


Abstract
To provide molecular-level insights into the spontaneous replication error and the mismatch discrimination mechanisms of human DNA polymerase β (polβ), we report four crystal structures of polβ complexed with dG•dTTP and dA•dCTP mismatches in the presence of Mg2+ or Mn2+. The Mg2+-bound ground-state structures show that the dA•dCTP-Mg2+ complex adopts an ‘intermediate’ protein conformation while the dG•dTTP-Mg2+ complex adopts an open protein conformation. The Mn2+-bound ‘pre-chemistry-state’ structures show that the dA•dCTP-Mn2+ complex is structurally very similar to the dA•dCTP-Mg2+ complex, whereas the dG•dTTP-Mn2+ complex undergoes a large-scale conformational change to adopt a Watson–Crick-like dG•dTTP base pair and a closed protein conformation. These structural differences, together with our molecular dynamics simulation studies, suggest that polβ increases replication fidelity via a two-stage mismatch discrimination mechanism, where one is in the ground state and the other in the closed conformation state. In the closed conformation state, polβ appears to allow only a Watson–Crick-like conformation for purine•pyrimidine base pairs, thereby discriminating the mismatched base pairs based on their ability to form the Watson–Crick-like conformation. Overall, the present studies provide new insights into the spontaneous replication error and the replication fidelity mechanisms of polβ.
INTRODUCTION
Replication errors made by DNA polymerases, if not corrected, result in spontaneous mutations, of which transition mutations are most common (1). The spontaneous transition mutations can occur through replication of endogenous DNA lesions that result, for example, from deamination of methylcytosine at CpG sites, deamination of cytosine by apolipoprotein B mRNA-editing catalytic polypeptide protein and oxidative metabolism of guanine (2). The spontaneous transition mutations can also occur through errors made during replication of intact DNA (3–6). Elucidating the mechanisms of spontaneous transition mutations at the molecular level would further our understanding of the spontaneous replication error mechanisms and provide new insights into the replication fidelity mechanism of DNA polymerases.
Structures of various DNA polymerases in complex with dG•dTTP, dT•dGTP, dA•dCTP or dC•dATP mismatch (here, for example, dG•dTTP denotes the templating base•incoming nucleotide) show large variations of the conformations of the mismatches in the polymerase active site, suggesting that the spontaneous replication errors occur through varying mechanisms. For example, crystal structures of Bacillus stearothermophilus DNA polymerase I fragment (BF) show wobble dG•dTTP and dA•dCTP base pairs in the presence of the active-site Mg2+ (7). On the other hand, the BF-dA•dCTP-Mn2+ ternary complex shows the formation of Watson–Crick-like dA•dCTP base pair via amine-imino tautomerization (Figure 1). Structures of the B-family DNA polymerase RB69 show wobble dG•dTTP and dA•dCTP base pairs in the active site (8). In the active site of Y-family DNA polymerases Sulfolobus solfataricus Dpo4 and human polη, dT•dGTP forms a staggered base pair and a wobble base pair, respectively (9,10). In the case of the X-family DNA polymerase polλ, an error-prone enzyme that does not undergo an open-to-closed conformational transition during the catalytic cycle, dT•dGTP forms a Watson–Crick-like base pair in the presence of the active-site Mg2+ (11). The short distance between O4 of dT and O6 of dGTP (2.7 Å) suggests that the dT•dGTP mismatch occurs via keto-enol tautomerization, but the kinetic studies indicate that the mismatch occurs via ionization.
Figure 1.

Base pairings of G•T and A•C mismatches. (A) The formation of G•T base pair via wobble, Watson–Crick and pseudo-Watson–Crick base pairings. Base pairings involving enolized or ionized thymine are not shown. (B) The formation of A•C base pair via wobble and Watson–Crick base pairings. Watson–Crick base pair involving the imino tautomer of adenine is not shown.
