

Background: Nucleophosmin is a nucleolar protein that interacts with G-quadruplexes.
Results: Site-directed mutagenesis and molecular dynamics unravel the role of single residues in complex formation.
Conclusion: A disordered segment of nucleophosmin contributes to G-quadruplex recognition by facilitating the formation of an encounter complex and transiently interacting with the G-quadruplex.
Significance: Understanding the role played by flanking fuzziness is an important issue in protein/DNA interactions.
Keywords: Intrinsically Disordered Protein, Leukemia, Molecular Dynamics, Protein-DNA Interaction, Surface Plasmon Resonance (SPR), Encounter Complex, Flanking Fuzziness
Abstract
Nucleophosmin (NPM1) is a nucleocytoplasmic shuttling protein, mainly localized at nucleoli, that plays a number of functions in ribosome biogenesis and export, cell cycle control, and response to stress stimuli. NPM1 is the most frequently mutated gene in acute myeloid leukemia; mutations map to the C-terminal domain of the protein and cause its denaturation and aberrant cytoplasmic translocation. NPM1 C-terminal domain binds G-quadruplex regions at ribosomal DNA and at gene promoters, including the well characterized sequence from the nuclease-hypersensitive element III region of the c-MYC promoter. These activities are lost by the leukemic variant. Here we analyze the NPM1/G-quadruplex interaction, focusing on residues belonging to both the NPM1 terminal three-helix bundle and a lysine-rich unstructured tail, which has been shown to be necessary for high affinity recognition. We performed extended site-directed mutagenesis and measured binding rate constants through surface plasmon resonance analysis. These data, supported by molecular dynamics simulations, suggest that the unstructured tail plays a double role in the reaction mechanism. On the one hand, it facilitates the formation of an encounter complex through long range electrostatic interactions; on the other hand, it directly contacts the G-quadruplex scaffold through multiple and transient electrostatic interactions, significantly enlarging the contact surface.
Introduction
Nucleophosmin (also named B23 and NPM13) was initially identified as a phosphoprotein mainly localized at nucleoli but continuously shuttling between the nucleus and cytoplasm (1, 2). NPM1 has been reported to function as a chaperone for both proteins and nucleic acids and to play a role in ribosome biogenesis and export (3–5) as well as to have ribonuclease activity on the preribosomal RNA (6). Additionally, NPM1 has been shown to participate in cell cycle control, centrosome duplication, DNA repair, and responses to a variety of stress stimuli (7, 8). The basis for such promiscuous behavior resides in the modular nature of the protein, which is composed of different functional domains (9, 10). Among these, the C-terminal domain is responsible for nucleic acid binding and is the target of several mutations associated to acute myeloid leukemia (11). Mutations are always heterozygous and alter the C-terminal domain of NPM1, leading to its complete or partial unfolding (12), which causes detachment from nucleoli, and to the acquisition of a novel nuclear export signal. As a consequence, the mutated protein is stably and aberrantly translocated in the cytoplasm of leukemic blasts (9, 11).
Although the C-terminal domain of NPM1 is able to recognize both DNA and RNA in a sequence-independent manner (13, 14), it was recently shown that the affinity may significantly vary depending on the nucleic acid conformation (15). In particular, a higher affinity for oligonucleotides with G-quadruplex structure was envisaged (15). Indeed, NPM1 was shown to bind G-quadruplex regions at ribosomal DNA both in vitro and in vivo. This activity correlates with the nucleolar localization of the protein and is lost by the leukemic variants (16). NPM1 was also shown to interact with G-quadruplex regions at oncogene promoters. Among them, the interaction of NPM1 with a G-quadruplex region present at the nuclease-hypersensitive element III of the c-MYC promoter was analyzed both with the isolated C-terminal domain (15, 17) and in the context of the full-length protein (18). Similar dissociation constants were obtained in the two cases, confirming that the G-quadruplex binding activity is peculiar to the C-terminal domain. This interaction was also structurally analyzed in detail by means of NMR spectroscopy coupled to docking simulations with experimental restraints (19). A region between helices H1 and H2 of the terminal three-helix bundle of the protein was shown to contact the G-quadruplex scaffold, engaging several backbone phosphates in salt bridges and hydrogen bonds with lysine and asparagine residues lining the surface of the domain (see Fig. 1) (19). Intriguingly, it was also shown that a lysine-rich unstructured tail preceding the terminal three-helix bundle, although previously shown to be necessary for high affinity recognition (15), does not contact the G-quadruplex in the final complex (see Fig. 1, inset).
FIGURE 1.

The figure shows the structure of NPM1-C70WT, highlighting the terminal three-helix bundle preceded by a lysine-rich unstructured tail (underlined residues in the alignment) that is necessary for high affinity recognition of G-quadruplex oligonucleotides. The three-helix bundle residues found to contact the pu24I oligo as well as the five lysine residues in the tail are shown in sticks. The inset shows the structure of the C70WT·pu24I complex as obtained by experimentally constrained molecular docking (19). The alignment shows that three tail lysines are completely conserved. When not conserved, the total charge of the tail is nevertheless maintained by the presence of nearby lysines.
Here, we further analyzed the NPM1/G-quadruplex interaction through extensive mutagenesis and binding assays, taking into consideration residues belonging both to the interaction surface and to the unstructured tail. Molecular dynamics simulations of the complex between NPM1 C-terminal domain and a c-MYC promoter-derived oligo were also performed to gain additional information on the role played by the flanking unstructured tail. Collectively, we report that multiple electrostatic and polar interactions cooperate to stabilize the complex with a major contribution of residues belonging to helix H2. Importantly, we suggest that the unstructured tail plays a double role in the interaction: it facilitates the formation of an encounter complex through long range electrostatic interactions but also directly contributes to stabilize the complex by multiple and transient electrostatic interactions with the G-quadruplex scaffold that significantly enlarge the contact surface. The data we provide enhance our understanding of the mechanism of NPM1-DNA recognition. More in general, they help to dissect the function of fuzzy protein stretches that flank the DNA recognition domain, a feature common to several DNA-binding proteins and transcription factors.
EXPERIMENTAL PROCEDURES
Preparation of Protein Samples
The genes coding for the NPM1 C70WT and C53WT constructs (Fig. 1) were cloned in pET28a(+) vectors and expressed in Escherichia coli, and the corresponding protein constructs were purified as reported previously (15). All NPM1 C70WT variant proteins were obtained by site-directed mutagenesis using the QuikChange II Lightning site-directed mutagenesis kit (Stratagene) following manufacturer's instructions. Mutants were expressed and purified using the same protocol adopted for C70WT.
Circular Dichroism
Circular dichroism (CD) experiments were performed using a Jasco J710 instrument (Jasco Inc., Easton, MD) equipped with a Peltier apparatus for temperature control. Static spectra of protein samples were collected at 20 °C in 0.01 m phosphate buffer, pH 7.0, 0.1 m NaCl. Protein concentration was 10 μm. Spectra were collected using a quartz cell with 1-mm optical path length (Hellma, Plainview, NY) and a scanning speed of 100 nm/min. The spectral contribution of buffers was subtracted as appropriate.
Thermal denaturation experiments on protein samples (10 μm) were performed using a quartz cell with 1-mm optical path length and by monitoring the variation of CD signal at 222 nm. Temperature was progressively increased in 1 °C/min steps from 20 to 95 °C. Thermal denaturation data were fitted as already reported (20) using Kaleidagraph software.
