

Abstract
Tremendous gains and novel methods are often developed when people are challenged to do something new or difficult. This process is enhanced when people compete against each other-this can be seen in sport as well as in science and technology (e.g. the space race). The SAMPL challenges, like the CASP challenges, aim to challenge modellers and software developers to develop new ways of looking at molecular interactions so the community as a whole can progress in the accurate prediction of these interactions. In order for this challenge to occur, data must be supplied so the prospective test can be done. We have supplied unpublished data related to a drug discovery program run several years ago on HIV integrase for the SAMPL4 challenge. This paper describes the methods used to obtain these data and the chemistry involved.
Introduction
The AIDS epidemic has caused over 32 million deaths and over 33 million people are currently infected with HIV (WHO data, http://UNAIDS.org). Small molecule therapeutics against several different protein targets of the HIV virus have been developed over the past two decades [1–4] culminating in a drug combination regimen referred to as Highly Active AntiRetroviral Therapies (HAART) used to treat AIDS. However, these therapies only slow the replication of the virus in patients and new forms of the virus have appeared that are resistant to all the drugs to date [5, 6], so there is a continuing need for new drugs. The integrase enzyme (IN) is critical to the viral life cycle as it is required for the integration of viral DNA into the host chromatin, which in turn is required for formation of new copies of the virus. Integrase performs two catalytic functions termed “3′ processing”-cleaving two nucleotides off of the viral cDNA in a sequence-specific manner to generate “sticky ends” and the “strand transfer reaction”-which covalently attaches, or integrates, the cleaved viral cDNA into human genomic DNA, in a non-sequence-specific manner. There are already three molecules [2] that block the catalytic site in the integrase catalytic core domain (CCD) that have been approved for human use. The structure of HIV integrase has been studied in detail [7–10], and it is most often found as a dimer or tetramer in solution, with the tetrameric form thought to be the active form in the cell. Several pockets in the CCD have been identified to which small molecules have been shown to bind and inhibit enzymatic activity [11–13]. In addition to the CCD, integrase also contains N-terminal and C-terminal domains that are important for DNA binding (to both the viral DNA and cellular DNA).
The integrase protein forms a complex called the pre-integration complex (PIC) with viral DNA and several cellular factors from the host [10] [14]. The cellular protein Lens Epithelium Derived Growth Factor (LEDGF/p75) is normally part of the PIC complex and LEDGF/p75 has a conserved integrase binding domain (IBD, residues 347-429) [15]. LEDGF/p75 facilitates association between the PIC complex with the host chromosome, and in-vitro measures have shown the enhancement of binding of HIV integrase to DNA in the presence of LEDGF/p75 to be 30 to 50 fold [16]. The PWWP domain of LEDGF/p75 has been shown to have a role in localizing the site of integration to genes, such that a PWWP domain deletion mutant leads to a loss of interaction with condensed chromatin and reduced viral replication [17]. When HIV integrase is in a dimeric form, a pocket is created that binds to the IBD loop (residues 362 to 369). Peptides derived from the sequence of the IBD loop have been shown to bind to HIV integrase with micromolar affinity [14] and to compete with IBD binding. The structures of several peptide complexes have been determined and several key interactions are shared with small molecules that bind in this pocket [15]. In addition, full length integrase and LEDGF/p75 make further interactions through the N-terminal domain of integrase [18, 19]. It has recently been shown, through time of addition studies, that blocking formation of the integrase–LEDGF/p75 complex may prevent effective viral maturation, blocking the formation of competent viral particles [20]. Consistent with the role of LEDGF/p75 in the progression and pathogenesis of HIV is an association that has been shown between polymorphisms in the psip1 gene that codes for the LEDGF/p75 protein and variation in serologic levels of HIV virus [21].
At the commencement of our project in 2007, we were interested in identifying molecules that bound to any site on the CCD. Indeed as a result of the fragment screen we identified molecules that bound to the ‘fragment’ pocket [22], another pocket that we described as the Y3 site [13] and the LEDGF/p75 binding site [23] (figure 1). This year’s SAMPL4 challenge is based on the compounds that were developed to bind to HIV integrase during the intermediate stage of the program conducted by CSIRO and Avexa Ltd. Various aspects of this program have been published previously [13, 22–24] but the compounds selected for this challenge are in general not incorporated in these previous publications. The compounds were developed from a fragment screen run against the Maybridge fragment library and compounds were found in multiple sites on HIV integrase.
Figure 1.

