

Abstract
Glucose‐dependent insulinotropic polypeptide (GIP) was the first incretin to be identified. In addition to stimulating insulin secretion, GIP plays regulatory roles in the maintenance, growth and survival of pancreatic islets, as well as impacting on adipocyte function. The current review focuses on the intracellular signaling pathways by which GIP contributes to the regulation of β‐cell secretion and survival, and adipocyte differentiation and lipogenesis. Studies on signaling underlying the insulinotropic actions of the incretin hormones have largely been carried out with glucagon‐like peptide‐1. They have provided evidence for contributions by both protein kinase A (PKA) and exchange protein directly activated by cyclic adenosine monophosphate (EPAC2), and their probable role in GIP signaling is discussed. Recent studies have shown that inhibition of the kinase apoptosis signal‐regulating kinase 1 (ASK1) by GIP plays a key role in reducing mitochondria‐induced apoptosis in β‐cells through protein kinase B (PKB)‐mediated pathways, and that GIP‐induced post‐translational modification of voltage‐ dependent K+ (Kv) channels also contributes to its prosurvival role. Through regulation of gene expression, GIP tips the balance between pro‐ and anti‐apoptotic members of the B‐cell lymphoma‐2 (Bcl‐2) protein family towards β‐cell survival. GIP also plays important roles in the differentiation of pre‐adipocytes to adipocytes, and in the regulation of lipoprotein lipase expression and lipogenesis. These events involve interactions between GIP, insulin and resistin signaling pathways. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2012.00196.x, 2012)
Keywords: Apoptosis, Glucose‐dependent insulinotropic polypeptide, Incretin
Introduction
The hormones, glucose‐dependent insulinotropic polypeptide (GIP) and glucagon‐like peptide‐1 (GLP‐1), are released from the intestine in response to nutrient ingestion and stimulate insulin secretion in a glucose‐dependent manner, as a result of which they have been classified as ‘incretins’1–5. Additionally, both hormones play regulatory roles in the maintenance, growth and survival of pancreatic islets, as well as acting in an integrative manner on processes involved in nutrient metabolism, including events underlying the passage of chyme through the gastrointestinal tract, and nutrient digestion, absorption and storage1–4. The biological actions of GIP and GLP‐1 are terminated by the prolyl endopeptidase, dipeptidyl peptidase‐4 (DPP‐4)2,6. DPP‐4 resistant analogs of GLP‐1 and DPP‐4 inhibitors are two recently introduced classes of type 2 diabetes therapeutics that mimic or potentiate, respectively, actions of the incretins2,6. Although these agents have proven to be extremely effective in improving glucose tolerance, it is unlikely that their therapeutic potential has been fully realized, and it is important to elucidate the mechanistic basis underlying incretin actions. In the present review, we focus on the current understanding of the signaling events involved in GIP actions on β‐cell function and the adipocyte, with an emphasis on studies carried out in the authors’ laboratory.
GIP Stimulation of Insulin Secretion
The GIP receptor (GIPR) is a member of the class B G protein‐coupled receptor family that has now been identified in chordates ranging from fish to mammals3,7, although little is known about cellular signaling events in lower species. β‐Cell responses to GIP are glucose‐dependent in rodents8,9 and humans10. There is extensive literature on the mode of action of GLP‐1 on the β‐cell that has been expertly reviewed11–15, and information that is relevant to GIP action will be integrated into the current review. Uptake and metabolism of glucose in the β‐cell increases the intracellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio, resulting in closure of ATP‐sensitive K+ (KATP) channels and membrane depolarization, with consequent activation of voltage‐dependent Ca2+ channels (VDCC), increases in intracellular Ca2+ (iCa2+) and triggering of insulin granule exocytosis16–18. Membrane repolarization is mediated by voltage‐dependent K+ (Kv) channels19 and Ca2+‐sensitive K+ (KCa) channels. The incretin hormones act at multiple levels within this complex series of events.
Both GIP and GLP‐1 have been shown to stimulate adenylyl cyclase (AC) through a stimulatory G protein (Gs) coupled process resulting in increased cyclic adenosine monophosphate (cAMP)20,21, and this is considered to be the major signaling pathway involved in their potentiation of glucose‐induced insulin secretion. Although GIP‐stimulated insulin secretion is glucose‐dependent, cAMP production and subsequent activation of downstream signaling modules are not22,23. As discussed further below, synergistic interaction between glucose and incretin action occurs when Ca2+ fluxes are increased through modulation of KATP channels and VDCC, as well as through release from the endoplasmic reticulum (Figure 1)15,17,24. Type VIII AC has been proposed to act as a ‘coincidence detector’ of signals from glucose and cAMP17, as it is activated by both Ca2+‐calmodulin and Gαs25. Ca2+‐calmodulin also modulates the activity of phosphodiesterase (PDE) 1C, and synergistic interactions between Gαs and Ca2+‐calmodulin have been suggested to drive synchronous, in‐phase oscillations of cAMP and Ca2+24,26.
Figure 1.

Signaling pathways proposed to be involved in proximal events in glucose‐dependent insulinotropic polypeptide (GIP)‐mediated potentiation of glucose‐induced insulin secretion. (a) Evidence has been presented supporting roles for both protein kinase A (PKA) and cyclic adenosine monophosphate (cAMP)‐activated guanine nucleotide exchange factor (cAMP‐GEF)/exchange protein directly activated by cAMP (Epac) in the modulation of adenosine triphosphate (ATP)‐sensitive K+ (KATP) channels. Dissociation of cAMP‐Epac2 from sulfonulurea receptor 1 (SUR1) binding has been proposed to activate phospholipase C‐ε (PLCε) through Ras‐related protein 1 (Rap1), resulting in phosphatidylinositol 4,5 bisphosphate (PIP2) metabolism and inhibition of adenosine triphosphate‐sensitive channel subunit, Kir6.2, membrane depolarization and activation of voltage‐dependent Ca2+ channels (VDCC). (b) Diacylglycerol‐activated PKCε potentiates calcium‐induced calcium release through ryanodine receptors and phosphatidylinositol trisphosphate (IP3) stimulates Ca2+ release from IP3 sensitive endoplasmic reticulum (ER) Ca2+ stores. PKA might act to sensitize the Ca2+ release channels (based on references 15, 20, 31, 36). AC, adenylyl cyclase; CAM, calmodulin; Gαs, stimulatory G protein α‐subunit, IP3R, inositol trisphosphate receptor; P, phosphate; RyR, ryanodine receptor; SERCA, sarco(endo)plasmic reticulum Ca2+‐ATPase.
Incretin‐induced increases in β‐cell cAMP result in the activation of both protein kinase A (PKA) and cAMP‐activated guanine nucleotide exchange factor (cAMP‐GEF)/exchange protein directly activated by cAMP (Epac) (Figure 1)15,20,21,27–32. Epac2A has been identified as the major splice‐variant involved in β‐cell incretin signaling32, but there are multiple PKA isoforms present in β‐cells and it is unclear as to which contribute to the modulation of insulin secretion. There is also uncertainty over the relative roles played by Epac2 and PKA in events leading to increases in insulin secretion, with KATP channel closure, Ca2+ influx and intracellular mobilization, and processes underlying granule movement and exocytosis, all being potential targets. A number of investigators have carried out sophisticated electrophysiological and imaging studies to identify the pathways targeted by GLP‐1 and/or GIP and, as these studies have been comprehensively reviewed15,21,30,32, only a summary of the main findings will be presented.
