

Abstract
Background
Many people with schizophrenia do not achieve a satisfactory treatment response with just antipsychotic drug treatment and various adjunct medications are used to promote additional response. The antiepileptic carbamazepine is one such drug.
Objectives
To examine whether carbamazepine or oxcarbazepine alone is an effective treatment for schizophrenia and schizoaffective psychoses and whether carbamazepine or oxcarbazepine augmentation of neuroleptic medication is an effective treatment for the same illnesses.
Search methods
For the original version we searched The Cochrane Schizophrenia Group's Register of Trials (December 2001), The Cochrane Library (Issue 3, 2001), MEDLINE (1966‐2001), EMBASE (1980‐2001), Biological Abstracts (1980‐2001), PsycLIT (1886‐2001) and PSYNDEX (1974‐2001). For the most recent update we searched the Cochrane Schizophrenia Group's Register of Trials in July 2012. We also inspected references of all identified studies for further trials and contacted relevant pharmaceutical companies and authors for additional data.
Selection criteria
We included all randomised controlled trials (RCTs) comparing carbamazepine or compounds of the carbamazepine family with placebo or no intervention, whether as sole treatment or as an adjunct to antipsychotic medication for the treatment of schizophrenia and/or schizoaffective psychoses.
Data collection and analysis
We extracted data independently. For homogenous dichotomous data we calculated fixed‐effect, risk ratio (RR), with 95% confidence intervals (CIs) on an intention‐to‐treat basis. For continuous data, we calculated mean differences (MD). We assessed the risk of bias for included studies and created a 'Summary of findings' table using GRADE.
Main results
The updated search did not reveal any further studies that met our inclusion criteria. The number of included studies therefore remains at 10 with the number of participants randomised still 283.
One study comparing carbamazepine with placebo as the sole treatment for schizophrenia was abandoned early due to high relapse rate with 26 out of 31 participants relapsing by three months. No effect of carbamazepine was evident with no difference in relapse between the two groups (1 RCT n = 31, RR 1.07 CI 0.78 to 1.45). Another study compared carbamazepine with antipsychotics as the sole treatment for schizophrenia. No differences in terms of mental state were found when comparing 50% reduction in Brief Psychiatric Rating Scale (BPRS) scores (1 RCT n = 38, RR 1.23 CI 0.78 to 1.92). A favourable effect for carbamazepine was found when more people who received the antipsychotic (perphenazine) had parkinsonism (1 RCT n = 38, RR 0.03 CI 0.00 to 0.043). Eight studies compared adjunctive carbamazepine versus adjunctive placebo, we were able use GRADE for quality of evidence for these results. Adding carbamazepine to antipsychotic treatment was as acceptable as adding placebo with no difference between the numbers leaving the study early from each group (8 RCTs n = 182, RR 0.47 CI 0.16 to 1.35, very low quality evidence). Carbamazepine augmentation was superior compared with antipsychotics alone in terms of overall global improvement, but participant numbers were low (2 RCTs n = 38, RR 0.57 CI 0.37 to 0.88). There were no differences for the mental state outcome of 50% reduction in BPRS scores (6 RCTs n = 147, RR 0.86 CI 0.67 to 1.12, low quality evidence). Less people in the carbamazepine augmentation group had movement disorders than those taking haloperidol alone (1 RCT n = 20, RR 0.38 CI 0.14 to 1.02). No data were available for the effects of carbamazepine on subgroups of people with schizophrenia and aggressive behaviour, negative symptoms or EEG abnormalities or with schizoaffective disorder.
Authors' conclusions
Based on currently available randomised trial‐derived evidence, carbamazepine cannot be recommended for routine clinical use for treatment or augmentation of antipsychotic treatment of schizophrenia. At present large, simple well‐designed and reported trials are justified ‐ especially if focusing on people with violent episodes and people with schizoaffective disorders or those with both schizophrenia and EEG abnormalities.
Plain language summary
Carbamazepine for schizophrenia
People with schizophrenia will often hear voices or see things (hallucinations) and have strange beliefs (delusions). They may also experience apathy, tiredness, lack of drive and disorganised thoughts and behaviour. These symptoms make schizophrenia a severe illness that affects many people throughout their life.
The main treatment for schizophrenia is antipsychotic medication. However, although this medication is successful in treating the majority of people, 5% to 15% will continue to suffer from debilitating symptoms. For these people, several treatment options are available: changing the dose of medication; switching to another antipsychotic drug; or taking additional drugs that are not antipsychotics. Carbamazepine is a drug first used to treat epilepsy in the 1950s. It is also used as a mood stabiliser when people change between ‘high’ and ‘low’ moods (for example bi‐polar affective disorder). Side effects of carbamazepine include: poor coordination, headaches and drowsiness.
This review focuses on the effectiveness of carbamazepine for people with schizophrenia. A search of the Cochrane Schizophrenia Group's trials register was carried out July 2012. Ten studies were found with 283 people. Carbamazepine was compared with no active medication (‘dummy’ or placebo treatment), versus an antipsychotic or when taken in addition to an antipsychotic. However, all of the 10 studies were small and information in them was of a poor standard. There is therefore a lack of evidence whether carbamazepine reduces symptoms and side effects in people with schizophrenia or similar mental health problems. Larger well‐designed trials are necessary to provide stronger evidence before carbamazepine can be recommended as a treatment for people with schizophrenia.
This plain language summary has been written by a consumer Ben Gray: Service User and Service User Expert. Rethink Mental Illness.
Summary of findings
Summary of findings for the main comparison. Adjunctive carbamazepine + antipsychotics compared with placebo/no adjunctive treatment + antipsychotics for schizophrenia.
| adjunctive Carbamazepine + antipsychotics compared to placebo/no adjunctive treatment + antipsychotics for schizophrenia | ||||||
| Patient or population: People with schizophrenia Settings: Outpatients/ inpatients Intervention: Adjunctive Carbamazepine + antipsychotics Comparison: Placebo/no adjunctive treatment + antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo/no adjunctive treatment + antipsychotics | Adjunctive Carbamazepine + antipsychotics | |||||
| Acceptability of treatment Leaving the study early due to any reason Follow‐up: mean 7.7 weeks | 108 per 1000 | 51 per 1000 (17 to 145) | RR 0.47 (0.16 to 1.35) | 182 (8 studies) | ⊕⊝⊝⊝ very low1,2,3 | |
| No clinically important change in general mental state Scale used: less than 50% BPRS reduction Follow‐up: mean 6.1 weeks | 623 per 1000 | 536 per 1000 (418 to 698) | RR 0.86 (0.67 to 1.12) | 147 (6 studies) | ⊕⊕⊝⊝ low1,4 | |
| Average endpoint general mental state score Scale used: Average BPRS endpoint score. Scale from: 0 to 108. Follow‐up: mean 5.7 weeks | The mean global mental state in the control groups was BPRS total score points | The mean global mental state in the intervention groups was 0.3 higher (12.49 lower to 13.09 higher) | 79 (3 studies) | ⊕⊕⊝⊝ low5,6 | ||
|
Average endpoint positive symptom score Scale used: Mean PANSS positive subscore at endpoint. Scale from: 7 to 49. Follow‐up: mean 4 weeks |
The mean positive symptoms in the control groups was PANSS positive subscore | The mean positive symptoms in the intervention groups was 4.22 higher (0.75 to 7.69 higher) | 18 (1 study) | ⊕⊕⊕⊝ moderate6 | ||
| Average endpoint depressive symptom score Scale used: Hamilton scale at endpoint. Scale from: 0 to 54. Follow‐up: mean 5 weeks | The mean depression in the control groups was Hamilton scale at endpoint | The mean depression in the intervention groups was 0.35 lower (2.2 lower to 1.5 higher) | 26 (1 study) | ⊕⊕⊝⊝ low6 | ||
| Aggression | See comment | See comment | Not estimable | 0 (0) | See comment | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: reporting bias is especially prevalent in this study pool with high risk being a problem in a majority of reports. 2 Inconsistency: differences between studies (number of events, effects size sometimes in favour of carbamazepine sometimes in favour of no treatment). 3 Indirectness: acceptability was measured by the number of participants leaving the studies for any reason which is an indirect measure of acceptability. 4 Imprecision: 95% confidence interval includes both benefit and harm. Moreover, the number of events was small. 5 Inconsistency: high heterogeneity of trial results 6 Imprecision: very small population size.
Background
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder. It afflicts approximately one per cent of the population world‐wide with little gender differences. Its typical manifestations are 'positive' symptoms such as fixed, false beliefs (delusions) and perceptions without cause (hallucinations), 'negative' symptoms such as apathy and lack of drive, disorganisation of behaviour and thought, and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable with 80% to 90% not working (Marvaha 2004) and up to 10% dying (Tsuang 1978).
Description of the intervention
Carbamazepine is an anticonvulsant and mood‐stabiliser with a tricyclic structure. It has been employed in treatment of a variety of psychiatric disorders, including schizophrenia, where it has been used as an antipsychotic agent. Its steric structure is similar to that of chlorpromazine or imipramine. Typical adverse effects of carbamazepine include: impaired co‐ordination, headaches and drowsiness (Pate 1986).
How the intervention might work
Carbamazepine (C15H12N2O, Figure 1) is a neutral lipophilic substance that can easily cross cell‐membranes and efficiently get into the nervous system. Carbamazepine mainly influences sodium channels by keeping them inactive as well as by potentiation of gamma‐aminobutyric acid (GABA)‐receptors (Granger 1995; Pate 1986).
1.

Carbamazepine
Why it is important to do this review
Despite the introduction of antipsychotic (neuroleptic) medication in the 1950s, there is still a sizeable minority of people with schizophrenia and related conditions that do not have complete remission of symptoms (Schooler 1993). Over the last 40 years a variety of adjunctive treatments have been used to treat schizophrenia (Christison 1991). These are often used in addition to antipsychotics, in order to augment any alleviation of symptoms of schizophrenia, but can be used instead of antipsychotics. Treatments such as lithium (indicated for bipolar affective disorder), carbamazepine (or related compounds such as oxcarbazepine), benzodiazepines, beta‐blockers (Cheine 2001) and electroconvulsive therapy (Tharyan 2002) have all been used for people whose psychoses did not respond to traditional therapy. The situation has improved somewhat in recent years with the re‐introduction of clozapine which has proven efficacy for those who have not responded to traditional medications (Essali 2009). However, many people with psychoses have sub‐optimal responses to treatment, and clinicians are faced with the choice of changing to alternate types of medication, or augmenting existing neuroleptics with other drugs or treatments.
Carbamazepine is used for the treatment of epilepsy, but is also used to prevent relapse, as a 'mood stabiliser', in bipolar affective illness in a similar fashion to lithium (Dardennes 1995). Oxcarbazepine is a related compound that is said to be an improvement on the older 'parent' drug (Tiihonen 1995). In this review we do not examine the efficacy of carbamazepine for mood disorders and the affective psychoses. However, in two companion reviews the impact of lithium and benzodiazepines as sole or adjunctive treatment for schizophrenia and schizoaffective psychoses is examined (Leucht 2007a; Dold 2012).
Objectives
To examine whether carbamazepine or oxcarbazepine alone is an effective treatment for schizophrenia and schizoaffective psychoses and whether carbamazepine or oxcarbazepine augmentation of neuroleptic medication is an effective treatment for the same illnesses.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. If a trial is described as 'double blind' but implies randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in statistically significant differences, we did not add the data from these lower quality studies to the results of the better trials, but presented such data within a subcategory. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. Where people were given additional treatments within carbamazepine/oxcarbazepine, we only included data if the adjunct treatment was evenly distributed between groups and it was only the carbamazepine/ oxcarbazepine that was randomised.
Types of participants
Adults, however defined, with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis.
We are interested in making sure that information is as relevant to the current care of people with schizophrenia as possible so propose to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and as to whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Experimental interventions
1.1 Carbamazepine/oxcarbazepine alone: any dose. 1.2 Carbamazepine/oxcarbazepine in combination with any antipsychotic treatment: any dose.
2. Control interventions
2.1 Placebo (or no intervention). 2.2 Placebo (or no intervention) in combination with any antipsychotic treatment. 2.3. Antipsychotics alone: any dose.
Types of outcome measures
We grouped all outcomes by time ‐ short term (up to 12 weeks), medium term (13 to 26 weeks) and long term (over 26 weeks).
Primary outcomes
No clinically important response to treatment (at least 50% reduction of overall symptoms or at least much improved)
Secondary outcomes
1. Leaving the study early
1.1 For specific reasons 1.2 Acceptability of treatment (leaving the study early for any reason)
2. Service utilisation
2.1 Hospital admission 2.2 Days in hospital 2.3 Change in hospital status
3. Global state
3.1 Relapse ‐ as defined by each of the studies 3.2 Time to relapse 3.3 No change in global state 3.4 Average endpoint global state score 3.5 Average change in global state scores
4. Mental state
4.1 General mental state
4.1.1 No clinically important change in general mental state ‐ as defined by each of the studies 4.1.2 No change in general mental state 4.1.3 Average endpoint general mental state score 4.1.4 Average change in general mental state scores
4.2 Specific aspects of mental state
4.2.1 No clinically significant response in positive symptoms ‐ as defined by each of the studies 4.2.2 No change in positive symptoms 4.2.3 Average endpoint positive symptom score 4.2.4 Average change in positive symptom scores 4.2.5 No clinically significant response in negative symptoms ‐ as defined by each of the studies 4.2.6 No change in negative symptoms 4.2.7 Average endpoint negative symptom score 4.2.8 Average change in negative symptom scores 4.2.9 No clinically significant response in depressive symptoms ‐ as defined by each of the studies 4.2.10 No change in depressive symptoms 4.2.11 Average endpoint depressive symptom score 4.2.12 Average change in depressive symptom scores 4.2.13 No clinically significant response in manic symptoms ‐ as defined by each of the studies 4.2.14 No change in manic symptoms 4.2.15 Average endpoint manic symptom score 4.2.16 Average change in manic symptom scores
5. Behaviour
5.1 General behaviour
5.1.1 No clinically important change in general behaviour 5.1.2 No change in general behaviour 5.1.3 Average endpoint general behaviour score 5.1.4 Average change in general behaviour scores 5.1.5 Compulsory administrations of treatment 5.1.6 Use of further doses of medication
5.2 Specific behaviours
5.2.1 Self‐harm, including suicide 5.2.2 Injury to others 5.2.3 Aggression 5.2.3.1 No clinically important change in aggression 5.2.3.2 No change in aggression 5.2.3.3 Average endpoint aggression score 5.2.3.4 Average change in aggression scores 5.2.4 Self‐care 5.2.4.1 No clinically important change in self‐care 5.2.4.2 No change in self‐care 5.2.4.3 Average endpoint self‐care score 5.2.4.4 Average change in self‐care scores 5.2.5 Compliance 5.2.5.1 No clinically important change in compliance 5.2.5.2 No change in compliance 5.2.5.3 Average endpoint compliance score 5.2.5.4 Average change in compliance scores
6. Social functioning
6.1 No clinically important effects for social function 6.2 No effects for social function 6.3 Average endpoint social functioning score 6.4 Average change social functioning scores 6.5 Employment status during trial (employed / unemployed)
7. Adverse effects
7.1 Clinically important general adverse effects 7.2 Any general adverse effects 7.3 Average endpoint general adverse effect score 7.4 Average change in general adverse effect scores 7.5 Clinically important change in specific adverse effects such as movement disorders 7.6 Any change in specific adverse effects 7.7 Average endpoint specific adverse effects 7.8 Average change in specific adverse effects 7.9 Use of antiparkinsonian treatment
8. Sudden and unexpected death
9. Economic outcomes
9.1 Direct costs 9.2 Indirect costs
10. Satisfaction with treatment
10.1 Recipient of care not satisfied with treatment 10.2 Recipient of care average satisfaction score 10.3 Recipient of care average change in satisfaction scores 10.4 Carer not satisfied with treatment 10.5 Carer average satisfaction score 10.6 Carer average change in satisfaction scores
11. Quality of life
11.1 No clinically important change in quality of life 11.2 No change in quality of life 11.3 Average endpoint quality of life score 11.4 Average change in quality of life scores 11.5 No clinically important change in specific aspects of quality of life 11.6 No change in specific aspects of quality of life 11.7 Average endpoint specific aspects of quality of life 11.8 Average change in specific aspects of quality of life
12. Pharmacokinetic interactions ‐ change of haloperidol plasma levels.
13. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADE profiler (GRADE Profiler) to import data from RevMan 5 (Review Manager (RevMan)) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Acceptability of treatment ‐ leaving the studies early for any reason
No clinically important response to treatment
General mental state
Specific mental state: positive symptoms
Specific mental state: depression
Aggressive behaviour
Search methods for identification of studies
Search methods for the 2012 update are below, for previous searches please see Appendix 1.
No language restriction was applied within the limitations of the search tools.
Electronic searches
1. Cochrane Schizophrenia Group Trials Register
The Trials Search Co‐ordinator searched the Cochrane Schizophrenia Group's Trials Register (July 2012)
1.1 Intervention search
The 'Intervention' field was searched using the phrase: (*carbama* or *amizepine* or *carbag* or *carbap* or *carbaz* or *carbymal* or *carpaz* or *cephalon* or *degranol* or *epitol* or *finlepsin* or *fokalepsin* or *hermolepsin* or *neurotol* or *neurotop* or *nordotol* or *sirtal* or *tardotol* or *tegret* or *teril* or *timonil* or *trimonil* or *trialeptal* or *trilpetal*) The Cochrane Schizophrenia Group's Trials Register is compiled by systematic searches of major databases, handsearches of relevant journals and conference proceedings (see Group Module). Incoming trials are assigned to existing or new review titles.
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Methods used in data collection and analysis for this 2012 update are below, for previous methods please see Appendix 2
Selection of studies
Review authors SL and MD independently inspected all citations from the searches and identified relevant abstracts. Where disputes arose, the full report was acquired for more detailed scrutiny. If citations met inclusion criteria, we obtained full reports of the papers for more detailed inspection. Again, all reports were independently inspected by SL and MD in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
Review authors MD and BH extracted data from all included studies. Again, any disagreement was discussed, decisions documented and, if necessary, authors of studies contacted for clarification. With remaining problems SL helped clarify issues and these final decisions were documented. If data were presented only in graphs and figures, we extracted whenever possible, but only included the data if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trial lists for that particular trial.
Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences (SMD) (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data (endpoint data only)
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all endpoint data before inclusion: a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution), (Altman 1996); c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS, Kay 1986), which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and S min is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. We presented skewed endpoint data from studies of less than 200 participants in other tables within the data and analyses section rather than enter them into a statistical analysis. Skewed data pose less of a problem when looking at mean if the sample size is large; we entered endpoint data from studies with over 200 participants into syntheses.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for carbamazepine.
Assessment of risk of bias in included studies
Review author BH assessed risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. Review author SL supervised the process. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted the authors of the studies in order to obtain further information.
