

Abstract
Previous reports described important role of hepatocyte growth factor (HGF) in mitigation of pulmonary endothelial barrier dysfunction and cell injury induced by pathologic agonists and mechanical forces. HGF protective effects have been associated with Rac-GTPase signaling pathway activated by Rac-specific guanine nucleotide exchange factor Tiam1 and leading to enhancement of intercellular adherens junctions. This study tested involvement of a novel Rac-specific activator, Asef, in endothelial barrier enhancement by HGF and investigated a mechanism of HGF-induced Asef activation. Si-RNA-based knockdown of Tiam1 and Asef had an additive effect on attenuation of HGF-induced Rac activation and endothelial cell (EC) barrier enhancement. Tiam1 and Asef activation was abolished by pharmacologic inhibitors of HGF receptor and PI3-kinase. In contrast to Tiam1, Asef interacted with APC and associated with microtubule fraction upon HGF stimulation. EC treatment by low dose nocodazole to inhibit peripheral microtubule dynamics partially attenuated HGF-induced Asef peripheral translocation, but had negligible effect on Tiam1 translocation. These effects were associated with attenuation of HGF-induced barrier enhancement in EC pretreated with low ND dose and activation of Rac and its cytoskeletal effectors PAK1 and cortactin. These data demonstrate, that in addition to microtubule-independent Tiam1 activation, HGF engages additional microtubule- and APC-dependent pathway of Asef activation. These mechanisms may complement each other to provide the fine tuning of Rac signaling and endothelial barrier enhancement in response to various agonists.
Keywords: HGF, Rac GTPase, guanine nucleotide exchange factor, endothelium, permeability, cytoskeleton
1. Introduction
The lung endothelium forms a semi-selective barrier between circulating blood and interstitial fluid, which is dynamically regulated by a counterbalance of barrier protective and barrier disruptive bioactive molecules present in the circulation. Mechanisms which govern increased vascular permeability have been actively investigated [1-6], while cellular mechanisms of endothelial barrier enhancement by circulating vasoactive agonists and growth factors are far less understood.
Hepatocyte growth factor (HGF) is a multifunctional mesenchyme-derived factor secreted by several cell types including vascular endothelium. Along with other bioactive substances HGF appears in lung circulation under pathological conditions, such as acute lung injury, sepsis, lung inflammation, and ventilator induced lung injury, and has been implicated in lung repair, cell survival, and restoration of lung barrier function [7-9]. Barrier protective effects of HGF have been observed in human pulmonary endothelial cells (EC) [10] and cerebral endothelium [11]. HGF stimulates multiple signaling pathways including activation of Src and c-Abl tyrosine kinases [12, 13], mitogen activated protein (MAP) kinases Erk1/2 and p38, protein kinase C, phosphatidylinositol-3-kinase (PI3-kinase) and its downstream effector GSK-3β [10] and small GTPase Rac [8, 9].
One mechanism of HGF-induced endothelial barrier enhancement involves activation of PI3-kinase causing stimulation of guanine nucleotide exchange factor (GEF) Tiam1, which facilitates exchange of GDP for GTP in the nucleotide-binding center of small GTPase Rac leading to Rac activation [14]. As result, activated Rac induces remodeling of the actin cytoskeleton and increases interaction between adherens junction proteins α,β,γ-catenin and VE-cadherin [9, 10].
Tiam1 belongs to the Dbl family of GEFs, and its nucleotide exchange activity is regulated by diverse mechanisms, including PI3-kinase-dependent, receptor tyrosine kinase-dependent, protein kinase A-dependent, and Epac-Rap1-dependent pathways [14-18]. Tiam1 is directly involved in Rac-mediated endothelial barrier protective effects by a number of agonists including sphingosine-1 phosphate, HGF, high molecular weight hyaluronan and protective oxidized phospholipids [19-21]. However, Tiam1-dependent mechanism does not fully explain the potent HGF-induced EC barrier enhancement and stimulation of Rac signaling, as inhibition of Tiam1 did not cause complete inhibition of HGF effects in the lung endothelium.
Another member of the Dbl family of Rac-specific GEFs, Asef has been recently implicated in the regulation of the actin cytoskeleton remodeling in epithelial and neuronal cells by activating Rac and Cdc42 GTPases [22]. Constitutive Asef activation by truncated APC or Asef overexpression decreased cell-cell adhesion and migration of colorectal tumor cells [23], but decreased E-cadherin-mediated cell-cell adhesion and promoted migration of kidney epithelial cells [24]. Asef contains Dbl homology (DH) domain exhibiting GEF activity, plekstrin homology (PH) domain which determines the subcellular localization and activity by interacting with phosphatidylinositol phosphate, Src homology 3 (SH3) autoinhibitory domain and a region that binds tumor suppressor Adenomatous Polyposis Coli Protein (APC), which also interacts with microtubules [24]. Constitutive Asef activation by truncated APC or Asef overexpression decreased cell-cell adhesion and migration of colorectal tumor cells [23], but decreased E-cadherin-mediated cell-cell adhesion and promoted migration of kidney epithelial cells [24]. Involvement of Asef in regulation of vascular endothelial barrier remains unknown.