GLP‐1 was first suggested to modulate β‐cell KATP channel activity through a PKA‐mediated pathway33, and it was later reported that phosphorylation of the β‐cell sulfonylurea 1 (SUR1) subunit resulted in KATP channel closure (Figure 1a)34. There is now controversy over the contribution of PKA to KATP channel regulation15,32,35, as compelling evidence points to Epac2 as the major factor linking cAMP to KATP channel closure and increasing Ca2+ influx15,20,31,32. Under non‐stimulated conditions, Epac2 interacts with the nucleotide‐binding fold‐1 of SUR1 (Figure 1a)30,36–39. On binding cAMP, Epac2 dissociates from SUR138,39, and activates the GTPase, Ras‐related protein 1 (Rap1). Holz et al. have proposed that Rap1 stimulates phospholipase C‐ε (PLCε)40, resulting in localized metabolism of plasma membrane phosphatidylinositol 4,5 bisphosphate (PIP2) to phosphatidylinositol trisphosphate (IP3) and diacylglycerol (DAG; Figure 1)34. As earlier studies showed that PIP2 reduces the sensitivity of KATP channels to ATP41,42, its depletion would be expected to result in potentiated ATP‐dependent channel closure and membrane depolarization.
β‐Cell stimulation by both GLP‐1 and GIP in the presence of elevated glucose has been shown to result in increased Ca2+ uptake through VDCC and non‐selective ion channels33,43,44, as well as stimulation of Ca2+ release from intracellular stores that, in the case of GLP‐1, has been shown to involve Epac2 activation15,31. GIP probably activates identical pathways (Figure 1b), although studies on KATP channel‐deficient mice showed that GIP actions on insulin secretion showed a greater dependency on KATP channels than GLP‐145. Phospholipase Cε has been proposed to link Epac and iCa2+ fluxes through the activation of endoplasmic reticulum (ER) inositol trisphosphate (IP3) channels and protein kinase Cε (PKCε)‐mediated potentiation of calcium‐induced calcium release (CICR) through ryanodine receptors (Figure 1b)15,46, possibly through activation of calcium‐calmodulin kinase II15. In addition, PKA is capable of sensitizing the intracellular Ca2+ release channels to the effects of IP3 and Ca2+15. GIP also activates an islet group VIA Ca2+‐independent phospholipase A2 (iPLA2), resulting in increased arachidonic acid (AA) production from membrane lipids47, and AA has been shown to increase release of Ca2+ from intracellular stores, suggesting that it might be coupled to insulin secretion.
β‐Cell repolarization involves closure of VDCC, as well as opening of delayed rectifier and A‐type Kv channels18,48. GIP and GLP‐1 both reduce Kv channel currents, prolonging β‐cell action potentials and potentiating Ca2+ signals19,48,49. Kv2.1 is the major delayed rectifier channel in rodent β‐cells, playing a dominant role in GLP‐119, and probably GIP, action. Post‐translational modification of Kv2.1 in response to GIP and GLP‐1 can modulate channel gating, promote inactivation and increase channel internalization through processes that involve phosphorylation by PKA and PKCζ19,50,51, and acetylation by cAMP‐response element binding protein (CREB) binding protein (CBP; see β‐Cell Prosurvival Effects of GIP section)51. GIP also stimulates endocytosis of Kv1.4 channels through PKA‐dependent phosphorylation49. As AA was also recently shown to increase the rate of inactivation of Kv2.1 channels52, GIP‐activation of iPLA2 might also be linked to β‐cell repolarization.
In addition to the ‘up‐stream’ events involved in insulin secretion, both incretins exert distal effects on secretory granule exocytosis through PKA‐53 and Epac‐28,30,32,36,46 dependent pathways. Protein kinase A phosphorylates proteins that are components of the exocytotic machinery21, including α‐soluble N‐ethylmaleimide‐sensitive fusion protein‐attachment protein (α‐SNAP) and mammalian uncoordinated homology 13‐1 (Munc 13‐1)4. A number of models have been proposed to explain the role of Epac2 in increasing the probability of granule exocytosis15,28,54. Eliasson et al.54 proposed that Epac2 interacts with SUR1 associated with both the secretory granule and the plasma membrane, resulting in activation of a secretory granule chloride channel CIC‐315,30, granule acidification and priming through a v‐type H+‐ATPase54. In a series of elegant experiments, Seino et al.32 showed that cAMP increases both readily releasable and reserve pools of secretory granules. Epac2A appears to be more important for regulating the readily‐releasable pool during the first phase of insulin secretion, whereas PKA might be critical for second phase release32. In addition to SUR1 and Rap1, Epac2 also interacts with Ras‐related in brain 3 (Rab3)‐interactive molecule 2 (Rim2), Piccolo and SNAP‐25, and a cAMP–Epac‐2–Rim2 complex was shown to play a central role in secretory granule dynamics20,21. Although the mechanisms involved in complex formation are still being clarified, dissociation of cAMP–Epac2A from Sur1 promotes Ca2+‐dependent heterodimerization of Rim2α and Piccolo, followed by interaction with Rab3A and Munc13‐1, core components of the exocytotic apparatus21,55. GIP‐induced insulin secretion was greatly impaired in Rim2α knockout mice and their isolated islets, establishing its importance in the secretory pathway55. Additionally, phosphorylation of snapin by PKA has been shown to be essential for incretin‐stimulated assembly of collectrin, SNAP‐25 and Epac256. Finally, we recently found that a selective Epac agonist was capable of activating protein kinase B (PKB; Akt)57, emphasizing the importance of interaction between kinase pathways. It is currently unclear as to whether this interaction is related to insulin secretion or restricted to β‐cell mitogenic and prosurvival effects of GIP (see next section).
β‐Cell Prosurvival Effects of GIP
β‐Cell dysfunction and reduced β‐cell mass are major factors in the etiology of type 1 diabetes and type 2 diabetes. Whereas autoimmune reactions are responsible for apoptotic loss of β‐cells in type 1 diabetes58, chronic hyperglycemia and hyperlipidemia, elevated cytokines, amyloid deposits, ER stress and other factors contribute to type 2 diabetes59. A number of procedures are being investigated for replenishing β‐cell mass in diabetes patients, including the production of surrogate β‐cells for transplantation and stimulation of residual β‐cell proliferation. However, it appears that human β‐cell regenerative capacity is limited to around the first three decades of life60,61. Therefore, prevention of β‐cell loss by inhibiting apoptotic processes is an attractive alternative target.
A role for incretin hormones in maintaining the normal integrity of pancreatic islets was first shown by the observation that GLP‐1R−/− mice have elevated levels of β‐cell apoptosis62. Both GIP and GLP‐1 have been shown to exert strong prosurvival effects on β‐cells in vitro3,4,11,12. Additionally, studies on a number of rodent models have shown that GIPR or GLP‐1R agonists exert marked anti‐apoptotic effects in vivo63–67. Our laboratory has focused on identifying mechanisms underlying the prosurvival actions of GIP and the following overview summarizes recent findings. GIP shows protective effects on cells that have been subjected to a number of apoptosis‐inducing stressors, including high glucose ± fatty acids (glucolipotoxicity), serum depletion and a low glucose environment or treatment with agents that induce genotoxic, mitochondrial or ER stress57,65,68,69. In studies on staurosporine‐induced apoptosis in insulinoma‐1 (INS‐1) β‐cells, GIP was shown to exert effects at multiple levels in the apoptotic pathway, with reduced mitochondrial translocation of B‐cell lymphoma‐2 (Bcl‐2) associated death promoter (Bad) and Bcl‐2 interacting mediator of cell deathEL (BimEL) and oligomerization of Bcl‐2‐associated X protein (Bax), release of cytochrome C and caspase 3 activation68. Ultimately, GIP reduces the influence of mitochondria‐associated pro‐apoptotic Bcl‐2 family members, thus restraining the effects of stressors on the β‐cell.
