

Abstract
Crosstalk between estrogen receptor (ER) and the inflammatory nuclear factor κB (NFκB) pathway in ER+ breast cancers may contribute to a more aggressive phenotype. PHLDA1 (Pleckstrin Homology-Like Domain, Family A, member 1), one target gene of ER-NFκB crosstalk, has been implicated in cell survival and stem cell properties. 17β-estradiol (E2), acting through ERα, and pro-inflammatory cytokines, acting through NFκB, increase the nascent transcript and PHLDA1 mRNA stability, indicating both transcriptional and post-transcriptional control of PHLDA1 expression. We show that PHLDA1 is a direct target of miR-181 and that mature miR-181a and b, as well as their host gene, are synergistically down-regulated by E2 and TNFα, also in an ER and NFκB-dependent manner. Thus, ER and NFκB work together to up-regulate PHLDA1 directly through enhanced transcription and indirectly through repression of miR-181a and b. Previous studies have suggested that PHLDA1 may be a stem cell marker in the human intestine that contributes to tumorigenesis. Our findings that PHLDA1 is up-regulated in mammospheres (MS) of ER+ breast cancer cells and that PHLDA1 knockdown impairs both MS formation and the expansion of aldehyde dehydrogenase (ALDH)-positive population, suggest that PHLDA1 may play a similar role in breast cancer cells. Up-regulation of PHLDA1 in MS is largely dependent on the NFκB pathway, with down-regulated miR-181 expression a contributing factor. Over-expression of miR-181 phenocopied PHLDA1 knockdown and significantly impaired MS formation, which was reversed, in part, by protection of the PHLDA1 3′UTR or overexpression of PHLDA1 lacking the 3′UTR. Furthermore, we find that elevated PHLDA1 expression is associated with a higher risk of distant metastasis in ER+ breast cancer patients. Altogether, these data suggest that high PHLDA1 expression is controlled through an ER-NFκB-miR-181 regulatory axis and may contribute to a poor clinical outcome in patients with ER+ breast tumors by enhancing stem-like properties in these tumors.
Keywords: breast cancer, estrogen receptor, nuclear factor κB, microRNA-181, cancer stem cells, mammosphere
Introduction
Nearly 75% of breast tumors express estrogen receptor α (ER) and will be treated with endocrine therapy, such as tamoxifen or aromatase inhibitors. Yet, about 50% of these tumors fail to respond and eventually recur as aggressive, metastatic cancers. Activation of nuclear factor κB (NFκB) is thought to be a potential driver of an aggressive phenotype, since it promotes tumor growth, cell survival, adhesion/migration/invasion, angiogenesis, and drug resistance. Indeed ER+ tumors with a high risk of recurrence have constitutive activation of the NFκB pathway1 and constitutive activation of NFκB in ER+ tumors is associated with endocrine and chemotherapy resistance2, 3. Furthermore, inhibition of NFκB signaling has been shown to restore sensitivity to endocrine therapy in several preclinical models of resistance4, 5. The underlying mechanisms on how NFκB activation in ER+ breast cancer influences poor outcome are not fully understood, but recent evidence has suggested that crosstalk between ER and NFκB may be a contributing factor. Until recently, the paradigm in the field was that ER and NFκB repress each other’s transcriptional activity, however, we have shown that these two factors can work together to up-regulate a gene signature associated with luminal B tumors and poor response to tamoxifen6. This is clinically significant because luminal B tumors tend to be aggressive, resistant to therapy, recur earlier, and are associated with an overall poor patient outcome.
One major feature of the gene signature regulated by ER-NFκB crosstalk is cell survival, as illustrated by BIRC3 (baculoviral IAP repeat containing 3) and PHLDA1 (Pleckstrin Homology-Like Domain, Family A, member 1). In previous studies, we established that BIRC3 is essential for estrogen-dependent breast cancer cell survival7. However, the function of PHLDA1 in ER+ breast cancer is less clear. PHLDA1 was described as a pro-survival factor by playing an important role in the anti-apoptotic effects of IGF-1 in breast cancer cells8. More recently, PHLDA1 has been described as an epithelial stem cell marker in the human small and large intestine9 that contributes to tumorigenesis. In breast cancer, factors and pathways that drive survival, maintenance, and propagation of stem-like cells, which are often termed cancer stem cells (CSCs), are biologically and therapeutically important, given that CSCs are thought to be responsible for therapy resistance and tumor recurrence. Interestingly, Luminal B tumors show the most overlap with CSC markers compared to other ER+ tumors10, 11. Therefore, PHLDA1’s position at the intersection of pro-survival signaling and stem-like cell properties prompted us to explore its regulation by ER-NFκB crosstalk and its function in ER+ breast cancer in greater detail.