This study tested an involvement of Asef in pulmonary EC barrier enhancement, relations between Asef and Tiam1 in stimulating HGF-induced Rac signaling and EC permeability response, and investigated a mechanism of HGF-induced Asef intracellular translocation and activation.
2. Materials and Methods
2.1. Cell culture and reagents
Human HGF was obtained from R&D Systems (Minneapolis, MN). Cell-permeable c-Met kinase inhibitor, N-(3-Fluoro-4-(7-methoxy-4-quinolinyl)phenyl)-1-(2-hydroxy-2-methylpropyl)-5-methyl-3-oxo-phenyl-2,3-dihydro-1H-pyrazole carboxamide, and PI3-kinase kinase inhibitor LY294002 were from EMD Millipore (Billerica, MA). Reagents for immunofluorescence were purchased form Molecular Probes (Eugene, OR). End-Binding protein-1 (EB1) and Rac1 antibodies were purchased from BD Transduction Laboratories (San Diego, CA); phospho-cortactin and phospho-PAK antibodies were from Cell Signaling (Beverly, MA); Asef, APC, Tiam1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA);. Unless otherwise specified, all biochemical reagents, including nocodazole, β-actin and β-tubulin antibodies, were obtained from Sigma (St. Louis, MO). Plasmid for bacterial expression of Rac (G15A) mutant used in GEF activation assays was a generous gift from Katalin Szaszi (St. Michael's Hospital, Toronto, Canada). Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (East Rutherford, NJ) and used for experiments at passages 5-7.
2.2. Si-RNA and DNA transfections
HPAEC were treated with pre-designed Asef-, Taim1-, or APC-specific siRNA. Set of three Stealth™ Select siRNA duplexes was purchased from Invitrogen (Carlsbad, CA) in ready to use, desalted, deprotected, annealed double-strand form. Transfection of EC with siRNA was performed using siPORT transfection reagent (Invitrogen, Carlsbad, CA) as previously described [25]. Non-specific, non-targeting RNA (Dharmacon, Lafayette, CO) was used as a control treatment. Seventy-two hours after transfection, cells were harvested and used for experiments.
2.3. Analysis of EC permeability
Endothelial permeability to macromolecules was monitored by express permeability testing assay (XPerT) [26, 27] available from Millipore (Vascular Permeability Imaging Assay, cat. #17-10398). This assay is based on high affinity binding of cell impermeable avidin-conjugated FITC-labeled tracer to the biotinylated extracellular matrix proteins immobilized on the surface covered with EC monolayers. In permeability visualization experiments, 15 min after EC stimulation with HGF, FITC-avidin solution was added directly to the culture medium for 3 min before termination of the experiment. Unbound FITC-avidin was washed out with PBS (pH 7.4, 37°C), cells were fixed with 3.7% formaldehyde in PBS (10 min, room temperature) and visualization of FITC-avidin on the bottoms of coverslips was performed using Nikon imaging system Eclipse TE 300 (Nikon, Tokyo, Japan) equipped with a digital camera (DKC 5000, Sony, Tokyo, Japan). Images were processed with Adobe Photoshop 7.0 software (Adobe Systems, San Jose, CA). For the permeability assay in the 96-well plates, cells were seeded on biotinylated gelatin-coated plates (3×104 cells/well). FITC-avidin solution was added directly to the culture medium at the final concentration 25 μg/ml for 3 min before termination of the experiment unless otherwise specified. Unbound FITC-avidin was washed out with 200 μl PBS, pH 7.4, 37°C (two cycles, 10 sec each). Finally, 100 μl PBS was added in each well, and the fluorescence of matrix-bound FITC-avidin was measured on Victor X5 Multilabel Plate Reader (Perkin Elmer, Waltham, MA) using an excitation wavelength of 485 nm and emission wavelength of 535 nm, 0.1 sec. Measurements of transendothelial electrical resistance (TER) across confluent HPAEC monolayers were performed using the electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described [28, 29].