SPR Measurements
The pu24I oligo of sequence 5′-TGAGGGTGGIGAGGGTGGGGAAGG-3′, biotinylated at the 5′-end and HPLC-purified, was purchased from Integrated DNA Technology Inc. (Coralville, IA). The lyophilized oligo was dissolved in 20 mm phosphate buffer, pH 7.0, 150 mm KCl. For annealing, the oligo was heated in the same buffer at 95 °C for 15 min and then allowed to gently cool down to room temperature overnight. The interactions between pu24I oligo and the purified proteins were all measured by SPR technique using a Biacore X100 instrument (Biacore AB, Uppsala, Sweden). The annealed pu24I oligo (ligand) was immobilized on a Sensor Chip SA precoated with streptavidin from Biacore AB. The capturing procedure on the biosensor surface was performed according to the manufacturer's instructions, setting the aim for ligand immobilization to 1000 resonance units. Running buffer was HEPES-buffered saline-EP, which contains 0.01 m HEPES, pH 7.4, 0.15 m NaCl, 0.003 m EDTA, 0.005% (v/v) Surfactant P20 (Biacore AB). All proteins (analytes) were dissolved in running buffer, and binding experiments were performed at 25 °C with a flow rate of 30 μl/min. The association phase (kon) was followed for 180 s, and the dissociation phase (koff) was followed for 300 s. The complete dissociation of active complex formed was achieved by addition of 10 mm HEPES, 2 m NaCl, 0.003 m EDTA, 0.005% (v/v) Surfactant P20, pH 7.4 for 60 s before each new cycle start as expected if the interaction formed is mainly electrostatic. Analytes were tested in a wide range of concentrations to reach at least a 2-fold increase from the lower concentration tested. When experimental data met quality criteria, kinetic parameters were estimated according to the simple 1:1 binding model using Biacore X100 Evaluation Software.
MD Simulations
Classical molecular dynamics simulations of the C70WT·Pu24I complex were performed with the GROMACS 4.6 package (21, 22) under the AMBER99 force field using a transferable intermolecular potential 3 point model water solvent. As a starting structure, the lowest energy model obtained by experimentally restrained molecular docking of the C70WT·pu24I complex (19) was chosen for the simulation. The protein·DNA complex structure was solvated in a triclinic water box (with 90-90-64 Å basis vectors) under periodic boundary conditions, resulting in a total number of 53,000 atoms and a charge of −21. This negative charge was neutralized by randomly substituting 21 water molecules with sodium atoms. Following a steepest descent minimization, the systems were then equilibrated in a canonical ensemble conditions for 300 ps, applying a temperature ramp from 70 to 300 K, and subsequently in the isothermal-isobaric ensemble for 300 ps, always applying position restraints to the heavy atoms of the protein·DNA complex. Finally, all restraints were removed, and 100-ns molecular dynamics runs were performed at 300 K. The temperature was maintained close to its reference value by applying the V-rescale (23) thermostat with a coupling constant of 0.1 ps. To maintain constant pressure (1 atm; isotropic coordinate scaling), the Berendsen barostat (24) was used with a relaxation time of 2.0 ps. van der Waals interactions were modeled using 6–12 Lennard-Jones potentials with a 1.4-nm cutoff. Long range electrostatic interactions were calculated using the particle mesh Ewald method with a cutoff for the real space term of 0.9 nm. All covalent bonds were constrained using the linear constraint solver algorithm. The time step used was 2 fs, and the coordinates were saved every 2 ps for analysis, which was performed using the standard GROMACS tools. The simulations of the C70-K229A,K230A,K236A mutant (obtained by “in silico” mutation of the three lysines) in complex with pu24I were performed using the same protocol as for the wild-type protein. Four independent 100-ns simulations were performed for both the wild-type and the mutant proteins. Analysis of the trajectories and of the distribution functions was performed using the standard GROMACS tools.
RESULTS
The c-MYC-derived Oligo Pu24I Is Recognized with Different Affinities by NPM1 C-terminal Constructs Differing for the Presence of an Unstructured Flanking Tail
Previous studies on the NPM1/G-quadruplex association were conducted with two different NPM1 constructs, one encompassing the terminal 70 residues (225–294), hereby named C70WT, and a shorter version encompassing the terminal 53 residues (243–294), hereby named C53WT (Fig. 1). It was shown that C70WT displays a higher affinity than C53WT for any oligo tested irrespective of their structure (15). Following this first observation, the structure of C70WT was determined, and it was shown that the region differentiating the two constructs (residues 225–242) is completely unstructured (19). Interestingly, NMR analysis of C70WT in complex with the pu24I G-quadruplex derived from the nuclease-hypersensitive element III region of the c-MYC promoter revealed that this fuzzy region remains unstructured after binding and apparently does not contribute to the final complex (see Fig. 1, inset).
Previous binding experiments were performed with a c-MYC-derived oligo, namely Pu27 (of sequence 5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′), which is different from pu24I (of sequence 5′-TGAGGGTGGIGAGGGTGGGGAAGG-3′). Stretches of three or more consecutive G are underlined because they contribute to G-quadruplex formation. Although Pu27 gives rise to an ensemble of different G-quadruplex topologies, pu24I was shown to be able to form a single homogenous parallel G-quadruplex assembly (19, 25). Therefore, we started this analysis by assessing the rate constants for the interaction with pu24I of the two NPM1 C-terminal constructs. A 5′-biotinylated version of the pu24I oligo was immobilized on a streptavidin chip and used as a bait in SPR analysis. Fig. 2, A and B, show the sensorgrams obtained when C70WT and C53WT were used as analytes, respectively. C70WT bound the pu24I oligo with 10-fold increased affinity with respect to C53WT (KD = 11 ± 2 μm versus KD = 102 ± 5 μm; see Table 1). This is in agreement with previous observations with the longer c-MYC-derived oligo Pu27 and with an oligo derived from the SOD2 promoter (15). Interestingly with pu24I and contrary to the other tested oligos, both kon and koff rate constants could be derived for the interaction with C53WT, allowing a comparison with C70WT. As shown in Table 1, the 10-fold higher dissociation constant for C53WT was determined by both a decrease of the association rate kon by 4–5-fold and an increase of the dissociation rate constant koff by 2-fold.
FIGURE 2.

Sensorgrams of the interaction between the immobilized biotinylated pu24I oligo and selected NPM1 C-terminal domain constructs used as analytes. Experimental data at increasing analyte concentrations are reported in circles, whereas fits to a 1:1 binding model are reported as black lines. A, C70WT was used as the analyte, and concentrations measured were 125, 62.5, 31.25, 15.625, 7.8125, and 3.9 μm. B, C53WT was used as the analyte, and concentrations measured were 125, 62.5, 31.25, 15.625, 7.8125, and 3.9 μm. C, the single bundle mutant C70-N274A was used as the analyte, and concentrations measured were 125, 62.5, 31.25, 15.625, and 7.8125 μm. D, the triple bundle mutant C70-K250A,K257A,K267A was used as the analyte, and concentrations measured were 125, 62.5, 31.25, 15.625, 7.8125, and 3.9 μm. RU, resonance units.
TABLE 1.