The X-family DNA polymerase β (polβ) is an error-prone polymerase that preferentially induces transition mutations over transversion mutations (12). Polβ is overexpressed in many cancer cells, and overexpression of the enzyme in mammalian cells has been shown to significantly increase spontaneous mutations (13,14). A crystal structure of polβ in complex with dC•dATP mismatch and the active-site Mn2+ shows a staggered base pair conformation and an upstream shift of the template strand. In addition, the dC•dATP-Mn2+ structure shows an ‘intermediate’ protein conformation, in which the α-helix N that contains minor groove interacting amino acid residues shifts 1.4–4.0 Å away from the position observed in the closed conformation (see Supplementary Figure S1). The dC•dATP-Mn2 structure is very different from a matched polβ structure with a coplanar Watson–Crick base pair conformation and a closed protein conformation. In addition, the polβ-dC•dATP-Mn2+ structure shows an incomplete coordination of the catalytic metal ion, coordination of which is critical for catalysis, indicating that this complex does not represent a mismatched structure with a catalytically competent conformation. Therefore, although the polβ-dC•dATP-Mn2+ structure has provided important insights into the misincorporation deterrence mechanism of polβ (15), lack of mismatched polβ structures with a catalytically competent conformation has limited our understanding of the misincorporation mechanism of the enzyme. Furthermore, the unavailability of polβ structures with other potential mismatches has precluded a comprehensive understanding of the spontaneous replication error and the mismatch discrimination mechanisms of the enzyme.
To gain a deeper insight into the mechanisms, we solved four ternary complex structures of polβ bound to DNA containing dG•dTTP and dA•dCTP mismatches in the presence of Mg2+ or Mn2+. In addition, we determined kinetic parameters for the formation of dG•dTTP and dA•dCTP mismatches, and evaluated the effect of pH and the active-site metal ion on the mismatch formation. These studies show that the dG•dTTP and dA•dCTP mismatches in the polβ active site occur through distinct mechanisms, and that polβ employs a two-stage mismatch discrimination mechanism to increase replication fidelity. Overall, our kinetic and structural studies provide new insights into the spontaneous replication error and the mismatch discrimination mechanisms of polβ.
MATERIALS AND METHODS
DNA sequences used for X-ray crystallographic studies
All the oligonucleotides used in this study were purchased from Integrated DNA Technologies. The template DNA sequence used for co-crystallization was 5′-CCGAC(X)TCGCATCAGC-3′ (X = dG or dA). The upstream primer sequence was 5′-GCTGATGCGA-3′. The sequence of downstream primer was 5′-phosphate/GTCGG-3′. The oligonucleotides were annealed to give a single-nucleotide gapped DNA.
Polβ-DNA co-crystallization
Polβ was expressed and purified from Escherichia coli with minor modifications of the method described previously (16). Polβ binary complex with a single-nucleotide gap opposite the templating dG or dA was prepared using conditions as described previously (16). Briefly, polβ was complexed with a single-nucleotide gapped DNA that contains the 16-mer template strand (5′-CCGAC(X)TCGCATCAGC-3′, X = dG or dA), the upstream primer (5′-GCTGATGCGA-3′) and the downstream primer (5′-phosphate/GTCGG-3′). The Mn2+-bound polβ ternary complex crystals with the dG•dTMPNPP (denoted as dG•dTTP*) and dA•dCMPNPP (dA•dCTP*) base pairs were grown over 2–4 weeks in a solution containing 50 mM imidazole pH 7.5, 14–23% PEG3400, 5 mM non-hydrolyzable nucleotide (dTMPNPP or dCMPNPP, purchased from Jena Biosciences), 20 mM MnCl2 and 350 mM NaOAc. The Mg2+-bound polβ ternary complex crystals with the dG•dTTP* and dA•dCTP* base pairs were obtained by soaking the polβ gapped binary complex crystal in a buffer solution containing 50 mM imidazole pH 7.5, 20% PEG3400, 12% ethylene glycol, 5 mM non-hydrolyzable nucleotide, 200 mM MgCl2 and 100 mM NaOAc (17). Crystals were cryoprotected in mother liquor supplemented with 12% ethylene glycol and were flash-frozen in liquid nitrogen. Diffraction data were collected at 100 K at the beamline 5.0.3 at the Advanced Light Source, Lawrence Berkeley National Laboratory. All diffraction data were processed using HKL 2000. Structures were solved by molecular replacement (18) with a gapped binary complex structure (PDB ID 1BPX) (19) and a ternary complex structure (PDB ID IBPY) as the search models. The model was built using COOT (20) and refined using CCP4 (21). MolProbity (22) was used to make Ramachandran plots. All the crystallographic figures were generated using PyMOL.