To the left is a surface representation of the HIV integrase CCD dimer with three of the binding sites highlighted in different colors: the fragment pocket in orange, the Y3 site in yellow and the LEDGF/p75 binding pocket in light blue. To the right in the same orientation is the HIV integrase CCD dimer represented with the residues as sticks, with the same three sites highlighted in the same colors, but with the individual protomers of the dimer colored in green and cyan. This shows that the fragment pocket and the LEDGF/p75 binding sites are at the dimer interface whereas the Y3 site is found in each of the individual protomers.
Experimental methods
N-terminally hexa-His tagged catalytic core domain (CCD) IN (residues 50 to 210) containing the mutations C56S, F139D and F185H (core3H) was cloned into the E. coli expression vector pET28b(+) (Novagen) and expressed and purified with the His-tag retained on the protein. Compounds were tested for affinity using SPR on a GE Healthcare Biacore T200 machine essentially as described previously [23]. Briefly, the ‘minimally biotinylated’ core3H protein was captured onto a Streptavidin chip surface in SPR capture buffer by injecting at 5 μL/min for 5 minutes over a single flow cell, typically resulting in immobilization of approximately 2300 RU of target protein. All SPR binding experiments were performed at 20 C in SPR binding buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.05 % (v/v) Tween20, 5 mM DTT, 10 mM MgCl2, 5% (v/v) DMSO). Small molecule stock solutions were serially diluted (2-fold) in SPR binding buffer and injected for 30 seconds contact time at 60 μL/min and then allowed to dissociate for 60 seconds. The entire concentration series for each compound was performed in triplicate.. Raw sensorgram data were processed, solvent corrected and double referenced using Scrubber software (BioLogic Software, Australia). For easy comparison between data sets, all experiments were normalized using a normalization formula of Giannetti et al. [25]. To determine the binding affinity (KD), responses at equilibrium for each analyte were fitted to a 1:1 steady state affinity model available within Scrubber.
All crystals were grown in the Collaborative Crystallisation Centre (C3, CSIRO) using the core3H CCD construct of the integrase protein, purified using a Ni-column, dialysis and gel filtration. The protein at 5.5 mg/mL concentration was crystallized in 1.6 to 2.0 M ammonium sulfate in sodium acetate buffer, pH 5.0 to 5.8 at 100 mM concentration. Crystals formed in 3 to 10 days at 20 C and were determined to be in spacegroup P312. Crystals were soaked with compounds for 12–48 hours prior to data collection at the Australian Synchrotron (at the MX-1 beamline) in a cryo-buffer containing 100 mM sodium acetate pH 5.5, 1.75 M ammonium sulfate, 25% (v/v) ethylene glycol and 5% (v/v) DMSO. Data were indexed using Mosflm [26], scaled using SCALA [27] and molecular replacement was done using Phaser [28]. The models were re-built manually using Coot [29] and refined using Refmac [30]. The compounds were fit into density using Afitt (OpenEye Scientific Software). Some compounds were found, based on inspection of OpenEye Shape toolkit shape overlays onto the refined structures, to be in higher energy conformations, and these were refit into the density and subsequently re-refined (using Refmac with dictionaries from Afitt).
Compounds were considered to be ‘non-binders’ based on two basic criteria: 1) if the binding affinity (KD) estimated by SPR was greater than 2 mM; 2) if there was no or ambiguous electron density, it was considered to be a non-binder for crystallography. True non-binders had both no electron density and KD > 2 mM. In reality, few compounds that did not bind by SPR were soaked into crystals as the SPR was almost always done prior to the crystallographic work and we tended to focus on the most promising compounds for the project.
Discussion
Background chemistry: Concurrent to our work, several groups designed small molecule inhibitors of the LEDGF-integrase interaction [12, 31–33].
Starting from a catechol scaffold that is well known to chelate metals in the active site of HIV integrase, a series of benzamides were synthesized and led to N-(cyclohexylmethyl)-2,3-dihydroxy-5-(piperidin-1-ylsulfonyl) benzamide, that had an IC50 of 8μM and CC50 >40μM [32]. Recently, activity against infection of whole cells was reported and this compound also bound at the active site and inhibited strand transfer with an IC50 of 19μM [32].
In one study, a pharmacophore based on the pocket of integrase to which the IBD binds was used to screen a virtual set of 160,000 compounds and from further docking results, 25 compounds were purchased and assayed in an AlphaScreen bio-assay [12], leading to the identification of a lead series of 2-(quinolin-3yl)acetic acids as inhibitors [12]. One compound, CHIBA-3003 [34] [figure 2, compound 4], which inhibited the interaction of LEDGF with integrase in an AlphaScreen assay with an IC50 of 35μM was identified from a pharmacophore based on the interactions of the residues of the IBD (I365, D366 and L368). It was predicted from docking that the phenolic hydroxyl of CHIBA-3003 could make a hydrogen bond to the backbone NH of Q168, while the carboxylate made hydrogen bonds to the backbone NHs of E170 and H171. This latter interaction is comparable to that of the side chain carboxylate of the IBD residue D366 as seen in the crystal structure [11].
Figure 2.