Effect of mutation of three-helix bundle residues on the interaction between NPM1 C-terminal domain and the pu24I G-quadruplex as determined by SPR analysis
| Analyte | kon | koff | KD |
|---|---|---|---|
| m−1 s−1 | s−1 | μm | |
| C70WT | 480 ± 60 | (5.1 ± 0.2) × 10−3 | 11 ± 2 |
| C53WT | 107 ± 3 | (10.9 ± 0.2) × 10−3 | 102 ± 5 |
| C70-K250A | 190 ± 20 | (10.5 ± 0.3) × 10−3 | 54 ± 8 |
| C70-K257A | 160 ± 40 | (9 ± 2) × 10−3 | 60 ± 30 |
| C70-K267A | 188 ± 7 | (13.9 ± 0.2) × 10−3 | 74 ± 3 |
| C70-K267Q | 130 ± 40 | (21 ± 2) × 10−3 | 160 ± 60 |
| C70-N270A | 180 ± 70 | (13.7 ± 0.3) × 10−3 | 75 ± 30 |
| C70-N274A | 360 ± 20 | (38.0 ± 0.7) × 10−3 | 105 ± 8 |
| C70-K250A,K257A | 180 ± 20 | (9.7 ± 0.2) × 10−3 | 54 ± 6 |
| C70-K250A,K267A | 70 ± 20 | (14.1 ± 0.8) × 10−3 | 190 ± 50 |
| C70-K257A,K267A | 116 ± 4 | (15.2 ± 0.1) × 10−3 | 132 ± 6 |
| C70-K250A,K257A,K267A | 46 ± 6 | (14.1 ± 0.2) × 10−3 | 310 ± 50 |
Analysis of C70WT Three-helix Bundle Residues
In Fig. 1, C70WT three-helix bundle residues that contact the pu24I oligonucleotide in the complex are highlighted (19). In particular, residues Lys-250 and Lys-257 belonging to helix H1 and residues Lys-267, Asn-270, and Asn-274 belonging to helix H2 of C70WT were found at the complex interface and interact with phosphate groups of the G-quadruplex scaffold (19). We mutated each of these residues to alanine, producing the single variant proteins C70-K250A, C70-K257A, C70-K267A, C70-N270A, and C70-N274A, and tested their role in the interaction with pu24I by SPR (Table 1).
A reduced affinity for pu24I with respect to C70WT was observed for all mutations. In particular, mutation of lysine residues located in the helix H1 led to KD values of 54 ± 8 and 60 ± 30 μm for C70-K250A and C70-K257A mutants, respectively (Table 1). The effect was more pronounced when mutating residues of helix H2: for the C70-K267A, C70-N270A (Table 1), and C70-N274A (Fig. 2C and Table 1) mutants, we obtained KD values of 74 ± 3, 75 ± 30, and 105 ± 8 μm, respectively.
Among helix H2 residues, Lys-267 was shown previously to be acetylated by p300 (26), and therefore we also designed and produced the acetylation-mimicking mutant C70-K267Q. This mutant displayed a KD of 160 ± 60 μm, further confirming its importance in the interaction (Table 1).
To further analyze the role of these residues in G-quadruplex binding, double and triple alanine mutants were also produced, and their affinity for pu24I was investigated by SPR. As reported in Table 1, KD values are 132 ± 6 and 190 ± 50 μm for the double mutants C70-K257A,K267A and C70-K250A,K267A, respectively. Conversely, the double mutant C70-K250A, K257A in which both residues belong to helix H1 has a KD of 54 ± 6 μm, a value comparable with those of single alanine mutants. These data suggest the presence of a synergistic effect when residues in both helices H1 and H2 are mutated. Finally, the triple mutant C70-K250A,K257A,K267A (Fig. 2D) in which contacts contributed by the helix H2 residue Lys-267 are also disrupted showed a much decreased affinity for the G-quadruplex as suggested by a KD of 310 ± 50 μm (Table 1). Overall, mutational data indicate a greater contribution of helix H2 residues to the binding energy but also suggest that to fully impair complex formation both helices have to be targeted. From a mechanistic point of view, both kon and koff variations were detected to different extents and depending on the number and position of mutations as shown in an isoaffinity plot in Fig. 3A. Here the kon value for any variant protein is reported as a function of its koff, and dashed lines represent points in the plot characterized by the same KD value. According to this representation, a decrease in the y axis value (kon) is associated to a less favorable encounter between the binding partners, whereas an increase in the x axis value (koff) is associated to a lower stability of the complex. Indeed, looking at the clustering of KD values, it appears that single alanine mutants as well as the double mutant with both residues located on the H1 helix reduce both the capacity of the protein to form an encounter complex and its stability once formed (the increase of KD is due to a decrease of the kon value and an increase in the koff one). Conversely, when double mutants occur with residues belonging to both H1 and H2 helices as well as when the triple mutant C70-K250A,K257A,K267A is considered, the additional increase of KD is associated prevalently with a further decrease in kon and therefore with a reduced capacity of the protein to efficaciously form the encounter complex with the oligo (Fig. 3A). Interestingly, the N274A mutant showed an opposite and unique effect with its 10-fold reduced affinity with respect to the wild-type protein (KD = 105 ± 8 μm) mainly due to an increased koff value (Table 1 and Fig. 3A). This suggests that an asparagine in this position is crucial for the stability of the complex. Such result is in good agreement with NMR data of the C70WT·Pu24I complex (19). Indeed, the intermolecular NOE signal due to the hydrogen bond between the amine group of Asn-274 and the sugar backbone of the oligo was the shortest contact distance between those experimentally identified (3.12 Å) (19).
FIGURE 3.
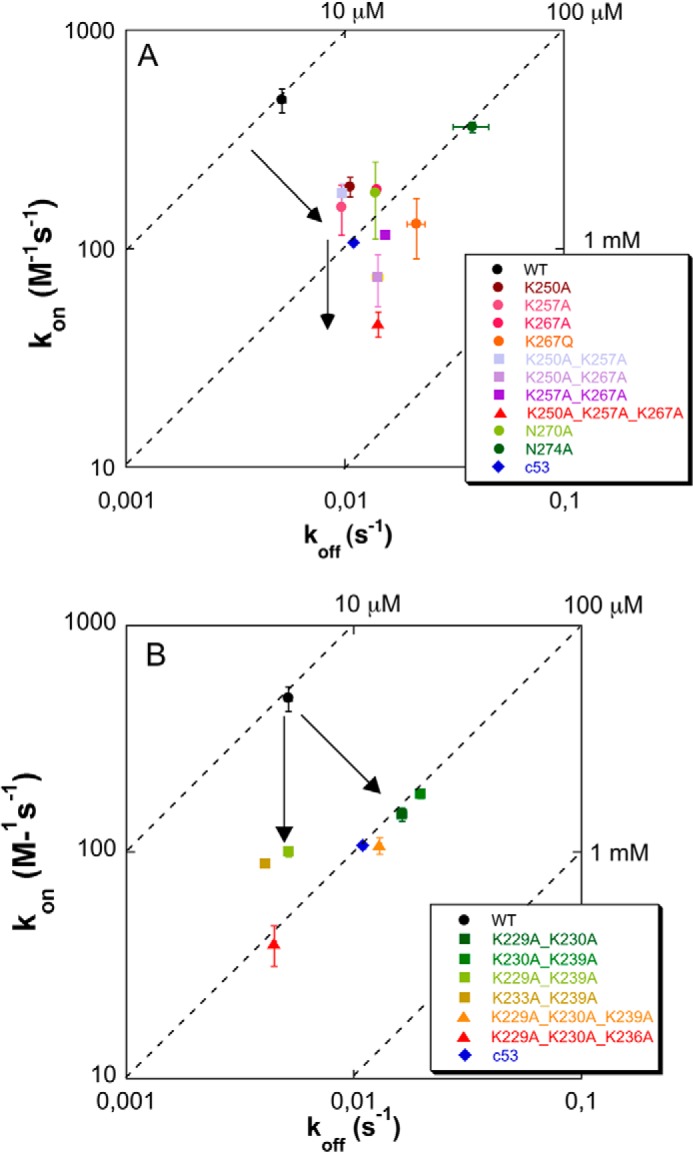
Isoaffinity graphs of the effect of C70 mutations on pu24I binding. For each mutant, the association rate constant kon is reported as a function of the corresponding koff value. Dashed lines represent isoaffinity points at the reported KD values. A, three-helix bundle mutants. Single alanine mutations exert their effect both in terms of reduced kon and increased koff (upper arrow), showing that they contribute to both the formation of the encounter complex and stabilization of the final complex. Residue Asn-274 constitutes a remarkable exception because its alanine mutant has a considerably reduced affinity for the G-quadruplex that is almost entirely due to an ∼10-fold higher koff with respect to WT. The additional effect played by double and triple mutants is exerted primarily in terms of further reduced kon (lower arrow), thus impacting complex formation rather than stabilization. B, tail mutants. For some of the mutants, variations are observed not only for the kon values, as expected in a classical fly casting mechanism, but also in terms of increased koff values, suggesting a direct involvement of the tail in complex stabilization. In both panels, error bars (S.E.) are explicitly shown; when error bars are not visible, it means that they are shorter than the size of the marker.