2.4. Immunofluorescence staining and imaging analysis
Endothelial cells plated on glass cover slips were treated with the agonist of interest, fixed in 3.7% formaldehyde solution in PBS for 10 min at 4° C, washed three times with PBS, permeabilized with 0.1% triton X-100 in PBS-Tween (PBST) for 30 min at room temperature, and blocked with 2% BSA in PBST for 30 min. Incubations with primary antibodies were performed in blocking solution (2% BSA in PBST) for 1 hr at room temperature followed by staining with Alexa 488-conjugated secondary antibodies. After immunostaining, slides were analyzed using a Nikon video imaging system (Nikon Instech Co., Japan) as described elsewhere [28, 30]. For microtubule analysis, cells were fixed with -20 °C methanol and immunostaining was carried out with β-tubulin or EB1 antibodies as described previously [25, 31]. In brief, the cell boundaries were outlined, and the concentric outline shapes reduced to 70% were applied to the images to mark peripheral (outer 30% of diameter) and central (inner 70%) regions. The integrated fluorescence density in the peripheral area was measured using MetaMorph software and was calculated as a percentage of the integrated fluorescence density in the total cell area. The results were normalized in each experiment.
2.5. Co-immunoprecipitation, subcellular fractionation and immunoblotting
After agonist stimulation, cells were washed in cold phosphate buffered saline (PBS) and lysed on ice with cold TBS-NP40 lysis buffer (20 mM Tris pH 7.4, 150 mM NaCl, 1% NP40) supplemented with protease and phosphatase inhibitor cocktails (Roche, Indianapolis, IN). Clarified lysates were then incubated with antibodies to APC or Asef overnight at 4°C, washed 3-4 times with TBS-NP40 lysis buffer, and the complexes were analyzed by Western blotting using appropriate antibodies. In fractionation studies, cytosolic (soluble) and membrane/cytoskeletal (particulate) fractions were isolated as described previously [32, 33]. For analysis of protein phosphorylation profile, cells were stimulated, then lysed, and protein extracts were separated by SDS-PAGE, transferred to polyvinylidene fluoride membrane, and probed with specific antibodies. Equal protein loading was verified by reprobing membranes with antibody to β-actin or specific protein of interest.
2.6. Rac and GEF activation assays
Rac activation was evaluated in pulldown assays using agarose beads with immobilized PAK1-PBD [28]. In brief, after stimulation, cell lysates were collected, and GTP-bound Rac was captured using pull-down assays with immobilized PAK1-PBD agarose. The levels of activated Rac as well as total Rac content were evaluated by western blot analysis. Active Asef or Tiam1 was affinity precipitated from cell lysates according to previously described protocol [34] using the Rac (G15A) mutant kindly provided by K. Szaszi (St. Michael's Hospital, Toronto, Canada). This mutant cannot bind nucleotide and therefore has high affinity for activated GEFs [35]. Activated Asef and Tiam1 in Rac (G15A) pulldowns were detected by Western blotting and normalized to total Asef or Tiam1 in cell lysates for each sample. Precipitation with glutathione–Sepharose beads containing no fusion proteins resulted in no guanine nucleotide exchange proteins precipitation.
2.7. Statistical analysis
Results are expressed as means ± SD of three to six independent experiments. Stimulated samples were compared to controls by unpaired Student's t-tests. For multiple-group comparisons, a one-way variance analysis (ANOVA), followed by the post hoc Fisher's test, were used. P<0.05 was considered statistically significant.
3. Results
3.1. HGF-induced activation of Asef and Tiam1 mediates barrier protective effects on pulmonary EC
Involvement of Asef and Tiam1 in HGF-induced EC barrier enhancement was first tested in experiments with measurements of permeability for macromolecules. Knockdown of Asef, Tiam1 or both proteins in human lung EC was performed using siRNA approach. Fresh EC monolayers with nearly established confluence were used to better illustrate barrier enhancing effects of HGF treatment and effects of protein knockdown. Bar graph (Figure 1A) represents quantitative analysis of EC permeability changes by measurements of fluorescence of accumulated FITC-avidin in 96-well plates using microplate reader, as described in Methods. Single knockdown of Tiam1 or Asef partially attenuated HGF-induced EC barrier enhancement indicated by increased binding of FITC-avidin to biotinylated substrate under EC monolayers. The double knockdown of Asef and Tiam1 exhibited additive effect on suppression of HGF-induced EC barrier enhancement.
Figure 1. Asef and Tiam1 knockdown attenuates HGF-induced EC barrier enhancement.

A - EC grown in 96-well plates were transfected with Asef-specific, Tiam1-specific siRNA or non-specific RNA were stimulated with HGF (50 ng/ml, 10 min). After unbound FITC-avidin was removed, the FITC fluorescence at the bottom of culture dish was measured as described in Methods; *P<0.05 vs. control; **P<0.05 vs. HGF-stimulated EC treated with nonspecific RNA; n=6. B - Pulmonary EC grown on glass coverslips with immobilized biotinylated gelatin (0.25 mg/ml) were transfected with specific siRNA or non-specific RNA. After cell stimulation with vehicle or HGF (50 ng/ml), FITC-avidin (25 μg/ml) was added for 3 min. Unbound FITC-avidin was removed, and FITC fluorescence signal was visualized by fluorescence microscopy.