The level of risk of bias was noted in both the text of the review (Risk of bias in included studies), the Characteristics of included studies table and Table 1.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The Number Needed to Treat/Harm (NNT/H) statistic with its CIs is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We prefer not to calculate effect size measures (standardised mean difference (SMD)). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Had we included cluster‐randomised trials and clustering had not been accounted for in primary studies, we planned to present data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, if we include cluster studies, we will seek to contact the first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). If clustering has been incorporated into the analysis of primary studies, we will present these data as if from a non‐cluster randomised study, but we will adjust for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC is not reported, it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant, the additional treatment arms were presented in comparisons. If data were binary, these were simply added and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions. Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
We share the concern that at some degree of loss of follow‐up, data must lose credibility (Xia 2009). However, from which degree of attrition onward this is a problem, is unclear. We therefore did not exclude studies on the basis of degree of attrition, but attrition was taken into account in the 'Risk of bias' assessment.
2. Binary
We presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were all assumed not to have changed in the given outcome. This rule is conservative for response, because it assumes that those who left the studies early would not have responded to treatment. It is not conservative concerning side effects, however, it would often have been an overestimation of the frequency of side effects if all participants who discontinued had been assumed to have experienced rare side effects.
3. Continuous
3.1 Intention‐to‐treat analysis
We used intention‐to‐treat datasets when possible, but included completer data if only these were available.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain the missing values from the authors. If not available, where there are missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals (CIs) available for group means, and either 'P' value or 't' values available for differences in mean, we can calculate them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics. Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values from the primary outcome.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). We reproduced these data, but readers should be aware of the problem.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, these were fully discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arise these were fully discussed.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of an I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, was interpreted as evidence of substantial levels of heterogeneity (Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for the heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes as there were less than 10 studies in all the analyses, and the numbers of participants were similar. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. In this review we therefore primarily chose the fixed‐effect model (following our original method). The reader is, however, able to choose to inspect the data using the random‐effects model, and we applied the random‐effects model in a sensitivity analysis of the primary outcome.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We proposed to undertake this review to provide an overview of the effects of carbamazepine for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems. The subgroups we planned to analyse included people with treatment‐resistant schizophrenia, people with EEG abnormalities, residual patients who were suffering predominantly from negative symptoms and people with schizoaffective disorder.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data had been entered correctly. Second, if data were correct, we looked at whether methodological reasons of the studies accounted for the heterogeneity. Finally, the graph was visually inspected and outlying studies successively removed to see if heterogeneity was restored. When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not undertake analyses relating to these.
Sensitivity analysis
We only applied sensitivity analyses to the primary outcome of this review.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcome, we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then all data were employed from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings of the primary outcome when we used our assumption compared with complete data only.
3. Imputed values
Had we included any cluster‐randomised trials, we would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
4. Fixed and random effects
All data were synthesised using a fixed‐effect model, however, we also synthesised data for the primary outcome using a random‐effects model to evaluate whether the greater weights assigned to larger trials with greater event rates, altered the significance of the results compared to the more evenly distributed weights in the random‐effects model.
Results
Description of studies
For substantive descriptions of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The original strategy identified hundreds of citations but only 10 studies met our inclusion criteria. In the update searches in 2005, 2007 and 2012 there were 24, 23 and eight new references respectively. We have not had any replies from the authors of studies that were classified as 'awaiting assessment' in previous versions of the review and we did not identify any ongoing studies (see Figure 2).
2.

Study flow diagram.
Included studies
Ten studies were included in analyses. No new studies were included in this update.
1. Design
Four of the included studies had a cross‐over design and six were parallel.
2. Length of trials
The longest study lasted for 95 days, the shortest, 18. Three out of 10 studies lasted six weeks, and four studies only five weeks. The mean duration of a study was approximately 41 days.
3. Participants
In the 10 included studies a total number of 283 participants were randomised. The two biggest studies involved 41 and 42 participants, the two smallest 18 and 13. On average there were about 28 participants in each study. The number of male and female participant is not indicated in two trials. In the remaining eight studies there is a proportion of about 5:2 of men and woman. Mean age of participants in not indicated in three studies. Based on the available data the mean age is around 26 years.
4. Setting
The majority of studies were set in hospitals with only one study involving outpatients.
5. Interventions
Two studies used carbamazepine as a sole treatment (one of them with a 800 to 120 mg dose range, the other with the mean dose of 1374 mg per day). The remaining eight studies used carbamazepine as an adjunctive treatment. In this case the doses were usually increasing and were at the level of 600 mg per day.
6.Outcomes
6.1 Leaving the study early
The numbers of participants leaving the study early were recorded for the categories any reason, adverse events and lack of efficacy.
6.2 Side effects
Side effects were reported in only two studies.
6.3 Scales
Details of scales that provided usable data are shown below.
6.3.1 Clinical Global Impression (CGI)
CGI (Guy 1976) is a rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery
6.3.2 Brief Psychiatric Rating Scale (BPRS)
BPRS (Overall 1962) is a brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The scale has 18 items, and each item can be defined on a seven‐point scale varying from 'not present' (one) to 'extremely severe' (seven). Scoring ranges from 18 ‐126.
6.3.3 Scale for the Assessment of Negative Symptoms (SANS)
SANS (Andreasen 1984) is a six‐point scale that gives a global rating of the following negative symptoms: alogia, affective blunting, avolition‐apathy, anhedonia‐asociality and attention impairment. Higher scores indicate more symptoms.
6.3.4 Positive and Negative Syndrome Scale (PANSS)
PANSS (Kay 1986) is a schizophrenia scale that has 30 items, each of which can be defined on a seven‐point scoring system varying from one ‐ absent to seven ‐extreme. It can be divided into three sub‐scales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P), and negative symptoms (PANSS‐N). A low score indicates lesser severity
6.3.5 Hamilton Depression Rating Scale (HAM‐D)
The HAM‐D (Hamilton 1960) is a scale developed to rate symptoms of depression. The original version contains 17 items rated on a three‐ or five‐point scale. Higher total scores mean more symptoms.
6.4 Missing outcomes
There were no data on aggression, service use such as 'duration of hospital stay', satisfaction with treatment or costs.
Excluded studies
In the 2012 update an additional two studies met some of the inclusion criteria closely enough to be listed as 'excluded studies' (Ohlmeier 2007; Ivkovi 2004). Thus, the category 'excluded studies' currently lists 91 trials. Sixty‐four trials were not randomised or the randomisation was undertaken inappropriately. Three studies were excluded due to participants not meeting the inclusion criteria, 19 studies due to type of intervention (typically there was no placebo group) and five because no usable outcomes were presented.
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 3 and Figure 4. Full details of judgements can be seen in the 'Risk of bias' tables.
3.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Four out of 10 of all the included studies were judged as having a low risk of potential selection bias because a specific method of generating a random sequence was provided (Llorca 1993; Mair 1990; Martin‐Munoz 1989; Neppe 1983). These studies typically used a random number list or a coin toss method. The remaining six studies state simply that the order was randomised without giving any further details about the procedure. All these studies are rated as having an unclear risk of selection bias. No studies were rated as possessing a high risk of selection bias.
Only two studies report on allocation concealment (Heßlinger 1998; Nachshoni 1994). In one of them sealed envelopes were used, in the other only a control psychiatrist had access to the identity of the interventions. The rest of the studies do not provide any informations that could be used to assess allocation concealment, thus they are rated as unclear in this respect.
As poor reporting of randomisation has consistently been associated with an overestimate of effect (Schulz 1995) all the trials rated as “unclear” could have a 30% to 40% overestimate of effect (Moher 2001; Schulz 1995).
Blinding
Objective and subjective outcomes were rated separately, because we consider blinding to be less important for objective as it is for subjective outcomes. Performance and detection bias was also rated separately resulting in four independent assessments of blinding.
1. Objective
For both performance and detection bias, the vast majority of studies were rated as having a low risk of bias (nine out of 10 studies), because a double‐blind procedure was employed. In only one study such an information was missing, thus is was rated as unclear (Svestka 1989).
2. Subjective
As for subjective outcomes, in terms of blinding of participants and personnel, only two studies provided enough information to be rated as having a low risk of bias (Llorca 1993; Nachshoni 1994). In both cases rhe use of identical capsules for drug and placebo was reported. The remaining eight studies did not provide any further details, hence they were rated as 'unclear'. The raters were described as blind only in three out of 10 studies (Carpenter 1991; Llorca 1993; Nachshoni 1994.
Incomplete outcome data
Eight out of 10 studies were assessed as having a low risk of attrition bias, meaning that the problem of incomplete outcome data was addressed in an appropriate way (the number of dropouts was not very high and was evenly distributed between groups). Only two studies were judged as unclear (Mair 1990; Neppe 1983). One of them did not report on dropouts at all (Mair 1990). The other one had an uneven distribution of early discontinuations between groups (Neppe 1983).
Selective reporting
The majority of studies (six out of 10) were judged as having a high risk of reporting bias due to lack of informations about some predefined outcomes. A very typical problem comprised not reporting about standard deviations. Out of the remaining four studies, one was rated as unclear due to the fact that only an abstract with a limited number of information was available (Simhandl 1996). The other three did not selectively report on any outcomes and thus were judged as having a low risk of bias in that respect (Carpenter 1991; Heßlinger 1998; Nachshoni 1994).
Other potential sources of bias
Nine out of 10 studies were rated as free of other potential sources of bias. Only one study was rated as unclear in this respect, because the dosages of rescue and additional medications differed between groups (no further details were provided) (Dose 1987).
Effects of interventions
See: Table 1
We used risk ratios (RR) for dichotomous data and mean differences (MD) for continuous data, with their respective 95% confidence intervals (CIs) throughout.
1. Comparison 1: Carbamazepine as sole treatment versus placebo as sole treatment
Only Carpenter 1991 compared carbamazepine as a sole agent with placebo in maintenance treatment.
1.1 Leaving the study early
One person receiving carbamazepine left early due to a rash and another due to leucopenia. Two people also left early from the placebo group, due to a conduction defect on the ECG and headache, respectively. No difference between groups was found (1 RCT n = 31, RR 1.07 CI 0.17 to 6.64).
1.2 Global state ‐ relapse
Data from the first 27 people included into this study showed that carbamazepine was no more effective than placebo in preventing relapse(1 RCT n = 31, RR 1.07 CI 0.78 to 1.45). As the majority of those in both groups (26 out of 31) did relapse, the study was stopped by three months.
1.3 Mental state
There was no significant difference in terms of mental state as measured by the number of patients with less than 20% BPRS reduction (1 RCT n = 31, RR 0.99 CI 0.75 to 1.30) or the mean BPRS at endpoint between both groups (1 RCT n = 27, MD ‐0.07 CI ‐0.46 to 0.32).
1.4 Adverse effects
Carpenter 1991 reported transient sedation and nausea in the carbamazepine group, although no figures were presented. Three people treated with carbamazepine developed a rash, this difference was not statistically significant (1 RCT n = 31, RR 7.44 CI 0.42 to 132.95), and one person developed leucopenia, again differences between treatment groups were not significant (1 RCT n = 31, RR 3.19 CI 0.14 to 72.69).
2. Comparison 2: Carbamazepine as sole treatment versus antipsychotics as sole treatment
Again, only one trial was found that compared carbamazepine with perphenazine in acutely ill patients with schizophrenia and schizoaffective disorder (Svestka 1989).
2.1 Leaving the study early
Two patients on carbamazepine versus none on perphenazine left the study before its end, this difference is not statistically significant (1 RCT n = 38, RR 4.52 CI 0.23 to 88.38).
2.2 Mental state
We found no significant differences in terms of mental state. A similar number of people treated with carbamazepine and perphenazine reached less than 20% (1 RCT n = 38, RR 1.29 CI 0.62 to 2.66), 35% (1 RCT n = 38, RR 1.67 CI 0.86 to 3.24) or 50% (1 RCT n = 38, RR 1.23 CI 0.78 to 1.92) BPRS reduction. Again, no significant difference in terms of mean BPRS at endpoint was found (1 RCT n = 38, MD 2.30 CI ‐3.84 to 8.44). However, when those with schizoaffective disorder were excluded, a statistically significant inferiority of carbamazepine in terms of 20% BPRS reduction (1 RCT n = 28, RR 3.09 CI 1.22 to 7.84) and 35% BPRS reduction (1 RCT n = 28, RR 2.32 CI 1.15 to 4.67) was found. This effect was not as evident for 50% BPRS reduction scores and the difference between groups just failed to reach significance (1 RCT n = 28, RR 1.40 CI 0.94 to 2.09). Since only 10 participants had schizoaffective disorder, an analysis of this subgroup was not thought to be meaningful.
2.3 Adverse effects
2.3.1 Movement disorders
Significantly more participants who received perphenazine needed antiparkinson medication (1 RCT n = 38, RR 0.23 CI 0.09 to 0.55, NNH 1 CI 1 to 2) or had parkinsonism (1 RCT n = 38, RR 0.03 CI 0.00 to 0.43, NNH 1 CI 0.9 to 1.4). No significant difference in terms of the number of participants with akathisia (1 RCT,n = 38, RR 0.13 CI 0.01 to 2.34) or tremor (1 RCT n = 38, RR 0.30 CI 0.01 to 6.97) was found.
2.3.2 Other adverse effects
The following other adverse effects were reported: collapse, dizziness, blurred vision, dryness of mouth, fatigue, nausea, constipation, salivation, tachycardia. Studies found no significant differences between groups.
3. Comparison 3: Adjunctive carbamazepine + antipsychotics versus placebo/no adjunctive treatment + antipsychotics
Eight studies compared adding carbamazepine to antipsychotic treatment with adding a placebo to just antipsychotic treatment alone.
3.1 Leaving the study early
Eight studies were able to contribute to the outcome of 'number leaving the study early', although four of these studies had no one leave early in either group. No difference was found (8 RCTs n = 182, RR 0.47 CI 0.16 to 1.35) between those allocated to the augmentation group and those taking placebo adjunctive therapy.
3.2 Global state
Only Neppe 1983 and Simhandl 1996 provided data on the outcome 'no general improvement'. Carbamazepine augmentation of neuroleptics was superior compared to various antipsychotics alone, but the number of patients included was very low (2 RCTs n = 38, RR 0.57 CI 0.37 to 0.88).
3.3 Mental state
3.3.1 General
The individual patient data from six studies could be used for the analysis of various degrees of BPRS reduction. No significant differences in terms of number of participants with less than 20% (6 RCTs n = 147, RR 0.69 CI 0.44 to 1.07), 35% (6 RCTs n = 147, RR 0.78 CI 0.57 to 1.05) or 50% BPRS reduction (6 RCTs n = 147, RR 0.86 CI 0.67 to 1.12) were found. The results at the 50% BPRS reduction level were significantly heterogeneous because two studies (Dose 1987; Heßlinger 1998) showed contrary results. No obvious reasons for this heterogeneity could be derived from the publications. This was a general problem of these two studies in a number of efficacy outcomes. Removing both studies did not change statistical significance, therefore we decided to leave them in. Similar equivocal results were found when the mean BPRS (3 RCTs n = 79, MD ‐3.21 CI ‐7.82 to 1.40) was analysed. The results based on the IMPS at endpoint showed a significant superiority of the control group (2 RCTs n = 50, MD 4.61 CI 1.30 to 7.91). However, the results of the two studies were highly heterogeneous and when a random‐effects model instead of the fixed‐effect model was used, there was no significant difference (2 RCTs n = 50, MD 5.2 CI ‐11.1 to 21.4).
3.3.2 Specific ‐ positive symptoms, negative symptoms and depression
Only very few data for specific symptoms of schizophrenia could be extracted. In the Heßlinger 1998 study, the participants in the carbamazepine group had, on average, more positive symptoms at endpoint than those in the control group (1 RCT n = 18, MD 4.22 CI 0.75 to 7.69). The Dose 1987 study showed oppositional results, but the data could only be presented in the 'other data' table because they were skewed (Analysis 3.8). No significant superiority of carbamazepine augmentation in terms of negative symptoms (2 RCTs n = 53, MD ‐2.75 CI ‐6.71 to 1.22) or depression (1 RCT n = 26, MD ‐0.35 CI ‐2.20 to 1.50) could be found.
3.8. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 8 Mental state: 2b. Specific ‐ positive symptoms (IMPS score at endpoint, high = poor).
| Mental state: 2b. Specific ‐ positive symptoms (IMPS score at endpoint, high = poor) | |
|---|---|
| Study | |
| Dose 1987 | 1. Carbamazepine + antipsychotics: mean 4.9, SD 6.4, N = 16. 2. Placebo + antipsychotics: mean 7.5, SD 8.1, N = 16. |
3.4 Behaviour
Two studies presented data on the average dose of additional medication needed for the treatment of agitated behaviour. In Dose 1987 people receiving carbamazepine augmentation needed less additional medication, whereas in Heßlinger 1998 they needed more additional medication than in the control group. Data were skewed and could therefore only be presented in the other data table (Analysis 3.11).
3.11. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 11 Behaviour: Average dose of medication used for agitation (chlorprothixene, skewed data).
| Behaviour: Average dose of medication used for agitation (chlorprothixene, skewed data) | |
|---|---|
| Study | |
| Dose 1987 | 1. Carbamazepine + antipsychotics: mean dose 40 mg/day, SD 36. N = 17. 2. Placebo + antipsychotics: mean dose 63 mg/day, SD 49. N = 17. |
| Heßlinger 1998 | 1. Carbamazepine + haloperidol: mean chlorprothixene dose 182 mg/day, SD 30.1. N = 9. 2. Placebo + haloperidol: mean dose 21.6 mg/day, SD 5.8. N = 9. |
3.5 Adverse effects
Side effects were not well reported in the studies.
3.5.1 Movement disorders
The effect of adjunctive carbamazepine on movement disorders is not clear. One small study (Martin‐Munoz 1989) reported on the binary outcome of 'movement disorder present'. Less people in the carbamazepine augmentation group had movement disorders than those taking haloperidol alone but the result just failed to reach significance (1 RCT n = 20, RR 0.38 CI 0.14 to 1.02). Skewed data from the Simpson‐Angus Scale were equivocal from three studies (Dose 1987; Nachshoni 1994; Simhandl 1996).
Three studies (Dose 1987; Heßlinger 1998; Simhandl 1996) presented data on the mean dose of antiparkinson medication used. These data are presented in the 'other data' tables, because they are skewed (Analysis 3.13). No consistent trend can be derived from these data.
