 1954;251:1006–1008. doi: 10.1056/NEJM195412162512504. [DOI] [PubMed] [Google Scholar]
- Glatt CE, DeYoung JA, Delgado S, Service SK, Giacomini KM, Edwards RH, et al. Screening a large reference sample to identify very low frequency sequence variants: Comparison between two genes. Nature Genetics. 2001;27:435–438. doi: 10.1038/86948. [DOI] [PubMed] [Google Scholar]
- Glatt CE, Wahner AD, White DJ, Ruiz-Linares A, Ritz B. Gain-of-function haplotypes in the vesicular monoamine transporter promoter are protective for Parkinson disease in women. Human Molecular Genetics. 2006;15:299–305. doi: 10.1093/hmg/ddi445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacology. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart CL, Ward AS, Haney M, Foltin RW, Fischman MW. Methamphetamine self-administration by humans. Psychopharmacology. 2001;157:75–81. doi: 10.1007/s002130100738. [DOI] [PubMed] [Google Scholar]
- Harrod SB, Dwoskin LP, Crooks PA, Klebaur JE, Bardo MT. Lobeline attenuates d-methamphetamine self-administration in rats. The Journal of Pharmacology and Experimental Therapeutics. 2001;298:172–179. [PubMed] [Google Scholar]
- Harrod SB, Dwoskin LP, Green TA, Gehrke BJ, Bardo MT. Lobeline does not serve as a reinforcer in rats. Psychopharmacology. 2003;165:397–404. doi: 10.1007/s00213-002-1289-6. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MF. Reactive dopamine metabolites and neurotoxicity: Implications for Parkinson’s disease. Advances in Experimental Medicine and Biology. 1996;387:97–106. doi: 10.1007/978-1-4757-9480-9_13. [DOI] [PubMed] [Google Scholar]
- Henry JP, Gasnier B, Roisin MP, Isamber MF, Scherman D. Molecular pharmacology of the monoamine transporter of the chromaffin granule membrane. Annals of the New York Academy of Sciences. 1987;493:194–206. doi: 10.1111/j.1749-6632.1987.tb27201.x. [DOI] [PubMed] [Google Scholar]
- Hillarp NA. The release of catechol amines from the amine containing granules of the adrenal medulla. Acta Physiologica Scandinavica. 1958;43:292–302. doi: 10.1111/j.1748-1716.1958.tb01596.x. [DOI] [PubMed] [Google Scholar]
- Hiroi N, White NM. The lateral nucleus of the amygdala mediates expression of the amphetamine produced conditioned place preference. The Journal of Neuroscience. 1991;11:2107–2116. doi: 10.1523/JNEUROSCI.11-07-02107.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebel BG, Monaco AP, Hernandez L, Aulisi EF, Stanley BG, Lenard L. Self-injection of amphetamine directly into the brain. Psychopharmacology. 1983;81:158–163. doi: 10.1007/BF00429012. [DOI] [PubMed] [Google Scholar]
- Hogan KA, Staal RG, Sonsalla PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: Disparity between homogenates and vesicle preparations. Journal of Neurochemistry. 2000;74:2217–2220. doi: 10.1046/j.1471-4159.2000.0742217.x. [DOI] [PubMed] [Google Scholar]
- Horton DB, Nickell JR, Zheng G, Crooks PA, Dwoskin LP. GZ-793A, a lobelane analog, interacts with the vesicular monoamine transporter-2 to inhibit the effect of methamphetamine. Journal of Neurochemistry. 2013;127:177–186. doi: 10.1111/jnc.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton DB, Siripurapu KB, Norrholm SD, Culver JP, Hojahmat M, Beckmann JS, et al. meso-Transdiene analogs inhibit vesicular monoamine transporter-2 function and methamphetamine-evoked dopamine release. The Journal of Pharmacology and Experimental Therapeutics. 2011;336(3):940–951. doi: 10.1124/jpet.110.175117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton DB, Siripurapu KB, Zheng G, Crooks PA, Dwoskin LP. Novel N-1,2-dihydroxypropyl analogs of lobelane inhibit vesicular monoamine transporter-2 function and methamphetamine-evoked dopamine release. The Journal of Pharmacology and Experimental Therapeutics. 