Results
Transcriptional and post-transcriptional regulation of PHLDA1 by ER and NFκB
To understand regulation of PHLDA1 by ER and the NFκB pathway, ER+ MCF-7 breast cancer cells were treated with 17β-estradiol (E2) in combination with pro-inflammatory cytokine tumor necrosis factor α (TNFα) to induce activation of the NFκB pathway. As previous data suggested6, the combination of E2 and TNFα results in the up-regulation of both PHLDA1 mRNA and protein (Figure 1a, 1b). Similarly, E2 combined with interleukin-1β (IL-1β), another pro-inflammatory cytokine that activates NFκB pathway, results in PHLDA1 up-regulation, while E2 in combination with IL-6, which cannot activate NFκB, has no effect on PHLDA1 expression (Supplemental Figure 1). The effect of E2+TNFα is mediated by both ER and the NFκB pathway since silencing ER via siRNA, blocking ER activity through the use of the antagonist ICI182,7808 (ICI), or inhibiting the NFκB pathway with a small molecule pan-IKK inhibitor (IKK7), which targets both IKKα and IKKβ, attenuated E2+TNFα action on both PHLDA1 mRNA (Figure 1a) and protein (Figure 1b).
Figure 1.

PHLDA1 is regulated by E2 and TNFα at both transcriptional and post-transcriptional level. (a) PHLDA1 mRNA expression was measured by QPCR in RNA from MCF-7 cells treated for 2 hrs with vehicle control (Veh) or the combination of E2 (10 nM) and TNFα (10 ng/mL). siERα (50 nM) was transfected 48 hrs prior to E2 and TNFα treatment or IKK7 (1 μM) was added 1 hr prior to treatment. ***P<0.001 for siERα and IKK7 groups vs. control E2+TNFα group. (b) PHLDA1 protein was measured by Western Blot in MCF-7 cells treated for 16 hrs with Veh, E2, TNFα or the combination of E2 and TNFα. The inhibitors ICI or IKK7 (1 μM each) were added 1 hr prior to treatment. β-actin served as a loading control. Densitometry was performed and the numbers above each PHlDA1 band indicate protein expression relative to both β-actin and control group. (c) The level of nascent PHLDA1 transcripts was measured in MCF-7 cells treated for 2 hrs with Veh or E2 and TNFα. ICI or IKK7 was added 1 hr prior to treatment. ***P<0.001 for ICI and IKK7 vs. none in the presence of E2+TNFα. (d) PHLDA1 mRNA stability was measured in MCF-7 cells in the presence of Act D (1 μg/mL) added alone or in combination with E2 and TNFα treatment for up to 2 hrs.
The role of the NFκB pathway was explored further in an alternative, cytokine-independent system that utilizes doxycycline (Dox)-inducible constitutively active IKKβ (CAIKKβ). Expression and activity of Dox-induced CA-IKKβ is shown in Supplemental Figure 2a and 2b, respectively. We find that combination of E2 and Dox-induced CA-IKKβ is sufficient to drive PHLDA1 expression to a greater extent than either E2 or Dox alone (Supplemental Figure 2c), suggesting that interaction between ER and the canonical NFκB pathway controls PHLDA1 expression in ER+ breast cancer cells.
Primers designed to detect nascent, unprocessed primary transcripts of PHLDA1 show that E2 and TNFα up-regulate PHLDA1 transcription (Figure 1c), also in an ER and NFκB dependent manner. However, we also find that the combination of E2 and TNFα increased the stability of PHLDA1 mRNA by extending its half-life from an estimated 1 hr to more than 2 hrs following Actinomycin D (Act D) treatment (Figure 1d). This suggests that E2 and TNFα regulate PHLDA1 expression through both transcriptional and post-transcriptional mechanisms.
Since microRNAs (miRs) can control mRNA stability, a bioinformatic search for putative miR-target gene pairs (TargetScan.org) was conducted. PHLDA1 was predicted to be a putative target of miR-181 family (Supplemental Figure 3a) because there is an exact match between the PHLDA1 3′UTR positions 357-364 and positions 2-8 of the mature miR-181. To determine whether miR-181 can target PHLDA1, miR-181a and b synthetic mimics were transfected into MCF-7 cells. Following E2 and TNFα treatment, we find that both mimics, either alone or in combination, attenuated the expression of both PHLDA1 mRNA (Figure 2a) and protein (Figure 2b). This effect was confirmed in another ER+ cell line T47D (Supplemental Figure 3b). To demonstrate that miR-181a and b specifically target the miR-181 site in PHLDA1’s 3′UTR, target protector technology was utilized12. Target protectors are single-stranded, modified RNAs that specifically interfere with the interaction of the miR with the 3′UTR of a single target, while leaving the regulation of other targets of the same miR unaffected. Numerous recent publications have successfully utilized protector technology to demonstrate the miR-target gene pair relationship13-16. As shown in Figure 2c, the miR target protector designed to prevent miR-181 binding to PHLDA1’s 3′UTR region reversed the inhibition of miR-181a+b mimics on PHLDA1 expression in a dose-dependent manner. This protector is specific for PHLDA1, because on another miR-181 target gene, Bcl-217, the PHLDA1-miR-181 protector has no effect (Supplemental Figure 3c). Together these results indicate that PHLDA1 is a direct target of miR-181a and b family members.
Figure 2.
