

Abstract
While Burkitt lymphoma (BL) has a well-known defect in HLA class I-mediated antigen presentation, the exact role of BL-associated HLA class II in generating a poor CD4+ T-cell response remains unresolved. Here, we found that BL cells are deficient in their ability to optimally stimulate CD4+ T cells via the HLA class II pathway. This defect in CD4+ T-cell recognition was not associated with low levels of co-stimulatory molecules on BL cells, as addition of external co-stimulation failed to elicit CD4+ T-cell activation by BL. Further, the defect was not caused by faulty antigen/class II interaction, because antigenic peptides bound with measurable affinity to BL-associated class II molecules. Interestingly, functional class II–peptide complexes were formed at acidic pH 5·5, which restored immune recognition. Acidic buffer (pH 5·5) eluate from BL cells contained molecules that impaired class II-mediated antigen presentation and CD4+ T-cell recognition. Biochemical analysis showed that these molecules were greater than 30 000 molecular weight in size, and proteinaceous in nature. In addition, BL was found to have decreased expression of a 47 000 molecular weight enolase-like molecule that enhances class II-mediated antigen presentation in B cells, macrophages and dendritic cells, but not in BL cells. These findings demonstrate that BL likely has multiple defects in HLA class II-mediated antigen presentation and immune recognition, which may be exploited for future immunotherapies.
Keywords: antigen presentation, Burkitt lymphoma, enolase-like molecules, HLA class II, immune escape
Introduction
Burkitt lymphoma (BL) is a high-grade B-cell malignancy and is one of the fastest growing malignancies in humans.1–3 It occurs most frequently in children in areas with holoendemic and hyperendemic malaria (endemic BL) and is found with lower frequency in all other parts of the world (sporadic BL), accounting for 1–2% of all lymphomas in western countries.4–6 In addition to geographic distribution, BL may vary in its clinical manifestation, with endemic BL presenting as tumours of the jaw and sporadic BL causing the formation of tumours in the gut and upper respiratory tract.1,7,8 In both adults and children, BL is frequently associated with immune deficiency.
A universal characteristic of all BL is the translocation of the MYC gene to an immunoglobulin locus, which results in its constitutive activation and over-expression.9–11 MYC encodes the oncogenic transcription factor c-myc, and studies indicate that up to 15% of all known genes may lie in its target gene network.8,12,13 Another feature of BL that is observed with varying frequencies depending on the geographical location is association with Epstein–Barr virus (EBV). EBV infection is seen in > 90% of endemic BL but only 10–15% of sporadic BL, and has received much attention as a possible co-factor for the development of BL.4,6,8 EBV products are involved in the transformation of BL cells and may also contribute to their decreased immunogenicity. In addition to BL, EBV has also been associated with other malignancies such as Hodgkin's lymphoma, transplant-related B-cell lymphomas, T-cell lymphomas, adult T-cell leukaemia, natural killer leukaemia, and other lymphoid diseases.14–17 While surgery, radiation, and chemotherapy have been used to treat lymphoma patients, immunotherapy has shown great promise with a positive long-term effect on overall survival.18,19
Studies suggest that BL cells are deficient in their ability to stimulate CD8+ T cells via the interaction of human leucocyte antigen (HLA) class I with T-cell receptors.20–22 While the antigen-specific lysis of tumours is predominantly a function of HLA class I-restricted CD8+ T-cell activation, HLA class II-restricted CD4+ T-cell function is crucial in maintaining sustained immune responses to tumours.23,24 Activation of natural killer cells has been shown to induce long-term cytotoxic T lymphocyte (CTL) memory, but CD4+ T cells remain essential for sustained immune responses and complete destruction of tumours by CTLs.25,26 The deficiency in the HLA class I-mediated stimulation of CD8+ T cells by BL has been well studied and stems from the poor immunogenicity of EBV nuclear antigen 1 (EBNA-1).27–29 However, it remains unclear why the immune system is unable to mount an effective HLA class II-restricted response against BL. It has been suggested that one EBV gene product, gp42, is involved in blocking the HLA class II/T-cell receptor interaction.30 Gp42 is an EBV envelope protein that mediates virus binding through its interaction with HLA class II, and there is evidence that it may also impair CD4+ T-cell activation by blocking interaction of the T-cell receptor with HLA class II on the B-cell surface.30–32 Previous research in our laboratory has also suggested that B-cell lymphomas are deficient in presenting antigen via HLA class II molecules.33 In the study presented here, we explore further the nature of this defect, specifically in BL.
The expression of co-stimulatory molecules (CD80/86) on antigen-presenting cells (APCs) is important to the activation of CD4+ T cells, yet BL cells have a decreased expression of these molecules.32 HLA class II antigen presentation to T cells in the absence of co-stimulation does not activate the T cell, but results in T-cell anergy. With this in mind, we investigated whether the addition of co-stimulatory signals, concomitant with HLA class II antigen presentation, could help BL cells to stimulate activation of CD4+ T cells.
Our current study suggests that there may be multiple defects in BL cells resulting in a failure to stimulate CD4+ T cells. We demonstrate that BL cells and EBV-immortalized B-lymphoblastoid cells (B-LCL) express similar detectable levels of a transfected HLA class II allele and that this HLA class II binds to exogenously delivered antigenic peptides to form class II–peptide complexes. However, in contrast to B-LCL, BL cells showed an impaired capacity to activate CD4+ T cells. This deficiency in BL was not overcome by the addition of external co-stimulation. We also show that class II-mediated antigen presentation capacity was restored in BL following incubation in an acidic buffer (pH 5·5), and acid eluate obtained from BL contained immunomodulatory molecules that impaired HLA class II-mediated antigen presentation in B-LCL. We further demonstrate the decreased expression of an immunostimulatory, 47 000 molecular weight (MW), enolase-like protein in BL which enhances antigen presentation in B cells, macrophages and dendritic cells, but not in BL. These findings suggest the presence of multiple defects in HLA class II-mediated antigen presentation in BL, which may provide novel targets for the future development of immunotherapies against lymphoid malignancies.
Materials and methods
Cell lines
Human BL cell lines (Ramos, Ous) and an acute lymphoblastic leukaemia line (Nalm-6) were maintained in complete RPMI-1640 supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 50 U/ml penicillin, 50 μg/ml streptomycin and 1% l-glutamine (Mediatech, Manassas, VA).33 Nalm-6 cells express cellular markers and growth patterns characteristic of BL. The 6.16 cells are a bare lymphocyte syndrome-like mutant line that is defective in a component of the regulatory factor X (RFX) transcription couple. Briefly, the 6.16 line is a sub-clone of the parental 6.1.6 bare lymphocyte syndrome-like line.34,35 The 6.16 line was transfected with DR4 to generate the 6.16.DR4 line, and was further transfected with DM to generate the 6.16.DR4.DM line.36 The 6.16.DR4.DM line expresses DR, DM and Ii molecules.37 The 6.16.DR4.DM line and a wild-type human B-lymphoblastoid cell line (Frev) were maintained in Iscove's modified Dulbecco's medium supplemented with 10% bovine growth serum (Hyclone, Logan, UT), 50 U/ml penicillin, 50 μg/ml streptomycin and 1% l-glutamine (Mediatech). The wild-type BL line Ous, and the wild type B-LCL line Frev constitutively express HLA-DR4 molecules. Nalm-6 and Ramos cells were retrovirally transfected for constitutive expression of HLA-DR4 (DRB1*0401) with linked drug selection markers for hygromycin and histidinol resistance to generate Nalm-6.DR4 and Ramos.DR4.36,38,39 HLA-DR4-expressing dendritic cells (FSDC.DR4) and monocytic macrophages (THP-1.DR4.DM) were also generated as described previously.36,38,39 Surface HLA-DR4 expression was confirmed by flow cytometric analysis using the DR4-specific monoclonal antibody, 359-F10.36,40,41 T-cell hybridomas 2.18a and 1.21 were generated by immunization of DR4-transgenic mice as described elsewhere39,42 and respond to IgG κ residues 188–203 and 145–159, respectively. T-cell hybridomas 4027/99 and 0921/98 (generously provided by Dr. Lars Fugger, Aarhus University Hospital, Aarhus, Denmark) are specific for DR4 and the immunodominant collagen peptide CII261–273. The T-cell hybridoma 17.9 responds to human serum albumin (HSA) residue 64–76K.43 T-cell hybridomas were cultured in RPMI-1640 with 10% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 50 μm β-mercaptoethanol (Invitrogen).