Steady-state kinetics of single-nucleotide incorporation by polβ
Steady-state kinetic parameters were determined using the conditions described previously (17). Oligonucleotides used for kinetic assays (the primer, 5′-FAM/CTGCAGCTGATGCG-3′; the downstream primer, 5′-phosphate/CGTACGGATC CCCGGGTAC-3′ and the template, 5′-GTACCCGGGGATCCGTACG(X)CGCATCAGCTGCAG-3′, X = dG or dA) were purchased from Integrated DNA Technologies.
Molecular dynamics simulations
The systems for molecular dynamics (MD) simulations were set up based on the ternary polβ-dG•dTTP* complexes in the open (PDB ID 4PGQ, Table 2) and closed conformations (PDB ID 4PGX, Table 2), respectively. Four systems were prepared for each conformation (open or closed). They are polβ-dG•dTTP with single or double Mg2+ ions and polβ-dG•dCTP with single or double Mg2+ ions, respectively, comprising a total of eight systems. The nucleotide-binding Mg2+ was included in all systems (i.e. both the single and double Mg2+ systems), and the catalytic Mg2+ was included in the double metal ion systems. For each system, a total of 150 ns MD simulations are carried out using the all-atom CHARMM27 force fields (23,24), the CMAP correction (25) and the fixed-geometry TIP3P water potential (26) and coordinates are saved at every 2 ps for the analysis. The particle mesh Ewald summation method (27) was used with the rhombic dodecahedron periodic boundary conditions with the lattice length parameter of 88.5 Å. The MD simulation was carried out with 2 fs integration time and the Langevin thermostat at 300 K. See Supporting Information for the details of system preparation and Supplementary Table S1 for the summary of the simulated systems.
Table 2. Data collection and refinement statistics.
| PDB code | dG•dTTP*-Mg2+ 4PGQ | dG•dTTP-Mn2+ 4PGX | dA•dCTP*-Mg2+ 4PHA | dA•dCTP*-Mn2+ 4PHD |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P21 | P21 | P21 | P21 |
| Cell Constants | ||||
| a (Å) | 54.898 | 50.969 | 54.440 | 54.245 |
| b | 80.087 | 79.796 | 80.710 | 79.954 |
| c | 55.077 | 55.635 | 54.834 | 54.403 |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 |
| β | 106.15 | 107.37 | 108.88 | 108.88 |
| γ | 90.00 | 90.00 | 90.00 | 90.00 |
| Resolution (Å)a | 20–2.30 (2.34–2.30) | 20–2.09 (2.13–2.09) | 20–2.52 (2.56–2.52) | 20–2.21 (2.25–2.21) |
| Rmergeb (%) | 0.066 (0.214) | 0.096 (0.441) | 0.138 (0.469) | 0.096 (0.495) |
| <I/σ> | 24.3 (4.61) | 21.0 (3.14) | 11.9 (2.03) | 17.3 (2.31) |
| Completeness (%) | 99.9 (99.7) | 100.0 (99.8) | 99.8 (99.6) | 99.9 (98.8) |
| Redundancy | 3.7 (3.6) | 5.6 (5.3) | 4.5 (4.2) | 4.7 (4.5) |
| Refinement | ||||
| Rworkc/Rfreed (%) | 21.6/25.9 | 18.5/22.6 | 22.0/27.8 | 24.5/29.5 |
| Unique reflections | 20377 | 25542 | 14862 | 22218 |
| Mean B Factor (Å2) | ||||
| Protein | 38.68 | 26.34 | 36.73 | 34.67 |
| Ligand | 35.97 | 33.00 | 28.02 | 24.23 |
| Solvent | 34.50 | 29.93 | 28.35 | 28.34 |
| Ramachandran Plot | ||||
| Most favored (%) | 97.2 | 99.1 | 94.7 | 95.9 |
| Add. allowed (%) | 2.5 | 0.8 | 5.0 | 3.8 |
| RMSD | ||||
| Bond lengths (Å) | 0.006 | 0.004 | 0.004 | 0.005 |
| Bond angles (degree) | 1.263 | 1.078 | 1.097 | 1.198 |
aValues in parentheses are for the highest resolution shell. bRmerge = Σ|I-<I>|/ ΣI where I is the integrated intensity of a given reflection. cRwork = Σ|F(obs)-F(calc)|/ΣF(obs). dRfree = Σ|F(obs)-F(calc)|/ΣF(obs), calculated using 5% of the data.