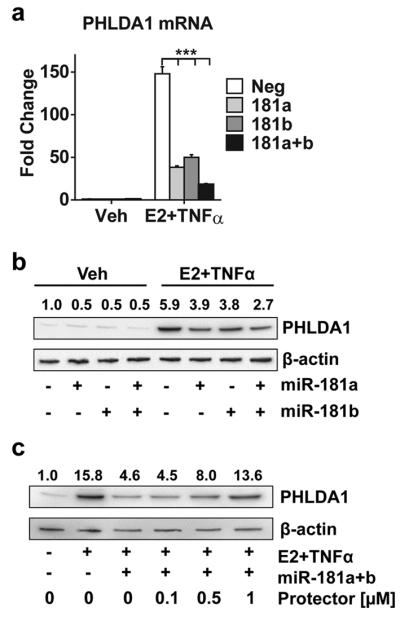
PHLDA1 is a direct target of miR-181a and b family members. (a) PHLDA1 mRNA expression was measured in MCF-7 cells transfected with siNeg or mimics for miR-181a, miR-181b, or both (20 nM each) followed by E2 and TNFα treatment for 2 hrs. ***P<0.001 for each of the miR-181 mimic groups compared to siNeg control. (b) PHLDA1 protein was measured in MCF-7 cells transfected as in (a), followed by E2 and TNFα treatment for 16 hrs. (c) PHLDA1 protein was measured in cells transfected with different concentrations of PHLDA1-miR-181 target protector (0-1 μM) together with miR-181a and b mimics, followed by E2 and TNFα treatment for 16 hrs.
E2 and TNFα repression of miR-181a and b contributes to PHLDA1 up-regulation
miR-181a and b are arranged in a bicistronic fashion as part of a non-protein coding RNA that was recently annotated as miR-181A1 host gene (HG) in chromosome 1. A previous report in the literature suggested that E2 down-regulates miR-181a and b expression18; however, regulation of the HG has not been explored. Our studies indicate that E2 treatment of MCF-7 cells reduces the expression of miR-181A1 HG, as well as mature miR-181a and b (Figure 3). Use of ICI (Figure 3b) or siERα (Supplemental Figure 4) to probe the role of ER in miR-181A1 HG expression, suggests that ER is not only required for E2-dependent down-regulation but that unliganded ER may be exerting a baseline repression of the gene.
Figure 3.

E2 and TNFα down-regulate miR-181A1 HG and mature miR-181a and b in an ER and NFκB dependent manner. (a) miR-181 HG mRNA was measured in MCF-7 cells treated with E2, TNFα, or the combination for 2 hrs. All treatment groups were significantly different than vehicle (Veh) control P<0.001. Treatment with E2+TNFα was significantly different from E2 or TNFα alone, *P<0.05, ***P<0.001. (b) ICI or (c) IKK7 was added 1 hr prior to E2 and TNFα treatment. *P<0.05, ***P<0.001, ns; not significant. miR-181 HG (d) and mature miR-181a (e) and mature miR-181b (f) were measured following treatment with E2 or E2+TNFα for up to 24 hrs. Significant differences between E2 and E2+TNFα are indicated. *P<0.05, **P<0.01, ***P<0.001.
Interestingly, TNFα also down-regulates miR-181A1 HG expression and the combination of E2+TNFα resulting in a further repression, compared to either E2 or TNFα alone (Figure 3a, 3d). A similar effect was observed in additional ER+ cell lines (Supplemental Figure 5). In addition, IKK7 prevented down-regulation of miR-181A1 HG indicating a role for the NFκB pathway as well (Figure 3c). While an effect of TNFα alone on miR-181a and b levels was not consistent or significant (data not shown), a more rapid and robust down-regulation of mature miR-181 levels was observed with E2+TNFα than with E2 alone (see 2 hrs time points in Figures 3e and 3f). The faster reduction of mature miR-181a and b by E2+TNFα is consistent with the timescale for PHLDA1 up-regulation and mRNA stabilization. This data suggests a model where E2 and TNFα repress transcription of the host gene, which leads to a reduction in both mature miR-181 family members.
To determine the extent to which the down-regulation of endogenous miR-181 may contribute to PHLDA1 up-regulation, two approaches were taken. First, cells were treated with Act D in the presence or absence of the PHLDA1-miR-181 target protector. As indicated in Figure 4a, PHLDA1 mRNA is elevated over 2-fold by the protector and confirms that PHLDA1 mRNA stability is controlled by endogenous miR-181. Second, over-expression of anti-miR-181a and b inhibitors elevates baseline PHLDA1, as well as TNFα-induced PHLDA1 expression (Figure 4b); this is similar to the protector effect shown in Figure 4a. Altogether, this data indicates that the down-regulation of endogenous miR-181a and b may contribute to the post-transcriptional regulation of PHLDA1 expression. Together, these findings suggest that E2 and TNFα act synergistically, via ER and NFκB, to up-regulate PHLDA1 expression not only at the transcriptional level but also at a post-transcriptional level by reducing miR-181a and b expression.
Figure 4.

B>. Post-transcriptional regulation of PHLDA1 by endogenous miR-181a and b. (a) PHLDA1 mRNA was measured in cells transfected with or without the target protector (1 μM, 48 hrs) prior to Act D treatment for 2 hrs. **P<0.01. (b) PHLDA1 mRNA expression was measured in MCF-7 cells transfected with siNeg or anti-miR-181a and b inhibitors (A-miR, 100 nM each) followed TNFα treatment for 2 hrs. **P<0.01.
