

Abstract
Background
Most Down syndrome children with acute myeloid leukemia (DS-AML) have an overall excellent prognosis, however, patients who suffer an induction failure or relapse, have an extremely poor prognosis. Hence, new therapies need to be developed for this subgroup of DS-AML patients. One new therapeutic approach is preventing cell cycle checkpoint activation by inhibiting the upstream kinase wee1 with the first-in-class inhibitor MK-1775 in combination with the standard genotoxic agent cytarabine (AraC).
Procedure
Using the clinically relevant DS-AML cell lines CMK and CMY, as well as ex vivo primary DS-AML patient samples, the ability of MK-1775 to enhance the cytotoxicity of AraC was investigated with MTT assays. The mechanism by which MK-1775 enhanced AraC cytotoxicity was investigated in the cell lines using Western blots to probe CDK1 and H2AX phosphorylation and flow cytometry to determine apoptosis, cell cycle arrest, DNA damage, and aberrant mitotic entry.
Results
MK-1775 alone had modest single-agent activity, however, MK-1775 was able to synergize with AraC in causing proliferation arrest in both cell lines and primary patient samples, and enhance AraC-induced apoptosis. MK-1775 was able to decrease inhibitory CDK1(Y15) phosphorylation at the relatively low concentration of 100 nM after only 4 hours. Furthermore, it was able to enhance DNA damage induced by AraC and partially abrogate cell cycle arrest. Importantly, the DNA damage enhancement appeared in early S-phase.
Conclusions
MK-1775 is able to enhance the cytotoxicity of AraC in DS-AML cells and presents a promising new treatment approach for DS-AML.
Keywords: AraC, Down syndrome, DNA damage, MK-1775, wee1
Introduction
Pediatric acute myeloid leukemia (AML) remains a difficult disease to treat and is associated with a relatively guarded prognosis. Patients with Down syndrome (DS), however, typically have much better outcomes, despite having an increased risk of developing AML [1]. This is due to enhanced sensitivity to cytarabine (AraC) and daunorubicin imparted by the unique biology of AML in the DS population (DS-AML), in which most cases are the megakaryocytic (AMKL) subtype and have somatic mutations in the X-linked transcription factor GATA1 gene [2–6].
Despite favorable outcomes, there are still challenges in treating this group of children. Patients with DS-AML who experience either an induction failure or relapse have dismal prognoses and very few options for salvage [7–9]. DS patients with relapsed AML treated on the Pediatric Oncology Group (POG) 9421 and Children's Cancer Group (CCG)-2891 AML studies had an overall survival (OS) rate of 12%, while a Japanese study reported an OS of 25.9% for relapsed and refractory DS AML patients [9]. DS-AML patients experience greater adverse toxicity, preventing the use of higher chemotherapy doses, and the high prevalence of congenital heart defects in the DS population makes anthracycline use especially challenging [10–13]. Following stem cell transplants (SCT), DS-AML patients only had an OS of 19% [14]. These studies all highlight that DS patients with refractory/relapsed AML have extremely chemotherapy-resistant disease. Thus, there is a clinical need for more effective therapies to be developed to treat this subgroup of DS-AML patients.
Inhibition of the wee1 kinase has recently been identified as a potential option for the treatment of several malignancies. The wee1 kinase is responsible for adding inhibitory phosphorylation to the tyrosine-15 residue of CDK1 [15,16]. This phosphorylation is required for the activation of S-phase and G2/M cell cycle checkpoints through the CHK1 pathway, inhibiting cdc25 phosphatases, which under normal circumstances constitutively remove the inhibitory phosphates, keeping CDKs active [17–20]. Therefore, preventing this initial phosphorylation event should abrogate these checkpoints that are induced by genotoxic chemotherapy drugs. Indeed, early studies have demonstrated a benefit of combining the first-in-class wee1 inhibitor, MK-1775 (currently in phase 1 and 2 clinical trials), with standard chemotherapy drugs in a variety of malignancies, including AML [21–28].
In this study, we investigated the potential role for the addition of MK-1775 to AraC for the treatment of DS-AML. Using the clinically relevant DS AMKL cell lines, CMK and CMY, and ex vivo primary DS-AML blast samples, we determined that MK-1775 was able to synergistically enhance the cytotoxicity of AraC. Furthermore, using the cell line models, we determined that MK-1775 enhanced AraC-induced apoptosis, likely by enhancing S-phase DNA damage caused by AraC. These results support the further development of the wee1 inhibitor MK-1775 for the treatment of DS-AML.