Western blot analysis
Cell lysates obtained from Frev, Nalm-6.DR4, Ramos.DR4, 6.16.DR4, 6.16.DR4.DM cell lines were separated on 4% to 12% Bis/Tris NuPage gels in MES buffer (Novex/Invitrogen), and analysed by Western blotting for HLA-DM (Santa Cruz Biotechnology, Santa Cruz, CA) protein expression as described.37,44 β-Actin was used a loading control.33
Antigens and peptides
Human IgG κ and cartilage bovine collagen type II (bCII) were purchased from Sigma (St Louis, MO). The human IgG immunodominant peptide κ188–203 (sequence: KHKVYACEVTHQGLSS), subdominant peptide κ145–159 (sequence: KVQWKVDNALQSGNS), type II collagen peptide CII261–273 (sequence: AGFKGEQGPKGEP), and human serum albumin peptide HSA64–76K (sequence: VKLVNEVTEFAKTK) were produced using Fmoc technology and an Applied Biosystems Synthesizer (Applied Biosystems, Foster City, CA) as described previously.39,42,43,45 The κ188–203 and κ145–159 peptides were labelled as indicated at the α N-termini by the sequential addition of two molecules of Fmoc-6-aminohexanoic acid followed by a single biotin to yield the sequence biotin-aminohexanoic acid–aminohexanoic acid–peptide. Mass spectrometric analysis confirmed that the peptide was tagged with a single biotin molecule at the N-terminus. Peptide purity (> 99%) and sequence were analysed by reverse-phase HPLC purification and mass spectroscopy. Peptides were dissolved in PBS and stored at −20° until used.
Antigen presentation assays
B-lymphoblastoid cells and BL cells were incubated with whole IgG κ antigen, κ synthetic peptide, whole CII antigen (40 μg/ml), CII synthetic peptide, or HSA peptide (0–20 μm) for 3–24 hr at 37° in the appropriate cell culture medium.36,39 Cells were then washed and co-cultured with the appropriate peptide-specific T-cell hybridoma for 24 hr at 37°. In parallel assays, T-cell hybridomas were stimulated with anti-CD3/CD28 before co-culture with BL or B-LCL that had been incubated with κ188–203 or κ145–159.43 Following co-culture, T-cell production of interleukin-2 (IL-2) was quantified by ELISA. Assays were repeated in triplicate with standard error for triplicate samples within a single experiment being reported. Additionally, 6.16.DR4.DM, Nalm-6.DR4, and Ramos.DR4 cells were washed once in citrate phosphate buffer (CPB; pH 7·4 and pH 5·5), and incubated with the biotinylated κ peptides (b-κ188–203 or b-κ145–159) (5–10 μm) in the respective CPB buffer for 4 hr. After incubation, cells were washed twice with complete RPMI-1640 medium, and co-cultured with the b-κ188–203 or b-κ145–159 peptide-specific T-cell hybridomas (2.18a for κ188–203, 1.21 for κ145–159) for 24 hr. T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA as described above.39 In separate T-cell assays, 6.16.DR4.DM, Nalm-6.DR4, THP-1.DR4, and FSDC.DR4 cells were pre-incubated with a 47 000 MW protein extract for 30 min, followed by the addition of HSA64–76K peptide (5–10 μm) for 4 hr. Cells were washed twice in complete RPMI-1640 and co-cultured with the HSA64–76K peptide-specific T-cell hybridoma (17·9) for 24 hr. Following incubation, T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA. Data are expressed as the mean ± SEM of triplicate wells.
ELISA and ELISArray
Interleukin-2 (IL-2) levels in antigen presentation assay supernatant were quantified by ELISA.46 A 96-well ELISA plate was coated overnight at 4° with purified rat anti-mouse IL-2 (Sigma). The plate was then washed and blocked with 2% BSA at room temperature for 30 min. After washing, standards and samples were plated in appropriate wells and incubated at room temperature for 2 hr. A standard curve was generated using recombinant IL-2 purchased from R&D (Minneapolis, MN). The plate was washed, and biotinylated rat anti-mouse IL-2 (Sigma) was added and incubated at room temperature for 1 hr. Following washing, avidin peroxidase (Pierce, Rockford, IL) was added to each well and incubated at room temperature for 30 min. The plate was washed and p-nitrophenyl phosphate, disodium salt (PNPP) substrate (Thermo Scientific, Rockford, IL) was added to each well and incubated at room temperature. Readings were taken every 30 min at 405 nm. Interleukin-2 levels in sample wells are expressed in pg/ml, calculated from the standard curve. Cytokines IL-4, IL-5, IL-10 and transforming growth factor-β (TGF-β) in the cell-free culture supernatants were analysed by commercially available ELISAarray kits (SABiosciences, Frederick, MD). Assays were repeated in triplicate, and data were expressed as mean pg/ml ± SEM.
RT-PCR
Semi-quantitative PCR was performed on Nalm-6.DR4, Ramos.DR4 and Frev cell lines for testing gene expression of HLA-DR, HLA-DM and HLA-DO following total RNA extraction using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. A reverse transcription reaction was performed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). β-actin mRNA was used as a control for each experiment using the primer sequence 5′-GACAGGATGCAGAAGGAGATTACT-3′ (sense) and 5′-TGATCCACATCTGCTGGAAGGT-3′ (anti-sense). Primer sequences were also used to test for HLA-DMβ 5′-GTCATGCCTCACAGCAGTGC-3′ (sense) and 5′-GGCTCAGGAGCCCCAA-3′ (anti-sense); HLA-DOβ 5′- ATGTCCACTGGCCCTATCAG-3′ (sense) and 5′-GCCACTCAGCATCTTTCTCC-3′ (anti-sense); and Ii 5′-GCTGTCGGGAAGATCAGAAG-3′ (sense) and 5′-GCCATACTTGGTGGCATTCT-3′ (anti-sense) gene expression. Thermal cycling parameters were 94° for 3 min, followed by 40 cycles of amplifications at 94° for 30 seconds, 60° for 1 min, 72° for 1 min and 72° for 5 min as the final elongation step. PCR products were subjected to electrophoresis by using 1·5% agarose gel and were visualized by ethidium bromide.
Peptide binding assays
Paraformaldehyde-fixed 6.16.DR4.DM, Nalm-6.DR4 and Ramos.DR4 cells were incubated overnight with biotinylated κI or κII peptides in 150 mm CPB (pH 5·5 and 7·4), washed with PBS, and lysed on ice for 20 min with 50 mm Tris buffer (pH 8) containing 0·15 m NaCl and 0·5% IGEPAL CA 630 (Sigma) as described previously.47,48 Cell supernatants were added to plates (Costar, Cambridge, MA) previously coated overnight with the anti-human class II antibody 37.1 (kindly provided by L. Wicker, Merck Research Laboratory, Rahway, NJ). The captured class II–peptide complexes were detected with europium-labelled streptavidin (Pharmacia Fine Chemicals, Piscataway, NJ) using a fluorescence plate reader (Delfia, Wallac, Turku, Finland). The number of total DR molecules within B-LCL/BL cells was quantified as described.39,42
Flow cytometric analysis
B-lymphoblastoid cells and BL cells were stained with monoclonal antibodies directed against DR (L243) and DR4 (359-F10), followed by secondary antibody labelled with FITC. Cells treated with neutral (CPB, pH 7·4) and acidic (CPB, pH 5·5) buffers as described in the binding assays were also washed and stained with L243, followed by secondary antibody labelled with FITC for cell surface class II molecules. Samples were analysed on a FACScan using CellQuest software (BD Bioscience, Mountain View, CA). Background fluorescence was evaluated using irrelevant isotype-matched monoclonal antibodies NN4 and IN-1 as described elsewhere.36,41,46
Protein extraction and digestion
Acid eluate was obtained from Nalm-6.DR4 and 6.16.DR4.DM as described previously.49 Extracts were passed through a 30 000 MW filter, concentrated, and protein concentrations were measured. Samples were then run on a non-reducing gel and stained with Coomassie blue.37 Gel plugs were excised and placed in an Eppendorf tube. Each plug was washed with 50 mm ammonium bicarbonate for 10 min. Next, the plugs were de-stained using 25 mm ammonium bicarbonate in 50% acetonitrile for 15 min, repeated twice. The plugs were dehydrated with 100% acetonitrile for 15 min, and dried in a speedvac. Each gel plug was covered with Proteomics Grade Trypsin (Sigma) and incubated at 37° overnight. The supernatant was collected in a clean dry Eppendorf tube. Peptides were further extracted with one wash of 25 mm ammonium bicarbonate for 20 min and three washes of 5% formic acid and 50% acetonitrile for 20 min each. The supernatant was collected and pooled after each wash then dried down in a speedvac to ∼ 1 μl. Before analysis, the samples were reconstituted with 10 μl of 0·1% trifluoroacetic acid. Samples were then concentrated with a C18 Ziptip (Millipore, Billerica, MA) and eluted with 0·1% trifluoroacetic acid, 50% acetonitrile, and 7·0 mg/ml α-cyano-4-hydroxycinnamic acid directly onto the matrix-assisted laser desorption/ionization target.
