

The crystal structure of KaiC from Thermosynechococcus elongatus allows insight into the different anatomies of the CI and CII ATPases.
Keywords: ATPase, circadian clock, KaiABC, kinase
Abstract
A post-translational oscillator (PTO) composed of the proteins KaiA, KaiB and KaiC is at the heart of the cyanobacterial circadian clock. KaiC interacts with KaiA and KaiB over the daily cycle, and CII domains undergo rhythmic phosphorylation/dephosphorylation with a 24 h period. Both the N-terminal (CI) and C-terminal (CII) rings of KaiC exhibit ATPase activity. The CI ATPase proceeds in an input-independent fashion, but the CII ATPase is subject to metabolic input signals. The crystal structure of KaiC from Thermosynechococcus elongatus allows insight into the different anatomies of the CI and CII ATPases. Four consecutive arginines in CI (Arg linker) that connect the P-loop, CI subunits and CI and CII at the ring interface are primary candidates for the coordination of the CI and CII activities. The mutation of linker residues alters the period or triggers arhythmic behavior. Comparison between the CI and CII structures also reveals differences in loop regions that are key to KaiA and KaiB binding and activation of CII ATPase and kinase. Common packing features in KaiC crystals shed light on the KaiB–KaiC interaction.
1. Introduction
Biological processes in organisms ranging from prokaryotic cyanobacteria to humans exhibit circadian rhythms with a period of ∼24 h that are controlled by an endogenous pacemaker (Dunlap et al., 2004 ▸). The hallmarks of circadian rhythms are (i) persistence of the oscillation under constant conditions (e.g. constant light or dark at constant temperature), (ii) temperature compensation (i.e. minimal changes in the clock period as a result of different constant temperatures within the physiological range) and (iii) entrainment of the endogenous pacemaker by the environmental light–dark cycle. In addition to these canonical properties, it is generally assumed that the central mechanism of the circadian clock in eukaryotic cells is comprised of a transcription/translation feedback loop (TTFL; Hardin et al., 1990 ▸; Aronson et al., 1994 ▸). Remarkably, the core pacemaker in cyanobacteria is not a TTFL but a biochemical oscillator that is coupled to a TTFL that regulates clock output genome-wide (Kitayama et al., 2008 ▸; Qin, Byrne, Xu et al., 2010 ▸; Zwicker et al., 2010 ▸; Teng et al., 2013 ▸). The former can be reconstituted in vitro from three proteins KaiA, KaiB and KaiC with ATP (Nakajima et al., 2005 ▸; Murakami et al., 2012 ▸) and functions as a post-translational oscillator (PTO) in vivo (Egli & Johnson, 2013 ▸). The PTO embedded within the TTFL appears to be particularly effective at promoting stability and robustness by protecting the clock against perturbations arising from cell division, temperature and metabolic fluctuations as well as transcriptional noise (reviewed in Johnson & Egli, 2014 ▸).
KaiC constitutes the central cog of the cyanobacterial PTO and is the only enzyme in the three-protein oscillator. It is the result of a gene duplication, and the N-terminal (CI) and C-terminal (CII) domains of the KaiC proteins of 519 (Synechococcus elongatus) and 518 (Thermosynechococcus elongatus) amino acids in length exhibit sequence similarities to helicases (e.g. DnaB) and recombinases (e.g. RecA) (Leipe et al., 2000 ▸). KaiC forms a homohexamer (Mori et al., 2002 ▸; Hayashi et al., 2003 ▸; Pattanayek et al., 2004 ▸) and undergoes regular cycles of phosphorylation and dephosphorylation in its CII half with a period of ∼24 h (Nakajima et al., 2005 ▸; Nishiwaki et al., 2007 ▸; Rust et al., 2007 ▸). KaiA stimulates phosphorylation at Thr432 and Ser431 (Nishiwaki et al., 2004 ▸; Xu et al., 2004 ▸) by binding to the KaiC C-terminal region and unraveling an activation loop (Vakonakis & LiWang, 2004 ▸; Pattanayek et al., 2006 ▸; Kim et al., 2008 ▸; Egli et al., 2013 ▸). KaiB antagonizes KaiA action (Kitayama et al., 2003 ▸) by forming a KaiCII6B6 complex (Villarreal et al., 2013 ▸) and sequestering KaiA in a ternary KaiABC complex (Clodong et al., 2007 ▸; van Zon et al., 2007 ▸; Brettschneider et al., 2010 ▸; Qin, Byrne, Mori et al., 2010 ▸; Pattanayek et al., 2011 ▸).
One of the fascinating properties of the KaiABC PTO is the low consumption of energy to complete one period of the circadian cycle, amounting to just 15 ATP molecules per KaiC monomer (Terauchi et al., 2007 ▸). The rate of ATP hydrolysis is thus estimated to be about 1800 times lower than that of RecA in the presence of DNA (Brenner et al., 1987 ▸; Murakami et al., 2008 ▸). A noteworthy aspect in the context of the ‘fuel’ efficiency of the clock is the mechanism of KaiC dephosphorylation that entails a phospho-transferase (ATP synthase); thus, phosphate groups on Thr432 and Ser431 are returned to ADP under formation of ATP (Egli et al., 2012 ▸; Nishiwaki & Kondo, 2012 ▸). Interestingly, the S431A/T432A KaiC mutant (KaiC-AA) that is unable to undergo phosphorylation was reported to consume 27 ATP molecules per day. This observation provides evidence that the kinase and ATPase activities are closely linked and was interpreted as phosphorylation at Thr432 and Ser431 providing negative feedback to the ATPase activity (Terauchi et al., 2007 ▸). However, the 15 ATPs hydrolyzed per day per KaiC subunit must reflect the net consumption, as the ATP synthase activity will generate at least two ATPs per subunit. The absolute number of ATPs consumed per day remains unknown, but it is clear that KaiC-AA still shows a weak ATP synthase activity, i.e. by generating ATP from ADP and Pi in a reversal of hydrolysis (Egli et al., 2012 ▸). A further fundamental property of the KaiC hexamer is that the ATPase is temperature-compensated (Terauchi et al., 2007 ▸). Because the KaiC-AA and S431D/T432E (KaiC-DE) mutants also exhibited temperature-compensated ATPase activity (S. elongatus), it follows that temperature compensation is not dependent on the KaiC phosphorylation state. However, an independent investigation of the ATPase in T. elongatus KaiC-AA found that the mutant displays a temperature-dependent activity (Murakami et al., 2008 ▸). Finally, it was established that ATPase activity and clock period in wild-type KaiC and a series of KaiC mutants are linearly correlated (Terauchi et al., 2007 ▸): the higher the ATPase activity, the faster the clock runs.
A fundamental difference despite similarities at the sequence level between the DnaB-type and RecA-type ATPases and KaiC lies in the dual domain subunit composition of the clock protein. Only the CII domain possesses kinase activity (absent in DnaB and RecA), whereas both CI and CII harbour ATPase activity, consistent with the presence of Walker A and B motifs and catalytic glutamates in both halves (Fig. 1 ▸). The sequences of the two domains in T. elongatus KaiC display 21% identity and 32% similarity (Hayashi et al., 2006 ▸). The ATPase activity of the CI half (KaiCI; residues 1–251) is 0.7 ± 0.3 molecules of ATP per hour per KaiCI hexamer at 25°C and is therefore about 20% of that of full-length KaiC (Murakami et al., 2008 ▸). In contrast, the activity of the CII half (KaiCII; residues 252–518) amounts to only about 30% of the KaiCI activity or about 6% of that of full-length KaiC. The ATPase activity of a mixture of KaiCI and KaiCII hexamers is still only about 30% of that of KaiC, providing evidence that only the fused CI and CII domains are capable of delivering the full ATPase activity. As in the case of S. elongatus KaiC, the ATPase activities of the AA mutants of T. elongatus KaiC and KaiCI are drastically increased compared with the wild-type proteins (by 350 and 270%, respectively; Murakami et al., 2008 ▸). KaiA is able to enhance the ATPase activity of KaiC and KaiCII, but has no effect on ATP hydrolysis in KaiCI. This is consistent with the KaiCII kinase/ATPase integrating input signals through the ATP/ADP ratio and the KaiCI ATPase acting as an input-independent timer (Phong et al., 2013 ▸). In this fashion, the ATPase in KaiCI acts as an hourglass timer that is then flipped by the rhythmic kinase (phosphorylation)/phosphotransferase (dephosphorylation) activities in KaiCII, thus generating a stable oscillator (Egli & Johnson, 2013 ▸).
Figure 1.

Sequence alignment of the CI (ThKaiCI) and CII (ThKaiCII) domains of T. elongatus KaiC. Conserved residues are indicated by gray bars and changes in the charge of corresponding side chains are marked with blue (CI neutral to CII positive or CI negative to CII neutral) or red bars (CI positive to CII neutral or CI neutral to CII negative). Functionally important residues are indicated by black bars and labeled (the phosphorylation sites Ser431 and Thr432 are marked with a P). Arginine linkers are residues 216–219 in CI and residue 451 in CII. The sequence alignment was generated with Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/; Sievers et al., 2011 ▸) and modified manually in UCSF Chimera (Pettersen et al., 2004 ▸).
Remarkably, the KaiABC PTO maintains a stable period over a wide range of ATP/ADP ratios (Rust et al., 2011 ▸) and has the ability to keep time in the dark (Tomita et al., 2005 ▸). On the other hand, a change in the ATP:ADP ratio can shift the phase of the oscillator. Thus, the phosphorylation level of KaiC is a marker of the circadian phase and a regulator of the interactions with KaiA and KaiB as well as other factors involved in the output pathway of the clock (reviewed in Johnson et al., 2008 ▸, 2011 ▸; Egli & Johnson, 2013 ▸). The partitioning of the ATPase activities in CI and CII is apparent from the observation that mutation of the catalytic glutamates in CI (Glu77 and Glu78; Fig. 1 ▸) does not affect the course of phosphorylation and dephosphorylation in CII (Phong et al., 2013 ▸). However, although the ATPase in CII must therefore fuel the kinase in this domain, the mutant enzyme with the ATPase in CI shut down is unable to generate rhythms in vitro and is trapped in a highly phosphorylated state (Phong et al., 2013 ▸). The CI ATPase activity is also required for formation of the KaiBC complex and ultimately sequestration of KaiA at the KaiBC interface that closes the negative-feedback loop (Phong et al., 2013 ▸). The consequences for the conformation of KaiCI of the ATPase action there and how a putative change is transmitted to KaiCII, the domain that KaiB contacts (Pattanayek et al., 2008 ▸, 2013 ▸; Villarreal et al., 2013 ▸), remain to be determined.