3.13. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 13 Adverse effects: 1b. Movement disorders ‐ average dose of antiparkinsonism drugs (biperiden, skewed data).
| Adverse effects: 1b. Movement disorders ‐ average dose of antiparkinsonism drugs (biperiden, skewed data) | |
|---|---|
| Study | |
| Dose 1987 | 1. Carbamazepine + antipsychotics: mean dose 1.3 mg/day, SD 1.6. N = 17. 2. Placebo + antipsychotics: mean dose 3.8 mg/day, SD 2.3. N = 17. |
| Heßlinger 1998 | 1. Carbamazepine + haloperidol: mean dose 3.9 mg/day, SD 0.8. N = 9. 2. Placebo + haloperidol: mean dose 2.9 mg/day, SD 1.0 N = 9. |
| Simhandl 1996 | 1. Carbamazepine + antipsychotics: mean dose 2.67 mg/day, SD 2.89. N = 15. 2. Placebo + antipsychotics: mean dose 2.67 mg/day, SD 4.62. N = 12. |
3.5.2 Other side effects
Two studies used scales to assess side effects (Mair 1990; Martin‐Munoz 1989) but data were reported in such a way as to be unusable for this review. Dose 1987 reported several carbamazepine‐associated adverse effects (allergic reactions, elevation of liver enzymes, leucopenia, EEG change). Although these tended to be more prevalent in the carbamazepine augmented group, none reached the level of statistical significance.
3.6 Physiological effects
Dose 1987 and Heßlinger 1998 described mean plasma haloperidol to be lower in the carbamazepine‐augmented group but again these data are in the 'other data' tables.
3.7 Missing outcomes
Carbamazepine is said to have an effect upon aggression. Neppe 1983 reported that overt aggression was rated twice as severe with placebo compared to carbamazepine but no quantitative data were reported. Llorca 1993 did not find between‐group differences in SAPS or BPRS hostility and aggressiveness items but only 'P' values were presented. No data were found for 'service' outcomes such as 'duration of hospital stay'. Nor were there data on satisfaction with treatment or costs.
3.8 Subgroup analyses
All subgroup analyses were only conducted on the primary outcome (less than 50% BPRS reduction).
3.8.1 People with treatment‐resistant schizophrenia
Simhandl 1996 included only those with schizophrenia who had fulfilled specific criteria of neuroleptic non‐response. There was no significant difference compared to the other studies which did no have this inclusion criterion (Test for subgroup differences: Chi² = 0.28, df = 1 (P = 0.60), I² = 0%). Investigating Simhandl 1996 alone we found that significantly more patients treated with adjunctive carbamazepine improved according to the CGI and reached at least 20% BPRS reduction. However, this result was not consistent, because there were not significantly more patients treated with carbamazepine augmentation than with placebo augmentation reaching 35% and 50% BPRS reduction (primary outcome). Llorca 1993 examined the effectiveness of adjunctive carbamazepine in those with treatment‐resistant schizophrenia (Kane 1988 criteria) using a cross‐over design. No mental state data were directly reported (P values only) but carbamazepine was not stated to be better than placebo in this small study (n = 12).
3.8.2 People with EEG abnormalities
Neppe 1983 examined a small group of 13 relatively non‐responsive patients with EEG abnormalities, of which nine had schizophrenia. In this cross‐over trial, more patients fared somewhat better in the carbamazepine than in the placebo phase for 'leaving the study earlier', 'no global clinical improvement' and the mental state ratings (BPRS). The patient population was quite heterogeneous and diagnostic criteria were not indicated. Overall, the effects in this study did not differ significantly from the rest of the studies (Test for subgroup differences: Chi² = 0.84, df = 1 (P = 0.36), I² = 0%).
3.8.3 People with negative symptoms
Nachshoni 1994 carried out a double‐blind randomised controlled trial in 28 residual patients who were suffering predominantly from negative symptoms. After five weeks no superiority of adjunctive carbamazepine compared with placebo on negative symptoms could be found. The results on the primary outcome were not different from the other studies (Test for subgroup differences: Chi² = 1.04, df = 1 (P = 0.31), I² = 3.7%).
3.8.4 People with schizoaffective disorder
Only 12 people included in this review had schizoaffective disorder so analyses of this subgroup did not appear to be meaningful.
3.9 Sensitivity analyses
All sensitivity analyses were only applied to the primary outcome (less than 50% BPRS reduction). Therefore, only the use of a random‐effects model instead of the fixed‐effect model was relevant. There was no difference between monotherapy with antipsychotics and augmentation with carbamazepine when a random‐effects model was used (6 RCTs n = 147, RR 0.93 CI 0.60 to 1.45).
3.10 Publication bias
Only six studies contributed to the primary outcome. Therefore, the funnel‐plot method was not meaningful as described in Assessment of reporting biases.
Discussion
Summary of main results
1. General
Although much original data were received from trial lists, a total of 258 participants is still a small base upon which to judge the effectiveness of carbamazepine. The included studies in this review, were therefore unable to provide sufficient data to clarify the role of carbamazepine for the treatment or augmentation of antipsychotic treatment of schizophrenia and schizoaffective disorder.
2. Comparison 1: Carbamazepine as a sole treatment versus placebo as a sole treatment
The little available data suggest that carbamazepine is no better than placebo for maintenance treatment. Considering that the single study contributing data (Carpenter 1991) was stopped early, because the majority of those in both groups relapsed, these data are unlikely to be supplemented.
3. Comparison 2: Carbamazepine as a sole treatment versus antipsychotics as a sole treatment
In the only small study available (Svestka 1989), carbamazepine was not inferior when compared with perphenazine in terms of improvement of mental state and carbamazepine was associated with fewer extrapyramidal side effects than perphenazine. However, due to the small sample size of this trial (n = 38), carbamazepine can not be considered as a reasonable alternative to antipsychotics, and in the subgroup analysis in which those with schizoaffective disorder were excluded, perphenazine was superior to carbamazepine in some efficacy outcomes.
4. Comparison 3: Adjunctive carbamazepine + antipsychotics versus placebo/no adjunctive treatment + antipsychotics
4.1 Acceptability of treatment ‐ leaving the study early for any reason
Only 13 out of 180 people left the studies before completion with no difference between groups. This very low rate of attrition is rare within trials relevant to the care of those with schizophrenia. Adjunctive therapy of this sort seems to be very acceptable to people with schizophrenia, at least within the confines of a trial.
4.2 General improvement
Two small trials (Neppe 1983; Simhandl 1996) presented data on the outcome of 'no general improvement', and found a slight, but statistically significant difference between groups favouring the carbamazepine group. Little can be concluded from two small trials including 38 schizophrenia patients. It is disappointing that more trials did not report this simple outcome.
4.3 Mental state
The interpretation of results on mental state has been improved by the analysis of individual patient data in a uniform way. The meta‐analysis of the data of six out of eight trials did not show a significant superiority of carbamazepine according to several levels of reduction of the Brief Psychiatric Rating Scale (BPRS) (Overall 1962). Furthermore, there was a significant heterogeneity of the study results with one study (Heßlinger 1998) showing especially bad results associated with carbamazepine augmentation. The inspection of the methods of each study did not reveal clear reasons for this heterogeneity. Therefore, current data suggest that carbamazepine augmentation of antipsychotic drugs for people with schizophrenia does not seem to have a clinically meaningful effect on mental state. However, since there was a non‐significant trend in terms of 20% BPRS reduction and since the total number of patients is still low, more trials are warranted. Specific symptoms of schizophrenia (positive symptoms, negative symptoms and depression) were only reported by one or two trials so that any meaningful statement is not possible.
4.4 Adverse effects
Most data about movement disorders were too skewed to summate and individual studies reported conflicting results. As a result, no firm conclusion can be drawn. The fact that some studies found that carbamazepine augmentation leads to fewer movement disorders might be explained by a reduction in haloperidol plasma levels. This lowering of plasma levels might be the expression of an induction of liver enzymes related to carbamazepine. Two of the included studies (Dose 1987; Heßlinger 1998), one trial excluded because it did not provide any usable data (Kidron 1985), and several uncontrolled trials (Jann 1985; Kahn 1990; Otani 1997) suggest that this enzyme induction occurs. This interaction must be carefully taken into account whenever carbamazepine augmentation is tried.
Carbamazepine augmentation may well cause more allergic reactions, elevation of liver enzymes, leucopenia, and deterioration in the EEG than placebo augmentation. Adverse effects were, however, poorly reported and the only small trial (Dose 1987, n = 41) that clearly reported these important events had limited power to investigate differences between groups.
4.5 Missing outcomes
Currently, there are no data relating to the effect of carbamazepine augmentation on aggression, 'service' outcomes such as 'duration of hospital stay', satisfaction with treatment or costs.
4.6 Schizophrenia sub‐types and subgroups
Carbamazepine augmentation was not more effective when subgroups of people with schizophrenia were the focus of the studies. People with a schizophrenic illness designated as resistant to treatment were not consistently better when they received carbamazepine augmentation. Those with negative symptoms were not different in their response to antipsychotic augmentation compared with people whose illness did not have a predominance of negative symptoms. The small Neppe 1983 study (n = 9) suggested that a relatively non‐responsive heterogeneous group of patients with EEG abnormalities did fare somewhat better with carbamazepine augmentation than with placebo. This should be considered as hypothesis‐generating only.
It is not clear whether it makes sense to use carbamazepine in schizophrenia(‐like) patients with 'excited states'. One randomised controlled study (Klein 1984) suggested that this could be useful, but data from this trial could not be used in this review as the treatment allocation of people who left the study early is unclear. In a letter the authors stated that they do not remember how to interpret the data sheets of the study. Furthermore, a large controlled study of adjunctive carbamazepine to antipsychotics in 'excited psychoses' (Okuma 1989a, n = 162) had to be excluded because of the potential for inclusion of bias at the point of randomisation. Forty‐three per cent of those in the carbamazepine augmentation group showed marked and moderate improvement compared to 27% in the placebo group (not statistically significant). A post hoc analysis of individual mental state scale items suggested that this was related to an effect on disturbances of affective or emotional functions, whereas other items such as hallucinatory behaviour worsened with adjunctive carbamazepine.
Finally, carbamazepine augmentation for those with schizoaffective disorder has been surprisingly poorly studied, although it is frequently used in the daily routine for this condition. Only 12 participants included in this review had schizoaffective disorder so any judgment on the effects of carbamazepine for this important subgroup is impossible.
Overall completeness and applicability of evidence
We believe that the data we gathered covered a broad spectrum of participants and problems, but the small number of included studies (due to poor quality of all available data) is too small to conclusively judge about the potential applicability of the obtained results. Trials with small sample sizes lack sufficient power to detect a small to moderate effect, and thus results from such trials are often inconclusive, even when a real effect does exist. A review has suggested that meta‐analyses based on summation of small trials should be interpreted as inconclusive, regardless of whether the combined estimate was significant (Davey Smith 1998).
Quality of the evidence
The quality of all identified relevant trials has to be judged as low. As many as 64 out of 91 studies that were eventually excluded form the final analysis failed to randomise the trial sequence. Due to this low quality, only 10 studies were included in the meta‐analysis and the quality of those studies is moderate at best. Almost all studies were randomised and double‐blind but for most of them detailed descriptions were not presented. Therefore, it is unclear whether these studies were adequately randomised and whether treatment allocation was really concealed and blinding assured through the whole procedure. Reporting bias is especially prevalent in this study pool with high risk being a problem in a majority of reports. Without original study protocols and raw data sets one cannot really use those trials to the full extent. However, to the advantage of the 10 included studies is has to be noted that attrition bias was consistently judged as representing a low level of risk. The threat of other possible biases was also typically absent. Nevertheless, these limitations led to low or very low quality judgements in several important outcomes in the 'Summary of findings' table.
Potential biases in the review process
The evidence presented here is, to our best knowledge, complete. However, one can never be certain whether some additional (unpublished) material exist that was not pooled into the analysis.
Agreements and disagreements with other studies or reviews
We are aware of only one previous unsystematic review concerning the use of carbamazepine in the treatment of schizophrenia (Simhandl 1992). The authors tended to a conclusion that carbamazepine could have a beneficial effect, especially when used as an adjunct treatment to other neuroleptics. We do not feel the evidence supports this stance.
Authors' conclusions
Implications for practice.
1. For clinicians
Based on currently available randomised trial‐derived evidence, carbamazepine cannot be recommended for routine clinical use for treatment or augmentation of antipsychotic treatment of schizophrenia. For patients with a past history of response to carbamazepine, a trial of the drug may be warranted. For healthcare professionals currently caring for patients who have been receiving carbamazepine as a putative treatment for schizophrenia, clinicians need to weigh up whether this treatment should be stopped. Carbamazepine is associated with a range of adverse effects. If there is no evidence that the treatment has been effective, then it should be gradually tapered off and then stopped altogether. The dose of concomitant antipsychotics may need to be revised in light of the potential pharmacokinetic interactions between carbamazepine and some antipsychotics as antipsychotic plasma levels may rise upon withdrawal.
2. For people with schizophrenia
People with schizophrenia should know of the lack of a strong empirical basis for the use of carbamazepine in their illness. If its recommendation is still perused, the recipient of this treatment should expect clear endpoints and duration of treatment to be agreed upon.
3. For managers and policy makers
Although idiosyncratic positive responses are always possible, there are no data to support the use of carbamazepine for those with schizophrenia as a routine measure.
Implications for research.
1. General
Any future studies should respect standards of measuring outcomes and of reporting data (CONSORT) in order to enhance the comparability of study results (Begg 1996). The fact that several authors (see Acknowledgements) shared their data with us very much improved the quality of this review. We would like to encourage similar collaboration in the future. The Cochrane Collaboration supports the ALLTRIALS initiative.
2. Specific
2.1 Reviews or additional comparisons
Table 2, derived from the excluded studies contains suggestions for further reviews or for further comparisons relevant to this review. Because there are so few compelling data suggesting a real effect of carbamazepine for people with schizophrenia it is difficult to know if some of the additional comparisons are warranted. However, they are added in Table 2 for the sake of completeness.
1. Reviews or additional comparisons.
| Category | Title | Excluded study |
| Additional comparison | Carbamazepine (+/‐ antipsychotics) versus lithium (+/‐ antipsychotics) | Bellaire 1990, Greil 1997, Kahn 1990, Lenzi 1986, Placidi 1986, Schulz 1990 |
| Carbamazepine plus one antipsychotic versus another antipsychotic | Ohlmeier 2007 | |
| Carbamazepine versus valproate | Mosca 1998 | |
| Different formulations of carbamazepine | Nijdam 1992 | |
| Oxcarbazepine versus lithium | Cabrera 1987 | |
| Existing review* | Clozapine versus typical antipsychotics | Covell 2004 |
| New review | Adrenochrome semicarbazone for schizophrenia | Sugerman 1970 |
| Hydroxyphenamate for schizophrenia | Miller 1965 | |
| Rimcazole for schizophrenia | Borison 1991, Munetz 1989 | |
| Tybamate for schizophrenia | Meshel 1967, Meshel 1968 | |
| Vitamin B6 for schizophrenia | Miodownik 2003 |
2.2 Trials
There seems to be little need to undertake randomised trials investigating the effects of carbamazepine augmentation for people with uncomplicated schizophrenia. Some special indications might, however, still be of research interest.
Despite the reintroduction of clozapine, the only drug proven to have superior efficacy than standard drugs for those with treatment‐resistant illness (Essali 2009), there is a need for the development of treatment strategies when clozapine does not work. The two randomised trials investigating the effects of carbamazepine augmentation for people with treatment‐resistant schizophrenia (Llorca 1993; Simhandl 1996) only randomised a total of 66 patients. Even the combined totals lack the power to identify anything but gross differences between groups. Even small differences in outcome may be of great importance in this subgroup and therefore a large simple trial is justified. Clarification of the role of carbamazepine for the treatment of people with both schizophrenia and EEG abnormalities may be warranted. Carbamazepine is used for those with aggressive or violent episodes and its evaluation within trials in this subgroup of people with schizophrenia would be valuable. Carbamazepine is also used for those with schizoaffective disorders but data from placebo‐controlled trials do not exist. The bipolar type of schizoaffective disorder especially warrants further studies.
For a suggested design of study please see Table 3.
2. Suggested design for future study.
| Methods | Allocation: random. Blinding: blind or independent raters. Duration: minimum one year follow‐up. |
| Participants | Diagnosis: people with schizophrenia whose illness is resistant to treatment; people with psychoses and EEG abnormalities; people with psychoses and aggressive behaviour; and people with schizoaffective disorders. Age: Sex: male and female. N = 750. |
| Interventions | 1. Carbamazepine/oxcarbazepine alone: any dose. 2. Placebo (or no intervention). 3. Carbamazepine/oxcarbazepine in combination with any antipsychotic treatment: any dose. 4. Placebo (or no intervention) in combination with any antipsychotic treatment. 5. Antipsychotics alone: any dose. |
| Outcomes | Leaving the study early. Service utilisation. Global state ‐ no clinically important change.* Relapse. Mental state ‐ no clinically important change. Behaviour ‐ no clinically important change. Social functioning ‐ no clinically important change. Adverse effects ‐ clinically important general adverse effects*; sudden and unexpected death. Economic outcomes. Satisfaction with treatment. Quality of life. Pharmacokinetic interactions. All outcomes by time ‐ short term (up to 12 weeks), medium term (13 to 26 weeks) and long term (over 26 weeks). |
| Notes | *Primary outcomes of interest. |
What's new
| Date | Event | Description |
|---|---|---|
| 14 January 2014 | New citation required but conclusions have not changed | Update completed, no new studies found with no overall changes to conclusion. |
| 15 October 2012 | New search has been performed | Results of 2012 update search added, the 'Risk of bias' tables and description of bias added; background information, results and discussion extended; structure of the review updated. |
History
Protocol first published: Issue 4, 1998 Review first published: Issue 4, 1999
| Date | Event | Description |
|---|---|---|
| 3 May 2012 | Amended | Additional table linked to text |
| 7 July 2010 | Amended | New plain language summary added |
| 26 April 2008 | Amended | Converted to new review format. |
| 21 May 2007 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The Cochrane Schizophrenia Group produces and maintains a template for the methods section of their reviews. We have used this and adapted it for our review.
We would like to thank very much Drs Carpenter, Svestka, Dose, Neppe, Martin‐Munoz, Simhandl, Normann and Nachshoni for sending us their original patient data. Without their contribution this update would not have been possible.
We would also like to thank Paul White who helped with original protocol development and data interpretation.
Appendices
Appendix 1. Previous searches
1. Update searches in March 2005 and in December 2006
For the update we searched the Cochrane Schizophrenia Group's Register of Trials (March 2005 and November 2006) using the phrase: [((*carbama* or *amizepine* or *carbag* or *carbap* or *carbaz* or *carbymal* or *carpaz* or *cephalon* or *degranol* or *epitol* or *finlepsin* or *fokalepsin* or *hermolepsin* or *neurotol* or *neurotop* or *nordotol* or *sirtal* or *tardotol* or *tegret* or *teril* or *timonil* or *trimonil* or *trialeptal* or *trilpetal*) in Ti, Ab and In fields in References) AND (carbama* in Intervention field in Study)] This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module)
2. Original search 2.1 Electronic searching 2.1.1 We searched Biological Abstracts (January 1980 ‐ August 2001) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP near1 (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide"]
2.1.2 We searched The Cochrane Library (Issue 3, 2001) using the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP and (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide" or explode CARBAMAZEPINE / all]
2.1.3 We searched the Cochrane Schizophrenia Group's Register of Trials (December 2001) using the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP and (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide"or #42 = 684 or #42 = 20]
#42 is the intervention field within the register and 684 and 20 are the codes for carbamazepine.