RESULTS
Steady-state kinetic studies
To investigate the spontaneous replication error mechanism of polβ, we performed steady-state kinetic measurements for the formation of dG•dTTP and dA•dCTP mismatches. Kinetic parameters were determined for dCTP/dTTP incorporation opposite the templating dG in the presence of Mg2+ or Mn2+ at pH 7 or pH 9 to evaluate the effect of pH and the active-site metal ion on the dG•dTTP formation (Table 1). The change of pH from 7.0 to 9.0 reduces the dCTP insertion efficiency 5-fold, but enhances the dTTP insertion efficiency 14-fold, thereby decreasing the replication fidelity 54-fold. Substituting Mn2+ for Mg2+ does not significantly change the dG•dCTP insertion efficiency, but increases the dG•dTTP insertion efficiency 35-fold, which results in the decrease of the replication fidelity by 29-fold. The dA•dCTP insertion efficiency is about half of the dG•dTTP insertion efficiency with Mg2+ and increases only 5-fold upon substituting Mn2+ for Mg2+.
Table 1. Steady-state kinetic parameters for misincorporation by polβ.
| Template-dNTP | pH | Metal ion | Km (μM) | kcat (10−3 s−1) | kcat/Km (10−3 s−1 μM−1) | fb |
|---|---|---|---|---|---|---|
| G-Ca | 7.0 | Mg | 0.6 ± 0.1 | 212.0 ± 19.9 | 353.3 | 1 |
| G-Ta | 7.0 | Mg | 56.1 ± 4.6 | 2.8 ± 0.4 | 0.049 | 1.4×10−4 |
| G-C | 9.0 | Mg | 1.6 ± 0.07 | 147.0 ± 6.0 | 91.88 | 1 |
| G-T | 9.0 | Mg | 18.4 ± 1.3 | 12.6 ± 0.6 | 0.69 | 7.5×10−3 |
| G-C | 7.0 | Mn | 0.08 ± 0.01 | 30.3 ± 1.5 | 383.7 | 1 |
| G-T | 7.0 | Mn | 11.2 ± 0.5 | 19.1 ± 0.8 | 1.71 | 4.4×10−3 |
| A-T | 7.0 | Mg | 0.85 ± 0.1 | 175.3 ± 4.7 | 206.9 | 1 |
| A-C | 7.0 | Mg | 74.6 ± 4.8 | 1.8 ± 0.7 | 0.024 | 1.2×10−4 |
| A-C | 7.0 | Mn | 25.7 ± 1.2 | 3.0 ± 0.1 | 0.12 | 5.8×10−4 |
The kinetic parameters with standard deviations represent an average of three to six independent determinations.
aData from (33). bRelative efficiency f = (kcat/Km)[incorrect base pair]/(kcat/Km)[correct base pair].
Structure of polβ incorporating a dTTP analogue opposite dG in the presence of Mg2+
To gain insight into the formation of dG•dTTP mismatch by polβ, we determined a ternary structure of polβ incorporating a nonhydrolyzable dTMPNPP (hereafter dTTP*) opposite the templating dG in the presence of the active-site Mg2+. dTTP* was used because it is isosteric to dTTP, yet is not processed by polβ for nucleotidyl transfer and has an active-site metal ion coordination indistinguishable from that of the natural nucleotide (17).
The resulting structure, which is denoted here as the polβ-dG•dTTP*-Mg2+ ternary complex, was refined to 2.3 Å resolution (Table 2). The polβ-dG•dTTP*-Mg2+ structure is significantly different from published polβ ternary structures with a matched base pair (e.g. PDB ID 2FMS (dA•dUTP*), RMSD = 1.208 Å) and a mismatched base pair (e.g. PDB ID 3C2L (dC•dATP*), RMSD = 1.558 Å) (Figure 2A). Polβ in the dG•dTTP*-Mg2+ ternary complex assumes an open protein conformation rather than a closed or an ‘intermediate’ conformation, which are observed in the polβ ternary structure with matched or mismatched base pairs, respectively. The open protein conformation is characterized by the orientation of the α-helix N, which contains the minor groove recognition amino acid residues Asn279 and Arg283. In the open protein conformation, the α-helix N moves away from the active site relative to the position observed in the closed conformation. Notably, the nascent base pair of the dG•dTTP*-Mg2+ ternary complex forms an unusual pseudo-propeller twist conformation. The O4 of the incoming dTTP* is H-bonded to N1 of the templating dG with a pseudo-propeller twist angle of ∼60o (Figure 2B and D). In addition, N2 of the templating dG is H-bonded to the hydroxyl group of Tyr271 (Figure 2D).