Methods
Cell Lines, Culture Conditions, and Reagents
CMK cells were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ; Braunschweig, Germany). The CMY cell line was a gift from Dr. A. Fuse (National Institute of Infectious Diseases, Tokyo, Japan). The DS-AMKL cell lines CMK and CMY were both cultured in RPMI 1640 with 10% FBS (Life Technologies, Carlsbad, CA) and 2 mM L-glutamine plus 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies), in a 37°C humidified atmosphere containing 5% CO2/95% air. AraC was purchased from Sigma (St. Louis, MO) and MK-1775 was purchased from Selleck Chemical (Houston, TX). Diagnostic blast cells from DS children with AMKL (n = 2) were obtained from the Children's Hospital of Michigan leukemia cell bank. Both patients remain in first remission. Written consent was obtained according to the Declaration of Helsinki. The research protocol was approved by the Human Investigation Committee of Wayne State University School of Medicine.
Antibodies
Rabbit antibodies directed against wee1, p-CDK1(Y15), total CDK1, PARP, phosphorylated histone H3(S10) (pH3) and γH2AX were purchased from Cell Signaling Technologies (Danvers, MA). Mouse anti-β-Actin was purchased from Sigma. Goat anti-rabbit IRDye 800CW antibody for Western blots was purchased from Licor (Lincoln, NE). Goat anti-rabbit-Alexa-488 for flow cytometry was purchased from Life Technologies.
In Vitro Cytotoxicity Assay
Viable cells were determined using a standard 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay as previously described [29]. Briefly, cells were added to a 96-well plate and cultured in the presence of increasing drug concentrations for 72 hours. After 72 hours, MTT (Sigma) was added to a final concentration of 1 mM. After 4 hours, formazan crystals were solubilized by addition of 10 mM HCl/10% sodium dodecyl sulfate (SDS). Crystals were allowed to dissolve overnight and plates were read using a microplate reader at 590 nm. For patient samples, 50,000 cells per well were cultured for 48 hours with ITS (Sigma) and conditioned media (20% supernatant from 5637 bladder cancer cell line—a source of GM-CSF) in the presence of various drug concentrations, after which MTT addition occurred as above. The presence or absence of synergistic drug interactions was determined using the CompuSyn software (ComboSyn, Parasmus, NJ) [30]. Error bars represent standard error on the mean, determined from at least three independent experiments. When indicated, IC50s were compared using non-parametric Mann–Whitney U test using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA).
Lentiviral shRNA Knockdown of wee1 Expression
Lentivirus mediated gene expression knockdown was performed as described previously [31]. Lentivirus shRNA constructs directed against WEE1 (designated sh-Wee1) or against no known gene (designated sh-NTC) were purchased from Sigma. Mixed cultures expressing the shRNA of interest were selected for by adding puromycin (Invivogen, San Diego, CA) to the standard culture media. Knockdown was confirmed by both quantitative reverse transcription polymerase chain reaction (qRT-PCR) and Western blot.
Western Blotting
Western blots were performed as previously described [32]. Briefly, cell lysates were prepared in the presence of protease and phosphatase inhibitors (cOmplete Mini and PhosStop, Roche, Indianapolis, IN). Cell lysates were subjected to SDS–PAGE and transferred to PVDF membranes (Pierce Scientific, Rockford, IL) for blotting with antibodies. Membranes were scanned using an Odyssey scanner (Licor). Images are representative of at least three identical, independent experiments.
qRT-PCR
qRT-PCR was performed as previously described [32]. Briefly, TaqMan probes from Life Technologies directed against either wee1 or GAPDH were used for amplification and detection. Relative transcript levels were determined using the delta–delta-CT method [33] and the experiment was performed on a LightCycler 480 instrument (Roche).
Flow Cytometry
Flow cytometry for apoptosis was performed using AnnexinV/Propidium iodide (PI) dual staining as described previously [34]. Analysis of γH2AX and pH3 versus PI was performed using a protocol from Huang and Darzynkiewicz [35], with the following modifications. One million cells per condition were spun at 300 rcf for 4 minutes and resuspended in 0.5 ml of 1% formaldehyde at 4°C for 15 minutes. Samples were spun and resuspended in residual volume, followed by dropwise addition of 1 ml of ice-cold methanol and storage at 4°C for at least 30 minutes. Samples were then processed according to protocol, using the antibody dilutions recommended by the manufacturer with overnight primary incubation. Samples were then counterstained with PI and analyzed as above. Histograms were analyzed with FlowJo (TreeStar Software, Ashland, OR). Images are representative of at least two identical, independent experiments. Where indicated, statistical analyses were performed using a paired t-test.