Visualization of paracellular permeability was performed by fluorescence microscopy of control and stimulated EC grown on glass coverslips after quick incubation with FITC-avidin permeability tracer (Figure 1B). In control conditions, low levels of basal accumulation of FITC-labeled tracer were observed at sites underlying the cell-cell junction area and reflecting basal levels of paracellular permeability. HGF significantly decreased the basal EC monolayer permeability for FITC-labelled avidin. Barrier enhancing effect of HGF was partially attenuated by single knockdown of Asef or Tiam1 and was completely abrogated in EC monolayers with double Asef/IQGAP1 knockdown.
3.2. HGF-induced activation of Rac is mediated by Asef and Tiam1
Single Asef or Tiam1 knockdown partially attenuated HGF-induced Rac activation evaluated by Rac-GTP pulldown assays. Rac activation was completely inhibited by double knockdown of Asef and Tiam1 (Figure 2A, left panel). siRNA-induced taget protein knockdown was confirmed by western blot analysis of EC lysates (Figure 2A, right panel). HGF activated Asef and Tiam1 guanine nucleotide exchange activities towards Rac evaluated by pulldown of activated GEFs on Rac(G15A) beads. These effects were abrogated by the cell pretreatment with pharmacologic inhibitors of HGF receptor c-Met and PI3-kinase (Figure 2B). These results demonstrate dual regulation of HGF-induced Rac activity by Asef and Tiam1.
Figure 2. Role of Asef and Tiam1 in HGF-induced activation of Rac.

A – Pulmonary EC were transfected with Asef-specific, Tiam1-specific siRNA, or their combination, or with non-specific RNA and stimulated with HGF (50 ng/ml, 5 min). Rac activation was determined by Rac-GTP pulldown assay. Content of activated Rac was normalized to the total Rac content in EC lysates. siRNA-induced target protein depletion was verified by western blot analysis. Bar graphs represent quantitative densitometry of western blot experiments. *P<0.05 vs. nsRNA; n=3. B – Cells were preincubated with vehicle, c-Met inhibitor (carboxamide 50 nM, 30 min) or PI3-kinase inhibitor (LY294002 20μM, 30 min) followed by stimulation with HGF (5 min). Asef and Tiam1 activation was determined by pulldown assay with immobilized RacG15A and evaluated by increased GEF association with RacG15A. Content of activated Asef or Tiam1 was normalized to the total GEF protein content in EC lysates. Bar graphs represent quantitative densitometry of western blot experiments. *P<0.05 vs. HGF treatment without inhibitors; n=3.
3.3. HGF induces selective recruitment of Asef to microtubules and association with APC
Asef interaction with microtubule-associated protein adenomatous polyposis coli (APC) is required for its activation and guanine nucleotide exchange activity towards Rac [22]. Analysis of potential Asef and Tiam1 recruitment to microtubules was examined in experiments with isolation of microtubule enriched fraction. HGF stimulation increased the levels of Asef and APC in EC microtubule fraction (Figure 3A). By contrast, the basal levels of Tiam1 detected in MT fraction under non-stimulated conditions were not affected by HGF treatment. siRNA-induced knockdown of APC abolished HGF-induced recruitment of Asef to microtubule fraction suggesting APC-facilitated mechanism of Asef association with microtubules (Figure 3B). Analysis of total cell lysates showed that APC knockdown did not affect total Asef levels in pulmonary EC. APC-Asef association was further tested in reciprocal co-immunoprecipitation assays using APC or Asef antibody. HGF stimulated interaction between Asef and APC, which was abolished by cell pretreatment with c-Met and PI3-kinase inhibitors (Figure 3CD). These data suggest that in addition to activation of nucleotide exchange activity, c-Met - PI3-kinase signaling is also important for Asef interaction with APC.
Figure 3. HGF effects on APC, Asef and Tiam1 association with MT fraction and APC-Asef interactions.

A – EC were stimulated with HGF (50 ng/ml) followed by isolation of MT-enriched fractionation. APC, Asef and Tiam1 were detected by western blot of MT fractions and normalized to tubulin content. Bar graph represents quantitative densitometry of western blot experiments. *P<0.05; n=3. B – EC were treated with nonspecific and APC-specific siRNA, and HGF-induced accumulation of Asef in MT fraction was assessed. Lower panels show Western blot detection of total Asef and APC protein levels in total cell lysates. Bar graphs represent quantitative densitometry of western blot data. *P<0.05 vs. nsRNA; n=3. C and D - EC pretreated with vehicle, c-Met inhibitor or PI3-kinase inhibitor were stimulated with HGF (50 ng/ml), and APC (C) and Asef (D) proteins were immunoprecipitated under non-denaturing conditions using appropriate antibody. Presence of APC, Asef and Tiam1 in immune complexes was tested by western blot. Results are representative of three to six independent experiments.