Although the pathways linking incretin‐induced production of cAMP and activation of PKA and Epac2A with the stimulation of insulin secretion have been extensively studied, the role of cAMP in prosurvival pathways has not been clearly defined. GIP‐mediated anti‐apoptotic signaling in INS‐1 β‐cells appears to be strongly dependent on cAMP production, as low concentrations of the adenylyl cyclase inhibitor MDL‐12,300A (Santa Cruz Biotechnology, Santa Cruz, CA, USA) ablated the protective effects of GIP on staurosporine‐induced cell death68. One major pathway by which GIPR‐ and GLP1‐R‐mediated increases in cAMP production promote survival is by increasing expression of anti‐apoptotic genes through PKA phosphorylation of CREB at Serine (Ser)13370–72. In the case of GIP, bcl‐2 gene expression in INS‐1 cells was also found to involve PKA‐stimulated dephosphorylation of AMP activated protein kinase (AMPK) and increased nuclear entry of cAMP‐responsive CREB coactivator 2 (TORC2)70. Incretins activate a number of other genes involved in prosurvival pathways; for example, expression of insulin receptor substrate 2 (IRS2) is stimulated by GLP‐1‐activated PKA phosphorylation of CREB. Recently, CREB was reported to be responsible for an acute phase of cAMP‐dependent gene expression in β‐cells, whereas a delayed phase involved induction of IRS2/PKB pathways, activation of mammalian target of rapamycin (mTOR) and increasing hypoxia‐inducible factor (HIF) activity73. This increase was shown to be associated with altered INS‐1 β‐cell metabolic activity and improved cell viability73. An additional effect of sustained β‐cell stimulation by GIP‐induced activation of PKB is the phosphorylation and nuclear exclusion of forkhead box protein O1 (Foxo1), that also promotes cell survival as a result of the requirement of nuclear Foxo1 for expression of pro‐apoptotic proteins, such as Bax65. It remains to be determined whether the HIF pathway also modulates expression of prosurvival bcl‐2 family proteins.
Activation of PKB plays a central role in additional anti‐apoptotic effects of GIP65,67,68,74,75. PKB phosphorylation on Threonine (Thr)308 and Ser473 has been shown to be essential for enzyme activation in many cell types. However, in β‐cells, GIP was found to produce rapid increases in PKB activity through a non‐PI3kinase‐activated pathway in the absence of detectable Thr308 phosphorylation. As 8‐(4‐chlorophenylthio)‐2′‐O‐methyl cAMP (8′‐CPT cAMP), an EPAC‐selective agonist, mimicked the effects of GIP, activation appears to be mediated by EPAC2, although it is currently unclear as to whether Rap1 is involved57,68.
Apoptosis signal‐regulating kinase 1 (ASK1) has been shown to operate as a redox sensor that, on exposure to excessive levels of reactive oxygen species (ROS), initiates the mitochondria‐mediated apoptotic pathway through activation of p38 mitogen‐activated protein kinase (p38 MAPK) and jun N‐terminal kinase (JNK). Among the stressors shown to activate ASK1 are oxidative and ER stress, Ca2+ overload and receptor mediated inflammatory signals, all of which could contribute to β‐cell death. Exposure of INS‐1 β‐cells transfected with human ASK1 to thapsigargin was found to increase phosphorylation of Thr845 and reduce phosphorylation of Ser83 in ASK1, changes that increase its enzyme activity76,77, resulting in phosphorylation and activation of p38 MAPK and JNK, through MAPK/extracellular signal‐regulated kinase (ERK) kinase (Mek) 3/6 and Mek 4/7, respectively. Activation of PKB with GIP treatment prevented these changes in ASK1 phosphorylation and downstream targets4,68. Suppressing ASK1 activation with GIP treatment inhibited apoptosis induced by all apoptosis‐inducing agents tested. As a result of the complexity of the system, the mechanism of GIP action is currently only open to speculation. In non‐stressed cells, ASK1 forms a high molecular weight complex that has been termed the ‘ASK1 signalosome’77, and, in non‐stressed cells when ASK1 activity is inhibited, the anti‐oxidative protein, thioredoxin (Trx), is a component of the signalosome. If the oxidative state of the cell is greatly increased, the oxidized form of Trx dissociates from ASK1 and tumor necrosis factor (TNF) receptor‐associated factor 2 (TRAF2) and TRAF6 binding activates ASK1 signaling77. Although GIP‐stimulated phosphorylation of Ser83 by Akt was clearly involved in inhibiting ASK1 activity, it is also possible that the binding of thioredoxin and/or TRAF2/6 to ASK1 were impacted on. Further details remain to be elucidated.
Additional gene regulatory actions of GIP and GLP‐1 involve modification of chromatin structure, thus altering the accessibility of transcription factors to target genes. A number of different post‐translational modifications of histone N‐termini have been identified, and we showed that both incretin hormones modulate β‐cell chromatin structure by increasing acetylation and phosphorylation of core H3 histones78, with both elevated histone H3 acetyltransferase and reduced histone deacetylase activities contributing to the former78. Responses to both incretins involved activation of PKA, and downstream Erk1/2 (p44/42 MAPK) and p38 MAPK signaling modules, ultimately resulting in the activation of mitogen‐ and stress‐activated kinase‐1 (MSK‐1) and CREB. Histone acetyltransferase (HAT) inhibition resulted in significant reductions in incretin‐stimulated, CREB‐activated, Bcl‐2 gene transcription, showing that histone H3 modification plays an important role in the regulation of apoptosis‐related proteins78. Both GIP and GLP‐1 also inhibit ER stress‐induced apoptosis68,79,80 through complex mechanisms4 that involve both reductions in the apoptotic pathway, leading to caspase activation, and altered expression or activity of a number of proteins involved in the unfolded protein response (UPR), including activating transcription factor 4 (ATF4), binding immunoglobin protein (Bip), CCAAT‐enhancer‐binding protein homologous protein (CHOP) and growth arrest and deoxyribonucleic acid damage‐inducible protein 34 (GADD34)79,80.
In recent studies, we identified an interesting link between incretin‐stimulated post‐translational modification of Kv channels and apoptosis (Figure 2). Cell shrinkage (apoptotic volume decrease; AVD) occurs early in the apoptotic process, before DNA fragmentation, cytochrome C release and caspase 3 activation, and it is a prerequisite for completion of programmed cell death. Efflux of K+ is a critical component of the AVD and, as activities of a number of caspases and endonucleases are suppressed at normal intracellular K+ concentrations, decreases in its intracellular concentration result in their activation. In different cell types, several types of K+ channels have been implicated in AVD, including Kv channels. We decided to examine a possible role for the most prevalent rodent β‐cell family member, Kv2.1, in the development of apoptosis, and determine whether regulation of this process by GIP and GLP‐1 contributes to their prosurvival effects51. INS‐1 β‐cells, in which Kv2.1 was overexpressed, showed potentiated apoptotic responses to mitochondrial and ER stress, whereas GIP or GLP‐1 reduced the potentiation. In studies designed to identify their mode of action, both GIP and GLP‐1 promoted phosphorylation and acetylation of Kv2.1 through pathways involving PKA, and/or MSK‐1 and HAT (Figure 2). This was associated with reduced cell surface expression of Kv2.1. We subsequently found that GIP and GLP‐1 promoted nuclear/cytoplasmic shuttling of CBP, resulting in its interaction with Kv2.1 (Figure 2). Downregulation of CBP ablated incretin‐induced acetylation of Kv2.1, suggesting that this HAT is primarily responsible for the acetylation51. As ASK1 has been shown to play a key role in the pro‐apoptotic modulation of Kv channels81, it is likely that the GIP‐activated pathway leading to phosphorylation of serine 83 in ASK1 interacts with events leading to Kv protein modification.
Figure 2.