Figure 2.

Ternary structure of polβ with dG•dTTP* mismatch and the active-site Mg2+ (PDB ID 4PGQ). (A) Overall structure of polβ with the templating dG paired with an incoming nonhydrolyzable dTMPNPP (dTTP*) in the presence of Mg2+. The template strand is shown in yellow, and the primer and downstream strands are shown in orange. The templating dG is shown in magenta, and the incoming nucleotide is shown in blue. The α-helix N, shown in red, adopts an open conformation. The DNA sequence used for the crystallographic studies is shown. (B) Close-up view of the active site of the dG•dTTP*-Mg2+ ternary structure. The O4 of the incoming dTTP* forms an H-bond with N1 of the templating dG. Tyr271 is H-bonded to N2 of the templating dG. The three catalytic aspartic acid residues and the minor groove recognition amino acids (Asn279 and Arg283) are indicated. A 2Fo−Fc map contoured at 1σ around dTTP* and the templating dG. (C) Close-up view of the metal ion-binding site. Note that Asp256 is not coordinated to the catalytic metal ion, but is coordinated to a water molecule, which is in turn coordinated to the catalytic Mg2+. (D) The H-bonding interactions of the templating dG with Tyr271 and the incoming dTTP*.
The dG•dTTP*-Mg2+ ternary structure unlikely represents a catalytically competent conformation for dG•dTTP misincorporation. Coordination of Asp256 to the catalytic metal ion, which is critical for the catalysis (28), is absent in this structure. Instead, Asp256 is H-bonded to an ordered water molecule, which, in turn, is liganded to the catalytic metal ion (Figure 2C). Furthermore, the lack of base stacking interaction between the incoming dTTP and the 3′ primer terminus base would considerably lower the binding affinity of the incoming nucleotide and thus decrease the insertion efficiency (29). The dG•dTTP*-Mg2+ ternary structure most likely represents a ground-state structure, which requires further conformational reorganization of protein and DNA to reach a conformation that is competent for nucleotidyl transfer (30).
Structure of polβ incorporating a dTTP analogue opposite dG in the presence of Mn2+
To gain structural insights into a ‘pre-chemistry’ state of the mismatched complex, we solved a crystal structure of the polβ-dG•dTTP* complex in the presence of Mn2+, of which ionic radii and coordination geometry are very similar to those of Mg2+. Substituting Mn2+ for Mg2+ has been shown to significantly increase misincorporation rate (Table 1) (15,31). In addition, the use of Mn2+ has been shown to promote the completion of coordination of the active-site metal ion and the formation of a closed protein conformation (32,33), which may stabilize an otherwise unstable ‘pre-chemistry’ state of the mismatched polβ complex.
The polβ-dG•dTTP*-Mn2+ ternary structure, refined to 2.1 Å resolution, substantially differs from the polβ-dG•dTTP*-Mg2+ ternary structure and the published mismatched polβ ternary structures (Figure 3). The protein adopts a closed conformation and the replicating base pair forms a coplanar conformation, overlaying well with the published dA•dUTP*-Mg2+ ternary structure (PDB ID 2FMS, RMSD = 0.394 Å) (Figure 3F). In the Mn2+-bound structure, the α-helix N moves ∼6–9 Å from the position observed in the dG•dTTP*-Mg2 structure, thereby sandwiching the nascent dG•dTTP* base pair between the primer terminus base pair and the α-helix N (Figure 3B and D). Tyr271, Asn279 and Arg283 engage in H-bonding interactions with the minor groove edges of the primer terminus, incoming nucleotide and the templating base, respectively, which is a characteristic of the recognition of the matched base pair by polβ in the closed conformation. Furthermore, the incoming dTTP* now stacks with the primer terminus base and forms a coplanar base pair with the templating dG in the active site (Figure 3B). The O3′ of the primer terminus is 2.8 Å away from the Pα of the incoming dTTP* and is poised for in-line nulcleophilic attack at the Pα of dTTP*. Asp256 is now coordinated to the catalytic metal ion, completing the coordination sphere of the catalytic metal ion. Overall, the dG•dTTP*-Mn2+ structure most likely represents a catalytically competent ‘pre-chemistry state’ of polβ catalyzing dG•dTTP misincorporation. Substituting the active-site Mn2+ for Mg2+, which increases the binding affinity of dTTP 5-fold (Table 1) and promotes the completion of the coordination sphere of the catalytic metal ion, appears to enable the capture of the dG•dTTP complex in a ‘pre-chemistry’ state.