Results
MK-1775 Has Single Agent Effect in DS-AML
To investigate the potential of targeting wee1 for the treatment of DS-AML, two clinically relevant cell lines were used: CMK and CMY (which were derived from a patient who initially responded to chemotherapy and a patient who had refractory disease, respectively) [36,37]. Each cell line was originally derived from a pediatric DS patient with AMKL and a confirmed GATA1 mutation. As can be seen in Figure 1A, CMK is much more sensitive (>50-fold) to AraC than is CMY. CMY was also significantly more resistant to AraC-induced apoptosis as measured by AnnexinV/PI staining by flow cytometry compared to the CMK line (Fig. 3D and E). CMY demonstrated (using clinically achievable drug concentrations) class-wide resistance to both nucleoside analogues and topoisomerase inhibitors (Table S1). Interestingly, the cross resistance demonstrated by CMY was not seen with MK-1775 (Fig. 1B) with CMK and CMY having IC50s of 291 and 316 nM, respectively (P = 1.0). MK-1775 was able to induce comparable, dose-dependent levels of apoptosis in both cell lines (Fig. 1C). Knockdown of wee1 expression using lentiviral delivery of shRNA (Fig. 1D and E) was able to further sensitize both cell lines to MK-1775, suggesting that the effects of this drug are on-target.
Fig.1.
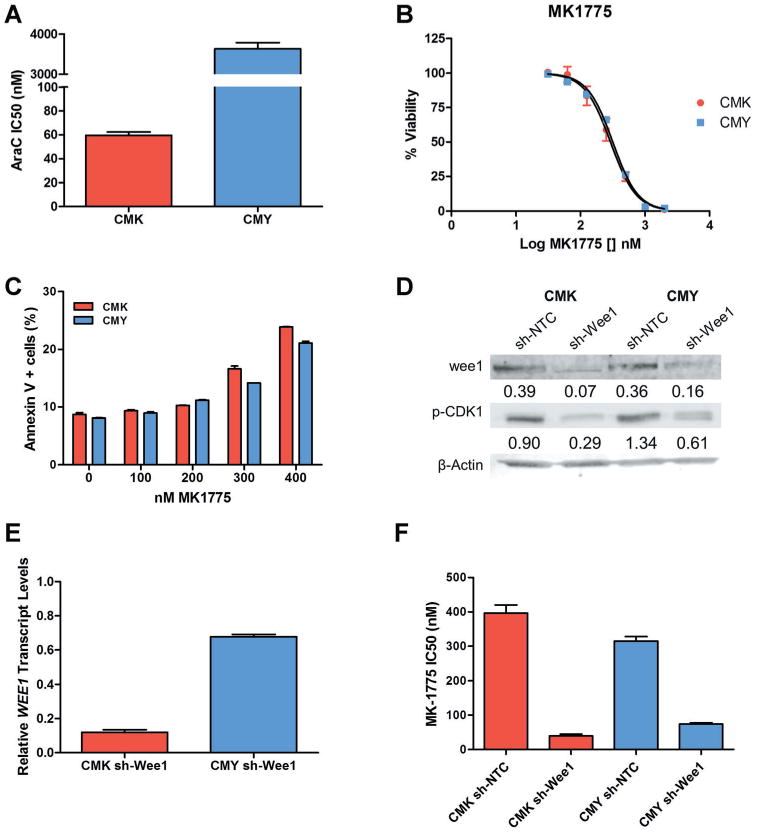
MK-1775 has single agent effect against DS-AML. A: AraC IC50s for CMK and CMY after 72-hour treatment as determined by MTT assay. B: Response of CMK and CMY to increasing concentrations of MK-1775 after 72-hour treatment as determined by MTT assay. C: Apoptosis induction in CMK and CMY cells after indicated 48-hour treatment, as determined by AnnexinV/PI staining and flow cytometry analysis. D and E: Expression of wee1 and p-CDK1(Y15), as determined by Western blots and qRT-PCR, after stable knockdown of wee1 using a lentivirus shRNA delivery system. F: Sensitivity of CMK and CMY cells to MK-1775 after knockdown of wee 1, as determined by a 72-hour MTT assay.
Fig. 3.