Mass spectrometric analysis matrix-assisted laser desorption/ionization time of flight/time of flight)
After the spots were dried completely, the plate was loaded into the Applied Biosystems 4800 Proteomics Analyzer.37 An external calibration was performed before analysing samples, using the manufacturer's standards and protocols. Samples were analysed in batch mode using 2000 laser shots per spectrum. First, peptide mass maps were acquired over the m/z range of 800–3500 in reflectron mode with a delayed extraction time optimized for m/z 2000 by averaging 2000 scans to locate peaks of peptide origin. The next batch run performed mass spectrometry (MS)-MS analyses to obtain sequence data on the 20 most abundant peaks from the MS analysis. Upon completion of the batch processing, the data were exported into the GPS Explorer data processing system for interpretation and identification. The MASCOT database-searching algorithm analysed the data, and summarized the results in report format. Database searches were performed using two missed cleavages and one differential modification of methionine oxidation. The top 20 matches were reviewed before assigning confident protein identifications. Proteins in acid eluate preparations were also separated on large, non-reducing 10% polyacrylamide gels. A ∼ 50 000 MW band was excised from these gels, and the protein was extracted by sonication in PBS on ice. The resulting extract was added to BL cells or B-LCL and incubated with HSA peptide for use in antigen presentation assays as described.
Statistical analysis
The data are expressed as the mean (± SD) and analysed using Student's t-test or one-way analysis of variance, with P ≤ 0·05 considered statistically significant.
Results
Diminished CD4+ T-cell response to HLA class II-mediated antigen presentation by BL cells
Burkitt lymphoma cells and B-LCL each express measurable levels of surface HLA class II molecules. However, to gain a more direct comparison of class II-mediated antigen presentation between these cell types, we expressed a common HLA class II allele in several BL and B-LCL cell lines. BL (Nalm-6 and Ramos) and B-LCL (6.16) were retrovirally transfected to express the DR4 allele, HLA DRB1*0401. Flow cytometric analysis showed that all three cell lines were successfully transfected and constitutively expressing the common DR4 allele (Fig. 1a). 6.16.DR4 cells were also transfected with HLA-DM to generate 6.16.DR4.DM cells that express similar levels of DM molecules when compared with Nalm-6.DR4 and Ramos.DR4 cells (Fig. 1b). HLA-DR4-expressing BL (Nalm-6.DR4 and Ramos.DR4) and B-LCL (6.16.DR4.DM) were then sorted, matched for surface DR4 expression and incubated in culture media with IgG κ synthetic epitopes (peptides κ145–159 and κ188–203) or whole antigen IgG. Cells were then washed and co-cultured with the appropriate peptide-specific T-cell hybridoma (1.21 for κ145–159 and 2.18a for κ188–203) for 24 hr. Interleukin-2 in the resulting supernatant was measured by ELISA as described.46 Data obtained from these assays revealed that the B-LCL 6.16.DR4.DM efficiently and dose-dependently presented the κ145–159 peptide to stimulate T cells (Fig. 1c). The 6.16.DR4.DM cells also functionally presented another synthetic peptide κ188–203 to CD4+ T cells (Fig. 1d). Although 6.16.DR4.DM cells elicited a strong CD4+ T-cell response when presenting both synthetic κ peptides, Nalm-6.DR4 and Ramos.DR4 cells were unable to stimulate the T-cell response (Fig. 1c,d). To examine whether BL cells are also unable to process and generate functional epitopes from the antigen IgG, we incubated cells with the whole antigen overnight and tested for T-cell responses. Again, 6.16.DR4.DM cells processed whole IgG and presented both class II-restricted IgG κ epitopes (κ188–203 and κ145–159) to T cells, whereas BL cells (Nalm-6.DR4 and Ramos.DR4) were deficient in their ability to stimulate CD4+ T cells with the same epitopes (Fig. 1e).
Figure 1.

Burkitt lymphoma (BL) and BL type cells are deficient in HLA class II-mediated antigen presentation and CD4+ T-cell recognition. (a) BL type cells (Nalm-6), BL (Ramos), and the B-lymphoblastoid cells (B-LCL) (6.16.DM) were retrovirally transfected to constitutively express a common allele of HLA-DR4 (DR4B*0401). Cells were stained with a pan-HLA-class II antibody (L-243, dotted line) and an HLA-DR4-specific antibody (359-F10, solid line). FACS plots are shown after retroviral transfection. Data are representative of at least three separate experiments. (b) Western blot analysis for HLA-DM proteins in Nalm-6.DR4, Ramos.DR4, and 6.16.DR4.DM cells. (c) Nalm-6.DR4, Ramos.DR4, and 6.16.DR4.DM cell lines were incubated with the synthetic κ145–159 peptide (0–20 μm) for 4 hr, washed, and co-cultured with the κ peptide-specific T-cell hybridoma (1.21) for 24 hr. (d) Cells were incubated with the synthetic κ188–203 peptide (20 μm) for 4 hr, washed and co-cultured with the peptide-specific T-cell hybridoma (2.18a) for 24 hr. (e) Cells were also incubated with the whole antigen IgG κ for overnight, washed, and co-cultured with the κ epitopes-specific T-cell hybridomas (2.18a for κ188–203, 1.21 for κ145–159) for 24 hr. Following incubation, T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA. Data are expressed as the mean ± SEM of triplicate experiments.
To examine whether wild-type BL cells constitutively expressing the same class II allele can functionally deliver κ145–159 epitopes/peptides to T cells, we employed a wild-type BL line Ous and a wild-type control B-cell line Frev. Interestingly, Frev cells induced strong T-cell responses and IL-2 production whereas Ous cells were unable to stimulate the same T cells (Fig. 2a,b). To evaluate if the same pattern is also true for other antigens, antigen presentation assays were carried out as described above, but using a whole CII antigen or its synthetic version of an epitope (CII261–273 peptide). As observed for whole IgG and the synthetic IgG κ peptides, 6.16.DR4.DM stimulated a strong CD4+ T-cell response while Nalm-6.DR4 and Ramos.DR4 failed to elicit significant T-cell activation (Fig. 2c,d). Peptide presentation can be modulated by HLA-DM molecules. To exclude the role of DM in differential antigen presentation, we tested DM-expressing 6.16.DR4.DM and Frev cells as well as DM-negative 6.16.DR4 cells for their ability to present antigen to T cells (Fig. 2e). All three cell lines efficiently presented the κ145–159 peptide to stimulate T cells regardless of DM expression. Taken together, these data suggest that a defect in HLA class II presentation is associated with BL cells.
Figure 2.
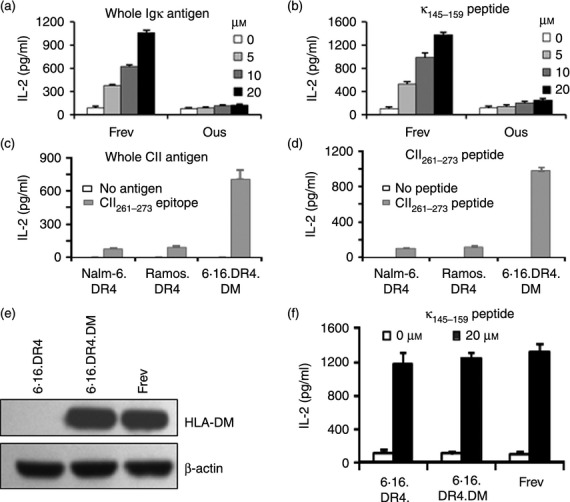
Disruption of class II–antigen presentation and CD4+ T-cell recognition of Burkitt lymphoma (BL). (a) A wild-type B-lymphoblastoid cell line (B-LCL) (Frev) and a wild-type BL line (Ous), which constitutively express HLA-DR4 (DR4B*0401) allele, were incubated with the whole antigen IgG κ (0–20 μm) for overnight. (b) Cells were also incubated with the synthetic κ145–159 peptide (0–20 μm) for 4 hr. After incubation, cells were washed and co-cultured with the κ145–159-specific T-cell hybridoma line (1.21) for 24 hr. (c) Cells were incubated with another self antigen type II collagen (CII; whole CII, 40 μg/ml) for overnight. (d) Cells were also incubated with the synthetic CII261–273 peptide (20 μm) for 4 hr. Cells were then washed and co-cultured with the CII261–273-specific T-cell hybridoma line 4027/99 for 24 hr. The production of IL-2 in the cell-free culture supernatant was quantified by ELISA. (e) Western blot analysis for HLA-DM proteins in 6.16.DR4, 6.16.DR4.DM, and Frev cells. (f) 6.16.DR4, 6.16.DR4.DM and Frev cells were incubated with the synthetic κ145–159 peptide (20 μm) for 4 hr. Cells were washed and co-cultured with the κ145–159-specific T-cell hybridoma line (1.21) for 24 hr. The T-cell production of IL-2 was quantified as described. Data are representative of the mean ± SEM of triplicate experiments.