3.4. Inhibition of peripheral microtubule dynamics attenuates HGF-induced barrier enhancement
Nocodazole (ND) is a MT depolymerizing agent capable of complete dissolution of microtubule network when used at high concentration. However, at low concentrations nocodazole has been shown to have no effect on gross MT cytoskeleton structure, but slow down peripheral MT dynamics [36]. In the test experiments we determined the ND concentration which had no considerable effect on basal EC permeability evaluated by measurements of transendothelial electrical resistance (TER) (Figure 4A) and microtubule arrangement under basal conditions (Figure 4B). At this low concentration (0.05 nM), ND attenuated HGF-induced barrier enhancement monitored by TER measurements in EC monolayers (Figure 5A). We evaluated effects of HGF and low dose ND pretreatment on peripheral MT density. EC after HGF stimulation were fixed with methanol and subjected to immunofluorescence staining with antibody to End-Binding protein-1 (EB1) which tracks the growing plus end of microtubules. Analysis of EB1 staining showed higher density of EB1-positive MT tips at the distal area of HGF-stimulated cells, as shown in Figure 5B and higher magnification insets. Pretreatment with low dose ND did not significantly affect the EB1-positive MT tips density in control cells, but abolished the HGF effects on peripheral MT expansion. Bar graph presents the quantitative analysis of immunofluorescence data.
Figure 4. Dose-dependent effects of nocodazole on EC permeability and microtubule arrangement.

A – EC were treated with the indicated concentrations of nocodazole, and changes in EC permeability were monitored by TER measurements. Shown are representative data from three independent measurements. B – Immunofluorescence staining of microtubule cytoskeleton in EC treated with various nocodazole concentrations was performed using β-tubulin antibody.
Figure 5. Effect of low dose nocodazole on HGF-induced EC barrier enhancement and microtubule peripheral growth.
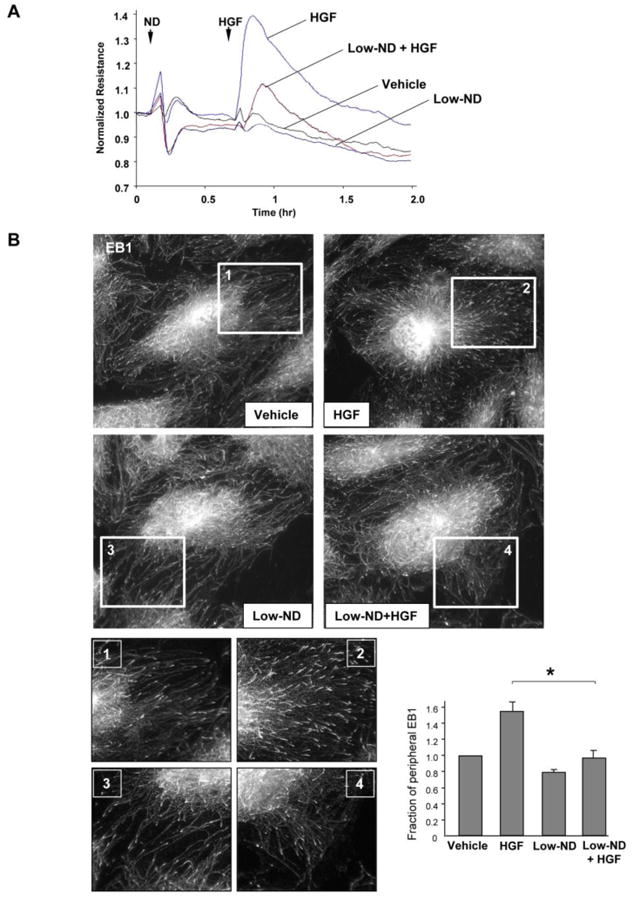
A – Human pulmonary EC were treated with low dose nocodazole (0.05 nM, marked by first arrow). At the time point indicated by second arrow, cells were stimulated with HGF (50 ng/ml), and TER was monitored over 2 hrs. Shown are representative data of four independent experiments. B - EC grown stimulated with HGF with or without pretreatment with low dose nocodazole (0.05 nM) followed by immunostaining with anti-EB1 antibody. Insets show high magnification images of cell peripheral areas with EB1-positive microtubule tips. Bar graph depicts quantitative analysis of peripheral EB1 in methanol-fixed HPAEC monolayers; *P<0.05, n=6.