2.1.4 We searched EMBASE (January 1980 ‐ August 2001) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP near1 (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide"or explode CARBAMAZEPINE / all]
2.1.5 We searched MEDLINE (January 1966 ‐ August 2001) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP near1 (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide"or explode CARBAMAZEPINE / all]
2.1.6 We searched PsycLIT (1886 ‐ August 2001) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP near1 (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide"or explode "CARBAMAZEPINE" / all]
2.1.7 We searched PSYNDEX (January 1974 ‐ August 2001) using the Cochrane Schizophrenia Group's phrase for both randomised controlled trials and schizophrenia (see Group search strategy) combined with the phrase:
[and carbamazepine* or tegretol or tardotol or tegretal or carbagamma or carbymal or carpaz or carbapin or degranol or finlepsin or fokalepsin or hermolepsin or neurotol or neurotop or nordotol or oxcarbazepine or sirtal or tardotol or timonil or trimonil or "10,11‐dihydro‐10‐oxo‐5H‐dobenz[b,f]azepine‐5‐carboxamide" or "5H‐dibenz[b,f]azepine‐5‐carboxamide" or (GP near1 (49.023 or 10.000 or 47.779 or 47.680)) or trialeptal or trileptal or "trans‐10,11‐dihydro‐10,11‐epoxy‐5h‐dibenz[b,f]azepine‐5‐carboxamide]
2.2 Reference lists We searched all references of articles selected for inclusion for further relevant trials.
2.3 Pharmaceutical companies We contacted companies performing trials with carbamazepine to obtain data on unpublished trials.
2.4 Personal contact We contacted the first author of each included study for more data of their study and any information regarding unpublished trials.
Appendix 2. Previous data collection methods
[For definitions of terms used in this, and other sections, please refer to the Glossary.]
1. Selection of trials SL independently inspected all reports identified by the search and JM re‐inspected these to ensure reliable selection. Where agreement could not be reached, we acquired the full report was for more detailed scrutiny. Once the full reports were obtained we independently inspected them to assess their relevance to this review. Again, if disagreement could not be resolved by discussion or from published information, we added the article to those awaiting assessment and contacted the authors of the study for clarification.
2. Assessment of methodological quality We assessed the methodological quality of included trials in this review using the criteria described in the Cochrane Handbook (Higgins 2005) and the Jadad Scale (Jadad 1996). The former is based on the evidence of a strong relationship among the potential for bias in the results and the allocation concealment (Schulz 1995) and is defined as below:
A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment)
The Jadad Scale measures a wider range of factors that impact on the quality of a trial. The scale includes three items:
1. Was the study described as randomised? 2. Was the study described as double‐blind? 3. Was there a description of withdrawals and drop outs?
Each item receives one point if the answer is positive. In addition, a point can be deducted if either the randomisation or the blinding/masking procedures described were inadequate or added if random number generation adequate or blinding appropriate. Scores on item 1 and 2 can therefore be 0, 1 or 2.
For the purpose of the analysis in this review, we included trials if they met the criteria A or B of the Cochrane Handbook. We did not use the Jadad scale to exclude trials in this review, but we used it to explore potential heterogeneity as a result of trial quality.
3. Data collection We independently extracted the data from included studies. Again, we discussed any disagreement and documented decisions. When this was not possible, we sought further information from authors of the studies and did not enter data from these trials but added them to the list of those awaiting assessment.
4. Data synthesis 4.1 Data types Outcomes are assessed using continuous (for example changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change') or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Currently RevMan does not support categorical data so they were presented only in the text of the review.
4.2 Incomplete data For studies that did not specify the reasons for people leaving the study early (dropped out), we assumed that these people had no change in the clinical outcome variables. If over 50% of people dropped out, and the study did not provide intention‐to‐treat results for continuous data, we excluded these data.
4.3 Cross‐over design We expected that some trials would use a cross‐over design. In order to exclude the potential additive effect in the second or more stages on these trials, we only analysed data from the first stage.
4.4 Dichotomous ‐ yes/no data We carried out an intention‐to‐treat analysis. On the condition that more than 50% of people completed the study, everyone allocated to the intervention were counted, whether they completed the follow‐up or not. We assumed that those who dropped out had the negative outcome, with the exception of death.
Where possible efforts were made to convert outcome measures to dichotomous data. This may be done by identifying cut‐off points on rating scales and dividing subjects accordingly into 'clinically improved' or 'not clinically improved'. If the authors of a study had used a designated cut‐off point for determining clinical effectiveness we used this where appropriate.
For dichotomous outcomes, a relative risk (RR) with the 95% confidence interval (CI) based on a fixed‐effect model was estimated. This is different to previous versions of this review. The reason for the change is that it has been shown that relative risks are more intuitive to clinicians than odds ratios (Boissel 1999). Furthermore, clinicians tend to interpret odds ratios as relative risks. This misinterpretation leads to an overestimate of effect (Deeks 2000). When overall results were significant we calculated the Number Needed to Treat (NNT) and/or the Number Needed to Harm (NNH) as the inverse of the absolute risk difference.
4.5 Continuous data 4.5.1 Normally distributed data: Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data the following standards were applied to data derived from continuous measures of endpoint ('state' data). The criteria were used before inclusion: i. standard deviations and means were reported in the paper or were obtainable from the authors and ii. the standard deviation (SD), when multiplied by 2, was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution) (Altman 1996). If a scale starts from a positive value (such as PANSS which can have values from 30 to 210) the calculation described above in ii) should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score. We did not enter data that did not meet the first or second standard into RevMan software for analysis, but reported the data in the text of the results section.
4.5.2 Scale derived data: A wide range of rating scales is available to measure outcomes in mental health trials. These scales vary in quality and many are questionably validated, or even ad hoc. It is generally accepted that measuring instruments should have the properties of reliability (the extent to which a test effectively measures anything at all) and validity (the extent to which a test measures that which it is supposed to measure). Before publication of an instrument, most scientific journals insist that reliability and validity be demonstrated to the satisfaction of referees. We therefore decided, as a minimum standard, not to include any data from a rating scale in this review unless its properties had been published in a peer‐reviewed journal. In addition, we set the following minimum standards for rating scales; the rating scale should either be i. a self‐report or ii. completed by an independent rater or relative. We may set more stringent standards for instruments in future updates of this review.
Whenever possible we took the opportunity to make direct comparisons between trials that used the same measurement instrument to quantify specific outcomes. Where continuous data were presented from different scales rating the same effect, we presented both sets of data and inspected the general direction of effect.
4.5.3 Endpoint versus change data: For continuous mean change data (endpoint minus baseline) the situation is even more problematic. In the absence of individual patient data it is impossible to know if change data is skewed. The RevMan meta‐analyses of continuous data are based on the assumption that the data are, at least to a reasonable degree, normally distributed. It is quite feasible that change data is skewed but, after consulting the ALLSTAT electronic statistics mailing list, it was entered into RevMan in order to summarise the available information. In doing this it is assumed that either data were not skewed or that the analyses within RevMan could cope with the unknown degree of skewness.
4.6 Individual patient data For this update we requested the individual patient data from the original authors. Most of these were data derived from the BPRS, a scale measuring mental state. We tried to convert these results to dichotomous data (see 4.3.1). As it seemed impossible to us to predefine which level of reduction of the total score is clinically meaningful, three levels were analysed: a relatively low level (at least 20% BPRS reduction), an intermediate level (at least 35% BPRS reduction) and a relatively high level (at least 50% BPRS reduction).
4.7 Data display We entered data into RevMan in such a way that the area to the left of the line of no effect indicated a favourable outcome for carbamazepine alone or carbamazepine augmentation.
4.8 Cluster trials Studies increasingly employ "cluster randomisation" (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a "unit of analysis" error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type 1 errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. in subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering was incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We sought statistical advice and were advised that the binary data as presented in a report should be divided by a "design effect". This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported, we assumed it to be 0.1 (Ukoumunne 1999). If cluster studies had been appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
5. Investigation for heterogeneity Firstly, we visually inspected graphs to investigate the possibility of statistical heterogeneity. This was supplemented using, primarily, the I‐squared statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I‐squared estimate was greater than or equal to 50% we interpreted this as indicating the presence of considerable levels of heterogeneity (Higgins 2005). The I‐squared statistic has been described to be a more appropriate indicator of heterogeneity than the Chi‐square test that was used in the previous version of the review (Higgins 2005). If either the I‐squared statistic was higher than 50% or the p‐value of the Chi‐square test, for reasons of consistency we did not deviate from the rule as to when the fixed and when the random‐effects model has to be applied, although we would now rather use the random‐effects model throughout.
6. Publication bias We entered data from all included trials into a funnel graph (trial effect versus trial size or 'precision') in an attempt to investigate the likelihood of overt publication bias. A formal test of funnel plot asymmetry (suggesting potential publication bias) was undertaken, where appropriate (Egger 1997). Significance levels of P < 0.1 were set a priori to accept the presence of asymmetry. Where only three or four studies reported an outcome or there was little variety in sample size (or precision estimate) between studies tests of asymmetry were not appropriate.
7. Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for carbamazepine.
Appendix 3. Previous plain language summary
Carbamazepine for schizophrenia
Schizophrenia is a serious mental illness which can cause people to change the way they sense and understand the world. It can be very serious and continue to affect some people throughout their life. Although most people with this illness can be helped by taking antipsychotic medication, 5% ‐15% will continue to suffer from debilitating symptoms. For this group of people a variety of medical options are available to reduce these symptoms, including adjusting the dose of medication, changing to another antipsychotic, or using drugs other than antipsychotics. Carbamazepine is a drug which has been used to treat epilepsy since the 1950s. It is also used as a mood stabiliser when people alternate between very ‘high’ and depressed moods (bi‐polar affective disorder). This review looks at the effectiveness of carbamazepine when compared to no active medication (placebo), an antipsychotic, or when it is used in addition to an antipsychotic in clinical trials on people who have schizophrenia both with additional mood problems (schizoaffective disorder), and without.Ten trials were found which included a total of 258 people. All except one of these studies were carried out in a hospital setting. One small trial compared carbamazepine treatment with placebo (31 people) and found that there was no significant difference between these in respect to preventing relapse, mental state or development of adverse effects. Another single study of 38 people compared carbamazepine (as a single treatment) with the antipsychotic perphenazine (as a single treatment) and found no significant difference between the people in the two groups except that those on perphenazine were more likely to have movement side effects and be taking medication for them.The remainder of the trials compared antipsychotic plus carbamazepine with antipsychotic plus placebo. Two of these trials (38 people) showed that when carbamazepine was taken with an antipsychotic, there was a general improvement in over half of these people. However, for the majority of the outcomes for which there was acceptable data, there was no significant difference between the two groups. Since all of these trials were small, there is not enough data to clarify whether carbamazepine reduces symptoms without giving too many adverse effects in people with schizophrenia and schizoaffective disorders who are resistant to treatment with antipsychotics alone. A larger well designed trial may provide more robust evidence to support the treatment options for these people. (Plain language summary prepared for this review by Janey Antoniou of RETHINK, UK www.rethink.org).
Data and analyses
Comparison 1. CARBAMAZEPINE AS SOLE TREATMENT vs PLACEBO AS SOLE TREATMENT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.17, 6.64] |
| 2 Relapse (by 3 months) | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.78, 1.45] |
| 3 Mental state: 1. Less than 20% BPRS reduction | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.75, 1.30] |
| 4 Mental state: 2. Average endpoint score of the BPRS at 3 months | 1 | 27 | Mean Difference (IV, Fixed, 95% CI) | ‐0.07 [‐0.46, 0.32] |
| 5 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 allergic reactions | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.44 [0.42, 132.95] |
| 5.2 blood dyscrasia | 1 | 31 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.19 [0.14, 72.69] |
1.1. Analysis.

Comparison 1 CARBAMAZEPINE AS SOLE TREATMENT vs PLACEBO AS SOLE TREATMENT, Outcome 1 Leaving the study early.
1.2. Analysis.

Comparison 1 CARBAMAZEPINE AS SOLE TREATMENT vs PLACEBO AS SOLE TREATMENT, Outcome 2 Relapse (by 3 months).
1.3. Analysis.

Comparison 1 CARBAMAZEPINE AS SOLE TREATMENT vs PLACEBO AS SOLE TREATMENT, Outcome 3 Mental state: 1. Less than 20% BPRS reduction.
1.4. Analysis.
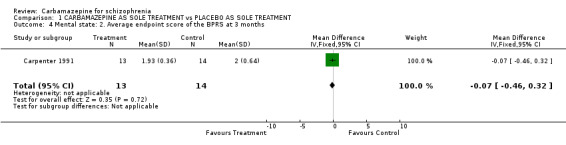
Comparison 1 CARBAMAZEPINE AS SOLE TREATMENT vs PLACEBO AS SOLE TREATMENT, Outcome 4 Mental state: 2. Average endpoint score of the BPRS at 3 months.
1.5. Analysis.
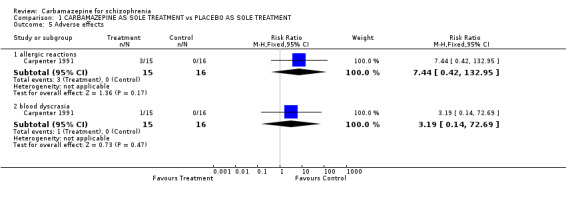
Comparison 1 CARBAMAZEPINE AS SOLE TREATMENT vs PLACEBO AS SOLE TREATMENT, Outcome 5 Adverse effects.
Comparison 2. CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.52 [0.23, 88.38] |
| 2 Mental state: 1. Categories of reduction on BPRS scores | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 less than 20% reduction | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.62, 2.66] |
| 2.2 less than 35% reduction | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.86, 3.24] |
| 2.3 less than 50% reduction | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.78, 1.92] |
| 3 Mental state: 2. Mean BPRS at endpoint | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | 2.30 [‐3.84, 8.44] |
| 4 Adverse effects: 1. Movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 akathisia | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.01, 2.34] |
| 4.2 parkinsonism | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.03 [0.00, 0.43] |
| 4.3 tremor | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.01, 6.97] |
| 4.4 use of anticholinergic drugs | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.09, 0.55] |
| 5 Adverse effects: 2. Others | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 blurred vision | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.04, 4.55] |
| 5.2 collapse | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.3 [0.03, 2.63] |
| 5.3 constipation | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.04, 4.55] |
| 5.4 dizziness | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.52 [0.23, 88.38] |
| 5.5 dry mouth | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.04, 4.55] |
| 5.6 fatigue | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.4 [0.72, 40.66] |
| 5.7 nausia | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.12, 62.70] |
| 5.8 salivation | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.12, 62.70] |
| 5.9 tachycardia | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.28, 2.04] |
| 6 Subgroup analysis ‐ schizoaffective disorder excluded ‐ Mental state: Categories of reduction on BPRS score | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 less than 20% reduction | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.09 [1.22, 7.84] |
| 6.2 less than 35% reduction | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [1.15, 4.67] |
| 6.3 less than 50% reduction | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.94, 2.09] |
2.1. Analysis.

Comparison 2 CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT, Outcome 1 Leaving the study early.
2.2. Analysis.
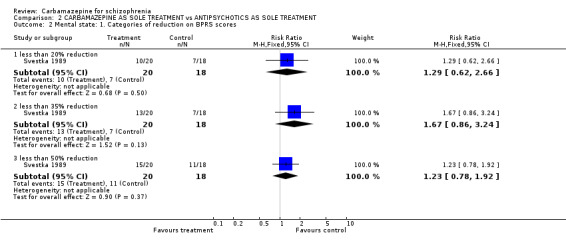
Comparison 2 CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT, Outcome 2 Mental state: 1. Categories of reduction on BPRS scores.
2.3. Analysis.

Comparison 2 CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT, Outcome 3 Mental state: 2. Mean BPRS at endpoint.
2.4. Analysis.

Comparison 2 CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT, Outcome 4 Adverse effects: 1. Movement disorders.
2.5. Analysis.

Comparison 2 CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT, Outcome 5 Adverse effects: 2. Others.
2.6. Analysis.

Comparison 2 CARBAMAZEPINE AS SOLE TREATMENT vs ANTIPSYCHOTICS AS SOLE TREATMENT, Outcome 6 Subgroup analysis ‐ schizoaffective disorder excluded ‐ Mental state: Categories of reduction on BPRS score.
Comparison 3. ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early | 8 | 182 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.16, 1.35] |
| 2 Global state: No improvement | 2 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.37, 0.88] |
| 3 Mental state: 1a. General ‐ categories of reduction on BPRS scores | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 less than 20% reduction | 6 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.44, 1.07] |
| 3.2 less than 35% reduction | 6 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.57, 1.05] |
| 3.3 less than 50% reduction | 6 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.67, 1.12] |
| 4 Mental state: 1b. General ‐ average BPRS endpoint score (high = poor) | 3 | 79 | Mean Difference (IV, Fixed, 95% CI) | ‐3.21 [‐7.82, 1.40] |
| 5 Mental state: 1c. General ‐ average BPRS endpoint score (high = poor, skewed data) | Other data | No numeric data | ||
| 6 Mental state: 1d. General ‐ average IMPS endpoint score (high = poor) | 2 | 50 | Mean Difference (IV, Fixed, 95% CI) | 4.61 [1.30, 7.91] |
| 7 Mental state: 2a. Specific ‐ positive symptoms (PANSS subscale at endpoint, high = poor) | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 4.22 [0.75, 7.69] |
| 8 Mental state: 2b. Specific ‐ positive symptoms (IMPS score at endpoint, high = poor) | Other data | No numeric data | ||
| 9 Mental state: 2c. Specific ‐ negative symptoms (SANS at endpoint, high = poor) | 2 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐2.75 [‐6.71, 1.22] |
| 10 Mental state: 2d. Specific ‐ depression (Hamilton scale at endpoint, high = poor) | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐2.20, 1.50] |
| 11 Behaviour: Average dose of medication used for agitation (chlorprothixene, skewed data) | Other data | No numeric data | ||
| 12 Adverse effects: 1a Movement disorders ‐ at least one movement disorder | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.14, 1.02] |
| 13 Adverse effects: 1b. Movement disorders ‐ average dose of antiparkinsonism drugs (biperiden, skewed data) | Other data | No numeric data | ||
| 14 Adverse effects: 1c. Movement disorders ‐ average endpoint score (SAS, high = poor, skewed data) | Other data | No numeric data | ||
| 15 Adverse effects: 1d. Movement disorders ‐ average endpoint TD rating (high = poor, skewed data) | Other data | No numeric data | ||
| 16 Adverse effects: 2. Others | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 allergic reaction | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.79 [0.16, 87.86] |
| 16.2 EEG deterioration | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.13 [0.59, 7.75] |
| 16.3 liver enzyme elevation | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.56 [0.53, 12.42] |
| 16.4 white blood cell decline (substantial) | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.09, 19.06] |
| 17 Physiological effect: Haloperidol plasma levels | Other data | No numeric data | ||
| 18 Subgroup analysis ‐ treatment‐resistant participants | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 treatment‐resistant | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.51, 1.18] |
| 18.2 not treatment‐resistant | 5 | 118 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.65, 1.23] |
| 19 Subgroup analysis ‐ EEG abnormalities | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 with EEG abnormalities | 1 | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.65, 1.90] |
| 19.2 without EEG abnormalities | 5 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.63, 1.11] |
| 20 Subgroup analysis ‐ negative symptoms | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 predominant negative symptoms | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.83, 1.21] |
| 20.2 no predominant negative symptoms | 5 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.55, 1.17] |
| 21 Sensitivity analysis: random effects model ‐ less than 50% BPRS reduction | 6 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.60, 1.45] |
3.1. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 1 Leaving the study early.