To visualize common features and differences in the architectures of the KaiCI and KaiCII ATPases and interactions of ATPs at the subunit interfaces in the two halves, we determined the crystal structure of full-length KaiC from T. elongatus to a resolution of 2.84 Å. Previous research had demonstrated that the CI domain of KaiC is largely responsible for hexamerization (Hayashi et al., 2006 ▸). Thus, under negative-stain EM conditions S. elongatus KaiCI readily hexamerized, whereas KaiCII did not, although it was still phosphorylated (Pattanayek et al., 2008 ▸). The structure of T. elongatus KaiC in conjunction with the deviating sequences of the CI and CII domains (Fig. 1 ▸) provide insights into the different stabilities of the hexamers formed by the two domains. A key difference between the two halves are the arginine residues that we term ‘Arg linkers’ in order to distinguish them from the ‘Arg fingers’ that commonly contact the ATP γ-phosphate in the active sites of ATPases. In the CII half the Arg linker entails a single residue; however, in the CI half it entails four consecutive arginines (Fig. 1 ▸). Arginine side chains in the latter motif establish important interactions with the P-loop among CI domains as well as between the CI and CII hexamers. The inter-hexamer contacts established by the Arg cluster in particular are outstanding candidates for transmitting across the CI–CII border conformational fluctuations as a result of enzymatic activities in the CI (ATPase) and CII halves (ATPase, kinase, phosphotransferase).
The KaiC proteins from T. elongatus and S. elongatus exhibit sequence differences in loop regions (e.g. CII A-loop and 422-loop) that undergo conformational transitions as a result of KaiA binding (Vakonakis & LiWang, 2004 ▸; Pattanayek et al., 2006 ▸) and stimulation of KaiC phosphorylation (Kim et al., 2008 ▸; Egli et al., 2013 ▸). Whereas T. elongatus KaiC monomers in the absence of ATP are stable, those from S. elongatus precipitate. Comparison between the T. elongatus and S. elongatus KaiC structures (Pattanayek et al., 2004 ▸) reveals subtle changes in conformation as a consequence of the variations in the sequence and differences in the electrostatic surface potentials that are indicative of higher polarity of the T. elongatus KaiC monomer, consistent with its improved solubility. Lastly, a conserved packing contact in all structures of full-length KaiC proteins hitherto determined displays a resemblance to the proposed KaiB–KaiC binding mode based on cryo-EM and computational simulations (Villarreal et al., 2013 ▸).
2. Materials and methods
2.1. Protein expression and purification
The full-length KaiC protein from T. elongatus comprising amino acids 1–518 and a C-terminal His6 tag was overexpressed in Escherichia coli as described previously (Pattanayek et al., 2006 ▸) and was purified in the presence of ATP with affinity chromatography (Qiagen Ni–NTA Superflow) following the manufacturer’s standard protocol. The S. elongatus GST-KaiC, GST-KaiCI and GST-KaiCII fusion proteins were expressed in E. coli following established protocols (Nishiwaki et al., 2004 ▸; Mori et al., 2007 ▸; Pattanayek et al., 2008 ▸) and were purified by affinity chromatography with glutathione agarose beads (GE Healthcare) and, following GST cleavage with PreScission protease (GE Healthcare), by gel-filtration chromatography on a Superdex 200 HR 10/30 column (GE Healthcare). The elution buffer was 50 mM Tris–HCl pH 7.8, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 5 mM MgCl2, 1 mM ATP. The DNA sequence of the S. elongatus KaiCI A192T/E197S/E198T triple mutant was codon-optimized for expression in E. coli, commercially synthesized (DNA2.0), cloned into the pAT109 vector (VU-CSB), transformed into E. coli BL21 (DE3) cells (Invitrogen) and expressed and purified following the above procedures for wild-type KaiCI. The purity of all proteins was analyzed by SDS–PAGE using mini-gels (Bio-Rad system) and their identities were confirmed by tryptic digestion followed by MALDI-TOF mass spectrometry.
2.2. Crystallization
Crystals of T. elongatus KaiC were grown by the hanging-drop vapor-diffusion technique at 22°C. Prior to crystallization ATP was replaced by ATPγS, a slowly hydrolyzing analogue of ATP. The protein concentration was around 10 mg ml−1 in a buffer consisting of 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 2 mM β-mercaptoethanol. Equal volumes of protein solution and reservoir solution (100 mM sodium acetate, 1 M sodium formate) were mixed and equilibrated against 1 ml reservoir solution. Crystals were mounted in nylon loops, cryoprotected in mother liquor containing 25%(v/v) glycerol and flash-cooled in liquid nitrogen.
2.3. Structure determination and refinement
X-ray diffraction data were collected on the LS-CAT 21-ID-G beamline at sector 21 of the Advanced Photon Source (Argonne, Illinois, USA). Data were processed with XDS and scaled with XSCALE (Kabsch, 2010 ▸). Selected crystal data and data-collection parameters are listed in Table 1 ▸. In space group C2221, the KaiC hexamer is located on a crystallographic dyad and the asymmetric unit consists of three subunits. Initial phases were obtained by the molecular-replacement method using Phaser (McCoy et al., 2007 ▸) in the CCP4 suite of programs (Winn et al., 2011 ▸) with the A, B and C subunits of the S. elongatus KaiC homohexamer crystal structure (PDB entry 3dvl; Pattanayek et al., 2004 ▸) as the search model. The initial model was refined with PHENIX (Adams et al., 2010 ▸), and Coot (Emsley & Cowtan, 2004 ▸) was used for model building. Water molecules and Mg2+ ions were added gradually and isotropic/TLS refinement continued with PHENIX, whereby side-chain torsion angles were adjusted intermittently. The final model comprises residues 11–504 of chain A, 12–501 of chain B and 11–500 of chain C and has been refined to an R factor of 21.5% (R free = 26.6%) at a resolution of 2.84 Å. A summary of the refinement parameters is provided in Table 1 ▸ and an example of the quality of the final Fourier 2F o − F c sum electron density is depicted in Fig. 2 ▸(a). Illustrations were generated with UCSF Chimera (Pettersen et al., 2004 ▸).
Table 1. T. elongatus KaiC crystal structure statistics.
The hexamer lies on a dyad and three subunits constitute the asymmetric unit. Values in parentheses are for the last shell.
| Crystal data | |
| Space group | C2221 |
| Unit-cell parameters (Å) | a = 130.81, b = 195.29, c = 136.64 |
| Resolution (Å) | 68.3–2.84 (2.91–2.84) |
| Unique reflections | 41378 (3024) |
| Completeness (%) | 99.5 (99.4) |
| R merge † | 0.118 (0.745) |
| 〈I/σ(I)〉 | 13.3 (2.6) |
| Refinement | |
| Working-set reflections | 41329 |
| Test-set reflections | 2078 [5%] |
| No. of protein non-H atoms | 11962 |
| No. of ATP molecules | 12 |
| No. of Mg2+ ions | 12 |
| No. of water molecules | 229 |
| R work ‡/R free § | 0.215/0.266 |
| Average B factors (Å2) | |
| Protein (all residues) | 54.9 |
| Solvent | 55.5 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.02 |
| Bond angles (°) | 0.9 |
| Ramachandran analysis (%) | |
| Favored | 91.9 |
| Allowed | 6.3 |
| Outliers | 1.8 |
R
merge = 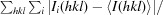
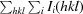 .
.
R
work = 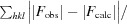
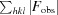 , where |F
o
hkl| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively.
, where |F
o
hkl| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively.
R free is calculated as for R work but using the set of reflections (5%) omitted from the refinement process.
Figure 2.

KaiC model quality and overall conformation. (a) Top: quality of the final Fourier 2F o − F c sum electron density drawn at the 1σ level in the region of Arg linker residue Arg218. Red crosses represent water molecules. Bottom: example of the quality of the OMIT electron density drawn at the 2.5σ level (residues Arg217–Thr220 were deleted). (b) Ribbon diagram of the overall structure of the T. elongatus KaiC hexamer. Residues of the CI (11–251) and CII (252–500) domains are colored gray and pink, respectively, and the N- and C-termini of subunits are labeled (# indicates a symmetry-related strand). ATPs and arginine linkers (light green C atoms; Arg216–Arg219 in CI and R451 in CII) are highlighted in CPK mode. C atoms of the six ATP molecules bound between the CI and CII subunits are colored black and purple, respectively, and C atoms of the phosphorylated residues Thr432 and Ser431 are colored yellow. A-loop residues (Glu487–Ile497) are cyan, 422-loop residues (Thr415–Ile425) are dark green and β8 strand residues (Thr436–Glu444) are brown. (c) Superimposition of the CI and CII domains from subunit b.