3.5. Differential role of peripheral microtubule dynamics in HGF-induced translocation of APC/Asef and Tiam1
Effects of MT peripheral dynamics on APC, Asef and Tiam1 recruitment to cell cortical compartment were examined using treatment with low ND dose, as described above. Subcellular fractionation experiments showed that low dose ND treatment abolished accumulation of APC and Asef in the membrane/cytoskeletal fraction. Of note, low dose ND treatment did not affect the HGF-induced membrane recruitment of Tiam1 (Figure 6A). Bar graphs represent quantitative analysis of Asef, APC and Tiam1 translocation in HGF-stimulated EC with and without ND pretreatment. Immunofluorescence analysis of intracellular Asef and Tiam1 localization shows increased accumulation of these proteins at the submembrane area at the cell periphery of HGF-stimulated EC. Pretreatment with low dose ND abolished the HGF-induced peripheral accumulation of Asef but did not affect accumulation of Tiam1 (Figure 6B).
Figure 6. Effect of low dose nocodazole pretreatment on HGF-induced Asef, Tiam1 and APC peripheral translocation.

Human pulmonary EC pretreated with low dose nocodazole (0.05 nM, 15 min) were stimulated with HGF (50 ng/ml). A – Asef, Tiam1 and APC accumulation in membrane/cytoskeletal fraction was monitored by western blot. The content of examined proteins in corresponding total cell lysates was used as a normalization control. Bar graph represents quantitative densitometry of western blot experiments. *P<0.05 vs. HGF without nocodazole treatment; n=4. B - Intracellular redistribution of endogenous Asef an Tiam1 in HGF-stimulated endothelial cells was examined by immunofluorescence staining with appropriate antibody. Shown are representative results of three to five independent experiments.
3.6. Inhibition of peripheral microtubule dynamics attenuates HGF-induced activation of Rac signaling and enhancement of EC barrier
EC pretreatment with low dose ND attenuated HGF-induced activation of Rac (Figure 7A) and phosphorylation of Rac effector PAK1 and regulator of actin remodeling, cortactin (Figure 7B). This parameter reflects activation of Rac signaling pathway, as phosphorylation of cortactin at Y421 and Y466 in response to Rac1 activation resulted in localization of phosphorylated cortactin with F-actin in lamellipodia and podosomes [37] and regulated actin dynamics. In turn, decreased cortactin tyrosine phosphorylation was observed in cells with inhibited Rac1 and was associated with inhibition of cortactin peripheral localization [38].
Figure 7. Pretreatment with low dose nocodazole suppresses HGF-induced activation of Rac pathway and EC barrier enhancement.

Human pulmonary EC pretreated with low dose nocodazole (0.05 nM, 15 min) were stimulated with HGF (50 ng/ml). A – Rac activation was assessed by pulldown of Rac-GTP using PAK-PBD beads. B – Rac-dependent PAK and cortactin phosphorylation was assessed by western blot analysis of total cell lysates. C and D – EC permeability was evaluated by XPerT permeability assay described in Methods. After unbound FITC-avidin was removed, the FITC fluorescence signal was visualized by fluorescence microscopy. The bar graph shows quantitative analysis of EC permeability of EC monolayers grown in 96-well plates; *P<0.05 vs. HGF without nocodazole treatment; n=6 (C). Permeability changes were monitored in pulmonary EC grown on glass coverslips with immobilized biotinylated gelatin (0.25 mg/ml) (D).
Effects of low ND dose on the HGF-induced EC barrier enhancement were further tested using visualization of local areas in the EC monolayers with increased EC permeability for macromolecules. Bar graph depicts results of quantitative analysis of permeability data (Figure 7C). HGF significantly decreased the basal EC monolayer permeability for FITC-labelled avidin. Barrier enhancing effect of HGF was attenuated in EC monolayers pretreated with low ND dose. Visualization of the sites of increased permeability showed that basal levels of FITC-labeled tracer accumulation at sites underlying the cell-cell junction area observed in control nonstimulated EC monolayers were reduced in HGF-treated EC, and HGF effect was attenuated by low dose ND pretreatment. (Figure 7D).
4. Discussion
The main finding of this study is a demonstration of a novel role of Asef in the HGF-induced enhancement of endothelial barrier. Our data also show different routes of HGF-induced Tiam1 and Asef translocation to cell periphery. While HGF-induced PI3-kinase activation was essential for activity of both GEFs, the HGF-induced Tiam1 activation and peripheral translocation was independent on microtubule dynamics. In contrast, HGF-induced microtubule peripheral growth was required for APC-assisted translocation of Asef to cell periphery, full activation of Rac signaling and EC barrier enhancement response. HGF increased association of Asef and APC with MT fraction and stimulated formation of Asef-APC complex detected by reciprocal co-immunoprecipitation assays. APC forms a complex with EB1 at microtubule plus-ends and colocalizes at cell junctions with adherens junction protein β-catenin [39]. On the other hand, APC binding to Asef via its armadillo repeat domain enhances its GEF activity [40]. These features suggest a potential role for APC in regulation of basal endothelial barrier and re-establishment of monolayer integrity via agonist-induced interactions with Asef and stimulation of Asef nucleotide exchange activity.