Addition of external co-stimulation fails to overcome diminished HLA class II-mediated antigen presentation in BL cells although they possess class II components and produce only negligible amounts of inhibitory cytokines
To examine whether B-LCL and BL cells express major components of the HLA class II pathway, we performed RT-PCR using Nalm-6.DR4 and Ramos.DR4 cells. A wild-type B-cell line Frev, which expresses all the components of the class II pathway, was used as a control. Data showed that both B-LCL and BL lines expressed similar levels of DM, DO, and Ii molecules (Fig. 3a). It is known that BL cells express lower levels of co-stimulatory molecules (CD 80/86) than B-LCL. To determine if the diminished CD4+ T-cell response to class II-mediated antigen presentation by BL cells was a result of this deficiency, antigen presentation assays were carried out as described, but using CD4+ T cells that had been provided with external co-stimulation. CD4+ T cells were stimulated with IgG cross-linked anti-CD28 before co-culture with BL cells or B-LCL presenting either κ145–159 or κ188–203 synthetic peptide. The results shown in Fig. 3(b) demonstrate that addition of external co-stimulation has little to no effect on the ability of BL cells to present antigen via HLA class II molecules. The B-LCL, 6.16.DR4.DM, induced similarly high levels of IL-2 production, both in the presence and absence of external co-stimulation, while Nalm-6.DR4 and Ramos.DR4 again failed to stimulate a CD4+ T-cell response under either condition. Analysis of BL/B-LCL culture supernatants by ELISArray detected negligible amounts of IL-4, IL-5, IL-10, and TGF-β (Fig. 3c), suggesting that these inhibitory cytokines were not responsible for the observed defect in antigen presentation by BL.
Figure 3.

Analysis of major class II pathway components, requirement of co-stimulation and production of inhibitory cytokines in Burkitt lymphoma (BL)/B-lymphoblastoid cells (B-LCL). (a) RT-PCR analyses of Nalm-6.DR4, Ramos.DR4, and Frev cells for HLA-DM, HLA-DO, and Ii gene expression. β-Actin was used as a loading control. Data are representative of at least three separate experiments. (b) Co-stimulation does not overcome the BL defect in HLA class II antigen presentation. Nalm-6.DR4, Ramos.DR4, and 6.16.DR4.DM cells expressing a common allele of HLA-DR4 (DR4B*0401) were incubated with synthetic κ peptides (κ188–203 or κ145–159), washed, and co-cultured with the appropriate κ peptide-specific T-cell hybridomas (2.18a for κ188–203, 1.21 for κ145–159) with or without external co-stimulation by IgG cross-linked anti-CD28. Following incubation, T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA. (c) Cell supernatants obtained from Nalm-6.DR4, Ramos.DR4, 6.16.DR4.DM and Frev cells, were analysed for IL-4, IL-5, IL-10, and TGF-β cytokines by ELISArray. Data are expressed as the mean ± SEM of triplicate wells.
Antigenic peptides bind to BL-expressed HLA class II molecules, and antigen presentation is restored in BL at acidic pH
To further assess the defect in HLA class II-mediated antigen presentation by BL cells, we evaluated the binding efficiency of antigenic peptides to HLA class II molecules expressed on the surface of BL cells and B-LCL. Nalm-6.DR4, Ramos.DR4, and 6.16.DR4.DM cells were fixed with 1% paraformaldehyde, washed and incubated with biotin-labelled synthetic κ peptides (b-κ145–159 or b-κ188–203) in neutral (pH 7·4) or acidic (pH 5·5) CPB for 4 hr. Cells were washed, lysed, and total class II–peptide complexes were captured by ELISA, followed by detection with europium-labelled streptavidin as described in the Materials and methods. Data obtained revealed that under neutral conditions, BL cells and B-LCL each bound measurable levels of b-κ145–159 or b-κ188–203 peptides (Fig. 4a). It was noted that the peptide binding to class II proteins was much better at acidic pH than the neutral pH (Fig. 4b), and the binding capacity was almost comparable for both BL cells and B-LCL. These data suggest that the impaired class II presentation by BL cells is not associated with a defect in peptide binding.
Figure 4.

Antigenic peptides bind to Burkitt lymphoma (BL) cell surface-expressed HLA class II proteins at both neutral and acidic conditions. DR4+ BL (Nalm-6.DR4 and Ramos.DR4) and B-lymphoblastoid cells (B-LCL) (6.16.DR4.DM) were incubated with biotin-labelled κ peptides (κ188–203 or κ145–159) in citrate phosphate buffer (CPB) at pH 7·4 (a) and pH 5·5 (b) for 4 hr. Cell lysate was collected and class II–peptide complexes were captured with anti-human HLA class II antibodies. Captured complexes were detected with europium-labelled streptavadin. Data are the mean fluorescence ± SEM of at least three separate experiments. (c,d) Nalm-6.DR4, Ramos.DR4 and 6.16.DR4.DM cells were also washed once in CPB (pH 7·4 and pH 5·5), and incubated with biotinylated κ peptides (b-κ188–203 or b-κ145–159) in CPB at pH 7·4 or pH 5·5 for 4 hr. Cells were washed twice with complete RPMI-1640 medium, and co-cultured with the appropriate κ peptide-specific T-cell hybridomas (2.18a for κ188–203, 1.21 for κ145–159) for 24 hr. Following incubation, T-cell production of IL-2 in the cell-free supernatant was measured by ELISA. Data are expressed as the mean ± SEM of triplicate experiments.
Having established that BL-expressed HLA class II protein is capable of binding antigenic peptides at pH 5·5 with a higher affinity, we sought to determine if this enhanced binding resulted in the formation of functional class II–peptide complexes. Antigen presentation assays were carried out using b-κ188––203 and b-κ145–159 peptides. Unlike regular antigen presentation assays, the initial incubation of peptide with BL cells or B-LCL was carried out under neutral and acidic pH conditions (7·4 and 5·5, respectively) to follow conditions used in binding assays. Briefly, cells were incubated with biotin-labelled synthetic κ peptides (b-κ188–203 or b-κ145–159) in neutral (pH 7·4) or acidic (pH 5·5) CPB for 4 hr. Cells were then washed and co-cultured with peptide-specific T cells in culture media for 24 hr. Analysis of IL-2 in the culture supernatants of these assays showed that when peptide binding was carried out at pH 7·4, antigen presentation by BL cells and B-LCL mirrored the results from previous assays, with Nalm-6.DR4 and Ramos.DR4 stimulating little to no IL-2 production, while 6.16.DR4.DM elicited a strong CD4+ T-cell response (Fig. 4c). However, when peptide binding was carried out at pH 5·5, antigen presentation by Nalm-6.DR4 and Ramos.DR4 was restored to levels nearly identical to 6.16.DR4.DM (Fig. 3d). These data indicated that functional class II–peptide complexes could be formed at low pH on BL cells, resulting in CD4+ T-cell stimulation.