PHLDA1 is up-regulated in mammospheres of ER+ breast cancer cells and is required for mammosphere formation and growth
Given the role of PHLDA1 in intestinal stem cells and tumorigenesis9, we decided to investigate the functional role of PHLDA1 in ER+ cancer cell mammospheres (MS), which are known to be enriched with breast CSCs19. The MS assay exploits the unique property of stem/progenitor cells to survive and grow in serum-free suspension, while more differentiated cells undergo anoikis and die in these conditions20. Additionally, MS culture enriches for cells with enhanced metastatic character, that are highly tumorigenic, and that are chemo- and radio-resistant21-24, and therefore represent a good model system to study breast CSCs. In a panel of ER+ breast cancer cells, we find that PHLDA1 mRNA (Figure 5a) and protein (Figure 5b) are up-regulated in MS cultures when compared to cells grown in standard, adherent monolayer cultures (2D). While it is possible that PHLDA1 may be regulated by MS media, we were unable to test this since culture of adherent monolayer of cells with MS media resulted in morphology changes and eventual cell death, possibly due to serum withdrawal (Supplemental Figure 6a). However, our finding that PHLDA1 is up-regulated in MS because of CSC content rather than the media formulation is consistent with the report by Murohashi et al. Their work showed that PHLDA1 gene expression is enriched in the CSC population, as identified by CD44high/CD24low surface marker expression, in multiple breast cancer cell lines, including MCF-7, HCC70 and HCC195425.
Figure 5.

PHLDA1 is up-regulated while miR-181A1 HG and mature miR-181b are down-regulated in ER+ breast cancer cell MS. (a) PHLDA1 mRNA expression in standard adherent 2D culture of ER+ breast cancer cells was compared to MS culture. (b) PHLDA1 protein was measured in MCF-7 2D and MS cultures. (c) PHLDA1 mRNA expression in MS was analyzed after 6 hrs IKK7 treatment or 24 hrs ER antagonist treatment (1 μM each). (d) Expression levels of miR-181A1 HG and mature miR-181a and b were compared in 2D vs. MS culture. *P<0.05 or ***P<0.001 compared to 2D or vehicle (Veh) treated controls.
Because the NFκB pathway is active and required for the survival of breast CSCs26, 27, we postulated that this pathway might also play a role in regulating PHLDA1 expression in MS. Treatment with IKK7 significantly reduced PHLDA1 expression in MS while ER-blockers such as ICI, 4-hydroxytamoxifen (4OHT), and desmethylarzoxifene (DMA) had no effect on PHLDA1 expression (Figure 5c). In addition, expression of miR-181A1 HG and mature miR-181b but not miR-181a was significantly lower in MS than 2D cultures (Figure 5d). Together, these findings suggest that high intrinsic NFκB activity and reduced miR-181b levels contribute to elevated PHLDA1 expression in ER+ breast cancer cells grown as MS.
To examine the potential role of PHLDA1 in MS formation and growth, we transfected MCF-7 cells with siRNA targeting PHLDA1 or control siRNA (siNeg) in standard 2D cultures and then seeded single cells in MS conditions. Efficiency of siPHLDA1 knockdown is shown in Supplemental Figure 6b. Silencing PHLDA1 results in attenuated MS formation and growth shown by a reduction in both the number of MS formed and MS size, respectively (Figure 6a, 6b and Supplemental Figure 6c). Yet, silencing PHLDA1 has no significant effect on cell viability in standard adherent monolayer cultures (Supplemental Figure 6d), suggesting that PHLDA1 might play an essential role in the survival and maintenance of ER+ breast CSCs. To corroborate this finding, an additional breast CSC marker was utilized. Aldehyde dehydrogenase (ALDH) activity assessed by the Aldefluor assay is used to identify cells with CSC-like properties28. E2 treatment is known to expand the breast CSC population29, hence we utilized E2 together with TNFα treatment on MCF-7 cells and probed for the role of PHLDA1. The increased ALDH-positive population resulting from E2+TNFα treatment is significantly attenuated with siPHLDA1 (Figure 6c and 6d), further supporting an essential function for PHLDA1 in ER+ breast CSCs.
Figure 6.

PHLDA1 is essential for MCF-7 MS formation and expansion of ALDH-positive cells. (a, b) MCF-7 cells were transfected with siNeg or siPHLDA1 (50 nM). After 48 hrs, cells were seeded at single-cell density in low attachment plates and MS were allowed to develop. After 7 days, the total number of MS ≥ 75 μm (a) and the average diameter of MS (b) were measured. ***P<0.001 compared to siNeg control. (c, d) MCF-7 cells transfected with siNeg or siPHLDA1 (50 nM each, 48 hrs) were treated with E2+TNFα for 2 hrs, washed and allowed to recover for 72 hrs prior to running the Aldefluor assay and FACS analysis. ***P<0.001 compared to siNeg control. In (c) quantitation of the ALDH-positive population is indicated. In (d) representative scatter plots from FACS are shown. (e) MCF-7 cells were transfected with miR-181a and b mimics (20 nM each), either alone or in combination with the PHLDA1-miR-181 target protector (1 μM). 48 hrs later, cells were seeded for MS and after 7 days the number of MS was measured. **P<0.01, ***P<0.001. (f) Stable cell lines overexpressing PHLDA1 or empty vector control were transfected with miR-181a and b mimics (20 nM each) and MS formation was measured. *P<0.05, ns, not significant.