MK-1775 synergizes with AraC in both cell lines and primary patient samples. A and B: CMK and CMY cells were treated with different combinations of AraC and MK-1775 for 72-hour and viability was determined by MTT assay. Standard, normalized isobolograms demonstrate the synergistic inhibition of cell viability by the combination of AraC and MK-1775. C: CMK and CMY-ntc and -shwee1 cells were treated for 72 hours with AraC and viability was determined by MTT. D and E: CMK (D) and CMY (E), were treated with varying doses of AraC in the presence or absence of 100 nM MK-1775 for48 hours and apoptosis was measured using AnnexinV/PI staining. F: Various combinations of AraC and MK-1775 were tested in two primary DS-AML samples ex vivo using 48-hour MTT. The CI-Fa plot shows the combination index versus the fraction affected, with CI < 1 representing synergy.
Pharmacodynamic Changes of CDK1 Phosphorylation After Treatment With MK-1775
Additional studies examined what dose schedule of MK-1775 would be the most effective for combination treatments. CMK and CMY cells were treated with increasing concentrations of MK-1775 for 24hours, and phosphorylation status of CDK1(Y15) was interrogated using Western blots (Fig. 2A and B). After 24 hour treatment, it was found that concentrations as low as 100 nM were able to cause near-maximal inhibition of CDK1 phosphorylation. To determine the temporal kinetics of this inhibition, cells were treated with 100 nM MK-1775, followed by drug washout (Fig. 2A and B). Four hours of treatment was sufficient to decrease CDK1 (Y15) phosphorylation, but the effect was transient, returning as soon as 1 hour post-drug washout. Based on these findings, it was determined that simultaneous treatment would be utilized for further experiments.
Fig. 2.

Pharmacodynamic changes in p-CDK1(Y15) after MK-1775 treatment. Top: Expression of CDK1 and p-CDK1(Y15) in CMK and CMY (A and B) cells treated for 24 hours with increasing concentrations of MK-1775, as determined by Western blots. Bottom: CMK and CMY (A and B) cells were treated with 100 nM MK-1775 for 4 hours followed by drug washout. Expression of CDK1 and p-CDK1(Y15) was determined by Western blots.
MK-1775 Enhances the Cytotoxic Effects of AraC
The combination of MK-1775 with AraC resulted in a synergistic reduction in viable cells (Fig. 3A and B), with CI values ranging from 0.57 to 0.74 and 0.7 to 0.77 for CMK and CMY, respectively (CI values <1 indicate synergism, =1 indicate additivity, and >1 indicate antagonism). Knockdown of wee1 was able to enhance AraC-induced cytotoxicity in both cell lines (Fig. 3C). To investigate whether MK-1775 could enhance AraC-induced apoptosis, flow cytometry analysis of AnnexinV/PI dual-staining was used. 100 nM MK-1775 was able to greatly enhance the apoptosis induced by AraC in both cell lines (Fig. 3D and E). Treatment of two diagnostic DS AMKL samples with varying doses of AraC and MK-1775 yielded synergistic inhibition of viable cells, with CIs ranging from 0.28 to 0.65 (Fig. 3F), confirming that these drug combinations could ultimately be incorporated into clinical protocols.
MK-1775 Enhances AraC-Induced DNA Damage in S-Phase
To begin elucidating the mechanism by which the combination of MK-1775 and AraC-induced apoptosis, CMK and CMY cells were treated for 24 hours with combinations of both agents and levels of p-CDK1(Y15), PARP cleavage, and γH2AX (a biomarker for DNA double strand breaks) were interrogated using Western blots. It is well-known that AraC can cause replication stress, which results in DNA damage and cell-cycle arrest through the ATR-CHK1 pathway [38]. Therefore, it was important to determine whether MK-1775 could abrogate the inhibition of CDK1 that results from activation of this checkpoint, preventing cell cycle arrest. We hypothesized that MK-1775 would be able to enhance AraC-induced DNA damage as inhibition of wee1 alone can cause replication stress as well as inhibition of DNA repair. Indeed, MK-1775 was able to abrogate the induction of p-CDK1(Y15) and increase γH2AX induction by AraC in a dose-dependent fashion that paralleled the induction of apoptosis (measured by PARP cleavage) (Fig. 4).
Fig. 4.