3.2. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 2 Global state: No improvement.
3.3. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 3 Mental state: 1a. General ‐ categories of reduction on BPRS scores.
3.4. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 4 Mental state: 1b. General ‐ average BPRS endpoint score (high = poor).
3.5. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 5 Mental state: 1c. General ‐ average BPRS endpoint score (high = poor, skewed data).
| Mental state: 1c. General ‐ average BPRS endpoint score (high = poor, skewed data) | |
|---|---|
| Study | |
| Martin‐Munoz 1989 | 1. Carbamazepine + antipsychotics: mean 8.6, SD 9.6, N = 10. 2. Placebo + antipsychotics: mean 7.4, SD 4.7, N = 10. |
| Nachshoni 1994 | 1. Carbamazepine + antipsychotics: mean 5.0, SD 2.70, N = 15. 2. Placebo + antipsychotics: mean 5.1, SD 1.6, N = 15. |
| Neppe 1983 | 1. Carbamazepine + antipsychotics: mean 14.7, SD 5.8, N = 3. 2. Placebo + antipsychotics: mean 22.0, SD 13.4, N = 4. |
3.6. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 6 Mental state: 1d. General ‐ average IMPS endpoint score (high = poor).
3.7. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 7 Mental state: 2a. Specific ‐ positive symptoms (PANSS subscale at endpoint, high = poor).
3.9. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 9 Mental state: 2c. Specific ‐ negative symptoms (SANS at endpoint, high = poor).
3.10. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 10 Mental state: 2d. Specific ‐ depression (Hamilton scale at endpoint, high = poor).
3.12. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 12 Adverse effects: 1a Movement disorders ‐ at least one movement disorder.
3.14. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 14 Adverse effects: 1c. Movement disorders ‐ average endpoint score (SAS, high = poor, skewed data).
| Adverse effects: 1c. Movement disorders ‐ average endpoint score (SAS, high = poor, skewed data) | |
|---|---|
| Study | |
| Dose 1987 | 1. Carbamazepine + antipsychotics: mean 1.03, SD 0.86. N = 17. 2. Placebo + antipsychotics: mean 2.84, SD 2.18. N = 17. |
| Nachshoni 1994 | 1. Carbamazepine + antipsychotics: mean 0.9, SD 0.9. N = 13. 2. Placebo + antipsychotics: mean 0.4, SD 0.5. N = 13. |
| Simhandl 1996 | 1. Carbamazepine + antipsychotics: mean 0.27, SD 0.19. N = 15. 2. Placebo + antipsychotics: mean 0.31, SD 0.35. N = 12. |
3.15. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 15 Adverse effects: 1d. Movement disorders ‐ average endpoint TD rating (high = poor, skewed data).
| Adverse effects: 1d. Movement disorders ‐ average endpoint TD rating (high = poor, skewed data) | |
|---|---|
| Study | |
| Simhandl 1996 | 1. Carbamazepine + antipsychotics: mean 7.1, SD 0.2. N = 15. 2. Placebo + antipsychotics: mean 13.1, SD 14.2. N = 12. |
3.16. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 16 Adverse effects: 2. Others.
3.17. Analysis.
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 17 Physiological effect: Haloperidol plasma levels.
| Physiological effect: Haloperidol plasma levels | |
|---|---|
| Study | |
| Dose 1987 | 1. Carbamazepine + antipsychotics: mean level 5.8 (mg haloperidol/d)/(microgms/L haloperidol), SEM 3.3. N = 17. 2. Placebo + antipsychotics: mean level 3.1 (mg haloperidol/d)/(microgms/L haloperidol), SEM 1.65. N = 17. |
| Heßlinger 1998 | 1. Carbamazepine + haloperidol: mean haloperidol level change ‐3.8 ng/mL, SD 3.0. N = 9 (˜45% decrease). 2. Placebo + haloperidol: mean haloperidol level change 1.9 ng/mL, SD 2.3. N = 9 (˜51% increase). |
3.18. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 18 Subgroup analysis ‐ treatment‐resistant participants.
3.19. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 19 Subgroup analysis ‐ EEG abnormalities.
3.20. Analysis.
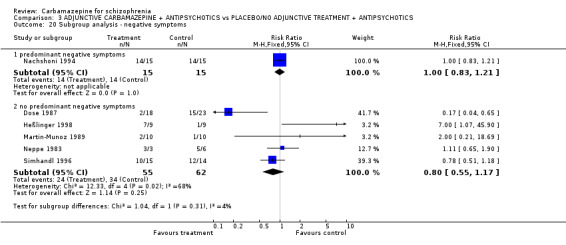
Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 20 Subgroup analysis ‐ negative symptoms.
3.21. Analysis.

Comparison 3 ADJUNCTIVE CARBAMAZEPINE + ANTIPSYCHOTICS vs PLACEBO/NO ADJUNCTIVE TREATMENT + ANTIPSYCHOTICS, Outcome 21 Sensitivity analysis: random effects model ‐ less than 50% BPRS reduction.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Carpenter 1991.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ no further information. Design: cross‐over. Duration: 95 days each phase. Setting: outpatient department. | |
| Participants | Diagnosis: schizophrenia (DSM‐III & RDC). N = 34.* Sex: 18 M, 9 F.* Age: mean ˜ 33 years. History: stabilised on neuroleptic maintenance, hospitalised ˜3 times, ill ˜10 years. | |
| Interventions | 1. Carbamazepine: dose 800‐1200 mg/day. N˜15.* 2. Placebo (neuroleptics withdrawn over 1‐5 days when study medication dose was reached). N˜16.* | |
| Outcomes | 1. Leaving the study early. 2. Relapse.* 3. Mental state: BPRS (mean and number with 20% reduction).* 4. Side effects ‐ allergic reactions, blood dyscrasia.* | |
| Notes | Data analysed on 31 patients, no information about treatment status of 3 people. Interim analyses showed high relapse rates in both arms of study ‐ study stopped. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned, within prognostic strata", no further details (p.69). |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind", "identical placebo tablets" (p.69), "number of carbamazepine tablets was adjusted by a nonblind psychiatrist" (p.70). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Double‐blind", "identical placebo tablets" (p.69), "number of carbamazepine tablets was adjusted by a nonblind psychiatrist" (p.70). |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Double‐blind", "identical placebo tablets" (p.69). |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | "Blind rater" (p.69). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Same dropout rate in each group. |
| Selective reporting (reporting bias) | Low risk | No selective reporting. |
| Other bias | Low risk | No other bias. |
Dose 1987.
| Methods | Allocation: randomised ‐ no further details. Blinding: double ‐ no further information. Design: parallel. Duration: 5 weeks (at week 4 interventions withdrawn). Setting: in hospital. | |
| Participants | Diagnosis: acute schizophrenia (ICD‐9 & DSM‐III). N = 41. Sex: not reported. Age: not reported. History: hospitalised ˜1.2 times, ill ˜6 years. | |
| Interventions | 1. Adjunctive carbamazepine: dose 200 mg, increased to 600‐1200 mg/day (target plasma level 8‐12 micrograms/dL) + haloperidol 6 mg/day then titrated to clinical judgement. N = 18. 2. Placebo additional treatment + haloperidol 6 mg/day then titrated to clinical judgement. N = 23. | |
| Outcomes | 1. Leaving the study early.
2. Mental state: (BPRS 20%, 35%, 50% reduction, mean at endpoint; mean IMPS at endpoint).
3. Side effects: allergic reactions, substantial white blood cell decline, increase of liver enzymes, worsening of EEG. Unable to use:. Medication use (mean haloperidol dose ‐ no SD, biperiden, chlorprothixene ‐ data skewed). Movement disorder (SAS ‐ data skewed). Haloperidol plasma‐levels ‐ data skewed. |
|
| Notes | ** Two people taking carbamazepine had falls in white cell counts, but not below usual reference range. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized", "randomly assigned" (p.304) ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind" ‐ no further details (p.304). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Double‐blind" ‐ no further details (p.304). |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Double‐blind" ‐ no further details (p.304). |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Double‐blind" ‐ no further details (p.304). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "[All patients] survived for the end‐of‐treatment‐analysis without any dropouts" (p.304). "No patients dropped out because of side effects or adverse reactions" (p.306). |
| Selective reporting (reporting bias) | High risk | CGI results not reported. |
| Other bias | Unclear risk | "Dosages of haloperidol, chlorprothixene and biperiden, as well as the occurrence of side effects, differed significantly (...) between the carbamazepine and placebo group" (p.307‐8). |
Heßlinger 1998.
| Methods | Allocation: randomised ‐ sealed envelopes. Blinding: single. Design: parallel. Duration: 4 weeks. Setting: in hospital. | |
| Participants | Diagnosis: schizophrenia (n = 16) or schizoaffective (n = 2) psychosis (ICD‐10). N = 18. Sex: 12 M, 6F. | |
| Interventions | 1. Adjunctive carbamazepine: dose mean 567mg/day, titrated in week 1 to plasma‐level of 6‐12 mg/mL + constant haloperidol dose: dose mean ˜ 15 mg/day, higher in this group to maintain effective plasma levels. N = 9. 2. No additional treatment + constant haloperidol dose. N = 9. | |
| Outcomes | 1. Leaving the study early.
2. Mental state (BPRS 20%, 35%, 50% reduction and mean at endpoint, PANSS positive score at endpoint). Unable to use:. Haloperidol plasma levels ‐ data skewed. Mean biperiden and chlorprothixen dose ‐ data skewed. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly assigned" (p.311). |
| Allocation concealment (selection bias) | Low risk | "Sealed envelopes". |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p.311). Patients were not aware of the treatment (information received by letter). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p.311) Patients were not aware of the treatment (information received by letter). |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p.311). Patients were not aware of the treatment (information received by letter). |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Psychiatrist (...) was blind to medication and drug levels. However, he had access to the nursing charts" (p.311). Patients were not aware of the treatment (information received by letter). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "25 out of 27 patients completed the study protocol", 2 CBZ dropouts, ITT analysis (p.311). |
| Selective reporting (reporting bias) | Low risk | No selective outcome reporting. |
| Other bias | Low risk | No other bias. |
Llorca 1993.
| Methods | Allocation: randomised ‐ Latin square procedure. Blindness: double ‐ identical capsules. Design: cross‐over, no wash‐out. Duration: 4 X 5 week cross‐over (preceded by 5 weeks no adjunctive treatment). Setting: in hospital. | |
| Participants | Diagnosis: treatment‐resistant schizophrenia patients (DSM‐III‐R, Kane criteria). N = 24. Sex: 18 M, 6 F. Age: mean ˜44 years. | |
| Interventions | 1. Adjunctive carbamazepine: dose 2 weeks 200 mg/day, 3 weeks 400 mg/day + constant haloperidol (15‐65 mg/day). N = 6. 2. Adjunctive bromocriptine: dose 2.5 mg/day + constant haloperidol (15‐65 mg/day). N = 6.* 3. Adjunctive cyproheptadine: 12‐24 mg/day + constant haloperidol (15‐65 mg/day). N = 6.* 4. Placebo additional treatment + constant haloperidol (15‐65 mg/day). N = 6. | |
| Outcomes | 1. Leaving the study early. Unable to use: . Mental state (BPRS, SAPS, SANS ‐ P values only). Movement disorder (SAS, AIMS ‐ P values only). |
|
| Notes | * Data from groups 2 & 3 not used in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomized succession using a latin square procedure" (p.565). |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Tout au long de l'étude, les patients sont à l'insu du traitement recu. Durant les 4 phase de l'expérimentation, patients et investigateurs sont à l'insu de la médication adjuvante recue". "Afin de maintenir les conditions de double aveugle, les résultats des dosages pratiqués sont collectés au fur et à mesure de l'étude par un médecin différent du cotateur (M.A.W.)". "capsules identiques". |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "Tout au long de l'étude, les patients sont à l'insu du traitement recu. Durant les 4 phase de l'expérimentation, patients et investigateurs sont à l'insu de la médication adjuvante recue". "Afin de maintenir les conditions de double aveugle, les résultats des dosages pratiqués sont collectés au fur et à mesure de l'étude par un médecin différent du cotateur (M.A.W.)" "capsules identiques". |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Tout au long de l'étude, les patients sont à l'insu du traitement recu. Durant les 4 phase de l'expérimentation, patients et investigateurs sont à l'insu de la médication adjuvante recue". "Afin de maintenir les conditions de double aveugle, les résultats des dosages pratiqués sont collectés au fur et à mesure de l'étude par un médecin différent du cotateur (M.A.W.)". "capsules identiques". |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | "Tout au long de l'étude, les patients sont à l'insu du traitement recu. Durant les 4 phase de l'expérimentation, patients et investigateurs sont à l'insu de la médication adjuvante recue". "Afin de maintenir les conditions de double aveugle, les résultats des dosages pratiqués sont collectés au fur et à mesure de l'étude par un médecin différent du cotateur (M.A.W.)". "capsules identiques". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant left the study early. |
| Selective reporting (reporting bias) | High risk | No SDs reported. |
| Other bias | Low risk | No clear evidence for other bias. |
Mair 1990.
| Methods | Allocation: randomised ‐ no further details. Blinding: open. Design: parallel. Duration: 5 weeks. Setting: in hospital. | |
| Participants | Diagnosis: schizophrenia & schizophrenia‐like psychoses (ICD‐9). N = 23. Age: range 31‐44 years. History: "acutely ill", unresponsive to 5 days clozapine or haloperidol, admitted 4‐9 times. | |
| Interventions | 1. Adjunctive carbamazepine: dose 600 mg/day + titrated dose of haloperidol or clozapine. N = 13. 2. No additional treatment + titrated dose of haloperidol or clozapine. N = 10. | |
| Outcomes | 1. Leaving the study early. Unable to use:. General functioning (CGI ‐ no SD). Mental state (BPRS ‐ no SD). Extrapyramidal side effects (Webster scale ‐ no data). Other side effects (FSUCL ‐ no data). Mean haloperidol/clozapine dose ‐ no SD. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "random number list" (p.79). |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Blinding not described (an open study) (p.79). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | Blinding not described (an open study) (p.79). |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | Blinding not described (an open study) (p.79). |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | Blinding not described (an open study) (p.79). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was not mentioned whether there were dropouts or not. |
| Selective reporting (reporting bias) | High risk | No SDs reported for CGI and BPRS, no data reported for FSUCL and Webster Scale. |
| Other bias | Low risk | No other bias. |
Martin‐Munoz 1989.
| Methods | Allocation: randomised ‐ no further information. Blinding: not stated. Design: parallel. Duration: 18 days. Setting: in hospital. | |
| Participants | Diagnosis: paranoid schizophrenia (RDC). N = 20. Age: mean ˜29 years. Sex: 18 M, 2 F. | |
| Interventions | 1. Adjunctive carbamazepine: dose initially 600 mg/day, adjusted to plasma levels of 8‐12 ng/mL + haloperidol, 30 mg/day, fixed dose. N = 10. 2. No additional treatment + haloperidol, 30 mg/day, fixed dose. N = 10. | |
| Outcomes | 1. Leaving the study early.
2. Mental state (20%, 35%, 50% BPRS reduction).
3. Movement disorder. Unable to use:. Side effects (UKU‐scale ‐ no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Allocated (...) randomly using a coin toss method" (letter). |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Doble ciego", no further details. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Doble ciego", no further details. |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Doble ciego", no further details. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Doble ciego", no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts (according to the letter). |
| Selective reporting (reporting bias) | High risk | No SD's in BPRS, UKU‐scale results not reported. |
| Other bias | Low risk | No clear evidence for other bias. |
Nachshoni 1994.
| Methods | Allocation: randomised ‐ random allocation list. Blinding: double ‐ identical capsules. Design: parallel. Duration: 5 weeks. Setting: in hospital. | |
| Participants | Diagnosis: residual schizophrenia (DSM‐III‐R). N = 30.* Age: mean ˜46 years. Sex: 15 M, 13 F. History: predominant negative symptoms, ill mean ˜19 years. | |
| Interventions | 1. Adjunctive carbamazepine: dose increased to 600 mg/day during week 1, then adjustment to plasma levels of 4‐12 ng/mL + 300‐800 chlorpromazine equivalent antipsychotic treatment. N = 15. 2. Placebo adjunctive treatment + 300‐800 chlorpromazine equivalent antipsychotic treatment. N = 15. | |
| Outcomes | 1. Leaving the study early.
2. Mental state (20%, 35%, 50% BPRS reduction, HRSD, SANS at endpoint). Unable to use:. EPS (SAS ‐ no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Random allocation list" (p.24). |
| Allocation concealment (selection bias) | Low risk | "Dr. Joseph Levin was the control psychiatrist who knew what the patients were receiving, according to a random allocation list" (letter from the authors). It appears that other members of the team were blind to allocation. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind design" (p.24), patients and personal were blind, tablets were identical (letter). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "Double‐blind design" (p.24), patients and personal were blind, tablets were identical (letter). |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Double‐blind design" (p.24), patients and personal were blind, tablets were identical (letter). |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Low risk | "Ratings were done by psychiatrist, who was blind to whether the patients were receiving placebo or carbamazepine" (p.23). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Two patients were withdrawn, one from each group" (p.24). |
| Selective reporting (reporting bias) | Low risk | No selective reporting. |
| Other bias | Low risk | No other bias. |
Neppe 1983.
| Methods | Allocation: randomised ‐ no further information. Blinding: double ‐ no further information. Design: cross‐over. Duration: 2 X 6 weeks (preceded by 3‐week baseline). Setting: in hospital. | |
| Participants | Diagnosis: schizophrenia (10), non‐progressive dementia (2), rapid cycling (1) (diagnostic criteria unclear). N = 13.* Age: mean ˜34 years. Sex: 8 M, 5 F. History: chronic, "poor‐responders", EEG abnormalities. | |
| Interventions | 1. Adjunctive carbamazepine: dose 600 mg/day + various antipsychotics (constant dose). N = 3. 2. Placebo adjunctive treatment + various antipsychotics (constant dose). N = 6. | |
| Outcomes | 1. Leaving the study early.