2.4. KaiC mutagenesis and in vivo rhythm assays
Mutations of specific residues of KaiC were performed by site-directed mutagenesis based on template plasmids harboring either wild-type kaiABC DNA. Briefly, the mutated kaiC strand DNA was synthesized using 18 thermal cycles with PfuUltra High-Fidelity DNA polymerase (Stratagene, San Diego, California, USA) and primers containing the desired mutations (Supplementary Table S21). After the non-mutated parental plasmids were digested at 37°C for 1 h with DpnI (New England Biolabs, Beverly, Massachusetts, USA), the circular, nicked mutant dsDNA was transformed into E. coli DH5a strain and the constructs were confirmed by DNA sequencing. S. elongatus PCC 7942 harboring a kaiBCp::luxAB reporter was used as a host strain for examination of the effects of mutant KaiCs on the clock-controlled rhythms, in which expression of the Vibrio harveyi luciferase structure genes luxAB was driven by the promoter of the kaiBC gene cluster. The wild-type kaiABC cluster was replaced by a kaiABC cluster containing wild-type or corresponding mutant kaiCs with a spectinomycin selection marker. Cyanobacterial strains were grown on BG11 agar plates supplemented with appropriate antibiotics (20 µg ml−1 spectinomycin and 7.5 µg ml−1 chloramphenicol) at 30°C under continuous cool-white illumination (LL; 40–50 µE m2 s−1). Before the cells were released into LL for the luminescence assay, a 12 h dark exposure was given to synchronize the rhythms of the individual cells in the population. The luminescence of the kaiBCp::luxAB reporter was monitored by real-time measurement of the luminescence as described previously (Xu et al., 2003 ▸). The period of the luminescence rhythms was analyzed with ChronoAnalysis II v.10.1 (courtesy of Dr Till Roenneberg).
2.5. KaiCI phosphorylation assays
The phosphorylation states of the S. elongatus wild-type and mutant KaiCI proteins as well as KaiCII were assayed using a 10% Tris SDS–PAGE gel. The gels were stained with 0.1% PhastGel Blue R solution in 10% acetic acid and 30% methanol. KaiC phosphorylation was assayed with an 8% Tris–HCl SDS–PAGE (pH 8.8) gel that was run using a Bio-Rad mini-gel apparatus. KaiC was incubated with λ protein phosphatase (New England BioLabs) for 2 h at room temperature following the manufacturer’s protocol.
2.6. Coordinates
Final coordinates and structure factors for the KaiC crystal structure have been deposited in the Protein Data Bank (http://www.rcsb.org) as entry 4o0m.
3. Results
3.1. Crystal structure of T. elongatus KaiC
We determined the crystal structure of the full-length T. elongatus KaiC hexamer with 518 amino acids per subunit and C-terminal His6 tags at 2.84 Å resolution (Table 1 ▸). The data were phased by molecular replacement using the crystal structure of S. elongatus KaiC as the search model (PDB entry 3vdl; Pattanayek et al., 2004 ▸; Johnson et al., 2008 ▸). The hexamer is positioned on a crystallographic dyad in space group C2221 and the asymmetric unit therefore consists of three KaiC subunits termed a, b and c from here on. The three individual KaiC molecules comprise residues Pro11–Glu504 (subunit a), Glu12–Glu501 (subunit b) and Pro11–Ala500 (subunit c). Representative examples of the quality of the final Fourier 2F o − F c sum and omit electron densities are depicted in Fig. 2 ▸(a). Along with 1474 amino acids (11 962 atoms), the final model of the KaiC trimer contains 229 water molecules and six ATPs with Mg2+ ions. As in the case of S. elongatus KaiC, the CI and CII rings harbor six ATP molecules, each at the subunit interfaces (Fig. 2 ▸ b). The root-mean-square deviation (r.m.s.d.) between the structures of KaiC hexamers from T. elongatus and S. elongatus based on 8694 atom pairs (backbone atoms) amounts to 2.9 Å; the sequences of the two enzymes are 80% identical (Supplementary Fig. S1). Interestingly, the r.m.s.d. between 4266 atom pairs from the CI domains (residues 15–251) of the two enzymes amounts to 3.7 Å, whereas the r.m.s.d. between 4446 atom pairs from their CII domains (residues 252–498) is just 0.7 Å. Regions omitted from the comparison include N-terminal residues that emerge from the side of the CI ring and C-terminal residues that protrude from the dome-shaped surface of the CII ring (Fig. 2 ▸ b). One clear difference between the conformations of the two hexamers is a somewhat different relative rotation between the CI and CII rings around the axis along the central channel, which amounts to ∼7° when the CII domains are superimposed (Supplementary Fig. S2). This observation suggests that rather than involving rhythmic stacking and unstacking (implying a concomitant change in the vertical spacing between rings; Chang et al., 2011 ▸), the potentially altered relative orientations of the CI and CII rings over the daily cycle may instead involve a twisting motion around the central axis.
The result of a gene duplication, the KaiC hexamer resembles a double doughnut with overall dimensions of 100 × 100 × 100 Å, a constricted waist region, a central channel and C-terminal tails that protrude from the dome-shaped surface of the CII ring (Fig. 2 ▸ b). The two rings are rotated relative to each other around the central axis such that ATP molecules bound between subunits in both the CI and CII halves are not aligned. Thr432 and Ser431 in the CII half were previously identified as phosphorylation sites (Nishiwaki et al., 2004 ▸; Xu et al., 2004 ▸; Fig. 1 ▸). Both carry phosphates in all three subunits and the structure is thus representative of the hyperphosphorylated form of T. elongatus KaiC. The enzyme was purified and crystallized in the presence of ATP, and subsequently ATP was replaced by the ATPγS analog, which undergoes slower hydrolysis. The hyperphosphorylated state is most likely to be preserved in the crystal as a result of the inability of KaiC to dephosphorylate the phosphorothioate moieties at pThr432 and pSer431, thereby producing a relatively homogeneous population of KaiC hexamers. The kinase/ATPase (CII) and ATPase (CI) active sites at the subunit interfaces exhibit a single Mg2+ ion whose coordination sphere includes catalytic glutamates and O atoms from the ATP β-phosphate and γ-phosphate groups (Supplementary Fig. S3). The active sites in both the CI and CII halves feature pairs of glutamates: Glu78 and Glu79 in CI and Glu318 and Glu319 in CII (Fig. 1 ▸ and Supplementary Fig. S3). Selected residues and regions, such as Arg fingers that contact the ATP γ-phosphate (Supplementary Fig. S3), the A-loop, the A422-loop and the β8 strand, that play an important role in the promotion of KaiC phosphorylation by KaiA (Vakonakis & LiWang, 2004 ▸; Pattanayek et al., 2006 ▸; Kim et al., 2008 ▸; Egli et al., 2013 ▸), phosphorylation sites as well as arginine residues that contact the P-loop (CI), the ATP ribose moiety (CII) and link subunits (termed Arg linkers here) are depicted in the context of the three-dimensional structure of T. elongatus KaiC in Figs. 2 ▸(b) and 2 ▸(c).
The N-terminal CI and the C-terminal CII domain both exhibit a RecA-like fold; Supplementary Fig. S4 depicts a secondary-structure diagram of the CI domain with individual α-helices and β-strands numbered. Despite the similarities in terms of the secondary-structural elements, the degree of amino-acid identity is just 21% and the two domains exhibit clear differences at the three-dimensional structural level (Fig. 2 ▸ c). Chief among them is an S-shaped loop in the C-terminal region of CII that is contacted by KaiA (A-loop; Pattanayek et al., 2004 ▸; Kim et al., 2008 ▸) and is missing entirely in CI. The N-terminal flexible tail of CI corresponds to the linker portion in CII that connects the two domains on the outside of the KaiC hexamer. Further notable differences concern the loop between the β8 and β9 strands (Fig. 2c and Supplementary Fig. S4) that contains four consecutive arginine residues in CI (Arg216–Arg219; Fig. 1 ▸) but just a single arginine (Arg451) in CII, and a tongue-like loop between β5 and α5 at the bottom of CI that is replaced by a tight α-helical curl in CII (Fig. 2 ▸ c). These examples of obvious differences are consistent with the divergent roles of the CI and CII domains in the daily clock cycle, in particular the kinase and ATP synthase activities of CII (Nishiwaki et al., 2004 ▸; Xu et al., 2004 ▸; Egli et al., 2012 ▸; Nishiwaki & Kondo, 2012 ▸) that are absent in CI, and the input-dependent (ATP/ADP ratio; KaiA) and input-independent ATPase activities of CII and CI, respectively (Terauchi et al., 2007 ▸; Murakami et al., 2008 ▸; Phong et al., 2013 ▸).
Separately expressed CI and CII domains of T. elongatus KaiC have previously been examined in terms of the stability of their hexamers in the presence of AMPPnP (Hayashi et al., 2006 ▸). Thermal melting experiments showed a T m for CI that was ∼15°C higher than that for CII (57 versus 42°C, respectively; 10 µM protein, 1 mM AMPPnP). Gel-filtration chromatography elution profiles in the presence of 0.1 mM ATP were consistent with the formation of hexameric CI but mostly trimer or tetramer as well as monomer in the case of CII. These data are in line with the outcome of a negative-stain EM analysis, which revealed hexamer formation for CI but not for CII (Pattanayek et al., 2008 ▸). Mixing CI and CII domains in the presence of ATP was observed to be insufficient for the formation of a stable hexamer (Hayashi et al., 2006 ▸), which demonstrates that covalently linked CI and CII domains are a prerequisite for the hexamerization of full-length KaiC (Fig. 2 ▸ b). Comparison of the CI and CII sequences (Fig. 1 ▸) readily discloses that the CII domain contains a greater number of charged residues, whereby the CII C-terminal tail (Ser498–Glu518) features no fewer than nine basic and acidic amino acids. In CII and CI (in parentheses), there are 20 (16) arginines, 12 (9) lysines, 24 (16) glutamates, 12 (13) aspartates and three (3) histidines. Assuming that most of the additional Arg, Lys and Glu residues in CII (15 in total) reside on or near the surface, an assumption that is supported by the significant number of charged residues in the KaiC C-terminal tail compared with the single lysine and single glutamate in the C-terminal tail of CI, this difference is likely to contribute to the higher stability of the CI hexamer and to the inability of CII to hexamerize without the CI ring acting as a structural platform.