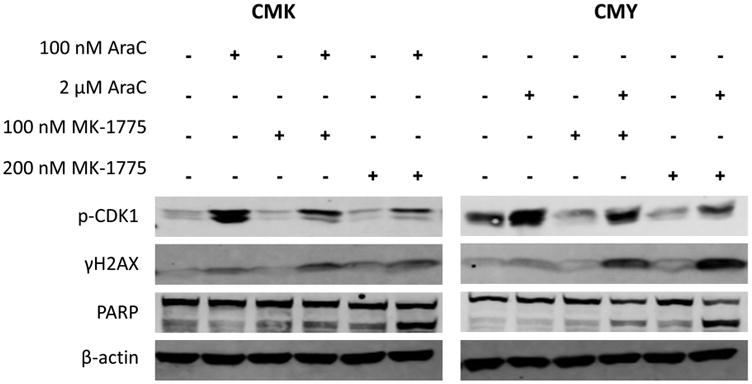
MK-1775 can abrogate AraC-induced CDK1(Y15) phosphorylation and enhance AraC-induced DNA damage. CMK and CMY cells were treated for 24 hours with the indicated drugs and levels of CDK1(Y15), γH2AX, and cleaved PARP were determined by Western blots.
There have been several reported mechanisms by which MK-1775 can enhance the cytotoxicity of different chemotherapeutic agents including wee1 inhibition preventing activation of the G2/M DNA damage checkpoint, allowing cells to progress aberrantly through the cell cycle resulting in either apoptosis or mitotic catastrophe. MK-1775 can also induce unscheduled mitosis, which typically results in cell death. To assess for these mechanisms, cells were treated with AraC, MK-1775, or the combination for 24 hours and p-H3 (a marker for mitosis) and γH2AX status were interrogated using flow cytometry.
AraC alone-induced S-phase arrest, while MK-1775 alone had little impact on cell cycle of CMY and CMK (Figs. 5 and S1, respectively). While the combination appeared to result in a slight rightward shift of the early-S peak seen from AraC alone, possibly indicating abrogation of an early-S arrest by MK-1775, the most notable change seen in the combination treatment was the significant increase in apoptosis as measured by cells with sub-G1 DNA content. Additionally, it does appear that MK-1775 was able to decrease the amount of G2/M and S-phase after AraC treatment in CMY and CMK, respectively (Figs. 5, S2, and S3). Though MK-1775 alone may have had a small impact on the number of mitotic cells (Figs. 5 and S1), it did not appear to have caused the degree of unscheduled mitoses reported previously in other cell types, and the modest nature of these changes is unlikely to account for the amount of cell death seen. Interestingly, MK-1775 was able to enhance AraC-induced γH2AX primarily in S-phase in CMY cells (Fig. 5). In CMK cells, the addition of MK-1775 to even low concentrations of AraC was able to primarily target cells in S-phase (Figs. S1 and S3), however, it is unclear whether the increase in DNA damage shown in Figure 4 occurred prior to or after the onset of apoptosis. It was expected that if MK-1775 primarily enhanced AraC-induced cell death by abrogation of G2/M checkpoint, there would have been an increase in γH2AX + cells in the G2/M compartment. The dearth of G2/M cells that stained positively for γH2AX therefore is suggestive that the addition of MK-1775 to AraC was able to cause sufficient insult to cells that they simply underwent apoptosis directly from S-phase.
Fig. 5.
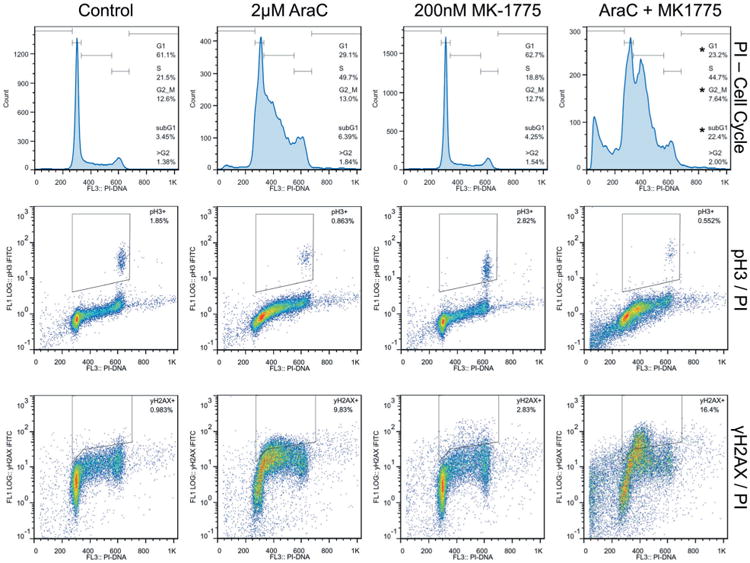
Effects of MK-1775 and AraC on cell cycle, mitosis, and DNA damage determined by flow cytometry analysis. CMY cells were treated with the indicated drugs for 24 hours and analyzed by flow cytometry. Top: The effects of AraC and MK-1775 on cell cycle were determined using PI staining. Middle: The effects of AraC and MK-1775 on mitosis versus cell cycle were determined using dual pH3/PI staining. Bottom: The effects of AraC and MK-1775 on DNA damage versus cell cycle were determined using dual γH2AX/PI staining. * Indicates P < 0.05 compared to AraC treatment.