BL-derived molecules disrupt HLA class II antigen presentation in B-LCL
To investigate whether incubation of cells in neutral and acidic conditions altered cell surface class II molecules, we stained Nalm-6.DR4 and 6.16.DR4.DM cells with a pan class II antibody L243 for surface class II proteins (Fig. 5a,b). Apparently, both cell lines expressed similar levels of class II molecules in neutral [mean fluorescence intensity (MFI) = 557·3 versus MFI = 568·1] and acidic (MFI = 385·8 versus MFI = 379·5) conditions as analysed by flow cytometry. Results from antigen presentation assays also suggested that at pH 5·5 molecules that inhibit HLA class II-mediated antigen presentation are eluted from the surface of BL cells, hence restoring the antigen-presenting capacity of these cells. Consequently, we sought to determine if the acid eluate obtained from BL cells contained HLA class II inhibitory activity. Acid eluate (pH 5·5) was collected from Nalm-6.DR4 and 6.16.DR4.DM and separated into < 30 000 and > 30 000 MW fractions. Antigen presentation assays were then conducted with 6.16.DR4.DM being incubated with κI or κII in the presence of the < 30 000 or > 30 000 MW fractions from 6.16.DR4.DM or Nalm-6.DR4. Subsequent steps of the antigen presentation and IL-2 ELISA quantification were the same as already described. Data from these experiments showed that the > 30 000 MW acid eluate fraction from Nalm-6.DR4 greatly diminished the capability of 6.16.DR4.DM to present κ188–203 or b-κ145–159 (Fig. 5c,d). The < 30 000 MW fraction had no effect. To further characterize the inhibitory component in the > 30 000 MW fraction, it was treated with proteinase K or trypsin (data not shown) and used in antigen presentation assays as above. Data shown in Fig. 5(e) revealed that following proteinase K treatment, any inhibitory activity associated with Nalm-6.DR4 acid eluate was almost completely lost. These data suggest that BL-associated protein molecules impair HLA class II antigen presentation.
Figure 5.

Burkitt lymphoma (BL) and B-lymphoblastoid cells (B-LCL) cells express comparable levels of class II proteins, but BL acid (pH 5·5) eluate contains HLA class II inhibitory molecules > 30 000 MW which are sensitive to proteinase K. (a) Nalm-6.DR4 (black line) and 6.16.DR4.DM (green line) cells were incubated in citrate phosphate buffer (CPB; pH 7·4), washed and stained with L-243 for surface class II molecules. (b) Cells were also incubated in CPB (pH 5·5), washed and stained for surface class II molecules. NN4 was used as an isotype control. Acid elutions (pH 5·5) obtained from Nalm-6.DR4 and 6.16.DR4.DM were passed through 30 000 MW cut-off filters. Both the retained fraction (> 30 000 MW) and the filtrate (< 30 000 MW) were collected. B-LCL cells (6.16.DR4.DM) were then incubated with κ peptides (κ188–203 or κ145–159) in acid eluates collected from Nalm-6.DR4 and 6.16.DR4.DM. Cells were washed and co-cultured with the appropriate κ peptide-specific T-cell hybridomas (2.18a for κ188–203, 1.21 for κ145–159). T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA. (c) < 30 000 MW fraction, (d) > 30 000 MW fraction. Data are expressed as the mean ± SEM of triplicate experiments. (e) The > 30 000 MW fraction from 6.16.DR4.DM and Nalm-6.DR4 was then subjected to proteinase K treatment, dialysed and concentrated, and incubated with 6.16.DR4.DM and κ peptides (κ188–203 or κ145–159). Cells were washed and co-cultured with the appropriate κ peptide-specific T-cell hybridomas as described. Following incubation, T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA. Data are expressed as the mean ± SEM of triplicate experiments. *P < 0·05.
A 47 000 MW enolase-like molecule enhances HLA class II antigen presentation in B-LCL, dendritic cells, and macrophages, but not in BL cells
Data collected in previous assays suggested the presence of a proteinaceous HLA class II inhibitory component in BL cell acid eluate, but not in B-LCL acid eluate. This led us to compare protein expression patterns between the two cell types at acidic pH. Acid eluate (pH 5·5) was collected from BL cells (Nalm-6.DR4) and B-LCL (6.16.DR4.DM), and the proteins were separated on a non-reducing gel, followed by staining with Coomassie blue. Data from this assay showed a ∼ 50 000 MW protein consistently expressed at high levels in 6.16.DR4.DM, but seen only in low amounts in Nalm-6.DR4 (Fig. 6a). A gel plug sample of this protein was cut and analysed by mass spectrometry, revealing a 47 000 MW enolase-like molecule (Accession number 4503571). Acid eluates from a wild-type BL cell line (Ous) and B-LCL (Frev) also showed a high level of the 47 000 MW molecule consistently found in B-LCL, not in BL cells (data not shown), suggesting that the enolase-like protein is being suppressed in BL.
Figure 6.

Burkitt lymphoma (BL) and B-lymphoblastoid cells (B-LCL) differentially express an acid labile 47 000, enolase-like molecule, which enhances HLA class II-mediated antigen presentation in B cells, macrophages and dendritic cells. Proteins in acid (pH 5·5) elutions from BL (Nalm-6.DR4) and B-LCL (6.16.DR4.DM) and a wild-type B-cell line (Frev) were separated on a non-reducing gel. (a) Proteins were detected and relative expression was determined by staining with Coomassie blue. Data shown are representative of at least three separate experiments. (b) A band corresponding to ∼ 50 000 MW was removed and analysed by matrix-assisted laser desorption/ionization time of flight/time of flight mass spectrometry. Proteins in acid (pH 5·5) elution from 6.16.DR4.DM cells also were separated on a non-reducing gel. Following staining with Coomassie blue, an approximately 50 000 MW band containing an enolase-like protein (∼ 47 000 MW) was cut and the protein was extracted by sonication in PBS on ice. The protein extract was added to HLA-DR4-expressing (c) BL (Nalm-6.DR4), (d) B-LCL (6.16.DR4.DM), (e) macrophages (THP-1.DR4.DM) or (f) dendritic cells (FSDC.DR4) for 30 min, followed by the addition of HSA64–76K peptide for 4 hr. Cells were washed and co-cultured with the HSA64–76K peptide-specific T-cell hybridoma (17.9) for 24 hr. Following incubation, T-cell production of IL-2 in the cell-free culture supernatant was measured by ELISA. Data are expressed as the mean ± SEM of triplicate experiments. *P < 0·01, **P < 0·05.
Results from protein expression pattern and mass spectral analysis raised the possibility that, in addition to expressing HLA class II inhibitory molecules, BL cells may be deficient in expressing immune stimulatory proteins. To further examine the role of the 47 000 MW enolase-like molecule, which is highly expressed in B-LCL but expressed at low levels in BL cells, proteins in acid eluate (pH 5·5) samples from 6.16.DR4.DM cells were separated on large, non-reducing gels. The band which corresponded to our 47 000 MW enolase-like molecule was excised and the protein was extracted by sonication in PBS on ice. To confirm the presence of the 47 000 MW protein in the gel extract, samples were separated on a non-reducing gel and stained with Coomassie blue. Results confirmed that only the 47 000 MW protein was present in the gel extract samples (Fig. 6b). Antigen presentation assays were then carried out with Nalm-6.DR4, 6.16.DR4.DM, dendritic cells (FSDC.DR4), and macrophages (THP-1.DR4.DM) being incubated with HSA64–76K synthetic peptide in the presence of the 47 000 MW enolase-like gel extract (Fig. 6c–f). Subsequent steps in the antigen presentation assay and IL-2 quantification were carried out as already described. Results from these assays showed that the 47 000 MW enolase-like molecule significantly enhanced antigen presentation by B cells, macrophages, and dendritic cells, but not by the BL cells.
Discussion
HLA-mediated antigen presentation is central to the immune system's ability to detect and mount responses to transformed cells. However, malignant B-cell tumours, including BL, are equipped with strategies to escape detection by the immune system.36,50 Studies have shown that BL cells are not recognized as foreign or malignant by CD8+ effector T cells that can directly kill transformed cells. This defect has been well-characterized and results from the poor immunogenicity of the EBNA-1 antigen, which weakly stimulates CD8+ T cells.20,51 As EBNA-1 is the lone EBV antigen synthesized in BL, few options are available for CD8+ T-cell activation. In contrast, much less is known about the role of HLA class II antigen presentation in the recognition of BL. While CD8+ T cells are capable of directly killing tumour cells, it is now understood that CD4+ T-cell activation is required for lasting anti-tumour responses.52,53
Although BL cells express HLA class II proteins on their cell surface and theoretically could prime CD4+ T cells against their self-antigens, we observed that they failed to do so. We found that BL-associated molecules may block the signals that effectively trigger the CD4+ T-cell activation. Specifically, we have shown that CD4+ T cells failed to recognize antigens in association with HLA class II molecules on BL cells, whereas B-LCL efficiently processed and presented antigens to T cells in the context of class II molecules. Second, BL cells incubated at pH 5·5 gained the ability to stimulate CD4+ T cells to produce IL-2 in an HLA class II specific manner. Third, acid-eluted molecules associated with BL cells suppressed B-LCL's ability to present antigen to CD4+ T cells. Fourth, BL/B-LCL cells produced extremely low levels of inhibitory cytokines TGF-β, IL-10, IL-5, or IL-4; which are unlikely to disrupt class II presentation. Finally, we show that BL cells have decreased expression of a 47 000 MW enolase-like molecule, which enhances class II-mediated antigen presentation in B cells, macrophages and dendritic cells, but not in BL cells. These important findings are directly applicable to other EBV-associated tumours such as Hodgkin's lymphoma, transplant-related B-cell lymphomas, and other malignant lymphoid diseases. Identification of BL-associated inhibitory molecules (BLAIM) that disrupt CD4+ T-cell recognition as well as deficiencies in expression of immunostimulatory molecules will spur future development of new therapies against B-cell lymphomas.