Proposed signaling pathways by which glucose‐dependent insulinotropic polypeptide (GIP) regulates endocytosis of voltage‐dependent K+ (Kv)2.1. Binding of GIP to its receptor (GIPR) activates protein kinase A (PKA) and, potentially, mitogen‐ and stress‐activated kinase‐1 (MSK‐1), resulting in phosphorylation of Kv2.1. Through an unknown mechanism, cyclic adenosine monophosphate‐response element binding protein binding protein (CBP) is translocated from the nucleus to the plasma membrane, where it acetylates Kv2.1. Potentiation of Kv2.1 internalization results in reduced K+ fluxes and reduced β‐cell apoptosis. AC, adenylyl cyclase; Ac, acetylated; CREB, cAMP‐response element binding protein; Gαs, stimulatory G protein α‐subunit; P, phosphorylated; TORC2, transducer of regulated cyclic adenosine monophosphate response element binding protein activity.
Effects of GIP on Adipose Tissue
Fat ingestion is a major stimulus for GIP secretion in humans, dogs and rodents3,82,83, and there is increasing evidence supporting a physiological role for GIP in promoting fat storage. There are two pathways by which GIP impacts on adipocyte metabolism: directly through interaction with GIP receptors on the adipocyte and through stimulation of insulin secretion. GIP infusion has been shown to promote the clearance of chylomicron‐associated triglyceride (TG) in dogs84, and to lower plasma TG responses to intraduodenal fat in rats85. However, GIP had no major effect on the rate of removal of intravenously administered TG86, suggesting that GIP stimulates release of TG from chylomicrons and uptake into adipose tissue. Support for a role for GIP in regulating adipose tissue mass came from rodent studies. Miyawaki et al.87 first showed that GIPR−/− mice were resistant to obesity when fed a high‐fat diet. Mice that were treated with GIPR peptide antagonists88, vaccinated against GIP89 or subjected to selective K‐cell ablation90 all showed increased resistance to high‐fat feeding‐induced obesity3, showing that GIP normally promotes lipid storage.
Early studies on direct adipocyte actions of GIP showed stimulatory effects on fatty acid (FA) synthesis from acetate in adipose tissue explants91, increased uptake and incorporation of glucose into lipids92, as well as enhanced free FA (FFA) incorporation into adipose tissue93. Adipocyte lipoprotein lipase (LPL) is responsible for the hydrolysis of TG in circulating chylomicrons, TG‐rich lipoproteins and very low‐density lipoproteins, resulting in adipocyte uptake of FFA and monoacylglycerol, and the promotion of lipogenesis. GIP was shown to increase LPL enzyme activity, in an insulin‐dependent manner, in cultured 3T3‐L1 adipocytes, rodent adipocytes and subcutaneous human adipocytes94–96. In view of the close correlation between human GIP responses and plasma post‐heparin LPL levels97, it has been suggested that GIP acts on adipocyte storage by matching adipose tissue uptake of FA with the triglyceride load98.
Suboptimal levels of circulating FFA result in greatly reduced β‐cell responsiveness to subsequent glucose stimulation99. In vitro studies showed that, in the absence of insulin, GIP stimulates adipocyte TG hydrolysis through PKA activation100,101, and we suggested that GIP primes β‐cells during fasting by releasing adipocyte FFA into the circulation3,100. Getty‐Kaushik et al.102 showed that GIP‐stimulated increases in glycerol production were accompanied by decreased FFA in perifused adipocytes. They interpreted these responses as reflecting GIP‐induced re‐esterification from excess FFA102,103. A similar response was recently reported in humans, with GIP infusion resulting in small increases in adipose tissue FFA re‐esterification104. Although, that study was carried out under hyperglycemic conditions. GIP might also have long‐term effects on lipid metabolism, as synthesis of pancreatic lipase and colipase were both stimulated by GIP105, an effect that should increase efficiency of lipid uptake.
In the presence of insulin, GIP signaling appears to play important roles in both the differentiation of preadipocytes and lipogenesis. A complex set of events is involved in preadipocyte to adipocyte development, including growth arrest, increased transcription factor and lipogenic enzyme expression, accumulation of lipid, and the development of sensitivity to regulatory hormones106. Expression of the GIPR is extremely low in preadipocytes103,107,108, but both messenger ribonucleic acid (mRNA) and protein expression increase during differentiation of 3T3‐L1 cell103,108 and human107 preadipocytes. GIP acted synergistically with insulin to increase neutral lipid accumulation during progression of 3T3‐L1 preadipocytes to the adipocyte phenotype108. However, it was unclear as to whether synergistic effects of GIP and insulin on GIPR mRNA levels were a result of direct effects on gene transcription or secondary to the progression of differentiation. 3T3‐L1 cell differentiation was associated with upregulation of nuclear levels of peroxisome proliferator‐activated receptor (PPAR)γ. Treatment with the PPARγ receptor agonists, LY171883 and rosiglitazone, increased GIPR expression in fully differentiated 3T3‐L1 adipocytes, whereas the antagonist, GW9662, ablated expression108. Acetylation of histone H3/H4 was also increased during differentiation, and both PPARγ and acetylated histone H3/H4 bound to a region of the GIPR promoter containing the peroxisome proliferator response element (PPRE). As RNA interference (RNAi) knockdown of PPARγ in differentiated 3T3‐L1 adipocytes greatly reduced GIPR levels, PPARγ appears to be a critical transcription factor in regulating adipocyte receptor expression, but it is unclear as to the role played by insulin.
Evidence has been presented for the involvement of both PPARα109,110 and PPARγ111 in the regulation of rodent β‐cell GIPR expression, and further studies are required to clarify whether there are cell‐selective differences in regulation. Additionally, in earlier studies, it was shown that the GIPR is downregulated in β‐cells of obese rodent models of diabetes109,110,112, but in studies on Vancouver Diabetic Fatty (VDF) Zucker rats, we recently found that, compared with lean controls, GIPR and PPARγ protein levels were increased in epididymal and retroperitoneal fat pads, decreased in the perirenal fat depot and unchanged in other fat deposits (Figure 3). In contrast, GIPR expression in subcutaneous adipose tissue from human obese females was reported to be lower than in lean control subjects113. However, these results are difficult to compare, because of the different fat depots studied. Additionally, the sensitivity of GIPR expression to the prevailing insulin concentration, the level of adipose tissue insulin resistance and the glycemic status of the subjects/animals could all contribute significantly to the level of GIPR expression.
Figure 3.
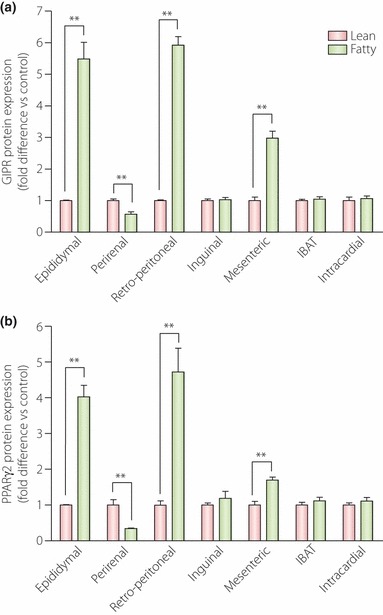
Glucose‐dependent insulinotropic polypeptide receptor (GIPR) and peroxisome proliferator‐activated receptor (PPAR)γ protein expression levels in adipose tissue depots from lean and obese Vancouver Diabetic Fatty (VDF) Zucker rats. Tissue was collected from 18‐week‐old rats and western blot analyses were carried out, with quantification by densitometry (n = 4–6 rats). Significance was tested using analysis of variance (anova) with Newman–Keuls post‐hoc test. **P < 0.05 vs lean control group. IBAT, interscapular brown adipose tissue.