Compounds described as binding at the LEDGF IBD binding site of HIV integrase, compound 1 CX04328 [12] crystal complex PDB 3LPT, compound 2 from De Luca 2009 [34] crystal complex PDB 3LPU, Compound 3 from Serrao 3013 [35], compound 4 CHIBA3003 [34].
One lead compound of this series (compound 6* [20] [12], also called CX04328 (compound 1, figure 2) had an IC50 of 1.37μM and an EC50 of 2.73 or 3.45μM for HIV-1 infection of MT-4 or PBMC cells, respectively. This compound was crystallized with the F185K mutant of the CCD domain of HIV integrase and shown to form hydrogen bonds with residues Glu170, His171 and Thr174 of HIV integrase (PDB: 3LPU) [20] [12]. It was observed that the side chain of A128 packs against the molecule. It is notable that these interactions are similar to those proposed by modelling for CHIBA3003. It is possible that this compound also binds to the active site of intergase since it was shown to have an IC50 of 19.5μM for inhibition of the strand transfer activity of HIV integrase.
A thieno[2,3]pyridine series was evolved from this work, with a lead that had an IC50 0.58μM in MT-4 cells, an EC50 0.76μM and a CC50 of 72.1μM. Further assay of these inhibitors with known mutants of HIV integrase, A128T in HIV-1 and A128M in HIV-2, showed more than 100 fold resistance to these compounds, suggesting that the binding of these involves interactions in addition to those of amino acids 170, 171 and 174 [20]. The compound CHI-1043 inhibited at 0.14μM in the active site directed strand transfer assay, but only 36μM in the AlphaScreen assay for binding at the LEDGF site [31].
High throughput screening identified a series of 5-carbonyl-1H-imidazole-4-carboxamide compounds, and follow-up led to compound 15 [figure 2, compound 3] with an IC50 of 400nM in an AlphaScreen assay, which had no effect on an active site (strand transfer) assay [35], although these compounds were inactive in whole cell assays up to 20μM. From docking studies, it was proposed that these compounds formed hydrogen bonding interactions with the backbone NH of E170 and the imidazole nitrogen of H171 and the backbone oxygen of Q168, as well hydrophobic interactions with a T125, A128 and W131 and W132.
History of the Avexa compounds
Based on the hits obtained from the initial fragment screen, several analogues were chosen from the CSIRO compound library and these were tested via SPR and crystallography for binding affinity and the location of binding, respectively (see Experimental methods). Compound 1 (figure 3) was found by SPR to have affinity 750μM for the core3H construct of the CCD. Based on compound 1 (figure 3) we decided to synthesize a series of 1H-inden-2(3H)-one (compounds of general formula 2, figure 3), and observed from the crystallography that in fact the aromatic ring does occupy the same hydrophobic pocket [PDB 3ZT3, table 2] as residue I365 in the LEDGF/p75 IBD loop [11]. To mimic the interaction of I365 even more closely we designed a series of compounds of formula 3 (figure 3).
Figure 3.