To confirm the role of PHLDA1 in CSCs, miR-181a and b mimics, which reduce PHLDA1 expression (see Figure 2), were transfected into MCF-7 cells prior to seeding in MS assays. Like siPHLDA1, miR-181a+b mimics attenuated MS formation and addition of the miR-181 target protector for PHLDA1 partially reversed this effect (Figure 6e). To further confirm that miR-181 targeting of PHLDA1’s 3′UTR contributes to MS formation, we performed a rescue experiment in which we over-expressed PHLDA1 lacking the 3′UTR (Supplemental Figure 6e); hence, miR-181a and b are unable to target it. MS formation of PHLDA1 over-expressing cells is not statistically different from that of vector control cells (Figure 6f). However, in the presence of miR-181a and b mimics, attenuated MS formation is partially restored by PHLDA1 overexpression (Figure 6f). The partial effect of the PHLDA1-miR-181 target protector and PHLDA1 overexpression may be explained by the fact that miR-181a and b have additional targets, such as Bcl-2 (Supplemental Figure 3c), which could also be involved in MS formation. Together, these findings suggest that targeting of PHLDA1 in MS by miR-181a and b leads to attenuated MS formation, similar to silencing of PHLDA1, and further supports a role for the PHLDA1-miR-181 axis in controlling MS formation of ER+ breast cancer cells.
High PHLDA1 expression predicts poor clinical outcome in patients with ER+ breast cancers
To examine the clinical relevance of PHLDA1 expression in breast tumors, we analyzed publically available patient datasets to determine whether PHLDA1 mRNA expression is correlated with clinical outcome. While there is no significant association between PHLDA1 and patient outcome when all breast cancer patients are examined (Figure 7a), elevated PHLDA1 expression is significantly associated with a higher risk of distant metastasis in patients with ER+ breast cancers (Figure 7b). The opposite is observed in patients with ER− breast cancers (Figure 7c). This finding suggests that not only is PHLDA1 expression a potential predictor of metastasis and aggressiveness in ER+ tumors but also that it may have different functions in ER+ vs. ER-breast tumors.
Figure 7.
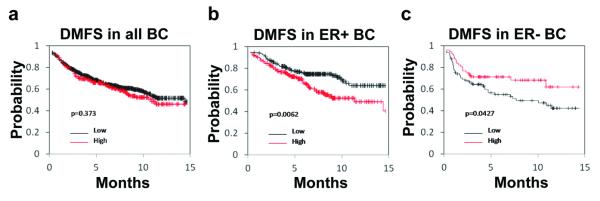
PHLDA1 is associated with increased risk of distant metastasis in ER+ breast cancers. (a, b, c) Kaplan-Meier analysis demonstrating the association between PHLDA1 expression and distant metastasis free survival (DMFS) are indicated for (a) all breast cancer (BC) patients, (b) only for ER+ BC, and (c) only for ER− BC patients.
Discussion
In this study, we have elucidated a unique ER-NFκB-miR181 regulatory loop that controls expression of PHLDA1 in ER+ breast cancer cells. More specifically, we showed that E2, acting via ER, and TNFα, acting via the NFκB pathway, work together to increase both transcription and stability of PHLDA1 mRNA (Figure 8). Furthermore, we have demonstrated that ER and NFκB working together to synergistically suppress expression of miR-181a and b, both of which directly target PHLDA1, to enhance PHLDA1 mRNA stability. Thus, a regulatory loop between ER, NFκB, and miR-181 demonstrates a concerted, highly coordinated mechanism to control the ultimate expression levels of the PHLDA1 gene. In addition to its regulation, we show that PHLDA1 plays an important role in the formation and growth of ER+ mammosphere, which enrich for cells with stem-like/progenitor properties, and the expansion of the ALDH-positive population. These findings indicate an essential function for PHLDA1 in bestowing CSC-like properties to ER+ cells and may contribute to the association between elevated PHLDA1 expression and poor outcome in patients with ER+ breast cancers.
Figure 8.

B>. Model for PHLDA1 regulation and function in ER+ breast cancer cells. ER and NFκB work together to regulate PHLDA1 through a direct transcriptional mechanism and an indirect, post-transcriptional mechanism that involves down-regulation of miR-181. High PHLDA1 expression and low miR-181a/b is essential for CSC-like properties. In turn, this may lead to an aggressive breast cancer phenotype and a poor patient outcome.
Previous work from our lab showed that positive crosstalk between ER and NFκB can up-regulate a number of genes associated with an aggressive breast cancer phenotype6, 30. In general, this crosstalk has been shown to involve cooperative ER and p65 recruitment to gene promoters resulting in transcriptional synergy31-33. Previous work has suggested that the upstream region of the PHLDA1 gene (also known as TDAG51) is complex with a bidirectional promoter arrangement34. Although this region contains an NFκB-RE, we have been unable to identify a clear transcriptional regulatory mechanism or consistent ER/NFκB binding within 50kb of the human PHLDA1 gene. Intriguingly, we have found that ER and NFκB also work together to synergistically down-regulate expression of the miR-181 host gene. To our knowledge, this is the first instance of these factors working cooperatively to repress gene expression and investigation into the mechanism is ongoing.