2011;339:286–297. doi: 10.1124/jpet.111.184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RG. Accumulation of biological amines into chromaffin granules: A model for hormone and neurotransmitter transport. Physiological Reviews. 1988;68:232–307. doi: 10.1152/physrev.1988.68.1.232. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Eshleman AJ, Meyers T, Neve KA, Janowsky A. Substrate-and cell-specific effects of uptake inhibitors on human dopamine and serotonin transporter-mediated efflux. Synapse. 1998;30:97–106. doi: 10.1002/(SICI)1098-2396(199809)30:1<97::AID-SYN12>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Research. Brain Research Reviews. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Critical Reviews in Biochemistry. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Kazumata K, Dhawan V, Chaly T, Antonini A, Marqouleff C, Belakhlef A, et al. Dopamine transporter imaging with fluorine-18-FPCIT and PET. Journal of Nuclear Medicine. 1998;39:1521–1530. [PubMed] [Google Scholar]
- Kilbourn MR, Butch ER, Desmond T, Sherman P, Harris PE, Frey KA. In vivo [11H]dihydrotetrabenazine binding in rat striatum: Sensitivity to dopamine concentrations. Nuclear Medicine and Biology. 2010;37:3–8. doi: 10.1016/j.nucmedbio.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbourn M, Lee L, Vander Borght T, Jewett D, Frey K. Binding of alpha-dihydrotetrabenazine to the vesicular monoamine transporter is stereospecific. European Journal of Pharmacology. 1995;278:249–252. doi: 10.1016/0014-2999(95)00162-e. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neural mechanisms of drug reinforcement. Annals of the New York Academy of Sciences. 1992;654:171–191. doi: 10.1111/j.1749-6632.1992.tb25966.x. [DOI] [PubMed] [Google Scholar]
- Liang NY, Rutledge CO. Comparison of the release of [3H]dopamine from isolated corpus striatum by amphetamine, fenfluramine and unlabelled dopamine. Biochemical Pharmacology. 1982;31:983–992. doi: 10.1016/0006-2952(82)90332-x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Edwards RH. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annual Review of Neuroscience. 1997;20:125–156. doi: 10.1146/annurev.neuro.20.1.125. [DOI] [PubMed] [Google Scholar]
- Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, et al. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- Liu Y, Roghani A, Edwards RH. Gene transfer of a reserpine-sensitive mechanism of resistance to N-methyl-4-phenylpyridinium. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9074–9078. doi: 10.1073/pnas.89.19.9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyness WH, Friedle NM, Moore KE. Destruction of dopaminergic nerve terminals in nucleus accumbens: Effect on d-amphetamine self-administration. Pharmacology, Biochemistry, and Behavior. 1979;11:553–556. doi: 10.1016/0091-3057(79)90040-6. [DOI] [PubMed] [Google Scholar]
- Madras BK, Meltzer PC, Liang AY, Elmaleh DR, Babich J, Fischman AJ. Altropane, a SPECT or PET imaging probe for dopamine neurons: I. Dopamine transporter binding in primate brain. Synapse. 1998;29:93–104. doi: 10.1002/(SICI)1098-2396(199806)29:2<93::AID-SYN1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Mantle TJ, Tipton KF, Garrett NJ. Inhibition of monoamine oxidase by amphetamine and related compounds. Biochemical Pharmacology. 1976;25:2073–2077. doi: 10.1016/0006-2952(76)90432-9. [DOI] [PubMed] [Google Scholar]