3.2. Distinct roles of arginines in the KaiCI and KaiCII domains
The sequences of the CI and CII domains exhibit a striking difference in the region where the loop between the β8 and β9 strands transitions into the latter (residues Arg216-Arg217-Arg218-Arg219 in CI versus Glu448-Met449-Ser450-Arg451 in CII; Fig. 1 ▸). In CII, the loop region maps to the surface (Figs. 2 ▸ b and 2 ▸ c), but the CI residues are buried at the constricted waist that marks the border between the CI and CII rings. When the CII ring is carved away, it becomes apparent that the four arginines in CI are prominently exposed in the vicinity of the central channel, forming a circular arrangement in the core of the KaiC hexamer (Fig. 3 ▸ a). The arginine conserved in CI (Arg219) and CII (Arg451) is situated nearest to ATP. However, only the arginine in CII engages in direct contacts with ATP (Fig. 3 ▸ f). Its guanidino moiety forms hydrogen bonds to both the 2′-hydroxyl and the 3′-hydroxyl groups of ATP across the subunit interface, but also to P-loop residues (Table 2 ▸). By comparison, Arg219 in CI is too far removed from ATP to contact the ribose, but does hydrogen-bond to P-loop residues Gly50 and Thr51 that correspond to Gly291 and Thr292 in CII (Figs. 1 ▸ and 3 ▸ e). It is important to distinguish these arginines from the so-called finger residues. Arg fingers contact the γ-phosphate group (Snider & Houry, 2008 ▸; Wendler et al., 2012 ▸), as observed for Arg227 in CI and Arg459 in CII (Fig. 1 ▸ and Supplementary Fig. S3). To clearly distinguish arginines in the β8/9 loop and β9 strand from the Arg fingers Arg227 and Arg459, we shall refer to them as Arg linkers. In the case of the Arg tetrad Arg216–Arg219 in CI this term is particularly apt, as these residues link not just CI subunits but also reach across the waist to tie together the CI and CII subunits (Fig. 3 ▸).
Figure 3.

Arginine-linker interactions in the CI and CII halves. (a) Axial view of the 24 CI Arg-linker residues (6 × Arg216–Arg219; green C atoms) located in the waist region of the KaiC hexamer. ATP molecules are highlighted in orange. Interactions are shown for (b) CI Arg216, (c) CI Arg217, (d) CI Arg218, (e) CI Arg219 and (f) CII Arg451. The color code for residues is identical to that in Fig. 2 ▸: C atoms of arginine side chains are highlighted in green, residues from CI domains are gray and those from CII domains are pink. Selected residues are labeled, with a, b and c designating the subunit; the dashed line indicates a cation–π interaction.
Table 2. Interactions of Arg-linker motifs in the T. elongatus KaiCI and KaiCII halves.
| Donor | Acceptor | Distance (Å) | ||||
|---|---|---|---|---|---|---|
| Amino acid | Atom | Atom | Amino acid | Subunit a † | Subunit b † | Subunit c † |
| Arg216 | NH1 | O=C | Lys233 | 2.4 (b ‡) | 2.4 (c ‡) | |
| OH | Tyr236 | 3.5 (c) | ||||
| NH2 | O=C | Glu235 | 2.3 (b) | 3.1 (c) | ||
| O=C | Glu215 | 3.2 (a) | 3.4 (b) | |||
| OE1 | Glu352 | 2.5 (c ‡) | ||||
| Arg217 | NH1 | O=C | Asn210 | 2.5 (a) | 2.5 (b) | 2.5 (c) |
| NH2 | OE1 | Glu222 | 3.4 (b) | 3.2 (c) | ||
| O (water) | 192 | 2.7 | ||||
| Arg218 | NH2 | OE1 | Gln394 | 2.6 (a) | 3.1 (b) | 2.6 (c) |
| Arg219 | NH2 | O=C | Gly50 | 2.7 (a) | 2.5 (b) | 2.5 (c) |
| Arg451 | NH1 | O=C | Gly291 | 3.2 (a) | 2.9 (b) | |
| O=C | Thr292 | 2.9 (a) | 3.1 (b) | 2.8 (c) | ||
| NH2 | O2′ | ATP | 2.7 (a) | 3.1 (b) | 3.4 (c) | |
| O3′ | ATP | 2.8 (b) | ||||
Subunit that the Arg-linker residue belongs to.
Subunit that the acceptor residue belongs to.
The first of the four linker residues in CI, Arg216, reaches across the subunit interface and forms a salt bridge with Glu235 and a hydrogen bond to the hydroxyl group of the neighboring Tyr236 (Fig. 3 ▸ b). The orientation of the tyrosine ring relative to the guanidino moiety of Arg216 is consistent with a cation–π interaction. Tyr236 establishes a further hydrogen bond via its hydroxyl group to Ser353 from the adjacent subunit, and Arg216 is thus able to stitch together distant residues at the subunit interface. The next residue, Arg217, also forms a salt bridge with a glutamate (Glu222) from the adjacent subunit and engages in a hydrogen bond to the backbone carbonyl of Asn210 from its own subunit (Fig. 3 ▸ c). The environment of the guanidino moiety of Arg217 besides Glu222 and Asn210 is quite remarkable, as it is wedged between the side chains of two leucines: Leu212 from its own subunit and Leu224 from an adjacent subunit (Fig. 3 ▸ c). Arg218 is the only one of the Arg-linker residues in CI that bridges the CI and CII domains; specifically, it forms a hydrogen bond via its guanidino moiety to Gln394 from the same subunit (Fig. 3 ▸ d). Thanks to a location near the subunit interfaces and the border between the CI and CII rings, each Arg linker thus mediates contacts between residues from three domains (CI and CII from its own subunit and CI from an adjacent one) and links them to the P-loop (Figs. 3 ▸ a–3 ▸ e). In CII, Arg451 lacks the tentacle-like reach of the CI Arg linker, but is nevertheless able to weld together ATP across the subunit interface, P-loop and Tyr442 from the β8-strand that links the KaiC A-loop to the ATPase/kinase active site (Figs. 2 ▸ b, 2 ▸ c and 3 ▸ f). Inspection of the previously determined crystal structure of S. elongatus KaiC indicates similar interactions of Arg-linker residues across CI subunit interfaces and CI and CII rings (Supplementary Table S1).
To assess the potential effects of mutations of Arg-finger and Arg-linker residues on the clock period, we prepared S. elongatus KaiC alanine mutants of Arg215, Arg216, Arg217, Arg218 and Arg226 in CI (note the different numberings of the CI residues for S. elongatus and T. elongatus KaiC in the CI half) and Arg451 and Arg459 in CII and measured the ensuing rhythm in vivo for individual mutant strains using an established luminescence assay (Ishiura et al., 1998 ▸). We used S. elongatus for these assays because the Johnson laboratory has exclusively relied on this mesophilic strain for all of their in vivo work. All mutant strains displayed significantly altered phenotypes relative to wild-type KaiC (Table 3 ▸, Fig. 4 ▸). These include dampened amplitudes (R215A and R218A), lack of rhythm (R216A and R217A) and very long periods and/or a single broad peak (R226A, R451A and R459A) (Supplementary Fig. S5). Although not every substantial alteration of the period and/or amplitude of the oscillation as a result of a mutation is necessarily an indication of an important functional role of the particular residue, it is certainly not unexpected for Arg226 and Arg459 (Arg fingers in CI and CII, respectively) that contact the γ-phosphate of ATP. That individual mutation of all of the Arg linker residues should also severely impair the oscillator is remarkable.
Table 3. Effect of S. elongatus KaiC arginine-linker and arginine-finger mutations on luminescence rhythms.
| Circadian phenotype | Overall expression of the kaiBCp-driven luxAB luminescence in LL† | |||
|---|---|---|---|---|
| KaiC mutation | Period‡ (h) | Characteristics of waveform | U min−1 colony−1 (mean ± SD) | Fold compared with WT |
| WT | 25.6 ± 0.1 | Robust | 2574 ± 566 | 1.00 |
| R215A§ | 19.0 ± 0.3 | Short period; quickly dampened | 1368 ± 175 | 0.53 |
| R216A§ | — | AR | 3794 ± 684 | 1.47 |
| R217A§ | — | A single regular peak then AR | 2758 ± 336 | 1.07 |
| R218A§ | 27.8 ± 0.1 | Long period; then quickly dampened | 4065 ± 264 | 1.58 |
| R226A | — | A single very broad peak | 2084 ± 201 | 0.81 |
| R451A§ | 73.5 ± 1.8 | Very long period | 3124 ± 869 | 1.21 |
| R459A | — | A single broad peak | 336 ± 11 | 0.13 |
Overall expression of the kaiBCp::luxAB luminescence was calculated from the total expression levels in constant light (LL) for 196 h and was converted to the average in luminescence units per minute per colony. The ratios of the luminescence values between wild type (WT) and mutants were calculated and the difference was analyzed by t-test.
Mean ± SD.
Arg linker.
p < 0.01.
Arhythmicity.
Arg finger.
Figure 4.

Phenotypes of S. elongatus KaiC arginine-linker and arginine-finger alanine mutants. Individual charts show regular rhythm (wild type) and the absence thereof for Arg-linker mutants (CI, R215A, R216A, R217A, R218A; CII, R451A) and Arg-finger mutants (CI, R226A; CII, 459A) under an LL regimen over the course of 8 d as assessed by luminescence. Error bars are shown in red.