Our results demonstrate that both PHLDA1 and miR-181 play a role in the formation and growth of mammospheres. Understanding the underlying mechanisms and molecular drivers of breast CSCs is important because CSCs are endowed with tumorigenic potential, are chemo- and radio-resistant, display an enhanced metastatic phenotype, and are thought to be responsible for recurrence21, 22, 24, 35-37. Hence, one way ER and NFκB may contribute to aggressive breast cancers is through up-regulation of PHLDA1 to enhance a CSC phenotype. Interestingly, down-regulation miR-181b also appears to be necessary for CSC phenotype, partially by targeting PHLDA1. However, our findings are somewhat contradictory to published roles for both PHLDA1 and miR-181 in breast cancer. PHLDA1 was identified as a direct substrate and mediator of Aurora A kinase action in ER− MDA-MB-231 breast cancer cells, and PHLDA1 loss was described as a bad prognostic factor38. Similarly, numerous publications showing elevated miR-181 family members is associated with a worse phenotype and worse prognosis in breast cancer. However, this typically applies to ER− breast cancers 39-41. For example, up-regulation of miR-181a by TGFβ was significantly associated with metastatic disease in ER− breast cancers39. However, ER+ and ER− tumors represent two very distinct disease states. Other functional partners of PHLDA1 that may differentially modulate its activity in ER+ vs. ER− remain to be identified. This type of context specific protein function is not unprecedented; other factors, such as Notch1 and c-Myb, were shown to behave as either tumor suppressors or oncogenes depending on the disease type or marker status42, 43. Since high expression of PHLDA1 is correlated with poor patient outcome in ER+ cancers and low expression is correlated with poor outcome in ER− cancers, our results suggest that these inconsistencies may be resolved if we consider the roles of PHLDA1 and miR-181 as context specific.
In conclusion, we determined that ER and NFκB factors work together to up-regulate PHLDA1 and simultaneously repress miR-181a and b expression in ER+ breast cancer cells, leading to amplified expression of the ER-NFκB-miR-181 target gene, PHLDA1. The inverse, dichotomous nature of low miR-181 and high PHLDA1 expression, in turn contributes to survival and growth of breast cancer stem-like cells. Together, these findings suggests that ER and NFκB crosstalk can mediate an aggressive phenotype by controlling gene regulatory loops that can impact on breast cancer stem cells.
Materials and Methods
Reagents
E2, 4OHT, and Act D were purchased from Sigma. TNFα, IL-1β, and IL6 were obtained from R&D Systems. ICI 182,780 (ICI) was purchased from Tocris. The NFκB inhibitor, IKK7, was purchased from EMD Millipore. The PHLDA1 antibody was purchased from Santa Cruz Biotechnology (sc-23866) and the β-actin antibody from Sigma (A5441). DMA was generously provided by Dr. Gregory Thatcher (UIC). siRNA targeting ERα or PHLDA1 or a nonspecific control (siNeg) was purchased from Ambion. PHLDA1 miScript target protector for miR-181 and miR-181a and b synthetic mimics were purchased from Qiagen. Anti-miR-181a and b inhibitors were purchased from Ambion.
Cell and Mammosphere Culture
Human ER+ breast cancer cell lines, MCF-7, T47D, and BT474, were obtained from Dr. Debra Tonetti (UIC) and routinely maintained in RPMI 1640 (Invitrogen Life Technologies) with phenol red supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 2 mM L-glutamine, 1% antibiotics penicillin-streptomycin, and 6 ng/mL insulin. Prior to treatment with ER ligands and/or cytokines, cells were cultured in phenol red-free media supplemented with 5% charcoal-dextran-stripped fetal bovine serum for 2–3 days prior to treatment. For mammospheres, breast cancer cells were seeded at single cell density in low attachment plates in media described by Dontu et al.44, supplemented with 1% methyl cellulose to prevent cellular aggregation45. After 7 days, the diameter of MS was measured and MS ≥ 75μm in diameter were counted. For RNA measurements, MS were grown for 7 days and inhibitors were added for the last 6-24 hrs prior to RNA isolation.
Plasmids, Lentiviral Transduction and Stable PHLDA1 Overexpressing Cell Line
The lentiviral expression vector for PHLDA1 or the empty vector control (pLX304) were purchased from DNASU46-48 and Addgene (plasmid 25890), respectively49. Briefly, 23 μg of PHLDA1 or vector plasmid, 15 μg of packaging plasmid (psPAX2) and 8 μg of envelope plasmid (pMD2.G) were used to transfect packaging cells (293FT) with polyethylenimine. Packaging plasmid and envelope plasmid were a generous gift from Dr. Chong Wee Liew (UIC). PHLDA1 overexpressing or vector control cell lines were generated by transducing MCF-7 cells with lentiviral particles and blasticidin (10 μg/mL) selection for two weeks.
siRNA and miRNA Transfections
siERα, siPHLDA1, miR-181a and b mimics or anti-miRs, and PHLDA1 target protector were transfected as previously described7. Experiments were carried out 48 hrs after transfection.