The expression levels of class II molecules, invariant chain (Ii), and HLA-DM/DO molecules in APCs may modulate antigen presentation to T cells.36,41,54 While HLA-DM plays a critical role in the peptide selection process, in the absence of DM editing, peptides may bind in different conformations and are recognized by different subsets of T cells.55–57 In this study, we did not observe any significant differences in class II, Ii, and DM protein expression in BL/B-LCL tested. Mutant B-LCL (6.16.DR4) lacking DM molecules also efficiently presented class II-restricted peptides to CD4+ T cells. In addition, accessory proteins on APCs, such as co-stimulatory molecules, are also necessary to optimally stimulate T cells. BL cells are known to express lower levels of co-stimulatory molecules than B-LCL,32 so it was possible that the defect we observed in BL-mediated CD4+ T-cell stimulation stemmed from this property. Defects in co-stimulation by APCs can be overcome by adding antibodies to co-stimulatory receptors on T cells. For example, anti-CD28 antibodies can serve as surrogates for the B7 co-stimulatory molecule expressed on APCs. While our T-cell hybridomas are not dependent on co-stimulatory molecules,39 we still tested whether external co-stimulation could help BL cells stimulate these T cells. Even in the presence of external co-stimulation, these tumours failed to functionally present antigens to CD4+ T cells. Other activating signals, such as cross-linking IgM on BL cells, also failed to stimulate CD4+ T cells (data not shown), indicating that co-stimulation was not sufficient to overcome the defect.
Efficient binding of antigen to HLA is a requisite for effective presentation to and activation of T cells. Having established that our BL cell lines were deficient in their ability to stimulate the activation of CD4+ T cells, and having determined that this deficiency was not the result of a defect in co-stimulation, we considered the possibility of a defect in antigen binding to HLA-DR4. Data from our binding assays suggest that peptides bind to DR4 molecules on BL cells at both acidic and neutral pH. Functional assays failed to detect CD4+ T-cell responses to these class II–peptide complexes when formed at neutral pH, suggesting a lack of CD4+ T-cell recognition of BL. In contrast, class II–peptide complexes formed on BL/B-LCL at acidic pH, were functional and restored T-cell recognition to BL. Under these conditions, BL/B-LCL also expressed similar levels of cell surface class II proteins. This suggests that the lack of T-cell recognition at neutral pH did not reflect a defect in peptide binding to class II molecules on BL cells, but rather a failure to form functional peptide–class II complexes on these tumours.
We have shown further that acid eluates from B-LCL cells did not influence synthetic κ peptide presentation by B-LCL, but those obtained from BL cells significantly inhibited peptide presentation by B-LCL, suggesting that BL-cell-derived acid-eluted molecules may disrupt HLA class II-restricted T-cell recognition. Acid eluate obtained from BL cells was passed through 30 000 MW cut-off filters and tested for inhibitory activity. Data showed that BL-derived molecules retained in the 30 000 MW cut-off filter (> 30 000 MW) significantly inhibited CD4+ T-cell production of IL-2, suggesting that BLAIM could be greater than 30 000 MW in size. Additionally, BLAIM were sensitive to proteolytic enzymes as proteinase K treatment negated their ability to block CD4+ T-cell recognition of BL cells. Treatment of samples with trypsin also inhibited the activity of BLAIM (data not shown), further implying that BLAIM may be proteinaceous in nature. However, it remains possible that BLAIM could be structure-dependent, domain-dependent, or even modified molecules.
Working under the premise that BLAIM may be proteinaceous in nature, we then compared protein expression patterns between BL cells and B-LCL. While initially looking for the over-expression of proteins in BL that may be BLAIM, we consistently observed the decreased expression of a ∼ 50 000 MW protein in BL. This led us to consider the possibility that, in addition to expressing molecules that may inhibit immune stimulation, BL cells may also be deficient in immunostimulatory molecules. Protein expression and mass spectrometry analyses revealed that the predominant protein was a 47 000 MW enolase-like molecule. When this protein was extracted from Coomassie-stained gels and used in antigen presentation assays, it significantly increased class II-mediated antigen presentation in macrophages and dendritic cells as well as B-LCL, but not in BL cells. Hence, in addition to expressing BLAIM, which impairs CD4+ T-cell activation, BL cells express lower levels of an immunostimulatory, 47 000 MW, enolase-like molecule. Enolase-1 protein has been reported to be a transcriptional repressor of c-myc, but can also function at the cell surface as a plasminogen receptor.58,59 The effect of our 47 000 MW extract on T cells was tested, and no significant differences in cell proliferation and IL-2 production were observed (data not shown). Recently, α-enolase-1 has been considered as a marker of pathological stress in a number of infectious and autoimmune diseases such as inflammatory bowel disease,60,61 autoimmune hepatitis,62 and membranous glomerulonephritis.63
Future studies will focus on further characterizing BLAIM and the 47 000 MW enolase-like molecule, as well as elucidating their mechanisms of action. The successful characterization of these molecules offers potential for the development of new immunotherapies to more efficiently treat BL, potentially lessening the dependence on harsh chemotherapeutic regimens. Although currently used chemotherapeutic treatments are successful, achieving high cure rates in both adults and children, treatment-associated toxicities can be severe and are not tolerated well by elderly or HIV-infected patients.64 Use of the anti-CD20 monoclonal antibody, rituximab, in conjunction with chemotherapy, has led to increased response rates, but treatment-associated toxicities remain problematic.65,66 Additionally, the use of the immunosuppressive rituximab in patients already immunocompromised with HIV infection is a debated issue.66 When considered together, these issues reveal the need for improvements in treatment options for BL patients as well as patients with other lymphoid malignancies. Exploiting BL's over-expression of BLAIM or decreased expression of the 47 000 MW enolase-like protein provides the potential to generate a more targeted immune response to these cells, possibly reducing bystander effects and resultant treatment-associated toxicities.
Acknowledgments
We thank Dr Janice Blum (Indiana University School of Medicine) for providing cell lines and reagents, and Dr Daniel Knapp (Department of Pharmacology) for mass spectrometry facility and technical assistance. We also thank Bently Doonan for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (R01 CA129560 and R01 CA129560-S1) to A. Haque. The research presented in this article was also supported in part by the Tissue Biorepository and Flow Cytometry Shared Resource as part of the Hollings Cancer Center at the Medical University of South Carolina, which is funded by a Cancer Center Support Grant P30 CA138313.
Glossary
- BL
Burkitt lymphoma
- B-LCL
B-lymphoblastoid cell line
- CII
type II collagen
- CPB
citrate phosphate buffer
- CTL
cytotoxic T lymphocyte
- EBNA-1
Epstein–Barr virus nuclear antigen 1
- EBV
Epstein–Barr virus
- HLA
human leucocyte antigen
- HSA
human serum albumin
- IL-2
interleukin-2
- MFI
mean fluorescence intensity
- MW
molecular weight
Disclosures
The authors have no financial conflict of interest.