The pathways involved in GIP‐stimulated lipogenesis are proving difficult to define, as a result of interactions between GIP, insulin and adipokine signaling. GIP stimulation of glucose uptake was shown to involve increasing plasma membrane glucose transporter‐4 (GLUT‐4) levels through a PKB‐mediated pathway103. Human LPL gene expression is also stimulated by GIP activation of PKB114, resulting in downstream reductions in LKB1 and AMPK phosphorylation, and increased translocation of TORC2 (cAMP‐responsive CREB coactivator 2 [CRTC2]) into the nucleus. Interaction between TORC2 and phospho‐CREB results in increased LPL gene expression114. Regulation of the phosphorylation state of TORC2 is complex and other members of the AMPK family (salt‐inducible kinases [SIK] and MARK2)115 are also capable of TORC2 phosphorylation, whereas calcineurin and a cAMP‐activated pathway induce dephosphorylation116. It has not been established as to which of these contribute to GIP‐mediated effects. GIP also enhances LPL enzyme activity in cultured 3T3‐L1 cells and subcutaneous human adipocytes by non‐transcriptional mechanisms95,96. In the 3T3‐L1 cell line, GIP induces transient activation of p38 MAPK and sustained activation of stress‐activated protein kinase (SAPK)/JNK, resulting in the release of resistin that, in turn activates PKB95. Somewhat surprisingly, subsequent events mimic those downstream of GIP in the human adipocyte, with decreases in LKB1 and AMPK phosphorylation linked to increased LPL secretion95. Human resistin (FIZZ3) shares only moderate sequence homology (∼53%) with the mouse peptide117. Additionally, FIZZ3/resistin is only weakly expressed in human adipocytes, and monocytes/macrophages are the major sites of FIZZ3/resistin production in adipose tissue118. It is currently unknown whether FIZZ3/resistin serves a paracrine function in adipocyte regulation or whether there is an entero–adipokine axis involving GIP and FIZZ3/resistin in humans. However, FIZZ3/resistin has been reported to increase FFA re‐esterification119.
In the majority of in vitro studies to date, the effects of GIP on adipogenesis and lipogenesis have been shown to involve synergistic actions with insulin, and there has been significant interest in GIPR antagonists as potential therapies for obesity88. However, adipose tissue expansion has been suggested to be an important adaptive response to increased food intake, as it protects against excess fat deposition in other sites, such as liver and muscle120,121. GIP could be an important contributor to this response and reducing its effect might not result in the anticipated benefits. Additionally, studies on both transgenic mice122 and pigs123 expressing a dominant‐negative GIPR showed greatly reduced β‐cell mass, with the mice becoming severely diabetic, supporting a critical role for GIP signaling in β‐cell development and proliferation.
Acknowledgements
Studies from the authors’ laboratory described in the review were generously supported by funding to CHSMc from the Canadian Institutes of Health Research, Canadian Diabetes Association and the Canadian Foundation for Innovation. The authors declare no conflict of interest.
References
- 1.Brubaker PL. The glucagon‐like peptides: pleiotropic regulators of nutrient homeostasis. Ann N Y Acad Sci 2006; 1070: 10–26 [DOI] [PubMed] [Google Scholar]
- 2.Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 2006; 368: 1696–1705 [DOI] [PubMed] [Google Scholar]
- 3.McIntosh CHS, Widenmaier S, Kim S‐J. Glucose‐dependent insulinotropic polypeptide (Gastric Inhibitory Polypeptide; GIP). Vitam Horm 2009; 80: 409–471 [DOI] [PubMed] [Google Scholar]
- 4.McIntosh CHS, Widenmaier S, Kim S‐J. Pleiotropic actions of the incretin hormones. Vitam Horm 2010; 84: 21–79 [DOI] [PubMed] [Google Scholar]
- 5.Holst JJ, Vilsbøll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol 2009; 297: 127–136 [DOI] [PubMed] [Google Scholar]
- 6.McIntosh CHS. Dipeptidyl peptidase IV inhibitors and diabetes therapy. Front Biosci 2008; 13: 1753–1773 [DOI] [PubMed] [Google Scholar]
- 7.Irwin D. Molecular evolution of mammalian incretin hormone genes. Regul Pept 2009; 155: 121–130 [DOI] [PubMed] [Google Scholar]
- 8.Jia X, Brown JC, Ma P, et al. Effects of glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐I‐(7‐36) on insulin secretion. Am J Physiol 1995; 268: E645–E651 [DOI] [PubMed] [Google Scholar]
- 9.Pederson RA, Brown JC. The insulinotropic action of gastric inhibitory polypeptide in the perfused isolated rat pancreas. Endocrinology 1976; 99: 780–785 [DOI] [PubMed] [Google Scholar]
- 10.Vilsbøll T, Krarup T, Madsbad S, et al. Both GLP‐1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept 2003; 114: 115–121 [DOI] [PubMed] [Google Scholar]
- 11.Drucker DJ. The biology of incretin hormones. Cell Metab 2006; 3: 153–165 [DOI] [PubMed] [Google Scholar]
- 12.Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007; 132: 2131–2157 [DOI] [PubMed] [Google Scholar]
- 13.Doyle ME, Egan JM. Mechanisms of action of glucagon‐like peptide 1 in the pancreas. Pharmacol Ther 2007; 113: 546–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Z, Jin T. New insights into the role of cAMP in the production and function of the incretin hormone glucagon‐like peptide‐1 (GLP‐1). Cell Signal 2010; 22: 1–8 [DOI] [PubMed] [Google Scholar]
- 15.Leech CA, Chepurny OG, Holz GG. Epac2‐dependent rap1 activation and the control of islet insulin secretion by glucagon‐like peptide‐1. Vitam Horm 2010; 84: 279–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashcroft F, Rorsman P. Molecular defects in insulin secretion in type‐2 diabetes. Rev Endocr Metab Disord 2004; 4: 135–142 [DOI] [PubMed] [Google Scholar]
- 17.Hinke SA, Hellemans K, Schuit FC. Plasticity of the beta cell insulin secretory competence: preparing the pancreatic beta cell for the next meal. J Physiol (Lond) 2004; 558: 369–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiriart M, Aguilar‐Bryan L. Channel regulation of glucose sensing in the pancreatic beta‐cell. Am J Physiol Endocrinol Metab 2008; 295: 1298–1306 [DOI] [PubMed] [Google Scholar]
- 19.MacDonald P, Wheeler MB. Voltage‐dependent K+ channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia 2003; 46: 1046–1062 [DOI] [PubMed] [Google Scholar]
- 20.Kashima Y, Miki T, Shibasaki T, et al. Critical role of cAMP‐GEFII‐‐Rim2 complex in incretin‐potentiated insulin secretion. J Biol Chem 2001; 276: 46046–46053 [DOI] [PubMed] [Google Scholar]
- 21.Seino S, Shibasaki T. PKA‐dependent and PKA‐independent pathways for cAMP‐regulated exocytosis. Physiol Rev 2005; 85: 1303–1342 [DOI] [PubMed] [Google Scholar]
- 22.Hinke SA, Pauly RP, Ehses J, et al. Role of glucose in chronic desensitization of isolated rat islets and mouse insulinoma (betaTC‐3) cells to glucose‐dependent insulinotropic polypeptide. J Endocrinol 2000; 165: 281–291 [DOI] [PubMed] [Google Scholar]
- 23.Ehses JA, Pelech SL, Pederson RA, et al. Glucose‐dependent insulinotropic polypeptide activates the Raf‐Mek1/2‐ERK1/2 module via a cyclic AMP/cAMP‐dependent protein kinase/Rap1‐mediated pathway. J Biol Chem 2002; 277: 37088–37097 [DOI] [PubMed] [Google Scholar]