Western Blot
Whole-cell extracts were prepared using M-PER (Thermo Scientific). Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes (Thermo Scientific), blocked for 1 hr in buffer containing 5% nonfat dry milk (Lab Scientific) or 5% BSA, and incubated with the appropriate primary antibody overnight. The next day, secondary antibody was applied and the signal was visualized on a Molecular Imager ChemidocXRS (Bio-Rad Laboratories) using the Pierce Supersignal West Pico chemiluminescent substrate (Thermo Scientific).
RT-Quantitative PCR (QPCR)
Total RNA was isolated and QPCR performed as described previously6. Fold change was calculated using the ΔΔCt method with 36B4 serving as the internal control. QPCR primer sequences are available upon request. For miRNA analysis, RNA was reverse transcribed using the miRCURY LNA Universal RT kit (Exiqon) and QPRC using LNA PCR primers sets (Exiqon) was run according to manufacturer’s guidelines. RNU44 and RNU48 served as internal controls.
MTS Cell Viability Assay
CellTiter96® AQueous One Solution (MTS) assay kit was purchased from Promega and MTS viability assay was run according to manufacturer’s guidelines.
Aldefluor Assay
Aldefluor assay (Stem Cell technologies) and FACS analysis were conducted as previously reported by Charafe-Jauffret et al.28.
PHLDA1 Gene Expression in Clinical Breast Cancer Specimens
The datasets used in survival analyses are publically available data sets and survival curves were generated using kmplot.com50; n=1609 for all breast tumors, n=1278 for ER+ tumors, and n=331 for ER− tumors.
Statistics
Data are presented as mean ± SEM from at least three independent determinations. Statistical analysis consisted of 1- or 2-way ANOVA followed by Tukey posttest, or t test, as appropriate. In all figures, asterisks denote significance levels as follows: * P<0.05, ** P<0.01, and *** P<0.001.
Supplementary Material
Acknowledgements
We thank Shuangping Zhao, Bryant Marure and Tuan-Ahn Tran for technical assistance. We are grateful for the financial support provided by the National Institute of Health (R01 CA130932-05 to JF), the University of Illinois at Chicago through the Chancellor’s Discovery Fund (JF), and by a postdoctoral fellowship grant from Susan G. Komen for the Cure® to IK (PDF12229484).
Financial support: Financial support was provided by the National Institute of Health (NIH), R01 CA130932-05 to JF, the University of Illinois at Chicago through the Chancellor’s Discovery Fund (JF), and by a postdoctoral fellowship grant from Susan G. Komen for the Cure® to IK (PDF12229484).
Footnotes
Conflict of interest: The authors report no conflicts of interest.
References
- 1.Zhou Y, Eppenberger-Castori S, Marx C, Yau C, Scott GK, Eppenberger U, et al. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int J Biochem Cell Biol. 2005;37:1130–1144. doi: 10.1016/j.biocel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007;7:59. doi: 10.1186/1471-2407-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones RL, Rojo F, A’Hern R, Villena N, Salter J, Corominas JM, et al. Nuclear NF-kappaB/p65 expression and response to neoadjuvant chemotherapy in breast cancer. J Clin Pathol. 2011;64:130–135. doi: 10.1136/jcp.2010.082966. [DOI] [PubMed] [Google Scholar]
- 4.deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, et al. NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann Oncol. 2004;15:885–890. doi: 10.1093/annonc/mdh232. [DOI] [PubMed] [Google Scholar]
- 5.Riggins RB, Zwart A, Nehra R, Clarke R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther. 2005;4:33–41. [PubMed] [Google Scholar]
- 6.Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, et al. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009;69:8918–8925. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanculescu A, Bembinster LA, Borgen K, Bergamaschi A, Wiley E, Frasor J. Estrogen promotes breast cancer cell survival in an inhibitor of apoptosis (IAP)-dependent manner. Horm Cancer. 2010;1:127–135. doi: 10.1007/s12672-010-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toyoshima Y, Karas M, Yakar S, Dupont J, Lee H, LeRoith D. TDAG51 mediates the effects of insulin-like growth factor I (IGF-I) on cell survival. J Biol Chem. 2004;279:25898–25904. doi: 10.1074/jbc.M400661200. [DOI] [PubMed] [Google Scholar]
- 9.Sakthianandeswaren A, Christie M, D’Andreti C, Tsui C, Jorissen RN, Li S, et al. PHLDA1 expression marks the putative epithelial stem cells and contributes to intestinal tumorigenesis. Cancer Res. 2011;71:3709–3719. doi: 10.1158/0008-5472.CAN-10-2342. [DOI] [PubMed] [Google Scholar]
- 10.Van Laere S, Limame R, Van Marck EA, Vermeulen PB, Dirix LY. Is there a role for mammary stem cells in inflammatory breast carcinoma?: a review of evidence from cell line, animal model, and human tissue sample experiments. Cancer. 2010;116:2794–2805. doi: 10.1002/cncr.25180. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- 13.Bridge G, Monteiro R, Henderson S, Emuss V, Lagos D, Georgopoulou D, et al. The microRNA-30 family targets DLL4 to modulate endothelial cell behavior during angiogenesis. Blood. 2012;120:5063–5072. doi: 10.1182/blood-2012-04-423004. [DOI] [PubMed] [Google Scholar]