Discussion
DS children with AML have an overall very favorable prognosis with event-free survival rates of ∼80% [1]. However, for patients with either refractory or relapsed disease, the prognosis is very poor highlighting the need to develop new effective therapeutic options. Targeting the wee1 kinase for treatment of malignancy is rapidly emerging with several phase 1 or 2 clinical trials currently underway (www.clinicaltrials.gov), including one pediatric brain tumor trial. In our study, we provide strong evidence to support the further development of combining the wee1 inhibitor MK-1775 with AraC for the treatment of DS-AML.
Early published results from phase 1 trials have indicated that the maximal plasma concentration of MK-1775 is approximately 400–500 nM, with an oral twice daily dosing regimen for five doses [39,40]. Though single-agent activity of MK-1775 at those levels appears to be modest, the concentrations used in this study (100–200 nM) for combination studies are well below that threshold. Therefore, even if substantial dose reductions are necessary in this population, our findings are more likely to have clinical relevance. Furthermore, based on the results from Figure 2 suggesting that AraC and MK-1775 should likely be dosed simultaneously, the MK-1775 dosing strategy used previously is likely compatible with the 4-day infusion schedule for AraC used in the most recent DS-AML clinical trial, Children's Oncology Group AAML0431. Although, this study lacks in vivo toxicity and efficacy data, a recently published study using a murine AML model found that the toxicity of the combination was similar to that of AraC alone [22].
Most of the prior studies investigating MK-1775 focused on its ability to bypass a G2/M cell cycle checkpoint to enhance cell death. It was initially reported that this abrogation was only effective in p53-mutant cells, though that notion has been challenged and may represent a cancer- or cell-type-specific phenomenon [21–28,41]. While similar results were expected in our study, it was surprising to see that G2/M checkpoint abrogation did not seem to contribute substantially to the cytotoxic effect of the combination of MK-1775 and AraC, and instead cells were likely dying in S-phase (Figs. 5, S1, and S3). Though it would be ideal if MK-1775 synergizes with anthracycline chemotherapy, the lack of substantial G2/M arrest abrogation, and unfavorable previously published results [22] suggest that this is likely not the case. It is important to note that these findings do not contradict those seen in previous studies (G2/M checkpoint abrogation, unscheduled mitosis), given that those studies were performed using mostly solid tumor lines. Instead, our results agree with a recently published study that suggests MK-1775 is able to abolish an intra-S checkpoint [22]. With this in mind, we propose a model (illustrated in Fig. 6) in which wee1 inhibition by MK-1775 prevents activation of the S-phase checkpoint normally activated by AraC. Subsequently, replication forks continue to fire [42,43] allowing for more AraC incorporation and subsequent fork collapse. Eventually, a catastrophic level of DNA damage is sustained, and the cell undergoes apoptosis without completing DNA replication. Though further work is necessary to fully confirm this hypothesis, if true, it could have important implications for the combination of MK-1775 with nucleoside analogues that rely on DNA synthesis for antitumor effect.
Fig. 6.
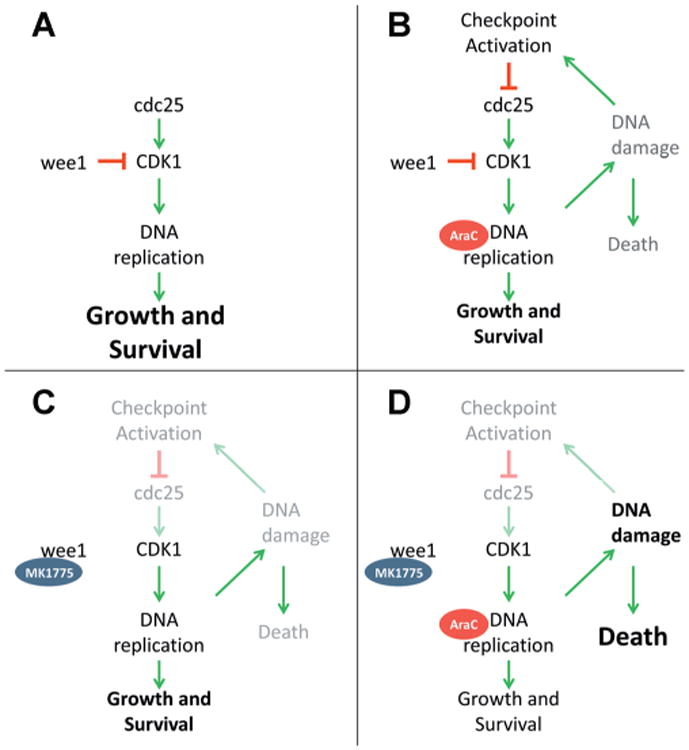
Schematic presentation of the effects of MK-1775 and AraC on cell survival. A: In untreated cells, DNA replication is successful and cell survival is primary endpoint. B: In AraC treated cells, S-phase arrest, and DNA damage occur, but damage can be repaired and cells can survive. C: In MK-1775 treated cells, some DNA damage occurs but cells primarily survive. D: In combination treated cells, MK-1775 overrides AraC induced checkpoint activation, resulting primarily in cell death.