- Matecka D, Lewis D, Rothman RB, Dersch M, Wojnicki FH, Glowa JR, et al. Heteroaromatic analogs of 1-[2-(diphenylmethoxy)ethyl]- and 1-[2-[bis(4-fluorophenyl)-methoxy] ethyl]-4-(3-phenylpropyl) piperazines (GBR 12935 and GBR 12909) as high-affinity dopamine reuptake inhibitors. Journal of Medicinal Chemistry. 1997;40:705–716. doi: 10.1021/jm9606599. [DOI] [PubMed] [Google Scholar]
- Merickel A, Rosandich P, Peter D, Edwards RH. Identification of residues involved in substrate recognition by a vesicular monoamine transporter. The Journal of Biological Chemistry. 1995;270:25798–25804. doi: 10.1074/jbc.270.43.25798. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Neugebauer NM, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. Effects of VMAT2 inhibitors lobeline and GZ-793A on methamphetamine-induced changes in dopamine release, metabolism and synthesis in vivo. Journal of Neurochemistry. 2013;127:187–198. doi: 10.1111/jnc.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DK, Crooks PA, Dwoskin LP. Lobeline inhibits nicotine-evoked [3H]dopamine overflow from rat striatal slices and nicotine-evoked 86RB+ efflux from thalamic synaptosomes. Neuropharmacology. 2000;39:2654–2662. doi: 10.1016/s0028-3908(00)00140-4. [DOI] [PubMed] [Google Scholar]
- Miller DK, Crooks PA, Teng LH, Witkin JM, Munzar P, Goldberg SR, et al. Lobeline inhibits the neuronal and behavioral effects of amphetamine. The Journal of Pharmacology and Experimental Therapeutics. 2001;296:1023–1034. [PubMed] [Google Scholar]
- Miller DK, Crooks PA, Zheng G, Grinevich VP, Norrholm SD, Dwoskin LP. Lobeline analogs with enhanced affinity and selectivity for plasmalemma and vesicular monoamine transporters and diminished affinity at α4β2* and α7* nicotinic receptors. The Journal of Pharmacology and Experimental Therapeutics. 2004;310:1035–1045. doi: 10.1124/jpet.104.068098. [DOI] [PubMed] [Google Scholar]
- Miller GW, Gainetdinov RR, Levey AI, Caron MG. Dopamine transporter and neuronal injury. Trends in Pharmacological Sciences. 1999;20:424–429. doi: 10.1016/s0165-6147(99)01379-6. [DOI] [PubMed] [Google Scholar]
- Miller DK, Green TA, Harrod SB, Bardo MT, Crooks PA, Dwoskin LP. Effects of lobeline on methamphetamine and nicotine-induced hyperactivity and sensitization. Drug and Alcohol Dependence. 2000;60:151. [Google Scholar]
- Naudon L, Raisman-Vozari R, Edwards RH, Leroux-Nicollet I, Peter D, Liu Y, et al. Reserpine affects differentially the density of the vesicular monoamine transporter and dihydrotetrabenazine binding sites. The European Journal of Neuroscience. 1996;8:842–846. doi: 10.1111/j.1460-9568.1996.tb01271.x. [DOI] [PubMed] [Google Scholar]
- Neugebauer NM, Harrod SB, Stairs DJ, Crooks PA, Dwoskin LP, Bardo MT. Lobelane decreases methamphetamine self-administration in rats. European Journal of Pharmacology. 2007;571:33–38. doi: 10.1016/j.ejphar.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell JR, Krishnamurthy S, Norrholm S, Deaciuc G, Siripurapu KB, Zheng G, et al. Lobelane inhibits methamphetamine-evoked dopamine release via inhibition of the vesicular monoamine transporter-2. The Journal of Pharmacology and Experimental Therapeutics. 2010;332:612–621. doi: 10.1124/jpet.109.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell JR, Zheng G, Deaciuc AG, Crooks PA, Dwoskin LP. Phenyl ring-substituted lobelane analogs: Inhibition of [3H]dopamine uptake at the vesicular monoamine transporter-2. The Journal of Pharmacology and Experimental Therapeutics. 2011;336(3):724–733. doi: 10.1124/jpet.110.172882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njus D, Kelley PM, Harnadek GJ. Bioenergetics of secretory vesicles. Biochimica et Biophysica Acta. 1986;853:237–265. doi: 10.1016/0304-4173(87)90003-6. [DOI] [PubMed] [Google Scholar]