Evolution of the SAMPL4 project compounds. Identification of dioxolanes from the fragment screen, led to selection of compound 1 by shape matching. A series of analogues of compound 1 of general formula 2 and 3 were synthesized and tested. Compounds of formula 3 were designed as analogues of compounds seen to bind in the fragment pocket.
Table 2.

| |||
|---|---|---|---|
| Compound # | R | Binder | |
| 40911 | H | yes | |
| 62772 | CH2-phtalimide | no | |
| 62777 | CH2-NHCO-(4-Pyr) | ||
| 62778 | CH2-NHCO-CH2-imidazole | no | |
SAMPL4 compounds
Most of the compounds put forward for the SAMPL challenge had both good electron density and consistent binding, as determined by multiple SPR measurements. For the challenge we have used data derived from using the core3H integrase construct, because the alternative core4H construct has an additional Y131D mutation in the wall of the LEDGF/p75 pocket, which we had previously shown can modulate binding of compounds at this site [23]. Those compounds that did not show binding at better than 2 mM in the SPR assay were determined to be ‘non-binding’ and used as the non-binding controls for the challenge. SPR binding was not perfectly correlated with crystallographic binding. The experiments are run under different conditions and at different pH’s, so we would not expect perfect correlation (see Experimental methods). We found that as the affinities improved (low micromolar instead of high micromolar or low millimolar affinities) that the correlation between the different methods improved. Some compounds had limited solubility, which can have different effects on the SPR and crystallographic experiments.
There were cases, particularly where an aliphatic moiety was present (e.g. AVX17557), when the density for part of the compound was not as strong as for the rest of the compound. Enantiomeric compounds were generally not separated prior to either SPR or crystallographic experiments. This has potential implications for the determined binding affinity (with a single stereocenter, it can be up to 2x better than stated if one enantiomer of a pair binds and the other has no or very low affinity), but it generally not for the crystallographic experiments as the resolution was high enough to determine which enantiomer bound or if there was binding of both.
We started off with about 70 compounds that had reasonable crystallographic density and that had been run through SPR and bioassays and hadn’t been previously published. This list was cut down to those that had the best data (multiple SPR experiments, the best electron density, and therefore the most confident data) leaving 58 compounds. Some of the compounds had different names but were in fact the same (e.g. compounds that were made as different salts were given different names, such as AVX17557 and AVX17587) giving 57 for the SAMPL4 challenge.
As generally observed in our work (and that from other groups), all small molecules found to date (see figure 2) have a carboxylic acid that makes a virtually identical interaction to E170 and H171 as is seen with D366 of the IBD [11]. This charge interaction is key to the series developed here, to the series of peptides that have been shown to interact with the LEDGF binding site on HIV integrase, and key for the binding of other small molecules that have been developed by other groups. This was the basis for our first analogue 3, which demonstrated similar affinity to 2 in the AlphaScreen assay (AS) (270μM and 200μM respectively, table 1 [23]), and the SPR core3H assay (1435μM vs 1375μM, table 1 [23]) and clear density in the LEDGF/p75 site.
Table 1.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound # | X | Y | Z | R1 | R | Binder | SPR KD (μM) | |
| 17558 | CH2 | CH2 | NHCO(CH2)3NH2 | - | - | yes | ||
| 17556 (GL524384) | CH2 | CH2 | NHCO(CH2)4NH2 | - | - | no | ||
| 17557-17587-101124 | CH2 | CH2 | NHCO(CH2)5NH2 | - | - | yes | 752 ± 158 (17557) 1460 ± 89 (17587) |
|
| 101124 | CH2 | CH2 | NHCO(CH2)5NH2; X= CH2 olefin | - | - | yes | ||
| 38747 | CH2 | CH2 | NHCO(CH2)5NH2 | CHCH2CH2CO2H | - | Yes | ||
| 38749 | CH2 | CH2 | NHCO(CH2)3NH2 | CHCH2CH2CO2H | - | yes | ||
| 38748 | CH2 | CH2 | NHCO(CH2)4NH2 | CHCH2CH2CO2H | - | yes | ||
| 17375 | CH2 | CH2 | CH-OH | - | - | yes | racemic | |
| 40868 | N | CH2 | C=O | - | 6-Cl | No | ||
| 40883 | N | CH2 | C=O | - | 7-Cl | No | ||
| 17377 | N | CH2 | C=O | - | H | yes | ||
| 17260 | CH2 | N | C=O | - | H | yes | ||
| 17257 | N | CH2 | C=O | - | H | yes | No dioxolane ring | |
| 101140 | N | C=O | C=O | CH2 | 6-Br | yes | ||
| 17286 | =CH | NH | C=O | - | H | yes | E and Z olefin | |
| 17285 | =CH | NH | C=O | - | H | yes | No dioxolane ring | |
| 38743 | =CH | CH2 | CH-OH | CHCH2CH2CO2H | H | yes | Racemic at OH | |
| 38742 | =CH | CH2 | C=NH-OH | CHCH2CH2CO2H | H | yes | ||
Modelling suggested that compounds of formula 2 (figure 3) with a methylene linker would tend to favor conformations with the plane of the two bicyclic rings at an angle to each other. To allow greater flexibility, a longer alkyl linker was introduced into the series of formula 3 (figure 3 and table 3). All of the compounds of this series tested were found to give a crystal complex with integrase (table 3) and some had measurable activity in the AS assay (e.g. <0.3mM for AVX17560, table 3).
Table 3.