2. Global impression (CGI).
3. Mental state (20%, 35%, 50% BPRS reduction). Unable to use: . General improvement (Global Assessment, OCR unpublished scales). |
|
| Notes | *Data extracted for 9 participants with schizophrenia from published data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomized" (p.326), "random number list" (letter). |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double blind" (p.326). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Double blind" (p.326). |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Double blind" (p.326). |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Double blind" (p.326). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Two out of six patients in the control group compared to zero out of three in the intervention group dropped out [out of 13]" (p.327). |
| Selective reporting (reporting bias) | High risk | No SDs for BPRS. |
| Other bias | Low risk | No other bias ("one [subject had] a history of contaminating cannabis use", p.327). |
Simhandl 1996.
| Methods | Allocation: randomised ‐ no further details. Blindness: double. Design: parallel. Duration: 8 weeks (intervention withdrawn at week 6). Setting: no information available. | |
| Participants | Diagnosis: schizophrenia (DSM‐III‐R). N = 42. Age: mean ˜35 years. Sex: 30 M, 12 F. History: "chronic", non‐response to > 3 neuroleptics (2 different chemical classes) in last 2 years, duration ill ˜ 10 years. | |
| Interventions | 1. Adjunctive carbamazepine: dose increased week 1‐2 until plasma levels = 15‐42 micromol/L + constant dose of antipsychotics. N = 15. 2. Lithium*: dose increased week 1‐2 until plasma level = 0.6‐1.2 mml/L) + constant dose of antipsychotics. N = 13. 3. Placebo + constant dose of antipsychotics. N = 14. | |
| Outcomes | 1. Leaving the study early.
2. Global impression (CGI).
3. Mental state (20%, 35%, 50% BPRS reduction, SANS at endpoint). Unable to use: . Plasma levels of antipsychotics ‐ skewed data. |
|
| Notes | Jadad = 4. *This group was not used in the analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Random", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind", no further details. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Double‐blind", no further details. |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Double‐blind", no further details. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Double‐blind", no further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two patients in each group dropped out, both because of non‐compliance. |
| Selective reporting (reporting bias) | Unclear risk | The study is only available as an abstract. |
| Other bias | Low risk | No clear other bias. |
Svestka 1989.
| Methods | Allocation: randomised ‐ no further details. Blindness: single (raters). Design: cross‐over. Duration: 6 weeks (2 weeks placebo ‐ 3 weeks carbamazepine/perphenazine ‐ 1 week placebo ‐ 3 weeks carbamazepine/perphenazine). Setting: hospital. | |
| Participants | Diagnosis: ICD‐9 schizophrenia (n = 28) or schizoaffective disorder (n = 10). History: "acutely ill", duration ill ˜9 years. N = 38. Age: mean ˜38 years. Sex: 30 M, 8 F (the gender of 2 people who left early is unknown). | |
| Interventions | 1. Carbamazepine, flexible dose, mean = 1374 SD = 334, N = 22. 2. Perphenazine, flexible dose, mean = 53, SD = 12. N = 18. Then 1 week placebo and 3 weeks cross‐over to other treatment. | |
| Outcomes | 1. Leaving the study early. 2. Mental state (20%, 35%, 50% BPRS reduction and BPRS at endpoint). 3. Various side effects. | |
| Notes | Jadad = 4. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomised" ‐ no further details ("intraindividual comparison", p.276). |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | No information available. |
| Blinding of outcome assessment (detection bias) Objective outcomes | Unclear risk | No information available. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | No information available. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Two out of 20 participants in the intervention group dropped out compared to zero out of 18 in the control group. |
| Selective reporting (reporting bias) | High risk | No SDs and global scores reported for BPRS. |
| Other bias | Low risk | No other bias. |
General abbreviations CBZ ‐ carbamazepine EPS ‐ Extrapyramidal side effects. EEG ‐ electroencephalogram M ‐ males F ‐ females ITT ‐ intention‐to‐treat N = number Mg = milligram SD ‐ standard deviation
Diagnostic tools DSM‐III‐R ‐ Diagnostic and Statistical Manual of Mental disorders, third edition, revised. ICD‐9/10 ‐ International Classification of Diseases, ninth/tenth revision. RDC ‐ Research Diagnostic Criteria
Global effect scales CGI ‐ Clinical Global Impression (Guy 1976) OCR ‐ Overall Clinical Rating
Mental state scales BPRS ‐ Brief Psychiatric Rating Scale (Overall 1962) HRSD ‐ Hamilton Rating Scale for Depression IMPS ‐ Inpatient Multidimensional Rating Scale (Lorr 1962) PANSS ‐ Positive and Negative Symptoms Scale (Kay 1987) SANS ‐ Scale for Assessment of Negative Symptoms (Andreasen 1982) SAPS ‐ Scale for Assessment of Positive Symptoms (Andreasen 1984)
Side effect scales CGI ‐ Clinical Global Impression, side effects (Guy 1976) UKU ‐ UKU Side effect Scale (Lingjaerde et al. 1987) AIMS ‐ Abnormal Involuntary Movement Scale (Guy 1976) SAS ‐ Simpson and Angus Scale (Simpson and Angus 1970) FSUCL ‐ Fischers Somatische Symptome oder Unerwünschte Effekte Check List (Fischer‐Cornellson 1986)
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arana 1986 | Allocation: not randomised, A‐B design. |
| Azorin 1986 | Allocation: not randomised, A‐B design. |
| Ballenger 1984 | Allocation: not randomised, case series. |
| Barnes 1996 | Allocation: not randomised, review. |
| Bellaire 1990 | Allocation: randomised. Participants: people with schizoaffective disorders or bipolar illness. Interventions: lithium versus carbamazepine, no placebo group. |
| Birkheimer 1985 | Allocation: not randomised, review. |
| Borison 1991 | Allocation: randomised. Participants: people with schizophrenia. Interventions: rimcazole, no carbamazepine. |
| Botte 1988 | Allocation: not randomised, A‐B design. |
| Cabrera 1987 | Allocation: randomised. Participants: people with schizoaffective disorders and bipolar illness. Interventions: oxcarbazepine versus lithium, no placebo group. |
| Cegalis 1984 | Allocation: not randomised, case report. |
| Chouinard 1990 | Allocation: not randomised, case series. |
| Costa 1986 | Allocation: not randomised, case report. |
| Covell 2004 | Allocation: randomised. Participants: people with schizophrenia. Interventions: clozapine versus typical antipsychotics, no carbamazepine group. |
| Dalby 1971 | Allocation: not randomised, A‐B design. |
| de Vogelaer 1981 | Allocation: randomised. Participants: very heterogeneous group of patients, some with "behavioral disorders", some with "psychotic" disorders. No clear diagnoses, study focuses on character disorders. Interventions: not entirely clear, but probably carbamazepine or placebo were added to antipsychotic drugs . Outcomes: impossible to extract data just for people with serious mental illness. As yet, no response from the author to a letter. |
| Dehing 1968 | Allocation: randomised (random number list). Participants: mixed group, no data on people with schizophrenia, the focus of the study was on 'character disorders'. Interventions: Carbamazepine or placebo added to ongoing treatment. Outcomes: according to the authors no original data are available and therefore no single outcome parameter can be used for this review. |
| Denicoff 1994 | Allocation: not randomised, clinical practice survey/audit. |
| Elphick 1985 | Allocation: not randomised, A‐B‐A design. |
| Frankenburg 1988 | Allocation: not randomised, case series. |
| Gadow 1992 | Allocation: not randomised, review. |
| Galletly 1997 | Allocation: not randomised, case series. |
| Ginestet 1996 | Allocation: not randomised, review. |
| Goncalves 1985 | Allocation: randomised. Participants: manic people, seven with bipolar disorder, five with schizoaffective disorder. Interventions: carbamazepine versus placebo, but most participants also received haloperidol. The trial could therefore not be classified according to the three comparisons analysed in this review and no data could be extracted. |
| Greil 1997 | Allocation: randomised. Participants: people with schizoaffective disorder. Interventions: carbamazepine versus lithium, no placebo group. |
| Hakola 1982 | Allocation: not randomised, A‐B design. |
| Hermle 1993 | Allocation: not randomised, case report. |
| Herrera 1987 | Allocation: not randomised, A‐B‐A design. |
| Ivkovi 2004 | Allocation: not randomised, controlled clinical trial. |
| Iwahashi 1995 | Allocation: not randomised, case‐control study. |
| Iwahashi 1996 | Allocation: not randomised, case series. |
| Jann 1985 | Allocation: not randomised, A‐B design. |
| Johns 1995 | Allocation: not randomised, review. |
| Kahn 1990 | Allocation: randomised. Participants: people with schizophrenia. Intervention: carbamazepine plus neuroleptics versus lithium plus neuroleptics, no placebo group. |
| Karper 1992 | Allocation: not randomised, case report. |
| Keck 1996 | Allocation: not randomised, review. |
| Kessler 1989 | Allocation: not randomised, case series. |
| Kidron 1985 | Allocation: randomised, cross‐over. Participants: people with schizophrenia or schizoaffective disorders and excited states. Interventions: haloperidol + carbamazepine versus haloperidol + placebo. Outcomes: no usable data. |
| Klein 1984 | Allocation: randomised, parallel (participants with poor response were crossed‐over at the end). Participants: those with schizophrenia or schizoaffective disorders and excited states. Interventions: haloperidol + carbamazepine versus haloperidol + placebo. Outcomes: no usable data. |
| Kraft 1984 | Allocation: not randomised, case report. |
| Lambert 1987 | Allocation: review. |
| Lapensee 1992 | Allocation: not randomised, review. |
| Lenzi 1986 | Allocation: randomised. Participants: mixed affective and nonaffective psychoses. Interventions: carbamazepine + chlorpromazine versus lithium + chlorpromazine, no placebo group. |
| Llorca 1992 | Allocation: not randomised, review. |
| Luchins 1983 | Allocation: not randomised, case series. |
| Luchins 1984 | Allocation: not randomised, case report. |
| Makaric 2000 | Allocation: controlled clinical trial, but not randomised. |
| McAllister 1985 | Allocation: not randomised, case series. |
| McKee 1989 | Allocation: not randomised, case report. |
| Meltzer 1992 | Allocation: not randomised, review. |
| Meshel 1967 | Allocation: randomised. Participants: people with psychosis. Interventions: tybamate versus placebo, no carbamazepine group. |
| Meshel 1968 | Allocation: randomised. Participants: people with schizophrenia. Interventions: tybamate versus placebo, no carbamazepine group. |
| Miceli 2000 | Allocation: randomised. Participants: healthy volunteers. |
| Miller 1965 | Allocation: unclear. Participants: people with schizophrenia. Interventions: hydroxyphenamate, no carbamazepine group. |
| Miodownik 2003 | Allocation: unclear, but probably randomised. Participants: people with schizophrenia. Interventions: vitamin B6, no carbamazepine group. |
| Mokrusch 1987 | Allocation: not randomised, review. |
| Morinigo 1992 | Allocation: randomised. Participants: people with mania. |
| Mosca 1998 | Allocation: randomised. Participants: people with violent behaviour or impulsivity. Interventions: carbamazepine versus valproate, no placebo. |
| Munetz 1989 | Allocation: randomised. Participants: people with schizophrenia. Interventions: rimcazole, no carbamazepine group. |
| Möller 1989 | Allocation: randomised, cross‐over. Participants: people with schizophrenia or schizoaffective disorders and excited states. Interventions: haloperidol + carbamazepine versus haloperidol + placebo. Outcomes: no usable data. |
| Möller 1996 | Allocation: not randomised, review. |
| Nasser 1990 | Allocation: not randomised, case series. |
| Nelson 1993 | Allocation: not randomised, review. |
| Neppe 1988a | Allocation: not randomised, review. |
| Neppe 1988b | Allocation: not randomised, review. |
| Neppe 1988c | Allocation: not randomised, case report. |
| Neppe 1991 | Allocation: not randomised, case series. |
| Nijdam 1992 | Allocation: unclear. Participants: people with mental retardation and psychoses. Interventions: comparison of two different formulations of carbamazepine, no placebo. |
| Ohlmeier 2007 | Allocation: randomised. Participants: people with schizophrenia. Interventions: carbamazepine plus perazine versus olanzapine, i.e. not the same antipsychotic in both groups. |
| Okuma 1989a | Allocation: alternate allocation, category C, inadequate randomisation. |
| Okuma 1989b | Allocation: not randomised, A‐B design. |
| Ortega 1991 | Allocation: randomised (aleatory numbers list generated with a computer program). Participants: inhalant‐induced psychotic disorders, not schizophrenia. Interventions: carbamazepine as a sole treatment versus haloperidol as a sole treatment. |
| Otani 1997 | Allocation: not randomised, A‐B design. |
| Pantelis 1996 | Allocation: not randomised, review. |
| Panu 1984 | Allocation: not randomised, case series. |
| Placidi 1986 | Allocation: randomised. Participants: mixed affective and nonaffective psychoses. Intervention: carbamazepine versus lithium, no placebo. |
| Raitasuo 1994 | Allocation: not randomised, case report. |
| Rankel 1988 | Allocation: not randomised, case series. |
| Rittmannsberger 1990 | Allocation: not randomised, review. |
| Scher 1983 | Allocation: not randomised, case series. |
| Schulz 1990 | Allocation: randomised. Participants: people with schizophrenia. Interventions: antipsychotics + carbamazepine versus antipsychotics versus lithium, no placebo. |
| Simhandl 1992 | Allocation: not randomised, review. |
| Siris 1993 | Allocation: not randomised, review. |
| Sramek 1988 | Allocation: not randomised, A‐B design. |
| Sugerman 1970 | Allocation: unclear, probably randomised. Participants: people with schizophrenia. Interventions: adrenochrome semicarbazone, no carbamazepine group. |
| Svestka 1985 | Allocation: not randomised, A‐B design. |
| Svestka 1988 | Allocation: not randomised, A‐B design. |
| Tohen 1994 | Allocation: not randomised, case series. |
| Walden 1996 | Allocation: not randomised, review. |
| Wetterling 1987 | Allocation: not randomised, A‐B‐A design. |
| Wunderlich 1983 | Allocation: not randomised, A‐B design. |
| Yassa 1983 | Allocation: not randomised, case report. |
Characteristics of studies awaiting assessment [ordered by study ID]
Kamisada 1988.
| Methods | Unclear whether study was randomised. |
| Participants | Schizophrenia. |
| Interventions | Probably either lithium or carbamazepine added to antipsychotic drugs. |
| Outcomes | Symptoms, side effects. |
| Notes | We wrote a letter but have not received an answer, we can not be sure whether the address was still correct. |
Lee 1996.
| Methods | Unclear whether study was randomised. |
| Participants | People with schizophrenia and negative symptoms. |
| Interventions | Probably carbamazepine and valproate added to antipsychotic drugs. |
| Outcomes | Probably symptoms, side effects. |
| Notes | We wrote a letter but have not received an answer, we can not be sure whether the address was still correct. |
Contributions of authors
Stefan Leucht ‐ protocol development, searching, data extraction, analysis, data interpretation and writing the final report.
Markus Dold ‐ searching, data extraction, analysis, data interpretation.
John McGrath ‐ protocol development, data checking, data interpretation.
Werner Kissling ‐ protocol development, data interpretation.
Bartosz Helfer ‐ updating, data extraction and writing parts of the report.
Sources of support
Internal sources
Queensland Health, Australia.
Freistaat Bayern, Germany.
External sources
No sources of support supplied
Declarations of interest
Stefan Leucht: In the last 3 years Stefan Leucht has received consulting/advisory board honoraria from Roche, Eli Lilly, Medavante, Bristol‐Myers Squibb, Alkermes, Janssen, Johnson & Johnson, and Lundbeck, Lecture honoraria from Abbvie, Astra Zeneca, Bristol‐Myers Squibb, ICON, Eli Lilly, Janssen, Johnson & Johnson, Roche, Sanofi‐Aventis, Lundbeck, and Pfizer, and Eli Lilly has provided medication for a trial with Stefan Leucht as the primary investigator.
Markus Dold: nothing to declare.
John McGrath is a member of the following advisory boards: Eli Lilly Australia, Lundbeck Australia and Pfizer Australia. In addition, JM has been a co‐investigator on studies of neuroleptic medications produced by the following companies: Astra, Janssen‐Cilag, Eli Lilly, Zeneca (ICI), Sandoz and Pfizer. The same companies have provided travel and accommodation expenses for John McGrath to attend relevant investigator meetings and scientific symposia. No funds have been paid directly to Dr McGrath. Payments related to participation in drug trials and board attendances have been paid to a Government‐audited trust account to support schizophrenia research.
Werner Kissling has received honoraria and/or research support from Janssen‐Cilag, SanofiAventis, Johnson & Johnson, Pfizer, BMS, Astra Zeneca, Lundbeck, Novartis and Eli Lilly. Dr Kissling is an investigator on one of the excluded trials in this review (Möller 1989).
Bartosz Helfer: nothing to declare.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Carpenter 1991 {published data only}
- Carpenter WT, Kurz R, Kirkpatrick B, Hanlon TE. Carbamazepine maintenance treatment in outpatient schizophrenics. Archives of General Psychiatry 1991;48(1):69‐72. [DOI] [PubMed] [Google Scholar]
Dose 1987 {published data only}
- Dose M. Die Bedeutung von Antikonvulsiva und Calciumantagonisten für die Pharmakotherapie von Psychosen. Habilitationsschrift. Technische Universität München, 1991. [Google Scholar]
- Dose M, Apelt S, Emrich HM. Carbamazepine as an adjunct of antipsychotic therapy. Psychiatry Research 1987;22(4):303‐10. [DOI] [PubMed] [Google Scholar]
- Dose M, Emrich HM. Carbamazepine as an adjunct to antipsychotic treatment. Schizophrenia Research 1988;1(2,3):207‐8. [Google Scholar]
- Dose M, Emrich HM. Combination of neuroleptics with carbamazepine. Application in the treatment of schizophrenic psychoses [Kombination von Neuroleptika mit Carbamazepin. Einsatz in der Behandlung schizophrener Psychosen]. Münchner Medizinische Wochenschrift 1990;132 (suppl 1):87‐90. [Google Scholar]
- Dose M, Garcia D, Weber M, Yassouridis A, Emrich HM. Combined treatment of schizophrenic psychoses with neuroleptics and anticonvulsants. Pharmacopsychiatry 1989;22(5):195. [Google Scholar]
Heßlinger 1998 {published and unpublished data}
- Hesslinger B, Klose P, Normann C, Langosch JM, Berger M, Walden J. Carbamazepine co‐treatment in schizophrenia. Fortschritte der Neurologie Psychiatrie 1998;66(4):145‐50. [DOI] [PubMed] [Google Scholar]
- Hesslinger B, Normann C, Langosch JM, Klose P, Berger M, Walden J. Effects of carbamazepine and valproate on haloperidol plasma levels and on psychopathologic outcome in schizophrenic patients. Journal of Clinical Psychopharmacology 1999;19(4):310‐5. [DOI] [PubMed] [Google Scholar]
- Normann C, Klose P, Hesslinger B, Langosch JM, Berger M, Walden J. Haloperidol plasma levels and psychopathology in schizophrenic patients with antiepileptic co‐medication: a clinical trial. Pharmacopsychiatry 1997;30:204. [Google Scholar]
- Walden J, Hesslinger B, Normann C, Langosch J, Berger M. Actions of carbamazepine and valproate on haloperidol plasma levels and psychopathological outcome. Proceedings of the 21st Congress of the Collegium Internationale Neuro‐psychopharmacologicum, Glasgow, Scotland. 1998.