References
- 1.Biko DM, Anupindi SA, Hernandez A, Kersun L, Bellah R. Childhood Burkitt lymphoma: abdominal and pelvic imaging findings. AJR Am J Roentgenol. 2009;192:1304–15. doi: 10.2214/AJR.08.1476. [DOI] [PubMed] [Google Scholar]
- 2.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122:3884–91. doi: 10.1182/blood-2013-05-498329. [DOI] [PubMed] [Google Scholar]
- 3.Gromminger S, Mautner J, Bornkamm GW. Burkitt lymphoma: the role of Epstein–Barr virus revisited. Br J Haematol. 2012;156:719–29. doi: 10.1111/j.1365-2141.2011.09007.x. [DOI] [PubMed] [Google Scholar]
- 4.Brady G, Macarthur GJ, Farrell PJ. Epstein–Barr virus and Burkitt lymphoma. Postgrad Med J. 2008;84:372–7. doi: 10.1136/jcp.2007.047977. [DOI] [PubMed] [Google Scholar]
- 5.Perkins AS, Friedberg JW. Burkitt lymphoma in adults. Hematology Am Soc Hematol Educ Program. 2008;2008:341–8. doi: 10.1182/asheducation-2008.1.341. [DOI] [PubMed] [Google Scholar]
- 6.Molyneux EM, Rochford R, Griffin B, et al. Burkitt's lymphoma. Lancet. 2012;379:1234–44. doi: 10.1016/S0140-6736(11)61177-X. [DOI] [PubMed] [Google Scholar]
- 7.Yustein JT, Dang CV. Biology and treatment of Burkitt's lymphoma. Curr Opin Hematol. 2007;14:375–81. doi: 10.1097/MOH.0b013e3281bccdee. [DOI] [PubMed] [Google Scholar]
- 8.Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156:744–56. doi: 10.1111/j.1365-2141.2011.09013.x. [DOI] [PubMed] [Google Scholar]
- 9.Bellan C, Stefano L, Giulia de F, Rogena EA, Lorenzo L. Burkitt lymphoma versus diffuse large B-cell lymphoma: a practical approach. Hematol Oncol. 2009;27:182–5. doi: 10.1002/hon.914. [DOI] [PubMed] [Google Scholar]
- 10.Liu D, Shimonov J, Primanneni S, Lai Y, Ahmed T, Seiter K. t(8;14;18): a 3-way chromosome translocation in two patients with Burkitt's lymphoma/leukemia. Mol Cancer. 2007;6:35. doi: 10.1186/1476-4598-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerbitz A, Mautner J, Geltinger C, et al. Deregulation of the proto-oncogene c-myc through t(8;22) translocation in Burkitt's lymphoma. Oncogene. 1999;18:1745–53. doi: 10.1038/sj.onc.1202468. [DOI] [PubMed] [Google Scholar]
- 12.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–64. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 13.Slack GW, Gascoyne RD. MYC and aggressive B-cell lymphomas. Adv Anat Pathol. 2011;18:219–28. doi: 10.1097/PAP.0b013e3182169948. [DOI] [PubMed] [Google Scholar]
- 14.Ambinder RF. Epstein–Barr virus and Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2007;2007:204–9. doi: 10.1182/asheducation-2007.1.204. [DOI] [PubMed] [Google Scholar]
- 15.Kawa K. Diagnosis and treatment of Epstein–Barr virus-associated natural killer cell lymphoproliferative disease. Int J Hematol. 2003;78:24–31. doi: 10.1007/BF02983236. [DOI] [PubMed] [Google Scholar]
- 16.Snow AL, Martinez OM. Epstein–Barr virus: evasive maneuvers in the development of PTLD. Am J Transplant. 2007;7:271–7. doi: 10.1111/j.1600-6143.2006.01650.x. [DOI] [PubMed] [Google Scholar]
- 17.Ohtsubo H, Arima N, Tei C. Epstein–Barr virus involvement in T-cell malignancy: significance in adult T-cell leukemia. Leuk Lymphoma. 1999;33:451–8. doi: 10.3109/10428199909058450. [DOI] [PubMed] [Google Scholar]
- 18.Griskevicius L, Stulpinas R, Vengalyte I, Saulyte-Trakymiene S, Mickys U, Pranys D, Jurgutis M. Favorable outcome with chemo-immunotherapy in Burkitt lymphoma and leukemia. Leuk Res. 2009;33:587–8. doi: 10.1016/j.leukres.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 19.Hesseling P, Israels T, Harif M, Chantada G, Molyneux E. Pediatric Oncology in Developing C. Practical recommendations for the management of children with endemic Burkitt lymphoma (BL) in a resource limited setting. Pediatr Blood Cancer. 2013;60:357–62. doi: 10.1002/pbc.24407. [DOI] [PubMed] [Google Scholar]
- 20.Sharipo A, Imreh M, Leonchiks A, Imreh S, Masucci MG. A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I κB α with the proteasome: a new mechanism for selective inhibition of proteolysis. Nat Med. 1998;4:939–44. doi: 10.1038/nm0898-939. [DOI] [PubMed] [Google Scholar]
- 21.Yin Y, Manoury B, Fahraeus R. Self-inhibition of synthesis and antigen presentation by Epstein–Barr virus-encoded EBNA1. Science. 2003;301:1371–4. doi: 10.1126/science.1088902. [DOI] [PubMed] [Google Scholar]
- 22.Frisan T, Zhang QJ, Levitskaya J, Coram M, Kurilla MG, Masucci MG. Defective presentation of MHC class I-restricted cytotoxic T-cell epitopes in Burkitt's lymphoma cells. Int J Cancer. 1996;68:251–8. doi: 10.1002/(SICI)1097-0215(19961009)68:2<251::AID-IJC19>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 23.Khanolkar A, Badovinac VP, Harty JT. CD8 T cell memory development: CD4 T cell help is appreciated. Immunol Res. 2007;39:94–104. doi: 10.1007/s12026-007-0081-4. [DOI] [PubMed] [Google Scholar]
- 24.Tel J, Sittig SP, Blom RA, Cruz LJ, Schreibelt G, Figdor CG, de Vries IJ. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J Immunol. 2013;191:5005–12. doi: 10.4049/jimmunol.1300787. [DOI] [PubMed] [Google Scholar]
- 25.Gao FG, Khammanivong V, Liu WJ, Leggatt GR, Frazer IH, Fernando GJ. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res. 2002;62:6438–41. [PubMed] [Google Scholar]
- 26.Sharma RK, Yolcu ES, Srivastava AK, Shirwan H. CD4+ T cells play a critical role in the generation of primary and memory antitumor immune responses elicited by SA-4-1BBL and TAA-based vaccines in mouse tumor models. PLoS ONE. 2013;8:e73145. doi: 10.1371/journal.pone.0073145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masucci MG, Torsteindottir S, Colombani J, Brautbar C, Klein E, Klein G. Down-regulation of class I HLA antigens and of the Epstein–Barr virus-encoded latent membrane protein in Burkitt lymphoma lines. Proc Natl Acad Sci U S A. 1987;84:4567–71. doi: 10.1073/pnas.84.13.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavioli R, De Campos-Lima PO, Kurilla MG, Kieff E, Klein G, Masucci MG. Recognition of the Epstein–Barr virus-encoded nuclear antigens EBNA-4 and EBNA-6 by HLA-A11-restricted cytotoxic T lymphocytes: implications for down-regulation of HLA-A11 in Burkitt lymphoma. Proc Natl Acad Sci U S A. 1992;89:5862–6. doi: 10.1073/pnas.89.13.5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jilg W, Voltz R, Markert-Hahn C, Mairhofer H, Munz I, Wolf H. Expression of class I major histocompatibility complex antigens in Epstein–Barr virus-carrying lymphoblastoid cell lines and Burkitt lymphoma cells. Cancer Res. 1991;51:27–32. [PubMed] [Google Scholar]
- 30.Ressing ME, van Leeuwen D, Verreck FA, et al. Epstein–Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J Virol. 2005;79:841–52. doi: 10.1128/JVI.79.2.841-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ressing ME, van Leeuwen D, Verreck FA, et al. Interference with T cell receptor-HLA-DR interactions by Epstein–Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U S A. 2003;100:11583–8. doi: 10.1073/pnas.2034960100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staege MS, Lee SP, Frisan T, et al. MYC overexpression imposes a nonimmunogenic phenotype on Epstein–Barr virus-infected B cells. Proc Natl Acad Sci U S A. 2002;99:4550–5. doi: 10.1073/pnas.072495599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radwan FF, Zhang L, Hossain A, Doonan BP, God JM, Haque A. Mechanisms regulating enhanced human leukocyte antigen class II-mediated CD4+ T cell recognition of human B-cell lymphoma by resveratrol. Leuk Lymphoma. 2012;53:305–14. doi: 10.3109/10428194.2011.615423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovats S, Whiteley PE, Concannon P, Rudensky AY, Blum JS. Presentation of abundant endogenous class II DR-restricted antigens by DM-negative B cell lines. Eur J Immunol. 1997;27:1014–21. doi: 10.1002/eji.1830270431. [DOI] [PubMed] [Google Scholar]