| |||
|---|---|---|---|
| Compound # | R | Binder | SPR KD (μM) |
| 40920 (GL5243-102) |
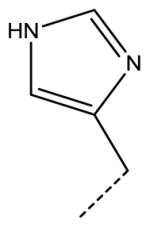
|
yes | |
| 40919 (GL5243-100) |

|
yes | |
| 40812 |

|
yes | 256 ± 12 |
| 17542 | ((CH2)4-NH2 | yes | |
| 17560 | (CH2)3-NH2 | yes | 268 ± 28 |
| GL5243104 |

|
yes | 432 ± 17 |
| GL5243106 | 4-(PhNH2) | yes | 200 ± 20 |
| GL5243100 |

|
yes | |
| 40811 |

|
yes | 481 ± 83 |
Based on fragments seen to bind in the fragment pocket of integrase [22], a series of compounds of formula 4 (figure 3 and table 6) were synthesized and tested. Some compounds of this series, such as AVX17287, had partial occupancy of both the fragment and LEDGF binding sites, whereas AVX17379 was observed in the crystals only in the LEDGF site. In comparison, AVX17285, which as an amide differs only from the amine compound AVX17379 by having the side chain carbonyl, was only observed with weak density at either site.
Table 6.

| ||
|---|---|---|
| Compound # | R | Binder |
| 17285 | X= NH2; R=H | yes |
| 17384 | X= NH2, R=tetrahydropyran | no |
| 17288 | X=OH, R=tetrahydropyran | no |
| 17287 |

|
yes |
| 17379 |

|
yes |
For those compounds with the 2,3-dihydrobenzo[b][1,4]dioxine ring (figure 4), the synthesis from the catechol gave a mixture of regioisomers, and these were tested as mixtures in the SPR assay. However in most cases where a crystal complex was formed, the 2-alkyl series was seen to preferentially bind (see figure 4 and table 1 and 4). The density was clear in several cases for the alkyl chain with carboxylic acid (e.g. AVX38747) but there were also cases where the density was unclear (e.g. AVX38743). We had hoped that a carboxylate in this position would interact with K173 but this was not observed.
Figure 4.

The 2,3-dihydrobenzo[b][1,4]dioxine series were synthesized as mixtures of regio-isomers with the propanoic acid chain in the 1 or 2 positions. These mixtures were tested in the bio-assay and soaked into the crystal, however the resulting crystal complexes only contained the 2-substituted compounds (left figure), which must preferentially bind in the crystal.
Table 4.