In polarized fibroblasts, APC accumulation was also observed at the actin-rich lamellopodia of migrating cells [41]. Importantly, the most frequent APC localization occurs at the tips of microtubule-dependent cellular protrusions which appear to be the areas with activated actin dynamics and migration activity in this direction [42]. These published data support the proposed role for Asef-APC complex in the microtubule-guided local regulation of Rac activity observed in our study.
The accumulation of APC at EC periphery observed in this study may be also mediated through direct interactions of APC with the Rac/Cdc42 effector protein, IQGAP1 [41]. Our unpublished data show presence of IQGAP1 in the Asef co-immunoprecipitates from HGF-stimulated EC. These observations support our hypothesis that HGF-induced EC barrier enhancement is mediated by local Rac signaling via MT-APC assisted delivery and IQGAP1 peripheral capturing of Asef leading to its activation and local stimulation of Rac siganling.
This study used EC treatment with low dose nocodazole to further dissecta role of peripheral MT polymerization/depolymerization dynamics in HGF-induced signaling by Asef and Tiam1. We determined the nocodazole concentration which, similarly to effects previously described in immortalized BSC-1 cell line [36], did not cause global changes in the pool of polymerized tubulin or MT network structure, but attenuated peripheral MT polymerization/depolymerization dynamics in pulmonary EC. At this concentration, nocodazole inhibited HGF-induced peripheral MT expansion and significantly attenuated HGF-induced peripheral accumulation of Asef without affecting peripheral translocation of Tiam1. “Freezing” of peripheral microtubule dynamics partially attenuated HGF-induced activation of Rac signaling and decreased HGF-induced barrier enhancement in EC monolayers which was detected by XPerT permeability assay. These results further support a novel role for Asef in local regulation of Rac signaling at cell periphery via cooperation with microtubule-dependent mechanisms.
In summary, the results of this study provide a new insight into barrier enhancing mechanisms stimulated by HGF and demonstrate an alternative mechanism of Rac stimulation via APC- and MT-mediated peripheral delivery and activation of Asef which acts in concert with PI3K-Tiam1-dependent axis of Rac signaling.
Highlights.
-
-
Asef and Tiam1 cooperatively regulate HGF-induced endothelial barrier enhancement
-
-
PI3-kinase activation was essential for activity of both GEFs
-
-
Tiam1 activation and peripheral translocation was independent on MT dynamics
-
-
HGF-induced MT peripheral growth was required for Asef-mediated Rac activation
Acknowledgments
This work was supported by the grants from National Heart, Lung, and Blood Institutes (HL89257 and HL107920).
Non-standard Abbreviations
- APC
Adenomatous Polyposis Coli Protein
- EB1
End-Binding protein-1
- EC
endothelial cells
- GEF
guanine nucleotide exchange factor
- HPAEC
human pulmonary artery endothelial cells
- MT
microtubules
- nsRNA
non-specific RNA
- TER
transendothelial electrical resistance
- XPerT
express permeability testing assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mochizuki N. Circ J. 2009;73(12):2183–2191. doi: 10.1253/circj.cj-09-0666. [DOI] [PubMed] [Google Scholar]
- 2.Dejana E, Tournier-Lasserve E, Weinstein BM. Dev Cell. 2009;16(2):209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Thromb Haemost. 2010;103(1):40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- 4.Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Vascul Pharmacol. 2002;39(4-5):173–185. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 5.Hirase T, Node K. Am J Physiol Heart Circ Physiol. 2012;302(3):H499–505. doi: 10.1152/ajpheart.00325.2011. [DOI] [PubMed] [Google Scholar]
- 6.Birukov KG, Zebda N, Birukova AA. Compr Physiol. 2013;3(1):429–484. doi: 10.1002/cphy.c100066. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto K, Nakamura T. J Biochem (Tokyo) 1996;119(4):591–600. doi: 10.1093/oxfordjournals.jbchem.a021283. [DOI] [PubMed] [Google Scholar]
- 8.Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG. J Biol Chem. 2007;282(42):30643–30657. doi: 10.1074/jbc.M702573200. [DOI] [PubMed] [Google Scholar]
- 9.Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. FASEB J. 2007;21(11):2776–2786. doi: 10.1096/fj.06-7660com. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. FASEB J. 2002;16(9):950–962. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- 11.Date I, Takagi N, Takagi K, Kago T, Matsumoto K, Nakamura T, Takeo S. Biochem Biophys Res Commun. 2004;319(4):1152–1158. doi: 10.1016/j.bbrc.2004.05.100. [DOI] [PubMed] [Google Scholar]