To demonstrate that the decreased affinities for the pu24I oligonucleotide of variant proteins are not due to reduced protein stability, CD spectroscopy was used as a diagnostic tool. CD spectra typical of α-helical proteins with diagnostic minima at 222 and 208 nm were recorded for all mutants (not shown). Thermal denaturation experiments (Fig. 4A) showed a clear transition from the folded to the unfolded species for all mutants. Melting temperatures (Tm) were never lower than that of the C70WT construct (Table 2). Indeed, in most cases and especially for double and triple mutants, higher Tm values were observed, suggesting that the removal of unpaired surface charges increases protein stability, an effect that may be ascribed to changes in the protein/solvent interactions.
FIGURE 4.

Circular dichroism analysis of C70 mutants. A, thermal denaturation profiles for C70WT and three-helix bundle mutants. Data highlight that thermodynamic stability is not decreased by mutations. Conversely, for double and triple mutants, an increase in stability is observed. The corresponding Tm values are reported in Table 2. B, thermal denaturation profiles for C70WT and tail mutants. Thermodynamic stability is not affected by mutations. The corresponding Tm values are reported in Table 4. In both panels, straight lines represent best fits of experimental data according to Swint and Robertson (20), and the color code is the same as in Fig. 3.
TABLE 2.
Melting temperatures for C70WT and three-helix bundle mutants
| Analyte | Tm |
|---|---|
| °C | |
| C70WT | 64.1 ± 0.1 |
| C70-K250A | 66.8 ± 0.1 |
| C70-K257A | 65.7 ± 0.1 |
| C70-K267A | 70.3 ± 0.2 |
| C70-K267Q | 71.9 ± 0.1 |
| C70-N270A | 69.3 ± 0.1 |
| C70-N274A | 66.7 ± 0.2 |
| C70-K250A,K257A | 67.4 ± 0.1 |
| C70-K250A,K267A | 75.1 ± 0.2 |
| C70-K257A,K267A | 74.3 ± 0.2 |
| C70-K250A,K257A,K267A | 75.7 ± 0.2 |
Analysis of C70WT Tail Residues
As shown in Fig. 2, A and B, and Table 1, C70WT displayed an ∼10-fold higher affinity for pu24I with respect to C53WT, raising the question of which is the exact role played by the 17-residue-long unstructured tail that distinguishes the two constructs (residues 225–242; Fig. 1, underlined). The tail is enriched in basic residues: two consecutive lysines are located at the beginning of the tail (Lys-229 and Lys-230), two are in the middle (Lys-233 and Lys-236), and one is at the end (Lys-239) close to the three-helix bundle (Fig. 1). We had previously mutated each of these lysines into alanine and reported variable but not substantial increases in the KD values for the complexes with the Pu27 and SOD2 oligos (15).
Here we prepared a new series of double and triple lysine to alanine mutants in the tail and tested them against the pu24I oligo. In particular, together with the C70-K229A,K230A mutant that was already available (15), we prepared the double C70-K229A,K239A, C70-K230A,K239A, and C70-K233A,K239A and the triple C70-K229A,K230A,K236A and C70-K229A,K230A,K239A mutants. The effect of mutations on the affinity for Pu24I was investigated by SPR as for the three-helix bundle mutants. In all cases, it was possible to fit the data with a 1:1 kinetic model and to determine rate constants. The sensorgrams for the interactions tested are shown in Fig. 5, and kinetic data and dissociation constants are listed in Table 3.
FIGURE 5.

Sensorgrams of the interaction between the immobilized biotinylated pu24I oligo and C70 tail mutants used as the analytes. Experimental data at increasing analyte concentrations are reported in circles, whereas fits to a 1:1 binding model are reported as black lines. Concentrations used were 125, 62.5, 31.25, 15.625, and 7.8125 μm for all mutants tested. A, C70-K229A,K230A mutant was used as the analyte. B, C70-K230A,K239A mutant was used as the analyte. C, C70-K229A,K239A mutant was used as the analyte. D, C70-K233A,K239A mutant was used as the analyte. E, the triple mutant C70-K229A,K230A,K239A was used as the analyte. F, the triple mutant C70-K229A,K230A,K236A was used as the analyte. RU, resonance units.
TABLE 3.
Effect of mutation of tail residues on the interaction between C70WT and the pu24I G-quadruplex as determined by SPR analysis
| Analyte | kon | koff | KD |
|---|---|---|---|
| m−1 s−1 | s−1 | μm | |
| C70-K229A,K230A | 150 ± 10 | (16.2 ± 0.7) × 10−3 | 110 ± 10 |
| C70-K229A,K239A | 101 ± 5 | (5.14 ± 0.05) × 10−3 | 51 ± 3 |
| C70-K230A,K239A | 182 ± 8 | (19.6 ± 0.3) × 10−3 | 108 ± 6 |
| C70-K233A,K239A | 89 ± 3 | (4.07 ± 0.03) × 10−3 | 46 ± 2 |
| C70-K229A,K230A,K236A | 40 ± 8 | (4.43 ± 0.09) × 10−3 | 110 ± 20 |
| C70-K229A,K230A,K239A | 108 ± 9 | (12.9 ± 0.2) × 10−3 | 120 ± 10 |
Mutants C70-K229A,K239A and C70-K233A,K239A (Fig. 5, C and D) displayed KD values of 51 ± 3 and 46 ± 2 μm, respectively, with a 5-fold increase with respect to C70WT. Interestingly, the double mutation C70-K230A,K239A (Fig. 5B) resulted in a more pronounced effect with a KD of 108 ± 6 μm, which is 10-fold higher than with wild type. Furthermore, the removal of both charges at the beginning of the tail in the C70-K229A,K230A mutant resulted in a KD of 110 ± 10 μm (Fig. 5A). By further deleting charges in the middle or at the end of the tail with mutants C70-K229A,K230A,K236A (Fig. 5F) and C70-K229A,K230A,K239A (Fig. 5E), no additional effect on affinity was detected as witnessed by KD values of 110 ± 20 and 120 ± 20 μm, respectively (Table 3). Overall, the more marked effect on KD values can be envisaged in all mutants with mutated Lys-230 (either double or triple), suggesting a major role for this residue in the C70WT/Pu24I interaction. Mechanistically, the mutations affected both the association rate constants and the dissociation rate constants as shown in Fig. 3B but with a predominant effect of reduced kon values. The effect of mutations on the stability of the various mutants was also assessed by CD analysis. Thermal denaturation profiles reported in Fig. 4B do not show any substantial difference in stability as reflected by the Tm values reported in Table 4.