| ||||||
|---|---|---|---|---|---|---|
| R2 | R | y | R3 | SPR KD (μM) | ||
| 101118 | Me | H | CH2 | yes | ||
| allyl | (CH2)2CO2H | CH2 | CH(Me)2-Ph | yes | ||
| 38781 | allyl | (CH2)2CO2H | CH2 | CH2-Chx | yes | |
| 38787 | Me | (CH2)2CO2H | CH2 | CH2-Chx | yes | |
| allyl | (CH2)2CO2H | CH2 | CH2-Chx | |||
| Me | - | - | CH2-Chx | 3ZSY | >2000 (core3H+/4H) | |
| 38780 | allyl | (CH2)2CO2H | CH2 | CH2Ph(4-OMe) | yes | 1080 ± 123 |
| allyl | (CH2)2CO2H | CH2 | CH2Ph(4-OMe) | 3ZSV | >2000(core3H+/4H) | |
| 38786 | Me | (CH2)2CO2H | CH2 | CH2Ph(4-OMe) | 3ZSR | NT |
| 17715 | H | - | - | CH2Ph(4-OMe) | yes | |
| H | - | - | CH2Ph(4-OMe) | 3ZT1 | >2000 (core3H+/4H) | |
| Me | - | - | CH2Ph(4-OMe) | 3ZTO | 595;1180 (core3H/4H) | |
| allyl | - | - | CH2Ph(4-OMe) | 3ZCM | 519;880(core3H/4H) | |
| 40859 | allyl | - | - | CH(Pr)Ph(4-OMe) | 3ZSQ | 11;74(core3H/4H) |
| 40860 | allyl | - | - | CH[Ph(4-OMe)]2 | 3ZSO | 8;76(core3H/4H) |
| 38782 | allyl | (CH2)2CO2H | CH2 | CH2-cyclo-Bu | yes | |
| 38788 | Me | (CH2)2CO2H | CH2 | CH2-cyclo-Bu | yes | |
| 38783 | allyl | (CH2)2CO2H | CH2 | CH2-(-2furyl) | yes | |
| 38789 | me | (CH2)2CO2H | CH2 | CH2-(-2furyl) | yes | |
| 38785 | Me | (CH2)2CO2H | CH2 | CH2Ph | yes | |
| 38784 | Me | (CH2)2CO2H | CH2 | CH(Me)CH2CH3 | yes | |
| allyl | (CH2)2CO2H | CH2 | CH(Me)CH2CH3 | 3ZSW | >2000(core3H/4H) | |
| Me | - | - | CH(Me)CH2CH3 | 3ZSZ | >2000(core3H/4H) | |
| 38779 | allyl | (CH2)2CO2H | CH2 | CH(Me)CH2CH3 | yes | |
| 38708 | Me | - | - | CH2Ph | yes | |
| allyl | - | - | CH2Ph | 3ZSX | 1755;1065(core3H/4H) | |
| 101121 |

|
yes | 214 ± 35 | |||
| 101122 |

|
yes | ||||
| 101119 |

|
yes | 793 ± 50 | |||
NOTE:red = PLOS one paper; blue means in PLOS one and SAMPL
As described previously [23] synthesis unexpectedly gave a series of compounds, including AVX17715, (of formula 6, figure 5 and table 4) that were observed to bind to the LEDGF/p75 site. It was observed that the R5 group (formula 6, figure 5) was now in proximity of the hydrophobic site formed by W131 in the LEDGF/p75 site. Also the amide nitrogen of these molecules typically took part in a hydrogen bond with the backbone carbonyl of Q168 of the HIV integrase. The SAR for this series indicated that higher affinity compounds had an alkyl R3 group and so we considered that modulating the pKa and/or hydrogen donating capability of this nitrogen might influence binding, so a series of urea linked molecules (formula 7, figure 5 and table 6) was prepared. Also the 1,2-disubstituted phenyl ring of the compounds of formula 6 (figure 5) sat above the protein backbone, not making any clear interactions and so we replaced this by an R4 group in the series of formula 7 (figure 5).
Figure 5.

Two further series of compounds were designed and synthesized of formula 6, based on the crystal complex found for compound 17715 (table 4). The phenyl linker of formula 6 was replaced to try to improve binding giving a series of compounds of formula 7 (table 5). The two series share a common linker length, as can be seen for the atoms highlighted in red.
It is worth noting that our original hypothesis was that a 7 membered ring could be modelled to occupy the hydrophobic site occupied by I366 of the IBD and subsequently compound AVX38741 was found to bind, however as observed before it was the ring open form [23] (figure 6, right hand side), that was found in the crystal structure.
Figure 6.