- 24.Holz GG, Heart E, Leech CA. Synchronizing Ca2+ and cAMP oscillations in pancreatic beta‐cells: a role for glucose metabolism and GLP‐1 receptors? Am J Physiol Cell Physiol 2008; 294: C4–C6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willoughby D, Cooper D. Organization and Ca2+ regulation of adenylyl cyclase in cAMP microdomains. Physiol Rev 2007; 87: 965–1010 [DOI] [PubMed] [Google Scholar]
- 26.Fridlyand LE, Harbeck MC, Roe MW, et al. Regulation of cAMP dynamics by Ca2+ and G protein‐coupled receptors in the pancreatic beta‐cell: a computational approach. Am J Physiol Cell Physiol 2007; 294: C1924–C1933 [DOI] [PubMed] [Google Scholar]
- 27.Holz GG. New insights concerning the glucose‐dependent insulin secretagogue action of glucagon‐like peptide‐1 in pancreatic beta‐cells. Horm Metab Res 2004; 36: 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seino S, Takahashi H, Fujimoto W, et al. Roles of cAMP signaling in insulin granule exocytosis. Diabetes Obes Metab 2009; 11(Suppl 4): 180–188 [DOI] [PubMed] [Google Scholar]
- 29.Holz GG. Epac: A new cAMP‐binding protein in support of glucagon‐like peptide‐1 receptor‐mediated signal transduction in the pancreatic beta‐cell. Diabetes 2004; 53: 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holz GG, Kang G, Harbeck M, et al. Cell physiology of cAMP sensor Epac. J Physiol (Lond) 2006; 577: 5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chepurny OG, Kelley GG, Dzhura I, et al. PKA‐dependent potentiation of glucose‐stimulated insulin secretion by Epac activator 8‐pCPT‐2′‐O‐Me‐cAMP‐AM in human islets of Langerhans. Am J Physiol Endocrinol Metab 2010; 298: E622–E633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest 2011; 121: 2118–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gromada J, Bokvist K, Ding WG, et al. Glucagon‐like peptide 1 (7‐36) amide stimulates exocytosis in human pancreatic beta‐cells by both proximal and distal regulatory steps in stimulus‐secretion coupling. Diabetes 1998; 47: 57–65 [DOI] [PubMed] [Google Scholar]
- 34.Light PE, Manning Fox JE, Riedel MJ, et al. Glucagon‐like peptide‐1 inhibits pancreatic ATP‐sensitive potassium channels via a protein kinase A‐ and ADP‐dependent mechanism. Mol Endocrinol 2002; 16: 2135–2144 [DOI] [PubMed] [Google Scholar]
- 35.Kasai H, Hatakeyama H, Ohno M, et al. Exocytosis in islet beta‐cells. Adv Exp Med Biol 2010; 654: 305–338 [DOI] [PubMed] [Google Scholar]
- 36.Niimura M, Miki T, Shibasaki T, et al. Critical role of the N‐terminal cyclic AMP‐binding domain of Epac2 in its subcellular localization and function. J Cell Physiol 2009; 219: 652–658 [DOI] [PubMed] [Google Scholar]
- 37.Kang G, Chepurny OG, Malester B, et al. cAMP sensor Epac as a determinant of ATP‐sensitive potassium channel activity in human pancreatic beta cells and rat INS‐1 cells. J Physiol (Lond) 2006; 573: 595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shibasaki T, Sunaga Y, Fujimoto K, et al. Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage‐dependent Ca2+ channel in insulin granule exocytosis. J Biol Chem 2004; 279: 7956–7961 [DOI] [PubMed] [Google Scholar]
- 39.Shibasaki T, Takahashi H, Miki T, et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 2007; 104: 19333–19338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dzhura I, Chepurny OG, Leech CA, et al. Phospholipase C‐epsilon links Epac2 activation to the potentiation of glucose‐stimulated insulin secretion from mouse islets of Langerhans. Islets 2011; 3: 121–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baukrowitz T, Schulte U, Oliver D, et al. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 1998; 282: 1141–1144 [DOI] [PubMed] [Google Scholar]
- 42.Shyng S‐L, Nichols C. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 1998; 282: 1138–1141 [DOI] [PubMed] [Google Scholar]
- 43.Lu M, Wheeler MB, Leng XH, et al. Stimulation of insulin secretion and insulin gene expression by gastric inhibitory polypeptide. Trans Assoc Am Phys 1993; 106: 42–53 [PubMed] [Google Scholar]
- 44.Wheeler MB, Gelling RW, McIntosh CHS, et al. Functional expression of the rat pancreatic islet glucose‐dependent insulinotropic polypeptide receptor: ligand binding and intracellular signaling properties. Endocrinology 1995; 136: 4629–4639 [DOI] [PubMed] [Google Scholar]
- 45.Miki T, Minami K, Shinozaki H, et al. Distinct effects of glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐1 on insulin secretion and gut motility. Diabetes 2005; 54: 1056–1063 [DOI] [PubMed] [Google Scholar]
- 46.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 2010; 50: 355–375 [DOI] [PubMed] [Google Scholar]
- 47.Ehses JA, Lee SS, Pederson RA, et al. A new pathway for glucose‐dependent insulinotropic polypeptide (GIP) receptor signaling: evidence for the involvement of phospholipase A2 in GIP‐stimulated insulin secretion. J Biol Chem 2001; 276: 23667–23673 [DOI] [PubMed] [Google Scholar]
- 48.MacDonald PE, El‐Kholy W, Riedel MJ, et al. The multiple actions of GLP‐1 on the process of glucose‐stimulated insulin secretion. Diabetes 2002; 51(Suppl 3): S434–S442 [DOI] [PubMed] [Google Scholar]
- 49.Kim S‐J, Choi WS, Han JSM, et al. A novel mechanism for the suppression of a voltage‐gated potassium channel by glucose‐dependent insulinotropic polypeptide: protein kinase A‐dependent endocytosis. J Biol Chem 2005; 280: 28692–28700 [DOI] [PubMed] [Google Scholar]
- 50.MacDonald PE, Salapatek AMF, Wheeler MB. Glucagon‐like peptide‐1 receptor activation antagonizes voltage‐dependent repolarizing K+ currents in beta‐cells: a possible glucose‐dependent insulinotropic mechanism. Diabetes 2002; 51(Suppl 3): S443–S447 [DOI] [PubMed] [Google Scholar]
- 51.Kim S‐J, Widenmaier SB, Choi WS, et al. Pancreatic β‐cell prosurvival effects of the incretin hormones involve post‐translational modification of Kv2.1 delayed rectifier channels. Cell Death Differ 2012; 19: 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacobson DA, Weber CR, Bao S, et al. Modulation of the pancreatic islet β‐cell‐delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem 2007; 282: 7442–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding WG, Gromada J. Protein kinase A‐dependent stimulation of exocytosis in mouse pancreatic beta‐cells by glucose‐dependent insulinotropic polypeptide. Diabetes 1997; 46: 615–621 [DOI] [PubMed] [Google Scholar]
- 54.Eliasson L, Ma X, Renstrom E, et al. SUR1 regulates PKA‐independent cAMP‐induced granule priming in mouse pancreatic B‐cells. J Gen Physiol 2003; 121: 181–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasuda T, Shibasaki T, Minami K, et al. Rim2alpha determines docking and priming states in insulin granule exocytosis. Cell Metab 2010; 12: 117–129 [DOI] [PubMed] [Google Scholar]