3.3. A-loop conformation and consequences of an Arg-to-Gly change between S. elongatus and T. elongatus KaiC
The C-terminal region of KaiCII contains the binding site for the KaiA dimer that stimulates KaiC phosphorylation (Vakonakis & LiWang, 2004 ▸; Pattanayek et al., 2006 ▸). The S-shaped loop at the C-terminus (Fig. 2 ▸ c), now termed the A-loop because it serves the interaction with KaiA, is a unique attribute of the CII domain. The C-terminal region of CI lacks this loop region (Figs. 1 ▸ and 2 ▸ c) as the 15-amino-acid stretch linking CI and CII on the surface of KaiC does not correspond to the A-loop but actually constitutes the N-terminal region of CII. The A-loop contains a β-stranded curl between residues Glu487 and Ile497 that becomes unraveled upon KaiA binding and is pulled out from the A-loop assembly in the KaiC hexamer that extends the central channel to the CII dome-shaped upper surface (Fig. 2 ▸ b; Vakonakis & LiWang, 2004 ▸; Egli et al., 2013 ▸). A-loops from adjacent subunits interact with each other in the hexamer. For example, the amide group of Arg496 from subunit A forms a hydrogen bond to the side chain of Glu487 from subunit B, and the backbone of Arg496 in the A-loop of subunit B in turn interacts with Glu487 from subunit C, and so forth (Fig. 5 ▸ a). Individual A-loops are stabilized by several internal hydrogen bonds, and additional hydrogen-bonding interactions to adjacent regions establish more long-range networks between A-loops and P-loops as well as the phosphorylation sites Thr432 and Ser431. The former network involves Glu444 from β8 (Figs. 2 ▸ c and 5 ▸ a), the carboxylate moiety of which is hydrogen-bonded to backbone amides from the A-loop residues Gly488, Ile489 and Ile490 (Fig. 5 ▸ a). The A-loop residue Gly492 then forms a hydrogen bond via its backbone carbonyl O atom to the amide N atom of Glu444 from an adjacent subunit. Tyr442, which is just one residue over from Glu444, establishes hydrogen bonds to the P-loop (Fig. 3 ▸ f). The long-range interaction between the A-loop and phosphorylation sites is mediated by the 422-loop residues (residue 422 sits at the apical position; Figs. 2 ▸ c and 6 ▸ a; Egli et al., 2013 ▸). Met420 and Gly421 in the 422-loop interact directly with A-loop residues, and the S-shaped 422-loop then continues to the phosphorylation sites Thr432 and Ser431 ten residues downstream (Fig. 6 ▸ a). By pulling out an A-loop KaiA destabilizes A-loops from neighboring subunits, and it is remarkable that the deletion of residues beyond Glu487 (A-loop removed) causes the KaiC mutant to be constitutively hyperphosphorylated. Conversely, the deletion of residues beyond Ile497 (A-loop present) causes the KaiC mutant to be constitutively hypophosphorylated (Kim et al., 2008 ▸). It is feasible that removal of the A-loop alters the dynamic behavior not just of the β8 strand and the associated P-loop, but also of the 422-loop and therefore the phosphorylation sites. Indeed, molecular-dynamics (MD) simulations of Δ487 and Δ497 mutant KaiC hexamers confirmed the increased mobility of CII residues including Glu444, P-loop and phosphorylation sites in the former and provided support for a concerted allosteric mechanism of the stimulation of KaiC phosphorylation by KaiA (Egli et al., 2013 ▸).
Figure 5.

KaiCII A-loop conformation and interactions. (a) Conformation of the A-loop, amino acids Glu487–Ile497, in subunit B of T. elongatus KaiC. C atoms of loop residues are highlighted in black and C atoms of additional B-subunit residues, E444b and N443b from the β8 strand, and residues from the adjacent A and C subunits that all interact with the loop are colored pink, brown and pale pink, respectively. Selected side chains are labeled (underlined in the case of loop residues), hydrogen bonds are shown as thin solid lines and hydrophobic contacts are shown as blue dashed lines. (b) Superimposition of the A-loops from the T. elongatus (black C atoms) and S. elongatus (cyan C atoms) KaiC hexamers (B subunit). Notice the very similar conformations and identical sequences with the exception of residue 488 (Gly, T. elongatus, green; Arg, S. elongatus, orange). Hydrogen bonds are shown as thin solid lines and an additional interaction between the side chains of Arg488 and Thr495 in some subunits of S. elongatus KaiC is indicated by a dashed line.
Figure 6.

Conformations of the extended phosphorylation site loop (422-loop) in KaiCII and the corresponding region in CI (B subunit). (a) Superimposition of the T. elongatus KaiCII loop region comprising residues Thr415–pThr432 (pink) and the KaiCI loop comprising residues Glu184–Glu199 (gray). Labels indicate equivalent residues based on the sequence alignment of the CI and CII halves (Fig. 1 ▸). The CI loop makes a tighter turn as it lacks amino acids corresponding to Ser422 and Asn423 in CII. CI residues Phe195, Gly196, Val197, Glu198 and Glu199 are part of a short α-helix. The r.m.s.d. for 16 Cα pairs amounts to 2.6 Å. (b) The superimposition of the extended 422-loops in T. elongatus (pink) and S. elongatus KaiC (cyan) reveals similar conformations in spite of deviating sequences in the apical region (Ala422-His423 versus Ser422-Asn423, respectively).
The sequences of T. elongatus and S. elongatus KaiC in the A-loop Glu487–Ile497 region differ at only a single position (Supplementary Fig. S1). In the former enzyme residue 488 is a glycine, while in the latter it is an arginine. At the three-dimensional structural level, the A-loops in the two enzymes display virtually identical backbone conformations and those of the side chains are very similar (Fig. 5 ▸ b). In the crystal structure of S. elongatus KaiC, Arg488 side chains from individual subunits jut into the central channel, thus virtually sealing the opening at the CII end (Pattanayek et al., 2004 ▸). In some subunits, the distance between the guanidino moiety and the hydroxyl group of Thr495 across the A-loop is consistent with a hydrogen bond (Fig. 5 ▸ b) that can be expected to stabilize the loop conformation and add to the resistance towards its unraveling by KaiA. In the thermophilic strain, this interaction is missing as a result of glycine occupying the position of the arginine. Perhaps one would have expected the variation of the amino acid at position 488 between the two KaiCs to be the opposite of the observed change, i.e. KaiC from the organism favoring environments of elevated temperature might benefit from increased stability of the A-loop (Arg488), whereas the loop conformation in the mesophilic strain does not need to be tied together further (Gly488). However, we found earlier that the R488A mutation in S. elongatus KaiC severely attenuates rhythmicity, although it does not disrupt hexamer formation (Pattanayek et al., 2006 ▸). The T495A mutation has previously been shown to abolish rhythmicity (Ishiura et al., 1998 ▸; Taniguchi et al., 2001 ▸).
3.4. Conformational deviations between the KaiCII 422-loop and the corresponding KaiCI loop
KaiC domains feature an extended mostly parallel-stranded β-sheet at their core, of which β7 constitutes the central strand (Supplementary Fig. S5). The β7 strand is followed by a long S-shaped loop that ends in a short α-helix (α8) in CI and the Ser431 and Thr432 phosphorylation sites in CII and eventually becomes β8. The loops rim the central channel in both CI and in CII, but only those in CII form close contacts via their Met420 and Gly421 residues to the C-terminal A-loops (Figs. 2 ▸ c and 5 ▸ a; A-loops are absent in CI). Comparison of the sequences in CI (Val186–Glu199) and CII (Asp417–pThr432; Fig. 1 ▸) shows a gap in CI at positions that correspond to Ser422 and Asn423 in CII. Indeed, the loop in CI describes a tighter turn than the 422-loop in CII, and superimposition of the two loops reveals significant conformational deviations (Fig. 6 ▸ a). Residues Val197, Glu198, Glu199, Phe200 and Val201 form the α8 curl in CI, and comparison with the corresponding region in CII that comprises pSer431 and pThr432 reveals considerably different conformations in that the latter region extends the loop to the beginning of the β8 strand.
The sequences of the T. elongatus and S. elongatus KaiC proteins in the 422-loop region are basically identical except for the residues at the very tip (Supplementary Fig. S1). Amino acids Ser422 and Asn423 in T. elongatus correspond to Ala422 and His423, respectively, in S. elongatus KaiC (Fig. 6 ▸ b). Despite this difference, the loop conformations in the crystal structures of these enzymes are nearly identical (Fig. 6 ▸ b). However, previous investigations uncovered drastic consequences of slight changes at the sequence level for the function of the central clock protein. In S. elongatus the conservative A422V mutation was found to result in a reduced sensitivity to resetting the phase of the clock (Kiyohara et al., 2005 ▸). Further work on the A422V mutant established a lack of phosphorylation at Ser431 and an altered dynamic behavior of the loop region, but only relatively minor differences in the conformation as a result of the mutation and no changes in terms of the thermodynamic stability (Egli et al., 2013 ▸). MD simulations of wild-type KaiC and A422V mutant hexamers provided support for an altered dynamic behavior of the loop and adjacent regions as a result of the mutation (Egli et al., 2013 ▸). Conversely, the A422S/H423N mutation appears not to affect the structure, dynamics or function of S. elongatus KaiC in a significant way. A closer look at the 422-loop in the T. elongatus KaiC crystal structure shows that the side chain of Ser422 is not involved in hydrogen bonds to other potential acceptors or donors in its vicinity within the loop (e.g. Thr416, Asp417, Gln418, Ser424 and Thr426; Fig. 6 ▸). The distances between the side chains of these residues across the loop are typically >4 Å, but could potentially be bridged by water molecules (Fig. 6 ▸ b). The crystal structure only revealed a small number of solvent molecules owing to the somewhat limited resolution, and none of them are associated with the 422-loop. However, we did observe water molecules linking side chains in the 422-loop in the crystal structure of the S. elongatus A422S KaiC mutant at 2.6 Å resolution (unpublished data).
3.5. Origins of the different enzymatic activities of KaiCI and KaiCII
The combined kinase and ATPase activity of the KaiCII hexamer and the lack of kinase activity of the KaiCI hexamer represent a striking difference between the two domains (Egli et al., 2012 ▸). One of reasons that there is no phosphoryl transfer at CI subunit interfaces is that target sites might be unavailable. In the CII ring the ATP γ-phosphate is transferred to Thr432 and Ser431 across the subunit interface, thus resulting in 12 phosphorylated residues in KaiC in the hyperphosphorylated state (Xu et al., 2004 ▸). The kinase is fueled by ATPase activity in the same ring (Phong et al., 2013 ▸) and dephosphorylation during the second half of the day proceeds via synthesis of ATP, i.e. phosphates on threonines and serines are handed back to ADP (Egli et al., 2012 ▸; Nishiwaki & Kondo, 2012 ▸). Thus, kinase, ATPase and ATP synthase all use the same active sites at CII subunit interfaces. We also identified a third phosphorylation site, Thr426, whose side-chain hydroxyl group is hydrogen-bonded to the phosphate of pSer431 (Xu et al., 2004 ▸). The residue at position 426 in KaiC needs to be phosphorylatable for the clock to exhibit proper rhythm, and the T426A, T426N and T426E mutations all abolish circadian rhythms (Xu et al., 2009 ▸). Crystal structures of S431A and S431A/T432E KaiC mutants exhibited phosphorylation at Thr426 in some subunits (Pattanayek et al., 2009 ▸).