AVX38741 was observed in the crystal complex with integrase to be the compound on the right hand side.
These data were given to the challenge organisers and were subsequently both manually and computationally vetted prior to inclusion in the challenge.
One of the more intriguing aspects to this particular challenge is that there were multiple binding sites for molecules to bind. This feature alone distinguishes this test from many others. Another distinguishing feature is that the non-binders looked very similar to the binders, many being in the same series and others using very similar scaffolds. We speculate that the fragment pocket was basically too small to bind much except fragments and a few promiscuous compounds. The Y3 pocket is a long shallow groove that picked up some promiscuous binders, but we couldn’t determine features that would give specificity to this site alone. The majority of the compounds bind in the LEDGF pocket as that is what we decided to target after the fragment screening and it was both large enough and had enough potential interactions that we could get specific binding of compounds.
Supplementary Material
Table 5.

| ||||
|---|---|---|---|---|
| Compound # | R2 | R3 | R4 | Binder |
| 38673 | Me | Ph | nBu | yes |
| 17628 | H | Ph | CH2Ph | no |
| 17629 (Et ester) | H | Ph | CH2Ph | no |
| 38669 | Me | Ph | H | no |
| 38670 | Me | Ph | CH2Ph(pOMe) | no |
| 38671 | Me | Ph | Me | no |
| 38672 | Me | Ph | tBu | yes |
| 38674 | Me | Ph | CH2Ph | yes |
Table 7.
assorted
| 17389 |