The data presented here support the further development of MK-1775 as a potential adjuvant to AraC for the treatment of DS-AML. MK-1775 is able to greatly enhance the anti-leukemic effects of AraC, primarily by enhancing AraC's DNA damaging effect in S-phase. The anti-leukemic effect was independent of sensitivity to AraC alone, and was validated ex vivo using primary DS-AML blasts. Based on our results, MK-1775 appears to bean exciting new option for the treatment of relapsed or refractory DS-AML and for further clinical trial development, which may also be relevant to non-DS AML patients as well.
Supplementary Material
Acknowledgments
National Cancer Institute; Grant numbers: R01 CA120772; Grant sponsor: Karmanos Cancer Institute; Grant sponsor: Children's Research Center of Michigan; Grant sponsor: Kids Without Cancer, Children's Leukemia Foundation of Michigan; Grant sponsor: Elana Fund; Grant sponsor: Herrick Foundation; Grant sponsor: Justin's Gift Charity; Grant sponsor: The Buric Family; Grant sponsor: Leukemia and Lymphoma Society; Grant sponsor: Sehn Family Foundation; Grant sponsor: Dale Meyer Memorial Endowment for Leukemia Research; Grant sponsor: Ring Screw Textron Endowed Chair for Pediatric Cancer Research
Footnotes
Conflict of interest: Nothing to declare.
References
- 1.Xavier AC, Ge Y, Taub JW. Down syndrome and malignancies: A unique clinical relationship: A paper from the 2008 William Beaumont hospital symposium on molecular pathology. JMD. 2009;11:371–380. doi: 10.2353/jmoldx.2009.080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ge Y, Jensen TL, Stout ML, et al. The role of cytidine deaminase and GATA1 mutations in the increased cytosine arabinoside sensitivity of Down syndrome myeloblasts and leukemia cell lines. Cancer Res. 2004;64:728–735. doi: 10.1158/0008-5472.can-03-2456. [DOI] [PubMed] [Google Scholar]
- 3.Ge Y, Stout ML, Tatman DA, et al. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005;97:226–231. doi: 10.1093/jnci/dji026. [DOI] [PubMed] [Google Scholar]
- 4.Taub JW, Huang X, Ge Y, et al. Cystathionine-beta-synthase cDNA transfection alters the sensitivity and metabolism of 1-beta-d-arabinofuranosylcytosine in CCRF-CEM leukemia cells in vitro and in vivo: A model of leukemia in Down syndrome. Cancer Res. 2000;60:6421–6426. [PubMed] [Google Scholar]
- 5.Taub JW, Matherly LH, Stout ML, et al. Enhanced metabolism of 1-beta-d-arabinofuranosylcytosine in Down syndrome cells: A contributing factor to the superior event free survival of Down syndrome children with acute myeloid leukemia. Blood. 1996;87:3395–3403. [PubMed] [Google Scholar]
- 6.Wechsler J, Greene M, McDevitt MA, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 7.Loew TW, Gamis A, Smith FO, et al. Induction therapy failures in Down syndrome patients with acute myelogenous leukemia. ASH Annu Meeting Abstr. 2004;104:4527. [Google Scholar]
- 8.Loew TW, Gamis A, Smith FO, et al. Down syndrome patients with relapsed acute myelogenous leukemia. ASH Annu Meeting Abstr. 2004;104:4526. [Google Scholar]
- 9.Taga T, Saito AM, Kudo K, et al. Clinical characteristics and outcome of refractory/relapsed myeloid leukemia in children with Down syndrome. Blood. 2012;120:1810–1815. doi: 10.1182/blood-2012-03-414755. [DOI] [PubMed] [Google Scholar]
- 10.Krischer JP, Epstein S, Cuthbertson DD, et al. Clinical cardiotoxicity following anthracycline treatment for childhood cancer: The Pediatric Oncology Group experience. J Clin Oncol. 1997;15:1544–1552. doi: 10.1200/JCO.1997.15.4.1544. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien MM, Taub JW, Chang MN, et al. Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: A report from the Children's Oncology Group Study POG 942. J Clin Oncol. 2008;26:414–420. doi: 10.1200/JCO.2007.13.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lange BJ, Kobrinsky N, Barnard DR, et al. Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood. 1998;91:608–615. [PubMed] [Google Scholar]
- 13.Rao A, Hills RK, Stiller C, et al. Treatment for myeloid leukaemia of Down syndrome: Population-based experience in the UK and results from the Medical Research Council AML 10 and AML 12 trials. Br J Haematol. 2006;132:576–583. doi: 10.1111/j.1365-2141.2005.05906.x. [DOI] [PubMed] [Google Scholar]