TABLE 4.
Melting temperatures for C70 tail mutants
| Analyte | Tm |
|---|---|
| °C | |
| C70-K229A,K230A | 65.9 ± 0.2 |
| C70-K229A,K239A | 65.4 ± 0.2 |
| C70-K230A,K239A | 64.7 ± 0.2 |
| C70-K233A,K239A | 64.1 ± 0.1 |
| C70-K229A,K230A,K236A | 66.2 ± 0.1 |
| C70-K229A,K230A,K239A | 65.1 ± 0.1 |
Molecular Dynamics Simulations
To further investigate the role played by the unstructured tail, molecular dynamics simulations of C70WT and of the triple mutant C70-K229A,K230A,K236A in complex with pu24I were undertaken. As a starting structure, the lowest energy model obtained by the experimentally restrained molecular docking of the C70WT·pu24I complex (19) was chosen.
In Fig. 6, the results of four independent MD runs are shown for both the wild type and the mutant. The total number of contacts are reported in Fig. 6, A (wild type) and D (mutant). The contributions of residues 225–242 (tail) are reported in Fig. 6, B (wild type) and E (mutant). The contributions of residues 243–294 (bundle) are reported in Fig. 6, C (wild type) and F (mutant). Moreover, the distributions of the number of contacts, along the entire set of simulation runs, are displayed in Fig. 7, A (total), B (tail), and C (bundle), using a continuous black line for the wild-type and a continuous red line for the mutant protein.
FIGURE 6.

Analysis of the trajectories obtained for C70WT and the triple mutant C70-K229A,K230A,K236A. Four independent MD simulations are shown in both cases. A–C, the number of contacts (distances below 6 Å) between C70WT and the pu24I G-quadruplex is reported as a function of time (A, total number of contacts; B, number of contacts from the tail, i.e. residues 225–242; C, number of contacts from the bundle, i.e. residues 243–294). D–F, the number of contacts formed by the triple mutant with the pu24I G-quadruplex are shown (D, total number of contacts; E, number of contacts from the tail; F, number of contacts from the bundle). The black straight lines in the panels represent the total number of contacts in the initial model (N0_tot; A and D), the number of contacts from the tail in the initial model (N0_tail; B and E), and the number of contacts from the bundle in the initial model (N0_bundle; C and F), respectively.
FIGURE 7.
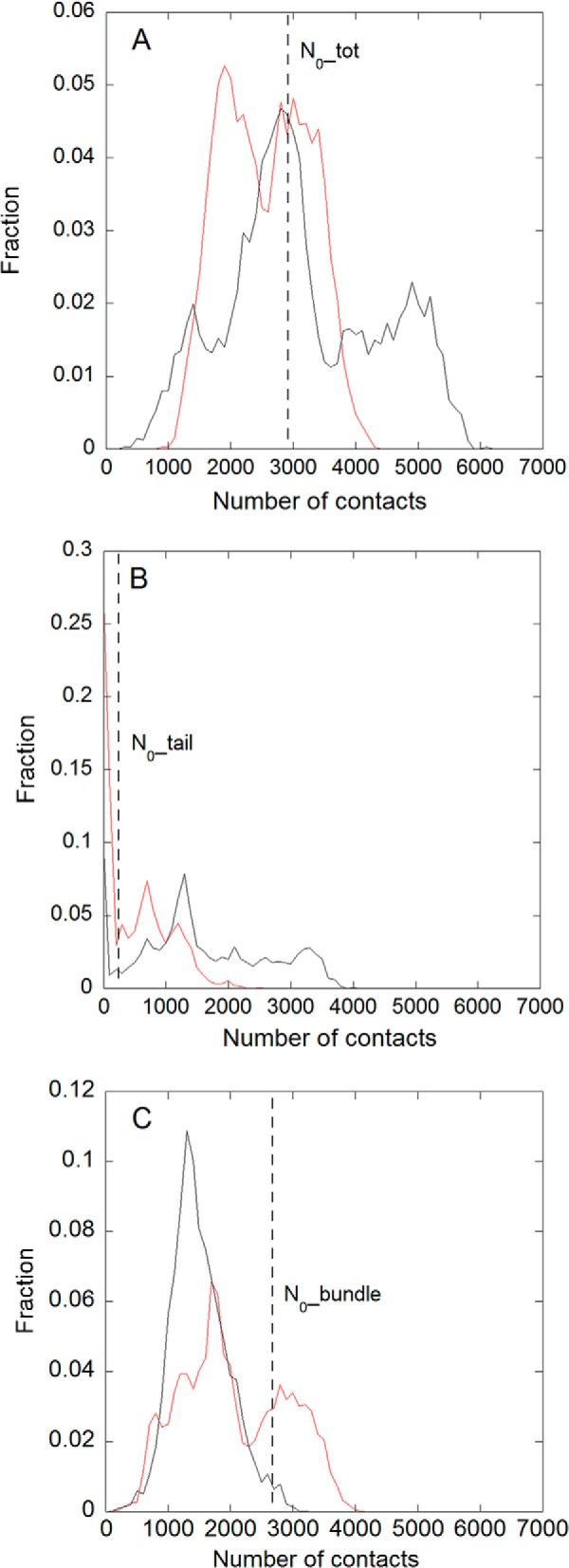
Distributions of the number of contacts in the overall set of MD simulations for the wild type (black line) and the C70-K229A,K230A,K236A mutant (red line). A, total number of contacts. B, number of contacts from the tail, i.e. residues 225–242. C, number of contacts from the bundle, i.e. residues 243–294. The dashed lines in the different panels represent the value of the corresponding number of contacts (A, total, B, tail; C, bundle) in the initial model.
As shown in Fig. 6A, the total number of contacts of the wild-type protein increased significantly in three of four cases explored with respect to the number of contacts of the initial model (19) (namely N0_tot = 2919 contacts; Fig. 6, A and D, straight black line). Conversely in the mutant (Fig. 6D), in all cases but one (red curve), the total number of contacts decreased. These results are summarized in Fig. 7A where the distribution of the number of contacts for the wild type and the mutant are compared. Interestingly, the distribution of the wild type has an average value of 3200 ± 1760, close to N0_tot, with a large standard deviation, being relevantly populated up to 6000, whereas the distribution of contacts of the mutant has an average value of 2050 ± 1212 and is clearly down-shifted with respect to N0_tot.
A detailed analysis of the relative contributions of the tail and the bundle contacts may be further informative. As is evident from Fig. 6, C (wild type) and F (mutant), the number of contacts along the trajectories due to the protein bundle is always lower than the initial value (N0_bundle = 2677 contacts; Fig. 6, C and F, straight black line) with the sole exception of one run in the mutant simulation (Fig. 6F, red line). This fact leads to average values in the distributions (see Fig. 7C) of 1700 ± 1009 for the wild type and 2100 ± 1240 for the mutant. It is reasonable that, along the room temperature MD trajectories performed, this number tends to be lower than that corresponding to the energy minimum of the starting model due to thermal motion. Moreover, in both cases, the main peak of the distributions is below N0_bundle. Although the wild type displays a sharp peak and a small fluctuation around the maximum, suggesting good stability for this protein/DNA interaction, the mutant shows a wider distribution with a consistent fraction above N0_bundle that is mainly due to the already mentioned unique trajectory (Fig. 6F, red line) in which a rearrangement of the protein/DNA interface occurs.