A look at the residues in T. elongatus CI corresponding to CII residues Thr426, Ser431 and Thr432 shows that they are Ala193, Glu198 and Glu199, respectively (Fig. 1 ▸). The change from TST (426/431/432) to AEE is certainly interesting as EE mimics the phosphorylated state and Ala cannot be the recipient of a phosphate group. In order to test whether mutation from AEE to TST at positions 193/198/199 in CI results in kinase activity, we expressed the separate CI domain triple mutant as a GST fusion protein and then subjected the protein with the GST tag cleaved off to incubation with ATP. However, the introduction of threonines and serine alone appears not to be sufficient to spark kinase activity in CI (Fig. 7 ▸). As pointed out above, the conformation of the CII chain in the vicinity of the two phosphorylation sites differs from the corresponding region in CI, where the loop is interrupted by a short α-helical section (Fig. 6 ▸ a). Therefore, the orientations of the newly introduced Ser198 and Thr199 side chains relative to the ATP γ-phosphate in the CI mutant may differ from those of the Ser431 and Thr432 residues in CII and ultimately prevent phosphoryl transfer. Perhaps a subtle change in sequence between the CI and CII (in parentheses) domains in the immediate vicinity of residues 198 (431) and 199 (432) constitutes a greater obstacle to phosphoryl transfer. In the CI half Glu199 (Thr199 in the triple mutant) is followed by Phe200 and Val201, but in CII the bulky Phe is missing and Ile433 follows directly after Thr432 (Fig. 1 ▸). Inspection of the ATPase active site in CI indicates that the Phe side chain could block transfer of the γ-phosphate towards the Ser198 and Thr199 targets in the CI mutant protein. We also produced the corresponding KaiCI quadruple mutant A192A/E197S/E198T/Δ199F (S. elongatus numbering) to establish its phosphorylation status and found no kinase activity (data not shown).
Figure 7.

Assessing a putative kinase activity of the KaiCI domain. SDS–PAGE assay of the phosphorylation status of full-length S. elongatus KaiC (KaiC), KaiC treated with λ phosphatase (λPP), S. elongatus KaiCI (wt-KaiCI), the S. elongatus KaiCI triple mutant A192T/E197S/E198T (TST-KaiCI) and S. elongatus KaiCII (KaiCII). Residue numberings between CI from S. elongatus and T. elongatus differ by one, i.e. S. elongatus mutations A192T/E197S/E198T correspond to T. elongatus mutations A193T/E198S/E199T. Multiple bands for KaiC and the double band for KaiCII represent the phosphorylated (top) and nonphosphorylated (bottom) states. In contrast, single bands for wt-KaiCI and TST-KaiCI indicate that neither protein is phosphorylated. That KaiCII expressed as a separate domain still exhibits phosphorylation despite its inability to form stable hexamers has previously been demonstrated (Pattanayek et al., 2008 ▸).
3.6. KaiC crystal lattice interactions and KaiB–KaiC binding
Crystals of KaiC feature a similar packing motif despite deviating crystal systems and/or space-group symmetries. In the T. elongatus KaiC crystal with space group C2221 (three subunits per asymmetric unit) and the S. elongatus crystal structure with space group P212121 (a single hexamer per asymmetric unit), the CII dome of a hexamer is approached by the CI portion of a subunit from a symmetry mate (KaiCI#; Figs. 8 ▸ a and 8 ▸ b). The CI region involved in the nearest approach comprises residues 100–185 (β5–α5–β6–α6–α7–β7; Supplementary Fig. S5), whereby the α-helical portion of the αβ sandwich is directed toward the CII surface of the neighboring hexamer (Fig. 8 ▸ d). In both structures the CI portion hovers above a CII domain interface, thus covering the ATP-binding site (Figs. 8 ▸ a, 8 ▸ b and 8 ▸ d). The two packing interactions differ in the orientation of the CI domain (KaiCI#) relative to the CII ring. In the T. elongatus crystal structure the β-sheet assumes a tangential orientation relative to the ring (Fig. 8 ▸ a), while in the S. elongatus crystal structure strands in the β-sheet point towards the central channel of the neighboring KaiC hexamer (Figs. 8 ▸ b and 8 ▸ d). The close approach between the CI domain from a subunit of one hexamer and the CII dome of a symmetry-related hexamer is replicated in the crystal structure of S. elongatus KaiC in space group P21 (two hexamers per asymmetric unit), with somewhat tighter packing between hexamers compared with the two other structures (not shown).
Figure 8.

Similarities between KaiC crystal-packing interactions and KaiC–KaiB binding. Packing interactions between the KaiCII ring and a KaiCI subunit from a symmetry-related KaiC hexamer (KaiC#) in the crystal structures of (a) T. elongatus KaiC and (b) S. elongatus KaiC. (c) The KaiCII–KaiB interaction (only a single KaiB is depicted) in the cryo-EM model of the S. elongatus KaiC6B6 complex. (d, e) The KaiCII–KaiCI# crystal-packing interactions shown in (b) and (c), respectively, rotated by 90° around the horizontal axis and viewed from the side. (f) Superimposition of the latch regions in T. elongatus KaiCI (residues 110–124) and CII (residues 350–356) and the adjacent α-helical and β-stranded portions.
The KaiC crystal-packing interactions bear some resemblance to the proposed interaction between KaiB and KaiC in the recently established cryo-EM model of the complex (Figs. 8 ▸ c and 8 ▸ e; Villarreal et al., 2013 ▸). Accordingly, six KaiB monomers form a ring on top of KaiCII such that the α-helical portions of the KaiB monomers are turned towards the CII dome. Similar to the KaiCI# symmetry mate covering two adjacent subunits in the KaiC crystals, each KaiB monomer lies across two KaiCII domains, thereby covering the ATP site at the subunit interface. This binding mode is consistent with the role of KaiB in promoting KaiC dephosphorylation, as it may prevent ATP access or interfere with the ATPase and/or kinase activities. The CII binding mode is supported by computational simulations that indicated a significantly larger buried surface for the association between the KaiB6 and KaiCII6 rings compared with KaiB6 bound to KaiCI6 at the opposite end. However, beyond the evidence from modeling studies and the close resemblance between the EM KaiB–KaiC binding and crystal KaiC–KaiC packing modes, strong support for an interaction between KaiB and CII also comes from further experimental data. (i) Native gel electrophoresis demonstrated a bandshift for mixtures of KaiB and KaiCII but not for mixtures of KaiB and KaiCI (Pattanayek et al., 2008 ▸). (ii) Negative-stain EM using C-terminally His6-tagged KaiC hexamer in combination with Ni–NTA-Nanogold provided direct support for location of KaiB on the same side as the tags with associated gold particles (CII; Pattanayek et al., 2013 ▸). (iii) SAXS data in solution indicated that the protrusions resulting from KaiB binding on one end of the KaiC hexamer are on the same side as the density attributed to CII terminal tails (Pattanayek et al., 2011 ▸). (iv) The locations of high electron-density regions in KaiC in the cryo-EM model of the KaiBC complex that are indicative of α-helices in the CI and CII halves support the correct orientation of KaiBs on the KaiC barrel (Villarreal et al., 2013 ▸). Finally, (v) a very recent investigation using native mass spectrometry in combination with modeling is supportive of our KaiC6B6 cryo-EM model and places KaiB monomers across adjacent KaiC subunits at the CII end (Snijder et al., 2014 ▸).
4. Discussion
KaiC is the only enzyme among the three Kai proteins and exhibits multiple activities. Despite over a decade of investigations directed at the structure and function of the cyanobacterial circadian timer, the coordination of these activities between the CI and CII halves has remained a mystery. The CI hexamer catalyzes ATP hydrolysis at subunit interfaces whereby the activity is input-independent. The CII hexamer harbors input-dependent ATPase, kinase and ATP synthase activities, such that the latter two result in a daily cycle of phosphorylation and dephosphorylation across subunit interfaces. Energy generated in CI does not fuel the kinase in CII. Rather, it is the CII ATPase that provides the energy for phosphotransfer in this hexamer (Phong et al., 2013 ▸). KaiC subunits consist of fused RecA-like domains that exhibit significantly different sequences (Fig. 1 ▸). The hexameric CI ring is more stable than the CII ring (Hayashi et al., 2006 ▸; Pattanayek et al., 2008 ▸), and the stability difference can be rationalized by the greater number of charged residues in CII and their location near the surface or in the C-terminal tail. Differences in the interactions with ATP at the subunit interfaces also provide an explanation for the higher stability of the CI hexamer. In this context, it is noteworthy that the CI ATPase is more active than the CII ATPase (∼30% of CI activity; Murakami et al., 2008 ▸). It is likely that the increased stability of CI and the higher activity relative to CII are somehow linked. A recent investigation of the structure and activity of the AAA domain from the C. elegans FIGL-1 protein found that the hexameric enzyme exhibits an unusually high ATPase activity (Peng et al., 2013 ▸). The crystal structure revealed a cluster of charged residues at the subunit interfaces, and disruption of this cluster by mutagenesis diminished both the ATPase activity and the oligomerization capacity of the protein.