- 12.Chen HT, Tsou HK, Chang CH, Tang CH. PLoS One. 2012;7(6):e38378. doi: 10.1371/journal.pone.0038378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cipres A, Abassi YA, Vuori K. Cell Signal. 2007;19(8):1662–1670. doi: 10.1016/j.cellsig.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Zheng Y. Trends Biochem Sci. 2001;26(12):724–732. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]
- 15.Welch HC, Coadwell WJ, Stephens LR, Hawkins PT. FEBS Lett. 2003;546(1):93–97. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 16.Arthur WT, Quilliam LA, Cooper JA. J Cell Biol. 2004;167(1):111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connor KL, Mercurio AM. J Biol Chem. 2001;276(51):47895–47900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- 18.Servitja JM, Marinissen MJ, Sodhi A, Bustelo XR, Gutkind JS. J Biol Chem. 2003;278(36):34339–34346. doi: 10.1074/jbc.M302960200. [DOI] [PubMed] [Google Scholar]
- 19.Singleton PA, Dudek SM, Chiang ET, Garcia JG. Faseb J. 2005;19(12):1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez E, Kou R, Michel T. J Biol Chem. 2006;281(6):3210–3216. doi: 10.1074/jbc.M510434200. [DOI] [PubMed] [Google Scholar]
- 21.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Circ Res. 2009;104(8):978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y, Higuchi O, Akiyama T. Science. 2000;289(5482):1194–1197. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- 23.Kawasaki Y, Furukawa S, Sato R, Akiyama T. Cancer Sci. 2013;104(8):1135–1138. doi: 10.1111/cas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawasaki Y, Sato R, Akiyama T. Nat Cell Biol. 2003;5(3):211–215. doi: 10.1038/ncb937. [DOI] [PubMed] [Google Scholar]
- 25.Tian X, Tian Y, Sarich N, Wu T, Birukova AA. Faseb J. 2012;26(9):3862–3874. doi: 10.1096/fj.12-207746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubrovskyi O, Birukova AA, Birukov KG. Lab Invest. 2013;93:254–263. doi: 10.1038/labinvest.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian X, Tian Y, Gawlak G, Sarich N, Wu T, Birukova AA. J Biol Chem. 2014;289(8):5168–5183. doi: 10.1074/jbc.M113.493924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. Circ Res. 2004;95(9):892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 29.Birukova AA, Birukov KG, Smurova K, Adyshev DM, Kaibuchi K, Alieva I, Garcia JG, Verin AD. FASEB J. 2004;18(15):1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 30.Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JGN, Verin AD. Microvasc Res. 2004;67(1):64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Komarova Y, De Groot CO, Grigoriev I, Gouveia SM, Munteanu EL, Schober JM, Honnappa S, Buey RM, Hoogenraad CC, Dogterom M, Borisy GG, Steinmetz MO, Akhmanova A. J Cell Biol. 2009;184(5):691–706. doi: 10.1083/jcb.200807179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birukova AA, Zagranichnaya T, Alekseeva E, Fu P, Chen W, Jacobson JR, Birukov KG. Exp Cell Res. 2007;313(11):2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birukova AA, Malyukova I, Poroyko V, Birukov KG. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L199–211. doi: 10.1152/ajplung.00020.2007. [DOI] [PubMed] [Google Scholar]
- 34.Waheed F, Dan Q, Amoozadeh Y, Zhang Y, Tanimura S, Speight P, Kapus A, Szaszi K. Mol Biol Cell. 2013;24(7):1068–1082. doi: 10.1091/mbc.E12-09-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- 36.Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L. Mol Biol Cell. 1997;8(6):973–985. doi: 10.1091/mbc.8.6.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Head JA, Jiang D, Li M, Zorn LJ, Schaefer EM, Parsons JT, Weed SA. Mol Biol Cell. 2003;14(8):3216–3229. doi: 10.1091/mbc.E02-11-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birukova AA, Tian Y, Dubrovskyi O, Zebda N, Sarich N, Tian X, Wang Y, Birukov KG. J Cell Physiol. 2012;227(10):3405–3416. doi: 10.1002/jcp.24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma M, Leung L, Brocardo M, Henderson J, Flegg C, Henderson BR. J Biol Chem. 2006;281(25):17140–17149. doi: 10.1074/jbc.M513027200. [DOI] [PubMed] [Google Scholar]
- 40.Akiyama T, Kawasaki Y. Oncogene. 2006;25(57):7538–7544. doi: 10.1038/sj.onc.1210063. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K. Dev Cell. 2004;7(6):871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 42.Nathke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. J Cell Biol. 1996;134(1):165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]