Therefore, a slight increase is observed on average for the total number of contacts of the wild-type protein despite the fact that the number of contacts due to the bundle is decreasing along the trajectories. This effect is due to the multiple transient contributions of the residues of the tail: indeed, the electrostatic forces between the positively charged lysine side chains and the negatively charged phosphate groups of the DNA backbone that occasionally form contacts during the large fluctuations of the tail can persist in the time window explored with a higher probability in the case of the wild-type protein (Fig. 6B) with respect to the mutant (Fig. 6E). There is still one exception even for the wild type (Fig. 6B, green curve) where these interactions are too weak, and the number of contacts remains in the same order as those formed in the MD simulations of the mutant. Looking at the corresponding distributions of Fig. 7B, the wild type shows a wide amplitude up to 4000 contacts that is absent in the mutant. Given the initial value for the number of contacts of the tail (N0_tail = 242), only 11.5% of the distribution is below this value in the wild type along the four simulations compared with a percentage of 43% in the mutant. This last result suggests that the tail in the wild-type protein may exert a flanking role, favoring the stabilization of the complex rather than a specific structured interaction, in agreement with the results obtained by the NMR data (19).
Pictures of two representative trajectories (Fig. 7, A and F, depicted in black) chosen by excluding the previously discussed extreme cases are displayed in Fig. 8, A and B, for the wild-type and the mutant proteins, respectively. Four snapshots are displayed at t = 0, 33, 66, and 100 ns in which the lysine residues of the protein tail (in yellow) and the DNA bases are explicitly shown (in blue). A movie of this trajectory for the wild type is included in the supplemental information (supplemental Movie 1). As shown by the corresponding trajectory in Fig. 6A (black curve), within about 40 ns, the long range electrostatic forces between the positively charged lysine-rich tail of NPM1-C70 and the negatively charged backbone of pu24I G-quadruplex, were able to trigger interactions that form a dynamic contact surface that persisted up to the end (100 ns) of the trajectory. The total interaction surface shown in red in the last snapshot of Fig. 8A corresponds to interatomic distances below 6 Å between the two molecules. Interestingly, in the first part of the simulation when the wider interaction area between the lysine-rich tail and Pu24I is not already formed, the adenine base of pu24I A10 nucleotide intercalates between Lys-239 (on the tail) and Lys-250 (of the H1 helix of the protein bundle), entering the bundle and making van der Waals contacts with non-polar Ile-247 and Phe-276 residues. These interactions persist until the end of the 100-ns trajectory and appear to cooperate with salt bridges established between Lys-239 and the DNA backbone to stabilize the complex.
FIGURE 8.

Ribbon representation of the complex structures along the molecular dynamics simulations. A, simulation of the C70WT·pu24I complex corresponding to the black curve of Fig. 6, A–C (wild type). As a starting structure, the structure reported in Gallo et al. (19) was used. Snapshots of the simulation at 0, 33, 66, and 100 ns (from left to right) are reported. The pu24I G-quadruplex is shown in blue. The C70WT three-helix bundle is shown in cyan, and its unstructured tail is shown in yellow (lysine side chains are in sticks). The contact surface at 100 ns between C70WT and pu24I is shown in red, highlighting the role of the tail in stabilizing the complex. B, simulations of the C70-K229A,K230A,K236A·pu24I complex corresponding to the black curve of Fig. 6, D–F. The C70-K229A,K230A,K236A mutant was obtained by in silico substitution, and its interaction with pu24I was simulated using the same starting structure as in A. Four snapshots at the same time points as in A are shown. The color code is the same as in A. The contact area at 100 ns (in red) is much reduced as compared with C70WT.
In Fig. 8B, four snapshots are displayed from the trajectory selected at the same times previously indicated (t = 0, 33, 66, and 100 ns) for the triple mutant C70-K229A,K230A,K236A, and the corresponding movie is included in the supplemental information (supplemental Movie 2). In this representative trajectory, within the first 40 ns, the still positively charged tail of the triple mutant moves toward the oligo backbone, exploring several different interactions. However, these interactions are not able to produce a stable increase of the interaction surface, and in the second part of the trajectory, the tail moves away from the oligo. The last snapshot of Fig. 8B depicts (in red) the final interacting surface, which is considerably smaller than that obtained with the wild-type protein (Fig. 8A).
DISCUSSION
NPM1 mutations in acute myeloid leukemia cause a profound destabilization of the protein C-terminal domain by altering its folding pathway (12, 27, 28). The mutated protein loses its ability to bind nucleoli (16) and is stably and aberrantly exported in the cytoplasm (10, 11). Moreover, mutations are always heterozygous in acute myeloid leukemia patients, and mutated NPM1 oligomerizes with the wild-type protein, which is also largely translocated in the cytosol. However, a small amount of wild-type NPM1 still resides at nucleoli of leukemic blasts and contributes to maintain nucleolar integrity and ribosome processing and export capabilities (29). These considerations have led to the suggestion that a possible strategy to specifically target NPM1 in acute myeloid leukemia patients with mutations at the NPM1 gene might be that of depriving leukemic blast nucleoli of residual NPM1, thus causing nucleolar stress and the activation of apoptotic cell death (29).
We have recently shown that NPM1 recognizes, through its C-terminal domain, several G-quadruplex regions at nucleolar ribosomal DNA. Nucleic acid binding activity is completely lost by the mutated protein and is necessary for nucleolar localization (16). Indeed, treatment of cells with TmPyP4, a G-quadruplex-selective ligand, completely displaced NPM1 from nucleoli to the nucleoplasm (16). However, G-quadruplex ligands are not specific for the NPM1/G-quadruplex interface; therefore a molecule designed to interfere with this complex should target the surface of NPM1 that interacts with the G-quadruplex rather than the G-quadruplex itself.
Starting from these premises, our first aim here was to define a specific surface in the C70WT three-helix bundle that may be targeted by small molecules. To this end, we mutated each residue found at the interface with the pu24I oligo alone or in combination. Single alanine mutants were all found to be detrimental for affinity with KD values increasing by 5–10-fold with respect to the wild-type domain. However, none of these residues may be considered as a “hot spot” for the interaction; i.e. none of the mutations resulted in a complete loss of affinity. Overall, a greater contribution of helix H2 residues was detected, and among them, residue Asn-274 is of particular interest. In fact, its mutation to alanine led to a 10-fold increase in KD that, contrary to mutation of lysine residues and of Asn-270, is almost entirely due to a marked increase of the dissociation rate constant (see Fig. 3A). This suggests that the interaction involving Asn-274 plays a pivotal role in stabilizing the complex once it is formed.
Interestingly, the effect on the KD of mutation of Lys-267 was 3-fold higher when the lysine was mutated to glutamine rather than alanine. This mutation was designed to mimic the acetylation of this residue by p300, whereas SIRT1 promotes its deacetylation (26). It was shown that acetylation of Lys-267 together with that of Lys-257 and the tail residues Lys-229 and Lys-230 causes dissociation of NPM1 from nucleoli (26). The higher effect measured for the K267Q mutation strongly suggests that acetylation modulates NPM1 cellular localization by interfering with its nucleic acid binding activity.