For the PTO to work properly and for it to liaise accurately with the TTFL the activities in the CI and CII rings have to be integrated, and since the former acts independently of KaiA and is not affected by the ATP:ADP ratio (Rust et al., 2011 ▸; Phong et al., 2013 ▸) the actions in CII have to be somehow transmitted to CI and vice versa. This is consistent with the observations that phosphorylation/dephosphorylation is the most consistent rhythm under different light conditions (Qin, Byrne, Xu et al., 2010 ▸) and that the interplay of protein–protein interactions in the PTO (i.e. KaiA–KaiC and KaiB–KaiC) is carefully timed (Mori et al., 2007 ▸). Looking at structural portions that could be of importance in mediating communication between the CI and CII rings, the linker region encompassing residues 245–260 comes to mind. However, beyond keeping the two rings in close proximity (mixing separate CI and CII rings abrogates proper function; Hayashi et al., 2006 ▸), it is hard to see how a simple tether could help to coordinate the activities of the two rings. However, the Arg linkers Arg216–Arg219 in CI are intriguing in this respect because of their location at the seam between the two hexamers and their ability to bridge far-flung regions including ATP and the P-loop and to tie together adjacent CI and adjacent CI and CII domains (Fig. 3 ▸). Mutation of individual linker residues has drastic consequences for period and/or amplitude in every case in both CI and CII (Fig. 4 ▸), a feature that Arg linkers share with the Arg-finger residues that are directly involved in the ATPase activity (Supplementary Fig. S3). Future work needs to be directed at the effect of mutations in the CI Arg linker on the ATPase activity there. It is interesting that a random mutation R215C in S. elongatus KaiC (corresponding to linker residue Arg216 in the T. elongatus enzyme) led to a drastically shortened period of 16 h (Ishiura et al., 1998 ▸). This observation supports the idea that the charges of the arginines may play a critical role in the mediation of interactions between the CI and CII rings and the integration of their activities.
The purposes of the CI and CII ATPase activities are very different. The activity in CII drives the kinase, whereas the activity in CI is a prerequisite for KaiB binding (Phong et al., 2013 ▸). The latter finding appears to cast doubt on the generally accepted role of KaiB in sensing the phosphorylation state of KaiC to act as an antagonist of KaiA (Nishiwaki et al., 2007 ▸; Pattanayek et al., 2011 ▸), although it remains unclear what activates/times the ATPase in CI in the first place. Along with the conflicting proposals as to the signal that triggers KaiB binding, there are now also two different models regarding the binding site of KaiB on KaiC. We have presented models of KaiB binding to the CII side of KaiC (Pattanayek et al., 2008 ▸, 2011 ▸, 2013 ▸; Villarreal et al., 2013 ▸). Others have placed KaiB on the CI side of KaiC (Mutoh et al., 2013 ▸; Tseng et al., 2014 ▸). KaiB apparently contacting both ends of the KaiC hexamer poses a conundrum. This is because the KaiCI and KaiCII surfaces at opposite ends of the hexamer display contrasting electrostatic surface potentials (ESPs; Fig. 9 ▸). The CI end is mostly negatively polarized (pink/red), while the CII end is mostly positively polarized (cyan/blue). Incidentally, inspection of the ESPs of T. elongatus KaiC (Fig. 9 ▸ a) and S. elongatus KaiC (Fig. 9 ▸ b) reveals a higher polarity of the surface of the former and may explain why its monomers are soluble in the absence of ATP or analogs such as AMPPnP, whereas S. elongatus KaiC without ATP aggregates. In addition to the differences in the ESPs of CI and CII, the topographies of the two ends are quite different (Fig. 2 ▸ b; Pattanayek et al., 2013 ▸). The N-terminal CI surface of the KaiC hexamer is relatively flat and features the highly flexible β5–α5 loop regions (referred to by some as B-loops or binding-loops; Tseng et al., 2014 ▸; Figs. 2 ▸ c and 8 ▸ f). The C-terminal CII surface is convex and interrupted in the center by protruding A-loops and adjacent C-termini (Figs. 2 ▸ b and 5 ▸).
Figure 9.

Electrostatic surface potentials (ESPs) of T. elongatus and S. elongatus KaiC monomers. ESP of (a) T. elongatus KaiC and (b) S. elongatus KaiC (PDB entry 3dvl). KaiC monomeric subunits are depicted in five different orientations, first viewed from the side and related by rotations of 120° around the vertical and then from the top (CII side) and the bottom (CI side). The minimum and maximum values of the electrostatic potential are −16kT/e and +16kT/e, respectively. ESPs were calculated using default parameters of APBS (Baker et al., 2001 ▸). ATP molecules are shown in ball-and-stick mode and are colored yellow.
It was observed some time ago that the C-terminal tail of KaiB is critical for its function and for proper rhythm of the KaiABC clock (Iwase et al., 2005 ▸). Shortening the tail or mutating individual Asp or Glu residues (the T. elongatus KaiB C-terminal tail features no fewer than seven acidic residues) to Asn or Gln, respectively, abolished rhythmicity. Taking into account the functionally important negatively polarized tail in KaiB lends further support to a binding mode involving the KaiCII side that is positively polarized, rather than the negatively polarized CI surface. The negative ESP of the latter is largely a consequence of an extended loop between β5 and α5 (Fig. 2 ▸ c) that harbors six aspartates and glutamates (Fig. 8 ▸ f). It is noteworthy that all experiments supporting KaiB binding to CI were carried out with a KaiB mutant that lacks the negatively charged C-terminal tail (Mutoh et al., 2013 ▸). Besides seemingly incompatible ESPs, modeling of KaiB monomers bound to either CI β5–α5 loops or the CII dome demonstrated a more limited interface in terms of the buried surface in the case of the former interaction (Villarreal et al., 2013 ▸). Another argument against the KaiB–KaiCI binding mode in the hexameric context is the fact that KaiB (if its role is to antagonize KaiA and stimulate dephosphorylation) would be far removed from the CII ATP-binding sites. Conversely, KaiBs interacting with the CII ring are in direct contact with ATP-binding clefts (Figs. 8 ▸ c and 8 ▸ e).
The combined evidence regarding the KaiB–KaiC binding mode gathered to date offers support for both binding on the CI and CII side. It remains possible that KaiB indeed contacts opposite ends of KaiC, but perhaps at different periods over the daily cycle. The CII binding mode presented by us (Pattanayek et al., 2008 ▸, 2013 ▸; Villarreal et al., 2013 ▸) is consistent with KaiB monomers engaging at ATP-binding sites, thus interfering with KaiA bound to the CII tails and actively promoting dephosphorylation in the PTO. On the other hand, the CI binding mode (Phong et al., 2013 ▸; Tseng et al., 2014 ▸), particularly if it results in destabilizing the CI subunit interfaces (Mutoh et al., 2013 ▸), is consistent with KaiC subunit exchange (Kageyama et al., 2006 ▸) that is crucial for maintaining the amplitude of the clock (Mori et al., 2007 ▸) and coincides with the KaiC dephosphorylation stage.
Supplementary Material
PDB reference: KaiC, 4o0m
Supporting Information. DOI: 10.1107/S1399004714003228/mn5052sup1.pdf
Acknowledgments
We would like to thank M. Said K. Sidiqi for help with protein expression and crystallization. This work was supported by National Institute of Health grants R01 GM073845 (to ME) and R01 GM067152 and R01 GM088595 (to CHJ). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Author contributions were as follows. RP and ME conceived the project. RP, YX and AL performed the experiments. RP, YX, AL, CHJ and ME analyzed the data. ME wrote and RP, YX and ME illustrated the paper. The authors declare that they have no conflict of interest.
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: MN5052).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aronson, B. D., Johnson, K. A., Loros, J. J. & Dunlap, J. C. (1994). Science, 263, 1578–1584. [DOI] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Brenner, S. L., Mitchell, R. S., Morrical, S. W., Neuendorf, S. K., Schutte, B. C. & Cox, M. M. (1987). J. Biol. Chem. 262, 4011–4016. [PubMed]
- Brettschneider, C., Rose, R. J., Hertel, S., Axmann, I. M., Heck, A. J. & Kollmann, M. (2010). Mol. Syst. Biol. 6, 389. [DOI] [PMC free article] [PubMed]
- Chang, Y.-G., Kuo, N.-W., Tseng, R. & LiWang, A. (2011). Proc. Natl Acad. Sci. USA, 108, 14431–14436. [DOI] [PMC free article] [PubMed]
- Clodong, S., Dühring, U., Kronk, L., Wilde, A., Axmann, I., Herzel, H. & Kollmann, M. (2007). Mol. Syst. Biol. 3, 90. [DOI] [PMC free article] [PubMed]
- Dunlap, J. C., Loros, J. J. & DeCoursey, P. J. (2004). Editors. Chronobiology: Biological Timekeeping. Sunderland: Sinauer.