- 56.Song W‐J, Seshadri M, Ashraf U, et al. Snapin mediates incretin action and augments glucose‐dependent insulin secretion. Cell Metab 2011; 13: 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Widenmaier SB, Sampaio AV, Underhill TM, et al. Noncanonical activation of Akt/protein kinase B in β‐cells by the incretin hormone glucose‐dependent insulinotropic polypeptide. J Biol Chem 2009; 284: 10764–10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mandrup‐Poulsen T. Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol 2003; 66: 1433–1440 [DOI] [PubMed] [Google Scholar]
- 59.Rhodes CJ. Type 2 diabetes‐a matter of beta‐cell life and death? Science 2005; 307: 380–384 [DOI] [PubMed] [Google Scholar]
- 60.Perl S, Kushner JA, Buchholz BA, et al. Significant human beta‐cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010; 95: E234–E249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cnop M, Igoillo‐Esteve M, Hughes SJ, et al. Longevity of human islet alpha‐ and beta‐cells. Diabetes Obes Metab 2011; 13(Suppl 1): 39–46 [DOI] [PubMed] [Google Scholar]
- 62.Li Y, Hansotia T, Yusta B, et al. Glucagon‐like peptide‐1 receptor signaling modulates beta cell apoptosis. J Biol Chem 2003; 278: 471–478 [DOI] [PubMed] [Google Scholar]
- 63.Wang Q, Brubaker PL. Glucagon‐like peptide‐1 treatment delays the onset of diabetes in 8 week‐old db/db mice. Diabetologia 2002; 45: 1263–1273 [DOI] [PubMed] [Google Scholar]
- 64.Farilla L, Hui H, Bertolotto C, et al. Glucagon‐like peptide‐1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 2002; 143: 4397–4408 [DOI] [PubMed] [Google Scholar]
- 65.Kim S‐J, Winter K, Nian C, et al. Glucose‐dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta‐cell survival is dependent upon phosphatidylinositol 3‐kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down‐regulation of bax expression. J Biol Chem 2005; 280: 22297–22307 [DOI] [PubMed] [Google Scholar]
- 66.Maida A, Hansotia T, Longuet C, et al. Differential importance of glucose‐dependent insulinotropic polypeptide vs glucagon‐like peptide 1 receptor signaling for beta cell survival in mice. Gastroenterology 2009; 137: 2146–2157 [DOI] [PubMed] [Google Scholar]
- 67.Widenmaier SB, Kim S‐J, Yang GK, et al. A GIP receptor agonist exhibits beta‐cell anti‐apoptotic actions in rat models of diabetes resulting in improved beta‐cell function and glycemic control. PLoS One 2010; 5: e9590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Widenmaier SB, Ao Z, Kim S‐J, et al. Suppression of p38 MAPK and JNK via Akt‐mediated inhibition of apoptosis signal‐regulating kinase 1 constitutes a core component of the beta‐cell pro‐survival effects of glucose‐dependent insulinotropic polypeptide. J Biol Chem 2009; 284: 30372–30382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ehses JA, Casilla VR, Doty T, et al. Glucose‐dependent insulinotropic polypeptide promotes beta‐(INS‐1) cell survival via cyclic adenosine monophosphate‐mediated caspase‐3 inhibition and regulation of p38 mitogen‐activated protein kinase. Endocrinology 2003; 144: 4433–4445 [DOI] [PubMed] [Google Scholar]
- 70.Kim S‐J, Nian C, Widenmaier S, et al. Glucose‐dependent insulinotropic polypeptide‐mediated up‐regulation of beta‐cell antiapoptotic Bcl‐2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP‐responsive CREB coactivator 2. Mol Cell Biol 2008; 28: 1644–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hui H, Nourparvar A, Zhao X, et al. Glucagon‐like peptide‐1 inhibits apoptosis of insulin‐secreting cells via a cyclic 5′‐adenosine monophosphate‐dependent protein kinase A‐ and a phosphatidylinositol 3‐kinase‐dependent pathway. Endocrinology 2003; 144: 1444–1455 [DOI] [PubMed] [Google Scholar]
- 72.Jhala US, Canettieri G, Screaton RA, et al. cAMP promotes pancreatic beta‐cell survival via CREB‐mediated induction of IRS2. Genes Dev 2003; 17: 1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van de Velde S, Hogan MF, Montminy M. mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proc Natl Acad Sci USA 2011; 108: 16876–16882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trümper K, Trümper A, Trusheim H, et al. Integrative mitogenic role of protein kinase B/Akt in beta‐cells. Ann N Y Acad Sci 2000; 921: 242–250 [DOI] [PubMed] [Google Scholar]
- 75.Trümper A, Trümper K, Trusheim H, et al. Glucose‐dependent insulinotropic polypeptide is a growth factor for beta (INS‐1) cells by pleiotropic signaling. Mol Endocrinol 2001; 15: 1559–1570 [DOI] [PubMed] [Google Scholar]
- 76.Tobiume T, Saitoh M, Ichijo H. Activation of Apoptosis Signal‐Regulating Kinase 1 by the stress‐induced activating phosphorylation of pre‐formed oligomer. J Cell Physiol 2002; 191: 95–104 [DOI] [PubMed] [Google Scholar]
- 77.Takeda K, Noguchi T, Naguro I, et al. Apoptosis signal‐regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol 2008; 48: 199–225 [DOI] [PubMed] [Google Scholar]
- 78.Kim S‐J, Nian C, McIntosh CHS. Glucose‐dependent insulinotropic polypeptide and glucagon‐like peptide‐1 modulate beta‐cell chromatin structure. J Biol Chem 2009; 284: 12896–12904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cunha DA, Ladriere L, Ortis F, et al. Glucagon‐like peptide‐1 agonists protect pancreatic beta‐cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 2009; 58: 2851–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yusta B, Baggio LL, Estall JL, et al. GLP‐1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 2006; 4: 391–406 [DOI] [PubMed] [Google Scholar]
- 81.Aras M, Aizenman E. Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci Lett 2005; 387: 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lardinois CK, Starich GH, Mazzaferri EL. The postprandial response of gastric inhibitory polypeptide to various dietary fats in man. J Am Coll Nutr 1988; 7: 241–247 [DOI] [PubMed] [Google Scholar]
- 83.Pederson RA. Gastric inhibitory polypeptide In: Walsh JH, Dockray GJ (eds). Gut Peptides: Biochemistry and Physiology. Raven Press, New York, 1994; 217–259 [Google Scholar]
- 84.Wasada T, McCorkle K, Harris V, et al. Effect of gastric inhibitory polypeptide on plasma levels of chylomicron triglycerides in dogs. J Clin Invest 1981; 68: 1106–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ebert R, Nauck M, Creutzfeldt W. Effect of exogenous or endogenous gastric inhibitory polypeptide (GIP) on plasma triglyceride responses in rats. Horm Metab Res 1991; 23: 517–521 [DOI] [PubMed] [Google Scholar]
- 86.Jorde R, Pettersen JE, Burhol PG. Lack of effect of exogenous or endogenous gastric inhibitory polypeptide on the elimination rate of Intralipid in man. Acta Med Scand 1984; 216: 19–23 [DOI] [PubMed] [Google Scholar]
- 87.Miyawaki K, Yamada Y, Ban N, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 2002; 8: 738–742 [DOI] [PubMed] [Google Scholar]
- 88.Irwin N, Flatt PR. Therapeutic potential for GIP receptor agonists and antagonists. Baillieres Best Pract Res Clin Endocrinol Metab 2009; 23: 499–512 [DOI] [PubMed] [Google Scholar]
- 89.Fulurija A, Lutz TA, Sladko K, et al. Vaccination against GIP for the treatment of obesity. PLoS One 2008; 3: e3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Althage MC, Ford EL, Wang S, et al. Targeted ablation of glucose‐dependent insulinotropic polypeptide‐producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J Biol Chem 2008; 283: 18365–18376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oben J, Morgan L, Fletcher J, et al. Effect of the entero‐pancreatic hormones, gastric inhibitory polypeptide and glucagon‐like polypeptide‐1(7‐36) amide, on fatty acid synthesis in explants of rat adipose tissue. J Endocrinol 1991; 130: 267–272 [DOI] [PubMed] [Google Scholar]