Llorca 1993 {published data only}
- Estorges JP, Llorca PM, Lancon C, Bougerol T, Scotto JC. Carbamazepine as adjuvant treatment to neuroleptics in schizophrenic patients. Encephale 1991;17(4):307. [Google Scholar]
- Llorca PM, Wolf MA, Lancon C, Bougerol T. Clinical efficacy of bromocriptine, carbamazepine, and cyproheptadine as adjuvant to neuroleptics in 24 chronic resistant schizophrenics [Efficacite comparee de la bromocriptine, de la carbamazepine et de la cyproheptadine en association aux neuroleptiques chez 24 patients schizophrenes chroniques resistants]. Encephale 1993;19(5):565‐71. [PubMed] [Google Scholar]
Mair 1990 {published data only}
- Mair M, Tschapeller I, Schubert H. Kombinationstherapie mit Neuroleptika und Carbamazepin. Eine kontrollierte Studie. In: Schonbeck G, Platz T editor(s). Schizophrenie erkennen, verstehen, behandeln.Beiträge aus Theorie und Praxis. Wien: Springer Verlag, 1990:77‐92. [Google Scholar]
Martin‐Munoz 1989 {published data only}
- Martin‐Munoz JC, Morinigo‐Dominguez AV, Mateo‐Martin I, Guajardo FI. Carbamazepine: An effective adjunct treatment for schizophrenia [La carbamacepina: un tratamiento adjunto eficaz en las esquizofrenias]. Actas Luso Espanolas de Neurologia, Psiquiatria y Ciencias Afines 1992;20(1):11‐6. [PubMed] [Google Scholar]
- Martin‐Munoz JC, Morinigo‐Dominguez AV, Mateo‐Martin I, Ibarra IG. Carbamazepine: An efficacious adjuvant treatment in schizophrenia [La carbamacepina: Un tratamiento adjunto eficaz en las esquizofrenias]. Actas Luso Espanolas de Neurologia, Psiquiatria y Ciencias Afines 1989;17(4):245‐50. [PubMed] [Google Scholar]
Nachshoni 1994 {published data only}
- Nachshoni T, Levin Y, Levy A, Kritz A. A double‐blind trial of carbamazepine in negative symptom schizophrenia. Biological Psychiatry 1994;35(1):22‐6. [DOI] [PubMed] [Google Scholar]
Neppe 1983 {published and unpublished data}
- Neppe VM. Carbamazepine as adjunctive treatment in nonepileptic chronic inpatients with EEG temporal lobe abnormalities. Journal of Clinical Psychiatry 1983;44(9):326‐31. [PubMed] [Google Scholar]
- Neppe VM. Carbamazepine in the psychiatric patient. Lancet 1982;2(8293):334. [DOI] [PubMed] [Google Scholar]
- Neppe VM. Non‐responsive psychosis ‐ a biochemical difference?. South African Medical Journal 1983;63(21):797‐8. [PubMed] [Google Scholar]
Simhandl 1996 {published data only}
- Meszaros K, Simhandl C, Denk E, Liechtenstein A, Topitz A, Thau K. A carbamazepine augmentation trial in chronic nonresponsive schizophrenia. NA 1996; Vol. NA:NA.
- Simhandl C, Meszaros K, Denk E, Thau K, Topitz A. Adjunctive carbamazepine or lithium carbonate in therapyresistant chronic schizophrenia. Canadian Journal of Psychiatry/Revue Canadienne de Psychiatrie 1996;41(5):317. [DOI] [PubMed] [Google Scholar]
Svestka 1989 {published data only}
- Svestka J, Ceskova E, Rysanek R, Nahunek K. Controlled cross‐over comparison of carbamazepine with perphenazine in schizophrenic psychoses. Activitas Nervosa Superior 1989;31(4):276‐7. [PubMed] [Google Scholar]
References to studies excluded from this review
Arana 1986 {published data only}
- Arana GW. Does carbamazepine‐induced reduction of plasma haloperidol levels worsen psychotic symptoms? 138th Annual Meeting of the American Psychiatric Association (1985, Dallas, Texas). American Journal of Psychiatry 1986;143(5):650‐1. [DOI] [PubMed] [Google Scholar]
Azorin 1986 {published data only}
- Azorin JM, Samuelian JC, Pringuey D, Donnet A. Place of carbamazepine in the treatment of endogenous psychoses: Results of an open trial [Place de la carbamazepine dans le traitement des psychoses endogenes: Resultats d'une etude ouverte]. Encephale 1986;12(3):115‐9. [PubMed] [Google Scholar]
Ballenger 1984 {published data only}
- Ballenger JC, Post RM. Carbamazepine in alcohol and withdrawal syndromes and schizophrenic psychoses. Psychopharmacology Bulletin 1984;20:572‐84. [PubMed] [Google Scholar]
Barnes 1996 {published data only}
- Barnes TR, McEvedy CJ. Pharmacological treatment strategies in the non‐responsive schizophrenic patient. International Clinical Psychopharmacology 1996;11(s2):67‐71. [DOI] [PubMed] [Google Scholar]
Bellaire 1990 {published data only}
- Bellaire W, Demisch K, Stoll K‐D. Carbamazepine vs. lithium. Application in the prophylaxis of recidivating affective and schizoaffective psychoses [Carbamazepin vs. Lithium. Einsatz in der Prophylaxe rezidivierender affektiver und schizoaffektiver Psychosen]. Münchner Medizinische Wochenschrift 1990;132:82‐6. [Google Scholar]
Birkheimer 1985 {published data only}
- Birkhimer LJ, Curtis JL, Jann MW. Use of carbamazepine in psychiatric disorders. Clinical Pharmacology 1985;4(4):425‐34. [PubMed] [Google Scholar]
Borison 1991 {published data only}
- Borison RL, Diamond BI, Dren AT. Does sigma receptor antagonism predict clinical antipsychotic efficacy?. Psychopharmacology Bulletin 1991;27(2):103‐6. [MEDLINE: ; PMID 1681560] [PubMed] [Google Scholar]
Botte 1988 {published data only}
- Botte L, Charles G. Clinical use of carbamazepine: Review from the literature and personal results [Utilisations cliniques de la carbamazepine: Revue de la litterature et resultats personnels]. Acta Psychiatrica Belgica 1988;88(3):181‐94. [PubMed] [Google Scholar]
Cabrera 1987 {published data only}
- Cabrera J, Albrecht J, Muller‐Oerlinghausen B. Combined preventive treatment of recurrent manic‐depressive disease with lithium and carbamazepine or oxcarbazepine. Nervenarzt 1987;58(4):245‐9. [PubMed] [Google Scholar]
Cegalis 1984 {published data only}
- Cegalis JA, Possick SG. Carbamazepine and psychotherapy in the treatment of schizoaffective psychosis. Yale Journal of Biology and Medicine 1985;58(4):327‐36. [PMC free article] [PubMed] [Google Scholar]
Chouinard 1990 {published data only}
- Chouinard G, Sultan S. Treatment of supersensitivity psychosis with antiepileptic drugs: Report of a series of 43 cases. Psychopharmacology Bulletin 1990;26(3):337. [PubMed] [Google Scholar]
Costa 1986 {published data only}
- Costa JF, Sramek J, Herrera JM. Hepatic reaction to carbamazepine. Journal of Clinical Psychopharmacology 1986;6(4):251‐2. [PubMed] [Google Scholar]
Covell 2004 {published data only}
- Covell NH, Weissman EM, Essock SM. Weight gain with clozapine compared to first generation antipsychotic medications. Schizophrenia Bulletin 2004;30(2):229‐40. [MEDLINE: ; EMBASE 2004274059] [DOI] [PubMed] [Google Scholar]
Dalby 1971 {published data only}
- Dalby MA. Antiepileptic and psychotropic effect of carbamazepine (Tegretol) in the treatment of psychomotor epilepsy. Epilepsia 1971;12(12):335‐40. [DOI] [PubMed] [Google Scholar]
Dehing 1968 {published data only}
- Dehing J. Studies on the psychotropic action of tegretol. Acta Neurologica Belgica 1968;68:895‐905. [PubMed] [Google Scholar]
Denicoff 1994 {published data only}
- Denicoff KD, Meglathery SB, Post RM, Tandeciarz SI. Efficacy of carbamazepine compared with other agents: A clinical practice survey. Journal of Clinical Psychiatry 1994;55(2):70. [PubMed] [Google Scholar]
de Vogelaer 1981 {published data only}
- Vogelaer J. Carbamazepine in the treatment of psychotic and behavioral disorders. A pilot study. Acta Psychiatrica Belgica 1981;81:532‐41. [PubMed] [Google Scholar]
Elphick 1985 {published data only}
- Elphick M. An open clinical trial of carbamazepine in treatment‐resistant bipolar and schizo‐affective psychotics. British Journal of Psychiatry 1985;147:198‐200. [DOI] [PubMed] [Google Scholar]
Frankenburg 1988 {published data only}
- Frankenburg FR, Tohen M, Cohen BM, Lipinski JF. Long‐term response to carbamazepine: A retrospective study. Journal of Clinical Psychopharmacology 1988;8(2):130‐2. [PubMed] [Google Scholar]
Gadow 1992 {published data only}
- Gadow KD. Pediatric psychopharmacotherapy: A review of recent research. Journal of Child Psychology and Psychiatry and Allied Disciplines 1992;33(1):153‐95. [DOI] [PubMed] [Google Scholar]
Galletly 1997 {published data only}
- Galletly CA, Tsourtos G. Antipsychotic drug doses and adjunctive drugs in the outpatient treatment of schizophrenia. Annals of Clinical Psychiatry 1997;9(2):77‐80. [DOI] [PubMed] [Google Scholar]
Ginestet 1996 {published data only}
- Ginestet D, Ghanem T, Slama M. The variety of indications for carbamazepine. Revue du Praticien Medecine Generale 1996;10(336):11. [Google Scholar]
Goncalves 1985 {published data only}
- Goncalves N, Stoll KD. A controlled double‐blind trial of carbamazepine in manic illness [Carbamazepin bei manischen Syndromen. Eine kontrollierte Doppelblind‐Studie]. Nervenarzt 1985;56(1):43‐7. [PubMed] [Google Scholar]
Greil 1997 {published data only}
- Greil W, Ludwig‐Mayerhofer W, Erazo N, Engel RR. Lithium vs cambamazepine in the maintenance treatment of schizoaffective disorder: A randomised study. European Archives of Psychiatry and Clinical Neuroscience 1997;247(1):42‐50. [DOI] [PubMed] [Google Scholar]
Hakola 1982 {published data only}
- Hakola HA, Laulumaa VA. Carbamazepine in treatment of violent schizophrenics. Lancet 1982;1(8285):1358. [DOI] [PubMed] [Google Scholar]
Hermle 1993 {published data only}
- Hermle L, Spitzer M. Succesful desensitization of a patient with schizoaffective psychosis and carbamazepin allergy. Nervenarzt 1993;64(3):208. [PubMed] [Google Scholar]
Herrera 1987 {published data only}
- Heh CW, Sramek J, Herrera J, Costa J. Exacerbation of psychosis after discontinuation of carbamazepine treatment. American Journal of Psychiatry 1988;145(7):878‐9. [DOI] [PubMed] [Google Scholar]
- Herrera JM, Sramek JJ, Costa JF. Efficacy of adjunctive carbamazepine in the treatment of chronic schizophrenia. Drug Intelligence & Clinical Pharmacy 1987;21(4):355‐8. [DOI] [PubMed] [Google Scholar]
Ivkovi 2004 {published data only}
- Ivkovi M, Damjanovi A, Marinkovi D, Paunovi RV. Carbamazepine for acute psychosis with EEG abnormalities. Military‐Medical and Pharmaceutical Review 2004;61(4):399‐403. [DOI] [PubMed] [Google Scholar]
Iwahashi 1995 {published data only}
- Iwahashi K, Miyatake R, Suwaki H, Hosokawa K. The drug‐drug interaction effects of haloperidol on plasma carbamazepine levels. Clinical Neuropharmacology 1995;18(3):233‐6. [DOI] [PubMed] [Google Scholar]
Iwahashi 1996 {published data only}
- Iwahashi K. Significantly higher plasma haloperidol level during cotreatment with carbamazepine may herald cardiac change [see comments]. Clinical Neuropharmacology 1996;19(3):267‐70. [DOI] [PubMed] [Google Scholar]
Jann 1985 {published data only}
- Jann MW, Ereshefsky L, Saklad SR, Seidel DR, Davis CM, Burch NR, et al. Effects of carbamazepine on plasma haloperidol levels. Journal of Clinical Psychopharmacology 1985;5(2):106‐9. [DOI] [PubMed] [Google Scholar]
Johns 1995 {published data only}
- Johns CA, Thompson JW. Adjunctive treatments in schizophrenia: Pharmacotherapies and electroconvulsive therapy. Schizophrenia Bulletin 1995;21(4):607‐19. [DOI] [PubMed] [Google Scholar]
Kahn 1990 {published data only}
- Kahn EM, Schulz SC, Perel JM, Alexander JE. Change in haloperidol level due to carbamazepine: A complicating factor in combined medication for schizophrenia. Journal of Clinical Psychopharmacology 1990;10(1):54‐7. [DOI] [PubMed] [Google Scholar]
Karper 1992 {published data only}
- Karper LP, Seibyl JP, Krystal JH. Valproate management of psychosis in a patient with carbamazepine‐induced hyponatremia. Journal of Clinical Psychopharmacology 1992;12(2):137‐9. [PubMed] [Google Scholar]
Keck 1996 {published data only}
- Keck PE, McElroy SL, Strakowski SM. New developments in the pharmacologic treatment of schizoaffective disorder. Journal of Clinical Psychiatry 1996;57(s9):41‐8. [PubMed] [Google Scholar]
Kessler 1989 {published data only}
- Kessler AJ, Barklage NE, Jefferson JW. Mood disorders in the psychoneurologic borderland: Three cases of responsiveness to carbamazepine. 29th Annual Meeting of the International Psychiatric Research Society (1987, Madison, Wisconsin). American Journal of Psychiatry 1989;146(1):81‐3. [DOI] [PubMed] [Google Scholar]
Kidron 1985 {published data only}
- Kidron R, Averbuch I, Klein E, Belmaker RH. Carbamazepine‐induced reduction of blood levels of haloperidol in chronic schizophrenia. Biological Psychiatry 1985;20(2):219‐22. [DOI] [PubMed] [Google Scholar]
Klein 1984 {published data only}
- Klein E, Bental E, Lerer B, Belmaker RH. Carbamazepine and haloperidol v placebo and haloperidol in excited psychoses. A controlled study. Archives of General Psychiatry 1984;41(2):165‐70. [DOI] [PubMed] [Google Scholar]
Kraft 1984 {published data only}
- Kraft AM, Hassenfeld IN, Zarr M. Response to functional hallucinations to carbamazepine. American Journal of Psychiatry 1984;141(8):1018. [DOI] [PubMed] [Google Scholar]
Lambert 1987 {published data only}
- Lambert PA, Venaud G. Use of valpromide in psychiatric therapeutics. Encephale 1987;13(6):367‐73. [CN‐00477270] [PubMed] [Google Scholar]
Lapensee 1992 {published data only}
- Lapensee MA. A review of schizoaffective disorder: II. Somatic treatment. Canadian Journal of Psychiatry Revue Canadienne de Psychiatrie 1992;37(5):347. [DOI] [PubMed] [Google Scholar]
Lenzi 1986 {published data only}
- Lenzi A, Lazzerini F, Grossi E, Massimetti G, Placidi GF. Use of carbamazepine in acute psychosis: A controlled study. Journal of International Medical Research 1986;14(2):78‐84. [DOI] [PubMed] [Google Scholar]
Llorca 1992 {published data only}
- Llorca PM, Wolf MA, Lancon C, Bougerol T. Adjuvant drugs for use in patients under neuroleptics. Annales de Psychiatrie 1992;7(4):195. [Google Scholar]
Luchins 1983 {published data only}
- Luchins DJ. Carbamazepine for the violent psychiatric patient. Lancet 1983;1(8327):766. [DOI] [PubMed] [Google Scholar]
- Luchins DJ. Carbamazepine in violent nonepileptic schizophrenics. Psychopharmacology Bulletin 1984;20(3):569‐571. [PubMed] [Google Scholar]
Luchins 1984 {published data only}
- Luchins DJ. Fatal agranulocytosis in a chronic schizophrenic patient treated with carbamazepine. American Journal of Psychiatry 1984;141(5):687‐8. [DOI] [PubMed] [Google Scholar]
Makaric 2000 {published data only}
- Makaric G, Folnegovic‐Smalc V, Folnegovic Z, Imica N. Agitation in acute episode of schizophrenia: carbamazepine treatment in combination with haloperidol versus combined neuroleptics. Schizophrenia Research 2000;41(1):210. [MEDLINE: ] [Google Scholar]
McAllister 1985 {published data only}
- McAllister TW. Carbamazepine in mixed frontal lobe and psychiatric disorders. Journal of Clinical Psychiatry 1985;46(9):393‐4. [PubMed] [Google Scholar]
McKee 1989 {published data only}
- McKee RW, Larkin JG, Brodie MJ. Acute psychosis with carbamazepine and sodium valproate. Lancet 1989;1(8630):167. [DOI] [PubMed] [Google Scholar]
Meltzer 1992 {published data only}
- Meltzer HY. Novel approaches to the pharmacotherapy of schizophrenia. Drug Development Research 1986;9(1):23‐40. [Google Scholar]
- Meltzer HY. Treatment of the neuroleptic‐nonresponsive schizophrenic patient. Schizophrenia Bulletin 1992;18(3):515‐42. [DOI] [PubMed] [Google Scholar]
Meshel 1967 {published data only}
- Meshel E, Denber HC. Double‐blind study of tybamate in psychotic patients. Diseases of the Nervous System 1967;28(5):311‐3. [MEDLINE: ; PMID 5338174] [PubMed] [Google Scholar]
Meshel 1968 {published data only}
- Meshel E, Denber HC. The use of tybamate in psychotic patients. (A further double blind study). Diseases of the Nervous System 1968;29(4):243‐5. [MEDLINE: ; PMID 4870945] [PubMed] [Google Scholar]
Miceli 2000 {published data only}
- Miceli JJ, Anziano RJ, Robarge L, Hansen RA, Laurent A. The effect of carbamazepine on the steady‐state pharmacokinetics of ziprasidone in healthy volunteers. British Journal of Clinical Pharmacology 2000;49 (suppl I):65S‐70S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miller 1965 {published data only}
- Miller MJ, Shettel R, Fiedler HT. Chronic toxicologic evaluation of hydroxyphenamate and possible synergism with phenothiazines. Psychosomatics 1965;6(5):340‐2. [MEDLINE: ; PMID 5319246] [DOI] [PubMed] [Google Scholar]
Miodownik 2003 {published data only}
- Miodownik C, Cohen H, Kotler M, Lerner V. Vitamin B6 add‐on therapy in treatment of schizophrenic patients with psychotic symptoms and movement disorders. Harefuah 2003;142(8‐9):592‐6, 647. [MEDLINE: ; EMBASE 2003374828; CN‐00440574.] [PubMed] [Google Scholar]
Mokrusch 1987 {published data only}
- Mokrusch T, Negele J, Kaschka WP. New aspects in the prophylaxis of affective and schizoaffective psychoses. Fortschritte der Medizin 1987;105(1):30‐4. [PubMed] [Google Scholar]