Importantly, when both lysines of helix H1 were mutated in the double mutant C70-K250A,K257A, the affinity for pu24I was comparable with that observed for the single mutants C70-K250A and C70-K257A. This suggests that the disruption of all contacts of helix H1 is not sufficient to severely impair complex formation. Conversely, the effect of double mutations was markedly higher with respect to single mutations for the mutants C70-K257A,K267A and C70-K250A,K267A with KD values increasing by 19- and 13-fold, respectively. Therefore only the simultaneous disruption of contacts on both helices H1 and H2 has an additive effect on the binding energy. In line with this suggestion, the triple mutant C70-K250A,K257A,K267A (in which contacts of both helix H1 lysines and the interactions of Lys-267 on helix H2 are affected) almost completely lost its affinity for the pu24I G-quadruplex (with a 31-fold increase of KD).
Overall, these data suggest that if we aim at interfering with the NPM1/G-quadruplex association we should design drugs capable of disrupting contacts made by both helices H1 and above all H2 of the three-helix bundle. Interestingly, such a drug may already have been described. In fact, it was shown that the antitumoral alkaloid (+)-avrainvillamide targets the NPM1 C-terminal domain by selectively alkylating Cys-275 (30), a residue that is located at the end of helix H2. Interestingly, Cys-275 is positioned at the interface of the C70WT·pu24I complex (19) and adjacent to Asn-274, which we have shown to be crucial for maintaining complex stability. We hypothesize that (+)-avrainvillamide may be bulky enough to impede the NPM1/G-quadruplex association, and therefore it will be very important in the future to test its effect on NPM1 localization and its cytotoxicity in relevant cell lines carrying NPM1 mutations.
In the second part of the study, we investigated an interesting issue that emerged from the structural and functional analysis of NPM1/G-quadruplex association. In fact, NMR data showed that a 17-residue unstructured segment (residues 225–242) that precedes the three-helix bundle does not participate in the final C70WT·pu24I complex, remaining unstructured after binding (19). However, SPR experiments on the C70WT interaction with different DNA oligos in comparison with the shorter NPM1 construct comprising the three-helix bundle alone, C53WT (residues 243–294), showed that this tail is always necessary to confer high affinity recognition (15). We confirmed this notion here by showing that C53WT has a 10-fold decreased affinity for pu24I with respect to C70WT.
The presence of flanking unstructured regions that do not bind the partner but contribute to overall affinity has been observed previously in other protein/protein and protein/DNA interactions and is termed “flanking fuzziness” (31, 32). It has been suggested that these regions might confer higher thermodynamic stability, thus indirectly contributing to binding energy (33), or provide a larger capture radius for long range electrostatic interactions that may facilitate the formation of an “encounter complex” or an ensemble of encounter complexes that later evolve into the final complex (34, 35). Indeed, it was shown previously that the C70WT construct is considerably more stable than the C53WT construct (33, 36). Furthermore, the unstructured tail is markedly positively charged with five lysine residues distributed along its length (see Fig. 1). Importantly, as shown in Fig. 1, three of the five lysines are perfectly conserved, and the remaining two are flanked by additional lysines so that the total charge of the tail is always conserved and markedly positive.
To analyze the role played by positive charges in the tail, we prepared a number of double and triple mutants and tested them against the pu24I oligo. Importantly, with this oligo, it was also possible for the first time to obtain kinetic values with the C53WT construct as the analyte (see Fig. 1B) showing that the ∼10-fold higher KD with respect to C70WT is mainly due to a pronounced decrease of the kon rate by ∼5-fold, whereas the koff increases by 2-fold (see Table 1).
Residues Lys-229, Lys-230, Lys-233, Lys-236, and Lys-239 were mutated in several combinations. Mutants C70-K229A,K239A and C70-K233A,K239A had KD values only ∼5-fold higher than that for C70WT. Mutants C70-K229A,K230A and C70-K230A,K239A had KD values ∼11-fold higher than that of wild type, fully reproducing the loss of affinity generated by the absence of the tail in the C53WT construct. The addition of one more mutation in the triple mutants C70-K229A,K230A,K239A and C70-K229A,K230A,K236A did not further contribute to the loss of affinity. Finally, thermal stability of all mutants was tested, and interestingly, whereas the Tm value obtained for C53WT is 5 °C lower than that of C70WT (33), the Tm values for all double and triple mutants were close to that of C70WT (see Table 4). This suggests that the additional stability provided by the tail does not really contribute to the complex binding energy.
Overall, our data suggest that, to mimic the absence of the tail as in C53WT, more than one positive charge has to be removed. Furthermore, the contribution of positive charges located at the beginning of the tail and in particular of Lys-230 appears higher than the contribution of lysine residues located closer to the three-helix bundle.
This conclusion is compatible with the previous mentioned hypothesis that the tail might facilitate the formation of an encounter complex through long range electrostatic interactions with the negatively charged G-quadruplex in a “fly casting” fashion. Furthermore, if this is the case, we might expect that the effect of lysine to alanine mutations should be primarily exerted in terms of a decreased association rate constant (kon) with respect to C70WT-Pu24I. Indeed, we observed kon values ranging from 3- to 10-fold lower than that of C70WT according to the different mutants (see Table 3). However, in a classical fly casting mechanism, the koff values should not be affected by mutations. Here we showed that, although with C53WT the koff value is 2-fold higher than that of wild type, with some of the mutants, in particular the K230A,K239A and the K229A,K230A,K239A mutants, koff values are 3–4-fold higher than with C70WT. The increase in koff values for some mutants suggests that the tail, most prominently its distal part, may directly contact the G-quadruplex and contribute to complex stabilization. This idea is corroborated by molecular dynamics simulations performed on the C70WT-pu24I complex. The positively charged tail is suggested to favor the formation of an additional interaction surface with pu24I that is dynamically flexible and, when present, persists in a time window of 100 ns. Conversely, simulations performed on the C70-K229A,K230A,K239A·pu24I complex suggest that the triple mutant is unable to form such a supporting surface within the same time window. It is worth mentioning that the tail residues did not produce a measurable chemical shift variation upon titration with pu24I in NMR spectra (19) possibly because their contribution is not well defined in the NMR time scale. Consistently, the additional surface highlighted by several repetitions of MD simulations appears transient and variable with lysine residues establishing different contacts with the oligo phosphates along the simulation time window.
In conclusion, the data we present here contribute to our understanding of the NPM1/G-quadruplex interaction. They help to define a specific surface at the interface of helices H1 and H2 in the three-helix bundle that may be targeted for leukemia treatment. Furthermore, they contribute to the understanding of the role played by flanking unstructured regions in recognition, an aspect that is relevant for a large number of protein·DNA and protein·protein complexes.
Supplementary Material
Acknowledgment
We thank Prof. Maurizio Brunori for useful advice in developing nucleophosmin research.
This work was supported by Associazione Italiana Ricerca sul Cancro Grant AIRC IG2011-11712 (to L. F.). and the Italian Ministry of University and Research (Linea D.1, 2012 Università Cattolica Sacro Cuore, Rome) (to A. A.).

This article contains supplemental Movies 1 and 2.
- NPM1
- nucleophosmin
- SPR
- surface plasmon resonance
- MD
- molecular dynamics.