- 14.Hitzler JK, He W, Doyle J, et al. Outcome of transplantation for acute myelogenous leukemia in children with Down syndrome. Biol Blood Marrow Transplant. 2013;19:893–897. doi: 10.1016/j.bbmt.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Develop. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 20.Peng CY, Graves PR, Thoma RS, et al. Mitotic and G2 checkpoint control: Regulation of 14–3–3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 21.Chaudhuri L, Vincelette ND, Koh BD, et al. Chk1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. 2013;99:688–696. doi: 10.3324/haematol.2013.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Linden AA, Baturin D, Ford JB, et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013;12:2675–2684. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bridges KA, Hirai H, Buser CA, et al. MK-1775 a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–5648. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK-1775 a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirai H, Arai T, Okada M, et al. MK-1775 a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 26.Guertin AD, Li J, Liu Y, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12:1442–1452. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 27.Hirai H, Iwasawa Y, Okada M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 28.Kreahling JM, Foroutan P, Reed D, et al. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PloS ONE. 2013;8:e57523. doi: 10.1371/journal.pone.0057523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge Y, Dombkowski AA, LaFiura KM, et al. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107:1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 31.Xie C, Drenberg C, Edwards H, et al. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PloS ONE. 2013;8:e79106. doi: 10.1371/journal.pone.0079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards H, Xie C, LaFiura KM, et al. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: Role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114:2744–2752. doi: 10.1182/blood-2008-09-179812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Xie C, Edwards H, Xu X, et al. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin Cancer Res. 2010;16:5499–5510. doi: 10.1158/1078-0432.CCR-10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang X, Darzynkiewicz Z. Cytometric assessment of histone H2AX phosphorylation: A reporter of DNA damage. Methods Mol Biol. 2006;314:73–80. doi: 10.1385/1-59259-973-7:073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato T, Fuse A, Eguchi M, et al. Establishment of a human leukaemic cell line (CMK) with megakaryocytic characteristics from a Down's syndrome patient with acute megakaryoblastic leukaemia. Br J Haematol. 1989;72:184–190. doi: 10.1111/j.1365-2141.1989.tb07681.x. [DOI] [PubMed] [Google Scholar]
- 37.Miura N, Sato T, Fuse A, et al. Establishment of a new human megakaryoblastic cell line, CMY, with chromosome 17p abnormalities. Int J Mol Med. 1998;1:559–563. doi: 10.3892/ijmm.1.3.559. [DOI] [PubMed] [Google Scholar]
- 38.Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene. 2003;22:9063–9074. doi: 10.1038/sj.onc.1207229. [DOI] [PubMed] [Google Scholar]
- 39.Leijen S, Schellens JH, Shapiro G, et al. A phase I pharmacological and pharmacodynamic study of MK-1775 a Wee1 tyrosine kinase inhibitor, in monotherapy and combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. ASCO Meeting Abstr. 2010;28:3067. [Google Scholar]
- 40.Tibes R, Bogenberger JM, Chaudhuri L, et al. RNAi screening of the kinome with cytarabine in leukemias. Blood. 2012;119:2863–2872. doi: 10.1182/blood-2011-07-367557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aarts M, Sharpe R, Garcia-Murillas I, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 42.Beck H, N¨ahse V, Larsen MSY, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188:629–638. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beck H, Nähse-Kumpf V, Larsen MSY, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.