- 35.Kovats S, Nepom GT, Coleman M, Nepom B, Kwok WW, Blum JS. Deficient antigen-presenting cell function in multiple genetic complementation groups of type II bare lymphocyte syndrome. J Clin Invest. 1995;96:217–23. doi: 10.1172/JCI118023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amria S, Hajiaghamohseni LM, Harbeson C, Zhao D, Goldstein O, Blum JS, Haque A. HLA-DM negatively regulates HLA-DR4-restricted collagen pathogenic peptide presentation and T cell recognition. Eur J Immunol. 2008;38:1961–70. doi: 10.1002/eji.200738100. [DOI] [PubMed] [Google Scholar]
- 37.Hossain A, God JM, Radwan FF, Amria S, Zhao D, Bethard JR, Haque A. HLA class II defects in Burkitt lymphoma: bryostatin-1-induced 17 kDa protein restores CD4+ T-cell recognition. Clin Dev Immunol. 2011;2011:780839. doi: 10.1155/2011/780839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovats S, Drover S, Marshall WH, Freed D, Whiteley PE, Nepom GT, Blum JS. Coordinate defects in human histocompatibility leukocyte antigen class II expression and antigen presentation in bare lymphocyte syndrome. J Exp Med. 1994;179:2017–22. doi: 10.1084/jem.179.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haque MA, Hawes JW, Blum JS. Cysteinylation of MHC class II ligands: peptide endocytosis and reduction within APC influences T cell recognition. J Immunol. 2001;166:4543–51. doi: 10.4049/jimmunol.166.7.4543. [DOI] [PubMed] [Google Scholar]
- 40.Hiraiwa A, Yamanaka K, Kwok WW, Mickelson EM, Masewicz S, Hansen JA, Radha SF, Nepom GT. Structural requirements for recognition of the HLA-Dw14 class II epitope: a key HLA determinant associated with rheumatoid arthritis. Proc Natl Acad Sci U S A. 1990;87:8051–5. doi: 10.1073/pnas.87.20.8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haque A, Hajiaghamohseni LM, Li P, Toomy K, Blum JS. Invariant chain modulates HLA class II protein recycling and peptide presentation in nonprofessional antigen presenting cells. Cell Immunol. 2007;249:20–9. doi: 10.1016/j.cellimm.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma C, Whiteley PE, Cameron PM, et al. Role of APC in the selection of immunodominant T cell epitopes. J Immunol. 1999;163:6413–23. [PubMed] [Google Scholar]
- 43.Pathak SS, Blum JS. Endocytic recycling is required for the presentation of an exogenous peptide via MHC class II molecules. Traffic. 2000;1:561–9. doi: 10.1034/j.1600-0854.2000.010706.x. [DOI] [PubMed] [Google Scholar]
- 44.Goldstein OG, Hajiaghamohseni LM, Amria S, Sundaram K, Reddy SV, Haque A. γ-IFN-inducible-lysosomal thiol reductase modulates acidic proteases and HLA class II antigen processing in melanoma. Cancer Immunol Immunother. 2008;57:1461–70. doi: 10.1007/s00262-008-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li P, Haque MA, Blum JS. Role of disulfide bonds in regulating antigen processing and epitope selection. J Immunol. 2002;169:2444–50. doi: 10.4049/jimmunol.169.5.2444. [DOI] [PubMed] [Google Scholar]
- 46.Younger AR, Amria S, Jeffrey WA, Mahdy AE, Goldstein OG, Norris JS, Haque A. HLA class II antigen presentation by prostate cancer cells. Prostate Cancer Prostatic Dis. 2008;11:334–41. doi: 10.1038/sj.pcan.4501021. [DOI] [PubMed] [Google Scholar]
- 47.Hill CM, Liu A, Marshall KW, et al. Exploration of requirements for peptide binding to HLA DRB1*0101 and DRB1*0401. J Immunol. 1994;152:2890–8. [PubMed] [Google Scholar]
- 48.Haque MA, Li P, Jackson SK, et al. Absence of γ-interferon-inducible lysosomal thiol reductase in melanomas disrupts T cell recognition of select immunodominant epitopes. J Exp Med. 2002;195:1267–77. doi: 10.1084/jem.20011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amria S, Cameron C, Stuart R, Haque A. Defects in HLA class II antigen presentation in B-cell lymphomas. Leuk Lymphoma. 2008;49:353–5. doi: 10.1080/10428190701814305. [DOI] [PubMed] [Google Scholar]
- 50.God JM, Haque A. Burkitt lymphoma: pathogenesis and immune evasion. J Oncol. 2010;2010:516047. doi: 10.1155/2010/516047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khanna R, Burrows SR, Steigerwald-Mullen PM, Moss DJ, Kurilla MG, Cooper L. Targeting Epstein–Barr virus nuclear antigen 1 (EBNA1) through the class II pathway restores immune recognition by EBNA1-specific cytotoxic T lymphocytes: evidence for HLA-DM-independent processing. Int Immunol. 1997;9:1537–43. doi: 10.1093/intimm/9.10.1537. [DOI] [PubMed] [Google Scholar]
- 52.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–63. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi JQ, Manches O, Wang J, et al. Development of autologous cytotoxic CD4+ T clones in a human model of B-cell non-Hodgkin follicular lymphoma. Br J Haematol. 2006;135:324–35. doi: 10.1111/j.1365-2141.2006.06294.x. [DOI] [PubMed] [Google Scholar]
- 54.Khalil H, Deshaies F, Bellemare-Pelletier A, Brunet A, Faubert A, Azar GA, Thibodeau J. Class II transactivator-induced expression of HLA-DOβ in HeLa cells. Tissue Antigens. 2002;60:372–82. doi: 10.1034/j.1399-0039.2002.600504.x. [DOI] [PubMed] [Google Scholar]
- 55.Lovitch SB, Petzold SJ, Unanue ER. Cutting edge: H-2DM is responsible for the large differences in presentation among peptides selected by I-Ak during antigen processing. J Immunol. 2003;171:2183–6. doi: 10.4049/jimmunol.171.5.2183. [DOI] [PubMed] [Google Scholar]
- 56.Pouyez J, Mayard A, Vandamme AM, Roussel G, Perpete EA, Wouters J, Housen I, Michaux C. First crystal structure of an endo-inulinase, INU2, from Aspergillus ficuum: discovery of an extra-pocket in the catalytic domain responsible for its endo-activity. Biochimie. 2012;94:2423–30. doi: 10.1016/j.biochi.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 57.Anders AK, Call MJ, Schulze MS, Fowler KD, Schubert DA, Seth NP, Sundberg EJ, Wucherpfennig KW. HLA-DM captures partially empty HLA-DR molecules for catalyzed removal of peptide. Nat Immunol. 2011;12:54–61. doi: 10.1038/ni.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feo S, Arcuri D, Piddini E, Passantino R, Giallongo A. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1) FEBS Lett. 2000;473:47–52. doi: 10.1016/s0014-5793(00)01494-0. [DOI] [PubMed] [Google Scholar]
- 59.Subramanian A, Miller DM. Structural analysis of α-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. J Biol Chem. 2000;275:5958–65. doi: 10.1074/jbc.275.8.5958. [DOI] [PubMed] [Google Scholar]
- 60.Roozendaal C, Zhao MH, Horst G, Lockwood CM, Kleibeuker JH, Limburg PC, Nelis GF, Kallenberg CG. Catalase and α-enolase: two novel granulocyte autoantigens in inflammatory bowel disease (IBD) Clin Exp Immunol. 1998;112:10–6. doi: 10.1046/j.1365-2249.1998.00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vermeulen N, Arijs I, Joossens S, et al. Anti-α-enolase antibodies in patients with inflammatory Bowel disease. Clin Chem. 2008;54:534–41. doi: 10.1373/clinchem.2007.098368. [DOI] [PubMed] [Google Scholar]
- 62.Ballot E, Bruneel A, Labas V, Johanet C. Identification of rat targets of anti-soluble liver antigen autoantibodies by serologic proteome analysis. Clin Chem. 2003;49:634–43. doi: 10.1373/49.4.634. [DOI] [PubMed] [Google Scholar]
- 63.Bruschi M, Carnevali ML, Murtas C, et al. Direct characterization of target podocyte antigens and auto-antibodies in human membranous glomerulonephritis: α-enolase and borderline antigens. J Proteomics. 2011;74:2008–17. doi: 10.1016/j.jprot.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 64.Aldoss IT, Weisenburger DD, Fu K, Chan WC, Vose JM, Bierman PJ, Bociek RG, Armitage JO. Adult Burkitt lymphoma: advances in diagnosis and treatment. Oncology (Williston Park) 2008;22:1508–17. [PubMed] [Google Scholar]
- 65.Thomas DA, Faderl S, O'Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106:1569–80. doi: 10.1002/cncr.21776. [DOI] [PubMed] [Google Scholar]
- 66.Oriol A, Ribera JM, Bergua J, et al. High-dose chemotherapy and immunotherapy in adult Burkitt lymphoma: comparison of results in human immunodeficiency virus-infected and noninfected patients. Cancer. 2008;113:117–25. doi: 10.1002/cncr.23522. [DOI] [PubMed] [Google Scholar]