|
yes |
| 17679 |

|
yes |
| 38741 |

|
yes |
| 15988 |

|
yes |
| 17631 |

|
yes |
| 17287 |

|
yes |
| pC2A03 |

|
yes |
Acknowledgments
DLM acknowledges the financial support of the National Institutes of Health (1R15GM096257-01A1), and computing support from the UCI GreenPlanet cluster, supported in part by NSF Grant CHE-0840513
Footnotes
In the Supporting Information, we provide tables with compound IDs and isomeric SMILES strings for all of the compounds examined here, as well as for a more extensive set of nonbinders which were tested.
Contributor Information
Thomas S Peat, Email: tompeat@hotmail.com, CSIRO, Parkville, VIC, Australia.
Olan Dolezal, CSIRO, Parkville, VIC, Australia.
Janet Newman, CSIRO, Parkville, VIC, Australia.
David Mobley, UC Irvine, Irvine, CA USA.
John J Deadman, Chemocopeia Pty Ltd, Melbourne, Vic, Australia.
References
- 1.Arribas JR, Eron J. Advances in antiretroviral therapy. Curr Opin HIV AIDS. 2013;8(4):341–9. doi: 10.1097/COH.0b013e328361fabd. [DOI] [PubMed] [Google Scholar]
- 2.Metifiot M, Marchand C, Pommier Y. HIV integrase inhibitors: 20-year landmark and challenges. Adv Pharmacol. 2013;67:75–105. doi: 10.1016/B978-0-12-405880-4.00003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Messiaen P, et al. Clinical use of HIV integrase inhibitors: a systematic review and meta-analysis. PLoS One. 2013;8(1):e52562. doi: 10.1371/journal.pone.0052562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flexner C, Saag M. The antiretroviral drug pipeline: prospects and implications for future treatment research. Curr Opin HIV AIDS. 2013;8(6):572–8. doi: 10.1097/COH.0000000000000011. [DOI] [PubMed] [Google Scholar]
- 5.Geretti AM, Armenia D, Ceccherini-Silberstein F. Emerging patterns and implications of HIV-1 integrase inhibitor resistance. Curr Opin Infect Dis. 2012;25(6):677–86. doi: 10.1097/QCO.0b013e32835a1de7. [DOI] [PubMed] [Google Scholar]
- 6.Quashie PK, Mesplede T, Wainberg MA. Evolution of HIV integrase resistance mutations. Curr Opin Infect Dis. 2013;26(1):43–9. doi: 10.1097/QCO.0b013e32835ba81c. [DOI] [PubMed] [Google Scholar]
- 7.Jenkins TM, et al. Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc Natl Acad Sci U S A. 1995;92(13):6057–61. doi: 10.1073/pnas.92.13.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldgur Y, et al. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc Natl Acad Sci U S A. 1998;95(16):9150–4. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JC, et al. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci U S A. 2000;97(15):8233–8. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherepanov P, et al. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol. 2005;12(6):526–32. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 11.Cherepanov P, et al. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A. 2005;102(48):17308–13. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christ F, et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol. 2010;6(6):442–8. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 13.Rhodes DI, et al. Structural basis for a new mechanism of inhibition of HIV-1 integrase identified by fragment screening and structure-based design. Antivir Chem Chemother. 2011;21(4):155–68. doi: 10.3851/IMP1716. [DOI] [PubMed] [Google Scholar]
- 14.Bukrinsky MI, et al. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc Natl Acad Sci U S A. 1992;89(14):6580–4. doi: 10.1073/pnas.89.14.6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsiang M, et al. Affinities between the binding partners of the HIV-1 integrase dimer-lens epithelium-derived growth factor (IN dimer-LEDGF) complex. J Biol Chem. 2009;284(48):33580–99. doi: 10.1074/jbc.M109.040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNeely M, et al. In vitro DNA tethering of HIV-1 integrase by the transcriptional coactivator LEDGF/p75. J Mol Biol. 2011;410(5):811–30. doi: 10.1016/j.jmb.2011.03.073. [DOI] [PubMed] [Google Scholar]
- 17.Gijsbers R, et al. Role of the PWWP domain of lens epithelium-derived growth factor (LEDGF)/p75 cofactor in lentiviral integration targeting. J Biol Chem. 2011;286(48):41812–25. doi: 10.1074/jbc.M111.255711. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Maertens G, et al. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J Biol Chem. 2003;278(35):33528–39. doi: 10.1074/jbc.M303594200. [DOI] [PubMed] [Google Scholar]
- 19.Hare S, et al. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009;5(1):e1000259. doi: 10.1371/journal.ppat.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balakrishnan M, et al. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PLoS One. 2013;8(9):e74163. doi: 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madlala P, et al. Association of polymorphisms in the LEDGF/p75 gene (PSIP1) with susceptibility to HIV-1 infection and disease progression. AIDS. 2011;25(14):1711–9. doi: 10.1097/QAD.0b013e328349c693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wielens J, et al. Parallel screening of low molecular weight fragment libraries: do differences in methodology affect hit identification? J Biomol Screen. 2013;18(2):147–59. doi: 10.1177/1087057112465979. [DOI] [PubMed] [Google Scholar]
- 23.Peat TS, et al. Small molecule inhibitors of the LEDGF site of human immunodeficiency virus integrase identified by fragment screening and structure based design. PLoS One. 2012;7(7):e40147. doi: 10.1371/journal.pone.0040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhodes DI, et al. Crystal structures of novel allosteric peptide inhibitors of HIV integrase identify new interactions at the LEDGF binding site. Chembiochem. 2011;12(15):2311–5. doi: 10.1002/cbic.201100350. [DOI] [PubMed] [Google Scholar]
- 25.Giannetti AM, Koch BD, Browner MF. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J Med Chem. 2008;51(3):574–80. doi: 10.1021/jm700952v. [DOI] [PubMed] [Google Scholar]
- 26.Leslie AW, Powell H. Processing diffraction data with mosflm. In: Read R, Sussman J, editors. Evolving Methods for Macromolecular Crystallography. Springer Netherlands; 2007. pp. 41–51. [Google Scholar]
- 27.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 28.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, et al. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.De Luca L, et al. 4-[1-(4-Fluorobenzyl)-4-hydroxy-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoic acid as a prototype to develop dual inhibitors of HIV-1 integration process. Antiviral Res. 2011;92(1):102–7. doi: 10.1016/j.antiviral.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 32.Fan X, et al. Design of HIV-1 integrase inhibitors targeting the catalytic domain as well as its interaction with LEDGF/p75: a scaffold hopping approach using salicylate and catechol groups. Bioorg Med Chem. 2011;19(16):4935–52. doi: 10.1016/j.bmc.2011.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Luca L, et al. Inhibitors of the interactions between HIV-1 IN and the cofactor LEDGF/p75. ChemMedChem. 2011;6(7):1184–91. doi: 10.1002/cmdc.201100071. [DOI] [PubMed] [Google Scholar]
- 34.De Luca L, et al. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem. 2009;4(8):1311–6. doi: 10.1002/cmdc.200900070. [DOI] [PubMed] [Google Scholar]
- 35.Serrao E, et al. Discovery of a novel 5-carbonyl-1H-imidazole-4-carboxamide class of inhibitors of the HIV-1 integrase-LEDGF/p75 interaction. Bioorg Med Chem. 2013;21(19):5963–72. doi: 10.1016/j.bmc.2013.07.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.