- Egli, M. & Johnson, C. H. (2013). Curr. Opin. Neurobiol. 23, 732–740. [DOI] [PMC free article] [PubMed]
- Egli, M., Mori, T., Pattanayek, R., Xu, Y., Qin, X. & Johnson, C. H. (2012). Biochemistry, 51, 1547–1558. [DOI] [PMC free article] [PubMed]
- Egli, M., Pattanayek, R., Sheehan, J. H., Xu, Y., Mori, T., Smith, J. A. & Johnson, C. H. (2013). Biochemistry, 52, 1208–1220. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Hardin, P. E., Hall, J. C. & Rosbash, M. (1990). Nature (London), 343, 536–540. [DOI] [PubMed]
- Hayashi, F., Iwase, R., Uzumaki, T. & Ishiura, M. (2006). Biochem. Biophys. Res. Commun. 318, 864–872. [DOI] [PubMed]
- Hayashi, F., Suzuki, H., Iwase, R., Uzumaki, T., Miyake, A., Shen, J.-R., Imada, K., Furukawa, Y., Yonekura, K., Namba, K. & Ishiura, M. (2003). Genes Cells, 8, 287–296. [DOI] [PubMed]
- Ishiura, M., Kutsuna, S., Aoki, S., Iwasaki, H., Andersson, C. R., Tanabe, A., Golden, S. S., Johnson, C. H. & Kondo, T. (1998). Science, 281, 1519–1523. [DOI] [PubMed]
- Iwase, R., Imada, K., Hayashi, F., Uzumaki, T., Morishita, M., Onai, K., Furukawa, Y., Namba, K. & Ishiura, M. (2005). J. Biol. Chem. 280, 43141–43149. [DOI] [PubMed]
- Johnson, C. H. & Egli, M. (2014). Annu. Rev. Biochem. In the press. [DOI] [PMC free article] [PubMed]
- Johnson, C. H., Egli, M. & Stewart, P. L. (2008). Science, 322, 697–701. [DOI] [PMC free article] [PubMed]
- Johnson, C. H., Stewart, P. L. & Egli, M. (2011). Annu. Rev. Biophys. 40, 143–167. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kageyama, H., Nishiwaki, T., Nakajima, M., Iwasaki, H., Oyama, T. & Kondo, T. (2006). Mol. Cell, 23, 161–171. [DOI] [PubMed]
- Kim, Y.-I., Dong, G., Carruthers, C. W. Jr, Golden, S. S. & LiWang, A. (2008). Proc. Natl Acad. Sci. USA, 105, 12825–12830. [DOI] [PMC free article] [PubMed]
- Kitayama, Y., Iwasaki, H., Nishiwaki, T. & Kondo, T. (2003). EMBO J. 22, 2127–2134. [DOI] [PMC free article] [PubMed]
- Kitayama, Y., Nishiwaki, T., Terauchi, K. & Kondo, T. (2008). Genes Dev. 22, 1513–1521. [DOI] [PMC free article] [PubMed]
- Kiyohara, Y. B., Katayama, M. & Kondo, T. (2005). J. Bacteriol. 187, 2559–2564. [DOI] [PMC free article] [PubMed]
- Leipe, D. D., Aravind, L., Grishin, N. V. & Koonin, E. V. (2000). Genome Res. 10, 5–16. [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mori, T., Saveliev, S. V., Xu, Y., Stafford, W. F., Cox, M. M., Inman, R. B. & Johnson, C. H. (2002). Proc. Natl Acad. Sci. USA, 99, 17203–17208. [DOI] [PMC free article] [PubMed]
- Mori, T., Williams, D. R., Byrne, M. O., Qin, X., Egli, M., Mchaourab, H. S., Stewart, P. L. & Johnson, C. H. (2007). PLoS Biol. 5, e93. [DOI] [PMC free article] [PubMed]
- Murakami, R., Miyake, A., Iwase, R., Hayashi, F., Uzumaki, T. & Ishiura, M. (2008). Genes Cells, 13, 387–395. [DOI] [PubMed]
- Murakami, R., Mutoh, R., Iwase, R., Furukawa, Y., Imada, K., Onai, K., Morishita, M., Yasui, S., Ishii, K., Valencia Swain, J. O., Uzumaki, T., Namba, K. & Ishiura, M. (2012). J. Biol. Chem. 287, 29506–29515. [DOI] [PMC free article] [PubMed]
- Mutoh, R., Nishimura, A., Yasui, S., Onai, K. & Ishiura, M. (2013). PLoS One, 8, e80200. [DOI] [PMC free article] [PubMed]
- Nakajima, M., Imai, K., Ito, H., Nishiwaki, T., Murayama, Y., Iwasaki, H., Oyama, T. & Kondo, T. (2005). Science, 308, 414–415. [DOI] [PubMed]
- Nishiwaki, T. & Kondo, T. (2012). J. Biol. Chem. 287, 18030–18035. [DOI] [PMC free article] [PubMed]
- Nishiwaki, T., Satomi, Y., Kitayama, Y., Terauchi, K., Kiyohara, R., Takao, T. & Kondo, T. (2007). EMBO J. 26, 4029–4037. [DOI] [PMC free article] [PubMed]
- Nishiwaki, T., Satomi, Y., Nakajima, M., Lee, C., Kiyohara, R., Kageyama, H., Kitayama, Y., Temamoto, M., Yamaguchi, A., Hijikata, A., Go, M., Iwasaki, H., Takao, T. & Kondo, T. (2004). Proc. Natl Acad. Sci. USA, 101, 13927–13932. [DOI] [PMC free article] [PubMed]
- Pattanayek, R., Mori, T., Xu, Y., Pattanayek, S., Johnson, C. H. & Egli, M. (2009). PLoS One, 4, e7529. [DOI] [PMC free article] [PubMed]
- Pattanayek, R., Wang, J., Mori, T., Xu, Y., Johnson, C. H. & Egli, M. (2004). Mol. Cell, 15, 375–388. [DOI] [PubMed]
- Pattanayek, R., Williams, D. R., Pattanayek, S., Mori, T., Johnson, C. H., Stewart, P. L. & Egli, M. (2008). EMBO J. 27, 1767–1778. [DOI] [PMC free article] [PubMed]
- Pattanayek, R., Williams, D. R., Pattanayek, S., Xu, Y., Mori, T., Johnson, C. H., Stewart, P. L. & Egli, M. (2006). EMBO J. 25, 2017–2028. [DOI] [PMC free article] [PubMed]
- Pattanayek, R., Williams, D. R., Rossi, G., Weigand, S., Mori, T., Johnson, C. H., Stewart, P. L. & Egli, M. (2011). PLoS One, 6, e23697. [DOI] [PMC free article] [PubMed]
- Pattanayek, R., Yadagiri, K. K., Ohi, M. D. & Egli, M. (2013). Cell Cycle, 12, 810–817. [DOI] [PMC free article] [PubMed]
- Peng, W., Lin, Z., Li, W., Lu, J., Shen, Y. & Wang, C. (2013). J. Biol. Chem. 288, 29305–29312. [DOI] [PMC free article] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Phong, C., Markson, J. S., Wilhoite, C. M. & Rust, M. J. (2013). Proc. Natl Acad. Sci. USA, 110, 1124–1129. [DOI] [PMC free article] [PubMed]
- Qin, X., Byrne, M., Mori, T., Zou, P., Williams, D. R., McHaourab, H. & Johnson, C. H. (2010). Proc. Natl Acad. Sci. USA, 107, 14805–14810. [DOI] [PMC free article] [PubMed]
- Qin, X., Byrne, M., Xu, Y., Mori, T. & Johnson, C. H. (2010). PLoS Biol. 8, e1000394. [DOI] [PMC free article] [PubMed]
- Rust, M. J., Golden, S. S. & O’Shea, E. K. (2011). Science, 331, 220–223. [DOI] [PMC free article] [PubMed]
- Rust, M. J., Markson, J. S., Lane, W. S., Fisher, D. S. & O’Shea, E. K. (2007). Science, 318, 809–812. [DOI] [PMC free article] [PubMed]
- Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J., Thompson, J. D. & Higgins, D. G. (2011). Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed]
- Snider, J. & Houry, W. A. (2008). Biochem. Soc. Trans. 36, 72–77. [DOI] [PubMed]
- Snijder, J., Burnley, R. J., Wiegard, A., Melquiond, A. S. J., Bonvin, A. M. J. J., Axmann, I. M. & Heck, A. J. R. (2014). Proc. Natl Acad. Sci. USA, 111, 1379–1384. [DOI] [PMC free article] [PubMed]
- Taniguchi, Y., Yamaguchi, A., Hijikata, A., Iwasaki, H., Kamagata, K., Ishiura, M., Go, M. & Kondo, T. (2001). FEBS Lett. 496, 86–90. [DOI] [PubMed]
- Teng, S.-W., Mukherji, S., Moffitt, J. R., de Buyl, S. & O’Shea, E. K. (2013). Science, 340, 737–740. [DOI] [PMC free article] [PubMed]
- Terauchi, K., Kitayama, Y., Nishiwaki, T., Miwa, K., Murayama, Y., Oyama, T. & Kondo, T. (2007). Proc. Natl Acad. Sci. USA, 104, 16377–16381. [DOI] [PMC free article] [PubMed]
- Tomita, J., Nakajima, M., Kondo, T. & Iwasaki, H. (2005). Science, 307, 251–254. [DOI] [PubMed]
- Tseng, R., Chang, Y.-G., Bravo, I., Latham, R., Chaudhary, A., Kuo, N.-W. & LiWang, A. (2014). J. Mol. Biol. 426, 389–402. [DOI] [PMC free article] [PubMed]
- Vakonakis, I. & LiWang, A. C. (2004). Proc. Natl Acad. Sci. USA, 101, 10925–10930. [DOI] [PMC free article] [PubMed]
- Villarreal, S. A., Pattanayek, R., Williams, D. R., Mori, T., Qin, X., Johnson, C. H., Egli, M. & Stewart, P. L. (2013). J. Mol. Biol. 425, 3311–3324. [DOI] [PMC free article] [PubMed]
- Wendler, P., Ciniawsky, S., Kock, M. & Kube, S. (2012). Biochem. Biophys. Acta, 1823, 2–14. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Xu, Y., Mori, T. & Johnson, C. H. (2003). EMBO J. 22, 2117–2126. [DOI] [PMC free article] [PubMed]
- Xu, Y., Mori, T., Pattanayek, R., Pattanayek, S., Egli, M. & Johnson, C. H. (2004). Proc. Natl Acad. Sci. USA, 101, 13933–13938. [DOI] [PMC free article] [PubMed]
- Xu, Y., Mori, T., Qin, X., Yan, H., Egli, M. & Johnson, C. H. (2009). PLoS One, 4, e7509. [DOI] [PMC free article] [PubMed]
- Zon, J. S. van, Lubensky, D. K., Altena, P. R. & ten Wolde, P. R. (2007). Proc. Natl Acad. Sci. USA, 104, 7420–7425. [DOI] [PMC free article] [PubMed]
- Zwicker, D., Lubensky, D. K. & ten Wolde, P. R. (2010). Proc. Natl Acad. Sci. USA, 107, 22540–22545. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: KaiC, 4o0m
Supporting Information. DOI: 10.1107/S1399004714003228/mn5052sup1.pdf