REFERENCES
- 1. Kang Y. J., Olson M. O., Jones C., Busch H. (1975) Nucleolar phosphoproteins of normal rat liver and Novikoff hepatoma ascites cells. Cancer Res. 35, 1470–1475 [PubMed] [Google Scholar]
- 2. Borer R. A., Lehner C. F., Eppenberger H. M., Nigg E. A. (1989) Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56, 379–390 [DOI] [PubMed] [Google Scholar]
- 3. Okuwaki M., Matsumoto K., Tsujimoto M., Nagata K. (2001) Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 506, 272–276 [DOI] [PubMed] [Google Scholar]
- 4. Murano K., Okuwaki M., Hisaoka M., Nagata K. (2008) Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol. Cell. Biol. 28, 3114–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maggi L. B., Jr., Kuchenruether M., Dadey D. Y., Schwope R. M., Grisendi S., Townsend R. R., Pandolfi P. P., Weber J. D. (2008) Nucleophosmin serves as a rate-limiting nuclear export chaperone for the mammalian ribosome. Mol. Cell. Biol. 28, 7050–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Savkur R. S., Olson M. O. (1998) Preferential cleavage in pre-ribosomal RNA by protein B23 endoribonuclease. Nucleic Acids Res. 26, 4508–4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang W., Budhu A., Forgues M., Wang X. W. (2005) Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat. Cell Biol. 7, 823–830 [DOI] [PubMed] [Google Scholar]
- 8. Colombo E., Alcalay M., Pelicci P. G. (2011) Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene 30, 2595–2609 [DOI] [PubMed] [Google Scholar]
- 9. Grisendi S., Mecucci C., Falini B., Pandolfi P. P. (2006) Nucleophosmin and cancer. Nat. Rev. Cancer 6, 493–505 [DOI] [PubMed] [Google Scholar]
- 10. Federici L., Falini B. (2013) Nucleophosmin mutations in acute myeloid leukemia: a tale of protein unfolding and mislocalization. Protein Sci. 22, 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Falini B., Mecucci C., Tiacci E., Alcalay M., Rosati R., Pasqualucci L., La Starza R., Diverio D., Colombo E., Santucci A., Bigerna B., Pacini R., Pucciarini A., Liso A., Vignetti M., Fazi P., Meani N., Pettirossi V., Saglio G., Mandelli F., Lo-Coco F., Pelicci P. G., Martelli M. F. for the GIMEMA Acute Leukemia Working Party (2005) Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352, 254–266 [DOI] [PubMed] [Google Scholar]
- 12. Grummitt C. G., Townsley F. M., Johnson C. M., Warren A. J., Bycroft M. (2008) Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J. Biol. Chem. 283, 23326–23332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hingorani K., Szebeni A., Olson M. O. (2000) Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 275, 24451–24457 [DOI] [PubMed] [Google Scholar]
- 14. Hisaoka M., Nagata K., Okuwaki M. (2014) Intrinsically disordered regions of nucleophosmin/B23 regulate its RNA binding activity through their inter- and intra-molecular association. Nucleic Acids Res. 42, 1180–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Federici L., Arcovito A., Scaglione G. L., Scaloni F., Lo Sterzo C., Di Matteo A., Falini B., Giardina B., Brunori M. (2010) Nucleophosmin C-terminal leukemia-associated domain interacts with G-rich quadruplex forming DNA. J. Biol. Chem. 285, 37138–37149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chiarella S., De Cola A., Scaglione G. L., Carletti E., Graziano V., Barcaroli D., Lo Sterzo C., Di Matteo A., Di Ilio C., Falini B., Arcovito A., De Laurenzi V., Federici L. (2013) Nucleophosmin mutations alter its nucleolar localization by impairing G-quadruplex binding at ribosomal DNA. Nucleic Acids Res. 41, 3228–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scognamiglio P. L., Di Natale C., Leone M., Poletto M., Vitagliano L., Tell G., Marasco D. (2014) G-quadruplex DNA recognition by nucleophosmin: new insights into protein dissection. Biochim. Biophys. Acta 1840, 2050–2059 [DOI] [PubMed] [Google Scholar]
- 18. Bañuelos S., Lectez B., Taneva S. G., Ormaza G., Alonso-Mariño M., Calle X., Urbaneja M. A. (2013) Recognition of intermolecular G-quadruplexes by full length nucleophosmin. Effect of a leukaemia-associated mutation. FEBS Lett. 587, 2254–2259 [DOI] [PubMed] [Google Scholar]
- 19. Gallo A., Lo Sterzo C., Mori M., Di Matteo A., Bertini I., Banci L., Brunori M., Federici L. (2012) Structure of nucleophosmin DNA-binding domain and analysis of its complex with a G-quadruplex sequence from the c-MYC promoter. J. Biol. Chem. 287, 26539–26548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swint L., Robertson A. D. (1993) Thermodynamics of unfolding for turkey ovo-mucoid third domain: thermal and chemical denaturation. Protein Sci. 2, 2037–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berendsen H. J., van der Spoel D., van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 22. Hess B., Kutzner C., Lindhal E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 23. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 24. Berendsen H. J., Postma J. P., van Gunsteren W. F., Di Nola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 25. Phan A. T., Kuryavyi V., Gaw H. Y., Patel D. J. (2005) Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 1, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shandilya J., Swaminathan V., Gadad S. S., Choudhari R., Kodaganur G. S., Kundu T. K. (2009) Acetylated NPM1 localizes in the nucleoplasm and regulates transcriptional activation of genes implicated in oral cancer manifestation. Mol. Cell. Biol. 29, 5115–5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scaloni F., Gianni S., Federici L., Falini B., Brunori M. (2009) Folding mechanism of the C-terminal domain of nucleophosmin: residual structure in the denatured state and its pathophysiological significance. FASEB J. 23, 2360–2365 [DOI] [PubMed] [Google Scholar]
- 28. Scaloni F., Federici L., Brunori M., Gianni S. (2010) Deciphering the folding transition state structure and denatured state properties of nucleophosmin C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 107, 5447–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Falini B., Gionfriddo I., Cecchetti F., Ballanti S., Pettirossi V., Martelli M. P. (2011) Acute myeloid leukemia with mutated nucleophosmin (NPM1): any hope for a targeted therapy? Blood Rev. 25, 247–254 [DOI] [PubMed] [Google Scholar]
- 30. Wulff J. E., Siegrist R., Myers A. G. (2007) The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J. Am. Chem. Soc. 129, 14444–14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tompa P., Fuxreiter M. (2008) Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8 [DOI] [PubMed] [Google Scholar]
- 32. Guo X., Bulyk M. L., Hartemink A. J. (2012) Intrinsic disorder within and flanking the DNA-binding domains of human transcription factors. Pac. Symp. Biocomput. 2012, 104–115 [PMC free article] [PubMed] [Google Scholar]
- 33. Marasco D., Ruggiero A., Vascotto C., Poletto M., Scognamiglio P. L., Tell G., Vitagliano L. (2013) Role of mutual interactions in the chemical and thermal stability of nucleophosmin NPM1 domains. Biochem. Biophys. Res. Commun. 430, 523–528 [DOI] [PubMed] [Google Scholar]
- 34. Ubbink M. (2009) The courtship of proteins: understanding the encounter complex. FEBS Lett. 583, 1060–1066 [DOI] [PubMed] [Google Scholar]
- 35. Schilder J., Ubbink M. (2013) Formation of transient protein complexes. Curr. Opin. Struct. Biol. 23, 911–918 [DOI] [PubMed] [Google Scholar]
- 36. Chiarella S., Federici L., Di Matteo A., Brunori M., Gianni S. (2013) The folding pathway of a functionally competent C-terminal domain of nucleophosmin: protein stability and denatured state residual structure. Biochem. Biophys. Res. Commun. 435, 64–68 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.