- 92.Hauner H, Glatting G, Kaminska D, et al. Effects of gastric inhibitory polypeptide on glucose and lipid metabolism of isolated rat adipocytes. Ann Nutr Metab 1988; 32: 282–288 [DOI] [PubMed] [Google Scholar]
- 93.Beck B, Max JP. Direct metabolic effects of gastric inhibitory polypeptide (GIP): dissociation at physiological levels of effects on insulin‐stimulated fatty acid and glucose incorporation in rat adipose tissue. Diabetologia 1986; 29: 68 [DOI] [PubMed] [Google Scholar]
- 94.Eckel RH, Fujimoto WY, Brunzell JD. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes 1979; 28: 1141–1142 [DOI] [PubMed] [Google Scholar]
- 95.Kim S‐J, Nian C, McIntosh CHS. Activation of lipoprotein lipase by glucose‐dependent insulinotropic polypeptide in adipocytes. A role for a protein kinase B, LKB1, and AMP‐activated protein kinase cascade. J Biol Chem 2007; 282: 8557–8567 [DOI] [PubMed] [Google Scholar]
- 96.Kim S‐J, Nian C, McIntosh CHS. Resistin is a key mediator of glucose‐dependent insulinotropic polypeptide (GIP) stimulation of lipoprotein lipase (LPL) activity in adipocytes. J Biol Chem 2007; 282: 34139–34147 [DOI] [PubMed] [Google Scholar]
- 97.Murphy MC, Isherwood SG, Sethi S, et al. Postprandial lipid and hormone responses to meals of varying fat contents: modulatory role of lipoprotein lipase? Eur J Clin Nutr 1995; 49: 578–588 [PubMed] [Google Scholar]
- 98.Morgan LM. The metabolic role of GIP: physiology and pathology. Biochem Soc Trans 1996; 24: 585–591 [DOI] [PubMed] [Google Scholar]
- 99.McGarry J. Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002; 51: 7–18 [DOI] [PubMed] [Google Scholar]
- 100.McIntosh CHS, Bremsak I, Lynn FC, et al. Glucose‐dependent insulinotropic polypeptide stimulation of lipolysis in differentiated 3T3‐L1 cells: wortmannin‐sensitive inhibition by insulin. Endocrinology 1999; 140: 398–404 [DOI] [PubMed] [Google Scholar]
- 101.Yip RG, Boylan MO, Kieffer TJ, et al. Functional GIP receptors are present on adipocytes. Endocrinology 1998; 139: 4004–4007 [DOI] [PubMed] [Google Scholar]
- 102.Getty‐Kaushik L, Song DH, Boylan MO, et al. Glucose‐dependent insulinotropic polypeptide modulates adipocyte lipolysis and reesterification. Obesity (Silver Spring) 2006; 14: 1124–1131 [DOI] [PubMed] [Google Scholar]
- 103.Song DH, Getty‐Kaushik L, Tseng E, et al. Glucose‐dependent insulinotropic polypeptide enhances adipocyte development and glucose uptake in part through Akt activation. Gastroenterology 2007; 133: 1796–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Asmar M, Simonsen L, Madsbad S, et al. Glucose‐dependent insulinotropic polypeptide may enhance fatty acid re‐esterification in subcutaneous abdominal adipose tissue in lean humans. Diabetes 2010; 59: 2160–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Duan RD, Erlanson‐Albertsson C. Gastric inhibitory polypeptide stimulates pancreatic lipase and colipase synthesis in rats. Am J Physiol 1992; 262: G779–G784 [DOI] [PubMed] [Google Scholar]
- 106.Rosen E, Spiegelman B. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 2000; 16: 145–171 [DOI] [PubMed] [Google Scholar]
- 107.Weaver RE, Donnelly D, Wabitsch M, et al. Functional expression of glucose‐dependent insulinotropic polypeptide receptors is coupled to differentiation in a human adipocyte model. Int J Obes (Lond) 2008; 32: 1705–1711 [DOI] [PubMed] [Google Scholar]
- 108.Kim S‐J, Nian C, McIntosh CHS. Adipocyte expression of the glucose‐dependent insulinotropic polypeptide receptor involves gene regulation by PPARgamma and histone acetylation. J Lipid Res 2011; 52: 759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lynn FC, Pamir N, Ng EH, et al. Defective glucose‐dependent insulinotropic polypeptide receptor expression in diabetic fatty Zucker rats. Diabetes 2001; 50: 1004–1011 [DOI] [PubMed] [Google Scholar]
- 110.Lynn FC, Thompson SA, Pospisilik JA, et al. A novel pathway for regulation of glucose‐dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J 2003; 17: 91–93 [DOI] [PubMed] [Google Scholar]
- 111.Gupta D, Peshavaria M, Monga N, et al. Physiologic and pharmacologic modulation of glucose‐dependent insulinotropic polypeptide (GIP) receptor expression in beta‐cells by peroxisome proliferator‐activated receptor (PPAR)‐gamma signaling: possible mechanism for the GIP resistance in type 2 diabetes. Diabetes 2010; 59: 1445–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu G, Kaneto H, Laybutt DR, et al. Downregulation of GLP‐1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes 2007; 56: 1551–1558 [DOI] [PubMed] [Google Scholar]
- 113.Rudovich N, Kaiser S, Engeli S, et al. GIP receptor mRNA expression in different fat tissue depots in postmenopausal non‐diabetic women. Regul Pept 2007; 142: 138–145 [DOI] [PubMed] [Google Scholar]
- 114.Kim S‐J, Nian C, McIntosh CHS. GIP increases human adipocyte LPL expression through CREB and TORC2‐mediated trans‐activation of the LPL gene. J Lipid Res 2010; 51: 3145–3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jansson D, Ng AC‐H, Fu A, et al. Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc Natl Acad Sci USA 2008; 105: 10161–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fu A, Screaton RA. Using kinomics to delineate signaling pathways: control of CRTC2/TORC2 by the AMPK family. Cell Cycle 2008; 7: 3823–3828 [DOI] [PubMed] [Google Scholar]
- 117.Yang R‐Z, Huang Q, Xu A, et al. Comparative studies of resistin expression and phylogenomics in human and mouse. Biochem Biophys Res Commun 2003; 310: 927–935 [DOI] [PubMed] [Google Scholar]
- 118.McTernan PG, Kusminski CM, Kumar S. Resistin. Curr Opin Lipidol 2006; 17: 170–175 [DOI] [PubMed] [Google Scholar]
- 119.Ort T, Arjona AA, MacDougall JR, et al. Recombinant human FIZZ3/resistin stimulates lipolysis in cultured human adipocytes, mouse adipose explants, and normal mice. Endocrinology 2005; 146: 2200–2209 [DOI] [PubMed] [Google Scholar]
- 120.Sethi J, Vudal‐Puig A. Adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res 2007; 48: 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim J‐Y, van de Wall E, Laplante M, et al. Obesity‐associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 2007; 117: 2621–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Herbach N, Goeke B, Schneider M, et al. Overexpression of a dominant negative GIP receptor in transgenic mice results in disturbed postnatal pancreatic islet and beta‐cell development. Regul Pept 2005; 125: 103–117 [DOI] [PubMed] [Google Scholar]
- 123.Renner S, Fehlings C, Herbach N, et al. Glucose intolerance and reduced proliferation of pancreatic beta‐cells in transgenic pigs with impaired glucose‐dependent insulinotropic polypeptide function. Diabetes 2010; 59: 1228–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]