Möller 1989 {published data only}
- Moller HJ, Kissling W, Riehl T, Bauml J. Double‐blind evaluation of the antimanic properties of carbamazepine as a comedication to haloperidol. Progress in Neuropsychopharmacology and Biological Psychiatry 1989;13(1‐2):127‐36. [DOI] [PubMed] [Google Scholar]
Möller 1996 {published data only}
- Möller H. Treatment of schizophrenia. State of the art. European Archives of Psychiatry and Clinical Neuroscience 1996;246(5):229. [DOI] [PubMed] [Google Scholar]
Morinigo 1992 {published data only}
- Morinigo A, Mateo I, Martin J, Noval D, Gonzalez S. Efectos terapeuticos del valproato sodico y la carbamacepina en la mania. Psiquis 1992;13(8):335‐8. [Google Scholar]
Mosca 1998 {published data only}
- Mosca LD//Licciardo JP//Coppola JL. A double‐blind carbamazepine‐controlled efficacy and safety study of valproate in impulsivity and violence. 11th Congress of The European College of Neuropsychopharmacology; Oct 31 ‐ Nov 4, Paris, France. 1998. [MEDLINE: ]
Munetz 1989 {published data only}
- Munetz MR, Schulz SC, Bellin M, Harty I. Rimcazole (BW234U) in the maintenance treatment of outpatients with schizophrenia. Drug Development Research 1989;16(1):79‐83. [MEDLINE: ; PMID 6342171] 6342171 [Google Scholar]
Nasser 1990 {published data only}
- Nasser D, Thomas B. Anticonvulsant treatment of psychoses. Australian and New Zealand Journal of Psychiatry 1990;24(2):164. [PubMed] [Google Scholar]
Nelson 1993 {published data only}
- Nelson JC. Combined treatment strategies in psychiatry. Journal of Clinical Psychiatry 1993;54(s9):42. [PubMed] [Google Scholar]
Neppe 1988a {published data only}
- Neppe VM. Carbamazepine in nonresponsive psychosis. Journal of Clinical Psychiatry 1988;49(s):22‐8. [PubMed] [Google Scholar]
Neppe 1988b {published data only}
- Neppe VM, Tucker GJ, Wilensky AJ. Fundamentals of carbamazepine use in neuropsychiatry. Journal of Clinical Psychiatry 1988;49(s):4‐6. [PubMed] [Google Scholar]
Neppe 1988c {published data only}
- Neppe VM. Carbamazepine for withdrawal pseudohallucinations. American Journal of Psychiatry 1988;145(12):1605. [DOI] [PubMed] [Google Scholar]
Neppe 1991 {published data only}
- Neppe VM, Bowman BR, Sawchuk KS. Carbamazepine for atypical psychosis with episodic hostility. Journal of Nervous and Mental Disease 1991;179(7):439‐41. [DOI] [PubMed] [Google Scholar]
Nijdam 1992 {published data only}
- Nijdam JR, Doorschot CH, Bavel LP, Loonen AJ. A comparison of carbamazepine Divitabs and a normal carbamazepine preparation in psychiatric and oligophrenic patients. Pharmacopsychiatry 1992;25(3):145‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ohlmeier 2007 {published data only}
- Ohlmeier MD, Jahn K, Wilhelm‐Gossling C, Godecke‐Koch T, Hoffmann J, Seifert J, et al. Perazine and carbamazepine in comparison to olanzapine in schizophrenia. Neuropsychobiology 2007;55(2):81‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Okuma 1989a {published and unpublished data}
- Okuma T, Yamashita I, Takahashi R, Itoh H. A double‐blind study of adjunctive carbamazepine versus placebo on excited states of schizophrenic and schizoaffective disorders. Acta Psychiatrica Scandinavica 1989;80(3):250‐9. [DOI] [PubMed] [Google Scholar]
- Okuma T, Yamashita I, Takahashi R, Itoh H, Otsuki S, Watanabe S, et al. Comparison of the therapeutic effect of carbamazepine and placebo in schizophrenia and atypical psychosis by double blind controlled study. Rinsho Hyoka 1988;16(2):327‐73. [Google Scholar]
Okuma 1989b {published data only}
- Okuma T, Yamashita I, Takahashi R, Itoh H. Clinical efficacy of carbamazepine in affective, schizoaffective, and schizophrenic disorders. Pharmacopsychiatry 1989;22(2):47‐53. [DOI] [PubMed] [Google Scholar]
Ortega 1991 {published data only}
- Hernandez‐Avila CA, Ortega‐Soto HA, Jasso A, Hasfura‐Buenaga CA, Kranzler HR. Treatment of inhalant‐induced psychotic disorder with carbamazepine versus haloperidol. Psychiatric Services 1998;49:812‐5. [DOI] [PubMed] [Google Scholar]
- Ortega SHA, Jasso A, Cecena G, Hernandez ACA. Validity and reproductivity of a scale for measuring neuroleptic‐induced extrapyramidal symptoms [La validez y la reproducibilidad de dos escalas para evaluar los sintomas extrapiramidales inducidos por neurolepticos]. Salud Mental 1991;14(3):1‐5. [Google Scholar]
Otani 1997 {published data only}
- Otani K, Ishida M, Yasui N, Kondo T, Mihara K, Suzuki A, et al. Interaction between carbamazepine and bromperidol. European Journal of Clinical Pharmacology 1997;52(3):219. [DOI] [PubMed] [Google Scholar]
Pantelis 1996 {published data only}
- Pantelis C, Barnes TR. Drug strategies and treatment‐resistant schizophrenia. Australian and New Zealand Journal of Psychiatry 1996;30(1):20‐37. [DOI] [PubMed] [Google Scholar]
Panu 1984 {published data only}
- Panu H, Hakola A, Laulumaa VA. Carbamazepine in violent schizophrenics. In: Emrich HM, Okuma T editor(s). Anticonvulsants in Affective Disorders. Amsterdam: Elsevier, 1984:204‐7. [Google Scholar]
Placidi 1986 {published data only}
- Placidi GF, Lenzi A, Lazzerini F, Cassano GB. The comparative efficacy and safety of carbamazepine versus lithium: A randomized, double‐blind 3‐year trial in 83 patients. Journal of Clinical Psychiatry 1986;47(10):490‐4. [PubMed] [Google Scholar]
- Placidi GF, Lenzi A, Rampello E, Andreani MF, Cassano GB, Grossi E. Long term‐double blind prospective study on carbamazepine versus lithium in bipolar and schizoaffective disorders. Preliminary results. In: Emrich HM, Okuma T editor(s). Anticonvulsants in Affective Disorders. Amsterdam: Elsevier, 1984:188‐97. [Google Scholar]
Raitasuo 1994 {published data only}
- Raitasuo V, Lehtovaara R, Huttunen MO. Effect of switching carbamazepine to oxcarbazepine on the plasma levels of neuroleptics: A case report. Psychopharmacology 1994;116(1):115‐6. [DOI] [PubMed] [Google Scholar]
Rankel 1988 {published data only}
- Rankel HW, Rankel LE. Carbamazepine in the treatment of catatonia. American Journal of Psychiatry 1988;145(3):361‐2. [DOI] [PubMed] [Google Scholar]
Rittmannsberger 1990 {published data only}
- Rittmannsberger H. Carbamazepine in the treatment of psychiatric illness: Effects and side effects. Wiener Medizinische Wochenschrift 1990;140(15):398. [PubMed] [Google Scholar]
Scher 1983 {published data only}
- Scher M, Neppe V. Carbamazepine adjunct for nonresponsive psychosis with prior hallucinogenic abuse. Journal of Nervous and Mental Disease 1989;177(12):755. [DOI] [PubMed] [Google Scholar]
Schulz 1990 {published data only}
- Schulz SC, Conley RR, Kahn EM, et al. Nonresponders to neuroleptics: a distinct subtype. Schizophrenia: scientific progress. New York: Oxford University Press, Date of publication and page numbers not indicated. [Google Scholar]
- Schulz SC, Kahn EM, Baker RW, et al. Lithium and carbamazepine augmentation in treatment‐refractory schizophrenia. The Neuroleptic‐Nonresponsive Patient: Characterization and Treatment. Washington DC: American Psychiatric Press, 1990:111‐36. [Google Scholar]
Simhandl 1992 {published data only}
- Simhandl C, Meszaros K. The use of carbamazepine in the treatment of schizophrenic and schizoaffective psychoses: A review. Journal of Psychiatry and Neuroscience 1992;17(1):1‐14. [PMC free article] [PubMed] [Google Scholar]
Siris 1993 {published data only}
- Siris SG. Adjunctive medication in the maintenance treatment of schizophrenia and its conceptual implications. British Journal of Psychiatry 1993;163(s22):22‐78. [PubMed] [Google Scholar]
Sramek 1988 {published data only}
- Heh CW, Potkin SG, Pickar D, Costa J, Herrera J, Sramek J, et al. Serum homovanillic acid concentrations in carbamazepine‐treated chronic schizophrenics. Biological Psychiatry 1989;25(5):639‐41. [DOI] [PubMed] [Google Scholar]
- Herrera J, Sramek J, Costa C, Heh C, Wernberg C. An evaluation of carbamazepine (Tegretol) in chronic treatment‐refractory schizophrenia. Proceedings of the 95th Annual Conference of the American Psychiatric Association. 1987:588‐9.
- Sramek J, Herrera J, Costa J, Heh C, Tran Johnson T, Simpson G. A carbamazepine trial in chronic, treatment‐refractory schizophrenia. American Journal of Psychiatry 1988;145(6):748‐50. [DOI] [PubMed] [Google Scholar]
- Tran‐Johnson T, Sramek JJ, Walker NR, Heh CD, Costa JF, Herrera JM. Effects of carbamazepine on serum calcium in schizophrenia. DICP 1989;23(12):1034. [DOI] [PubMed] [Google Scholar]
Sugerman 1970 {published data only}
- Sugerman AA, Hyams L. Electroencephalographic effects of adrenochrome semicarbazone in schizophrenia: quantitative amplitude analysis. Research Communications in Chemical Pathology and Pharmacology 1970;1(1):86‐98. [MEDLINE: ; PMID 4944698] [PubMed] [Google Scholar]
Svestka 1985 {published data only}
- Svestka J, Nahunek K, Ceskova E, Korbicka J. Carbamazepine prophylaxis of affective psychoses: Intraindividual comparison with lithium carbonate. 27th Annual Psychopharmacology Meeting (1985, Jesenik, Czechoslovakia). Activitas Nervosa Superior 1985;27(4):261‐2. [Google Scholar]
Svestka 1988 {published data only}
- Svestka J, Nahunek K, Ceskova E. Carbamazepine in the treatment and prophylaxis of affective psychoses [Pouziti carbamazepinu v lecbe a profylaxi afektivnich psychoz]. Ceskoslovenska Psychiatrie 1988;84(3):145‐55. [PubMed] [Google Scholar]
Tohen 1994 {published data only}
- Tohen M, Castillo J, Pope HG, Herbstein J. Concomitant use of valproate and carbamazepine in bipolar and schizoaffective disorders. Journal of Clinical Psychopharmacology 1994;14(1):67. [PubMed] [Google Scholar]
Walden 1996 {published data only}
- Walden J, Von WJ, Berger M, Grunze H. Efficacy of antiepileptic drugs in the treatment of psychiatric diseases. EEG Labor 1996;18(1):32. [Google Scholar]
Wetterling 1987 {published data only}
- Wetterling T. Open clinical trial of carbamazepine in chronic schizophrenic inpatients. Pharmacopsychiatry 1987;20(3):127‐30. [DOI] [PubMed] [Google Scholar]
Wunderlich 1983 {published data only}
- Wunderlich HP, Grunes JU, Neumann J, Zahlten W. Carbamazepine (Finlepsin(TM)) for manicdepressive and schizophrenic diseases. Deutsche Gesundheitswesen 1983;38(35):1352‐6. [Google Scholar]
Yassa 1983 {published data only}
- Yassa R, Dupont D. Carbamazepine in the treatment of aggressive behavior in schizophrenic patients: A case report. Canadian Journal of Psychiatry 1983;28(7):566‐8. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Kamisada 1988 {published data only}
- Kamisada M, Tateyama M, Nakano Y, Kawachi Y, Fujii Y, Tanoue A, et al. Comparison of the clinical effects of lithium carbonate and carbamazepine on excited schizophrenics. Psychopharmacology 1988;96(Suppl):348. [MEDLINE: ; PMID 4944698] 4944698 [Google Scholar]
Lee 1996 {published data only}
- Lee MS, Choi BH, Kim SH. Combined use of carbamazepine and valproic acid in negative symptom schizophrenia. 9th Congress of the European College of Neuropsychopharmacology; September 21‐25, Amsterdam, The Netherlands. 1996. [MEDLINE: ]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1982
- Andreasen NC. Negative symptoms in schizophrenia: definition and reliability. Archives of General Psychiatry 1982;39:784‐8. [DOI] [PubMed] [Google Scholar]
Andreasen 1984
- Andreasen NC. The scale for assessment of positive symptoms. University of Iowa 1984.
Begg 1996
- Beg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. JAMA 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use. Therapie 1999;54(4):405‐11. [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Cheine 2001
- Cheine M, Ahonen J, Wahlbeck K. Beta‐blocker supplementation of standard drug treatment for schizophrenia. Cochrane Database of Systematic Reviews 2001, Issue Issue 3. [DOI: 10.1002/14651858.CD000234] [DOI] [PubMed] [Google Scholar]
Christison 1991
- Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophrenia Bulletin 1991;17:217‐45. [DOI] [PubMed] [Google Scholar]
Dardennes 1995
- Dardennes R, Even C, Bange F, Heim A. Comparison of carbamazepine and lithium in the prophylaxis of bipolar disorder: a meta‐analysis. British Journal of Psychiatry 1995;166:378‐81. [DOI] [PubMed] [Google Scholar]
Davey Smith 1998
- Davey Smith G, Egger M. Meta‐analysis: Unresolved issues and future developments. BMJ 1998;16:221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Abstracts of 8th International Cochrane Colloquium; 2000 Oct 25‐28th; Cape Town, South Africa. 2000.
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Dold 2012
- Dold M, Li C, Tardy M, Khorsand V, Gillies D, Leucht S. Benzodiazepines for schizophrenia. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD006391.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2009
- Essali A, Al‐Haj Haasan N, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD000059.pub2; PUBMED: 19160174] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE Profiler [Computer program]
- GRADE Working Group. GRADE Profiler. Version 3.2. GRADE Working Group, 2004.
Granger 1995
- Granger P, Biton B, Faure C, Vige X, Depoortere H, Graham D, et al. Modulation of the gamma‐aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Molecular Pharmacology 1995;47:1189–96. [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU assessment manual for psychopharmacology‐revised. Washington, D.C: DHEW. National Institute of Mental Health 1976.
Hamilton 1960
- Hamilton M. A rating scale of depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad A, Moore A, Carroll D, Jenkinson C, Reynolds DJM, Gavanagh DJ, et al. Assessing the quality of reports of randomized clinical trials: Is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
Kane 1988
- Kane JM, Honigfeld G, Singer J, Meltzer H, and the Clozaril Collaborative study group. Clozapine for the treatment‐resistant schizophrenic. A double‐blind comparison with chlorpromazine. Archives of General Psychiatry 1988;45:789‐96. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and negative syndrome scale (PANSS) manual. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A. The positive and negative symptome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13(2):261‐75. [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2007a
- Leucht S, Kissling W, McGrath J. Lithium for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD003834.pub2] [DOI] [PubMed] [Google Scholar]
Lorr 1962
- Lorr M, McNair DM, Klett CJ, Lasky JJ. Evidence of ten psychotic symptoms. Journal of Consulting Psychology 1962;26:185. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams CE, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marwaha S, Johnson S. Schizophrenia and employment ? A review. Social Psychiatry and Psychiatric Epidemiology 2004;39:337‐49. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D, CONSORT Group (Consolidated Standards of Reporting Trials). The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;258:1987‐91. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:790‐812. [Google Scholar]
Pate 1986
- Pate LA. Carbamazepine in treatment of the violent psychotic patients. Jefferson Journal of Psychiatry 1986;4(1):28‐36. [Google Scholar]
Review Manager (RevMan) [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Schooler 1993
- Schooler NR, Keith SJ. Clinical research for the treatment of schizophrenia. Psychopharmacology Bulletin 1993;29:431‐46. [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Tharyan 2002
- Tharyan P, Adams CE. Electroconvulsive therapy for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD000076.pub2; CD000076] [DOI] [PubMed] [Google Scholar]
Tiihonen 1995
- Tiihonen J, Vartiainen H, Hakola P. Carbamazepine‐induced changes in plasma levels of neuroleptics. Pharmacopsychiatry 1995;28:26‐8. [DOI] [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality. Archives of General Psychiatry 1978;35:153‐5. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii‐92. [MEDLINE: ] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Leucht 2002
- Leucht S, McGrath J, White P, Kissling W. Carbamazepine augmentation for schizophrenia: how good is the evidence?. Journal of Clinical Psychiatry 2002;63(3):218‐24. [DOI] [PubMed] [Google Scholar]
Leucht 2002b
- Leucht S, McGrath J, White P, Kissling W. Carbamazepine for schizophrenia and schizoaffective psychoses. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD001258] [DOI] [PubMed] [Google Scholar]
Leucht 2007b
- Leucht S, Kissling W, McGrath J, White P. Carbamazepine for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD001258.pub2] [DOI] [PubMed] [Google Scholar]