- 14.Kim K, Madak-Erdogan Z, Ventrella R, Katzenellenbogen BS. A MicroRNA196a2* and TP63 circuit regulated by estrogen receptor-alpha and ERK2 that controls breast cancer proliferation and invasiveness properties. Horm Cancer. 2013;4:78–91. doi: 10.1007/s12672-012-0129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knauss JL, Bian S, Sun T. Plasmid-based target protectors allow specific blockade of miRNA silencing activity in mammalian developmental systems. Front Cell Neurosci. 2013;7:163. doi: 10.3389/fncel.2013.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makino K, Jinnin M, Aoi J, Hirano A, Kajihara I, Makino T, et al. Discoidin domain receptor 2-microRNA 196a-mediated negative feedback against excess type I collagen expression is impaired in scleroderma dermal fibroblasts. J Invest Dermatol. 2013;133:110–119. doi: 10.1038/jid.2012.252. [DOI] [PubMed] [Google Scholar]
- 17.Ouyang YB, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12:213–219. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maillot G, Lacroix-Triki M, Pierredon S, Gratadou L, Schmidt S, Benes V, et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009;69:8332–8340. doi: 10.1158/0008-5472.CAN-09-2206. [DOI] [PubMed] [Google Scholar]
- 19.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 20.Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. 2008;75:75–84. doi: 10.1159/000123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien CS, Howell SJ, Farnie G, Clarke RB. Resistance to endocrine therapy: are breast cancer stem cells the culprits? J Mammary Gland Biol Neoplasia. 2009;14:45–54. doi: 10.1007/s10911-009-9115-y. [DOI] [PubMed] [Google Scholar]
- 23.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 24.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murohashi M, Hinohara K, Kuroda M, Isagawa T, Tsuji S, Kobayashi S, et al. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br J Cancer. 2010;102:206–212. doi: 10.1038/sj.bjc.6605468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc Natl Acad Sci U S A. 2007;104:15852–15857. doi: 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16:45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumgarten SC, Frasor J. Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol. 2012;26:360–371. doi: 10.1210/me.2011-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pradhan M, Baumgarten SC, Bembinster LA, Frasor J. CBP mediates NF-kappaB-dependent histone acetylation and estrogen receptor recruitment to an estrogen response element in the BIRC3 promoter. Mol Cell Biol. 2012;32:569–575. doi: 10.1128/MCB.05869-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pradhan M, Bembinster LA, Baumgarten SC, Frasor J. Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NF{kappa}B cooperativity at adjacent response elements. J Biol Chem. 2010;285:31100–31106. doi: 10.1074/jbc.M110.155309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frasor J, Weaver AE, Pradhan M, Mehta K. Synergistic up-regulation of prostaglandin E synthase expression in breast cancer cells by 17beta-estradiol and proinflammatory cytokines. Endocrinology. 2008;149:6272–6279. doi: 10.1210/en.2008-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meier-Noorden M, Flindt S, Kalinke U, Hinz T. A CpG-rich bidirectional promoter induces the T-cell death-associated gene 51 and downregulates an inversely oriented transcript during early T-cell activation. Gene. 2004;338:197–207. doi: 10.1016/j.gene.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 35.Croker AK, Allan AL. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med. 2008;12:374–390. doi: 10.1111/j.1582-4934.2007.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velasco-Velazquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011;179:2–11. doi: 10.1016/j.ajpath.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14:29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 38.Johnson EO, Chang KH, de Pablo Y, Ghosh S, Mehta R, Badve S, et al. PHLDA1 is a crucial negative regulator and effector of Aurora A kinase in breast cancer. J Cell Sci. 2011;124:2711–2722. doi: 10.1242/jcs.084970. [DOI] [PubMed] [Google Scholar]
- 39.Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123:150–163. doi: 10.1172/JCI64946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neel JC, Lebrun JJ. Activin and TGFbeta regulate expression of the microRNA-181 family to promote cell migration and invasion in breast cancer cells. Cell Signal. 2013;25:1556–1566. doi: 10.1016/j.cellsig.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 41.Bisso A, Faleschini M, Zampa F, Capaci V, De Santa J, Santarpia L, et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell Cycle. 2013;12:1679–1687. doi: 10.4161/cc.24757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood. 2014 doi: 10.1182/blood-2013-08-355818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorner AR, Parker JS, Hoadley KA, Perou CM. Potential tumor suppressor role for the c-Myb oncogene in luminal breast cancer. PLoS One. 2010;5:e13073. doi: 10.1371/journal.pone.0013073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014;42:D1253–1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cormier CY, Park JG, Fiacco M, Steel J, Hunter P, Kramer J, et al. PSI:Biology-materials repository: a biologist’s resource for protein expression plasmids. J Struct Funct Genomics. 2011;12:55–62. doi: 10.1007/s10969-011-9100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cormier CY, Mohr SE, Zuo D, Hu Y, Rolfs A, Kramer J, et al. Protein Structure Initiative Material Repository: an open shared public resource of structural genomics plasmids for the biological community. Nucleic Acids Res. 2010;38:D743–749. doi: 10.1093/nar/gkp999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19:197–208. doi: 10.1530/ERC-11-0329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.