- Nordahl TE, Salo R, Leamon M. Neuropsychological effects of chronic methamphetamine use on neurotransmitters and cognition: A review. The Journal of Neuropsychiatry and Clinical Neurosciences. 2003;15:317–325. doi: 10.1176/jnp.15.3.317. [DOI] [PubMed] [Google Scholar]
- Partilla JS, Dempsey AG, Nagpal AS, Blough BE, Baumann MH, Rothman RB. Interaction of amphetamines and related compounds at the vesicular monoamine transporter. The Journal of Pharmacology and Experimental Therapeutics. 2006;319:237–246. doi: 10.1124/jpet.106.103622. [DOI] [PubMed] [Google Scholar]
- Peter D, Jimenez J, Liu Y, Kim J, Edwards RH. The chromaffin granule and synaptic vesicle amine transporters differ in substrate recognition and sensitivity to inhibitors. The Journal of Biological Chemistry. 1994;269:7231–7237. [PubMed] [Google Scholar]
- Peter D, Liu Y, Sternini C, de Giorgio R, Brecha N, Edwards RH. Differential expression of two vesicular monoamine transporters. The Journal of Neuroscience. 1995;15:6179–6188. doi: 10.1523/JNEUROSCI.15-09-06179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippu A, Beyer J. Dopamine and noradrenaline transport into subcellular vesicles of the striatum. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1973;278:387–402. doi: 10.1007/BF00501482. [DOI] [PubMed] [Google Scholar]
- Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: Plasmalemmal dopamine transporter versus vesicular monoamine transporter. Molecular Pharmacology. 1995;47:368–373. [PubMed] [Google Scholar]
- Riddle EL, Fleckenstein AE, Hanson GR. Role of monoamine transporters in mediating psychostimulant effects. The AAPS Journal. 2005;7:847–851. doi: 10.1208/aapsj070481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates: Implications for primary pulmonary hypertension. Circulation. 1999;100:869–875. doi: 10.1161/01.cir.100.8.869. [DOI] [PubMed] [Google Scholar]
- Sagne C, Isambert MF, Vandekerckhove J, Henry JP, Gasnier B. The photoactivatable inhibitor 7-azido-8-iodoketanserin labels the N terminus of the vesicular monoamine transporter from bovine chromaffin granules. Biochemistry. 1997;36:3345–3352. doi: 10.1021/bi9623439. [DOI] [PubMed] [Google Scholar]
- Schuldiner S, Liu Y, Edwards RH. Reserpine binding to a vesicular amine transporter expressed in Chinese hamster ovary fibroblasts. The Journal of Biological Chemistry. 1993;268:29–34. [PubMed] [Google Scholar]
- Sievert MK, Ruoho AE. Peptide mapping of the [125I] Iodoazidoketanserin and [125I]2-N-[(3′-iodo-4′-azidophenyl) propionyl] tetrabenazine binding sites for the synaptic vesicle monoamine transporter. The Journal of Biological Chemistry. 1997;272:26049–26055. doi: 10.1074/jbc.272.41.26049. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. The Journal of Neuroscience. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, et al. VMAT2 knockout mice: Heterozygotes display reduced locomotion, and enhanced MPTP toxicity. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng LH, Crooks PA, Dwoskin LP. Lobeline displaces [3H] dihydrotetrabenazine binding and releases [3H]dopamine from rat striatal synaptic vesicles: Comparison with d-amphetamine. Journal of Neurochemistry. 1998;71:258–265. doi: 10.1046/j.1471-4159.1998.71010258.x. [DOI] [PubMed] [Google Scholar]
- Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. Lobeline and nicotine evoke [3H]overflow from rat striatal slices preloaded with [3H]dopamine: Differential inhibition of synaptosomal and vesicular [3H]dopamine uptake. The Journal of Pharmacology and Experimental Therapeutics. 1997;280:1432–1444. [PubMed] [Google Scholar]
- Thiriot DS, Ruoho AE. Mutagenesis and derivatization of human monoamine vesicle transporter-2 (VMAT2) cysteines identifies transporter domains involved in tetrabenazine binding and substrate transport. The Journal of Biological Chemistry. 2001;276:27304–27315. doi: 10.1074/jbc.M103947200. [DOI] [PubMed] [Google Scholar]
- Uhl GR. Hypothesis: The role of dopaminergic transporters in selective vulnerability of cells in Parkinson’s disease. Annals of Neurology. 1998;43:555–560. doi: 10.1002/ana.410430503. [DOI] [PubMed] [Google Scholar]
- Vezina P. D1 dopamine receptor activation is necessary for the induction of sensitization by amphetamine in the ventral tegmental area. The Journal of Neuroscience. 1996;16:2411–2420. doi: 10.1523/JNEUROSCI.16-07-02411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, et al. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- Weaver JA, Deupree JD. Conditions required for reserpine binding to the catecholamine transporter on chromaffin granule ghosts. European Journal of Pharmacology. 1982;80:437–438. doi: 10.1016/0014-2999(82)90093-0. [DOI] [PubMed] [Google Scholar]
- Weihe E, Schafer MK, Erickson JD, Eiden LE. Localization of vesicular monoamine transporter isoforms (VMAT1 and VMAT2) to endocrine cells and neurons in rat. Journal of Molecular Neuroscience. 1995;5:149–164. doi: 10.1007/BF02736730. [DOI] [PubMed] [Google Scholar]
- Wilhelm CJ, Johnson RA, Eshleman AJ, Janowsky A. Lobeline effects on tonic and methamphetamine-induced dopamine release. Biochemical Pharmacology. 2008;75:1411–1415. doi: 10.1016/j.bcp.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmouth CE, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. Oral administration of GZ-793A, a VMAT2 inhibitor, decreases methamphetamine self-administration in rats. Pharmacology, Biochemistry, and Behavior. 2013;112:29–33. doi: 10.1016/j.pbb.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimalasena K. Vesicular monoamine transporters: Structure-function, pharmacology and medicinal chemistry. Medicinal Research Reviews. 2010;31:483–519. doi: 10.1002/med.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychological Review. 1987;94:469–492. [PubMed] [Google Scholar]
- Wise RA, Hoffman DC. Localization of drug reward mechanisms by intracranial injections. Synapse. 1992;10:247–263. doi: 10.1002/syn.890100307. [DOI] [PubMed] [Google Scholar]
- Xu DD, Mo ZX, Yung KK, Yang Y, Leung AW. Individual and combined effects of methamphetamine and ketamine on conditioned place preference and NR1 receptor phosphorylation in rats. Neurosignals. 2008;15:322–331. doi: 10.1159/000127492. [DOI] [PubMed] [Google Scholar]
- Yokel RA, Pickens R. Self-administration of optical isomers of amphetamine and methylamphetamine by rats. The Journal of Pharmacology and Experimental Therapeutics. 1973;187:27–33. [PubMed] [Google Scholar]
- Zheng G, Dwoskin LP, Deaciuc AG, Norrholm SD, Crooks PA. Defunctionalized lobeline analogues: Structure-activity of novel ligands for the vesicular monoamine transporter. Journal of Medicinal Chemistry. 2005;48:5551–5560. doi: 10.1021/jm0501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dwoskin LP, Deaciuc AG, Zhu J, Jones MD, Crooks PA. Lobelane analogues as novel ligands for the vesicular monoamine transporter-2. Bioorganic & Medicinal Chemistry Letters. 2005;13:3899–3909. doi: 10.1016/j.bmc.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]