Figure 3.
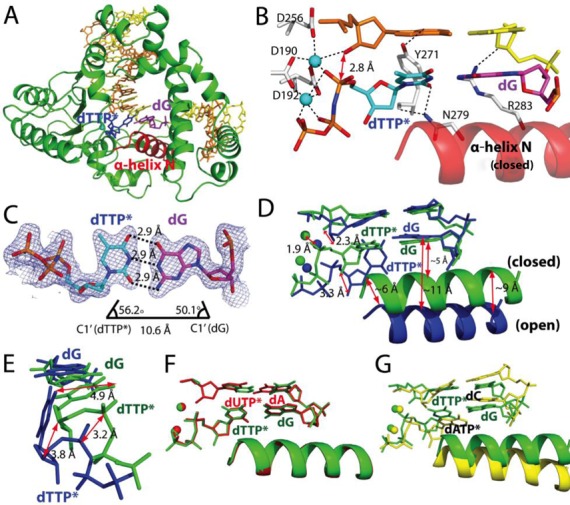
Ternary structure of polβ with dG•dTTP* mismatch and the active-site Mn2+ (PDB ID 4PGX). (A) Overall structure of the dG•dTTP*-Mn2+ ternary structure. Polβ adopts a closed conformation, and dTTP* and dG form a coplanar base pair conformation. (B) Close-up view of the active site of the dG•dTTP*-Mn2+ ternary structure. (C) Base-pairing properties of dTTP* and dG. (D) Comparison of the dG•dTTP*-Mn2+ ternary structure (green) with the dG•dTTP*-Mg2+ ternary structure (blue). (E) Comparison of the dG•dTTP* base pairs in the Mn2+ (green) and the Mg2+ (blue) complexes. (F) Comparison of the dG•dTTP*-Mn2+ structure (green) with the published dA•dUTP*-Mn2+ structure (PDB ID 2FMS, shown in red). (G) Comparison of the dG•dTTP*-Mn2+ structure (green) with the published dC•dATP*-Mn2+ structure (PDB ID 3C2L, yellow).
The most striking feature of the dG•dTTP*-Mn2+ structure is the formation of the Watson–Crick-like base pair between the templating dG and the incoming dTTP* (Figure 3C). The geometry of dG•dTTP* base pair in the pre-chemistry-state structure is essentially identical to that of the matched base pair, with an average H-bond distance of 2.9 Å and the C1′(dG)-C1′(dTTP*) distance of 10.6 Å. In principle, the Watson–Crick-like dG•dTTP* base pair can result from either ionization or tautomerization (enolization, Figure 1) (34,35). To elucidate the mechanism of Watson–Crick-like dG•dTTP* base pairing, we evaluated the effect of pH change on the efficiency of dTTP insertion opposite the templating dG by polβ.
Since the population of the ionized form of N1-H of guanine or N3-H of thymine will increase at high pH, if the Watson–Crick-like dG•dTTP base pairing involves ionization, the insertion efficiency for dG•dTTP is expected to increase as pH increases. On the other hand, if the Watson–Crick-like dG•dTTP base pairing involves tautomerization, pH will not affect much on the insertion efficiency. As mentioned above, the dG•dTTP insertion efficiency increased 14-fold as pH increased from 7 to 9 (Table 1). This strong pH dependence thus suggests that the base pairing between dG and dTTP* in the dG•dTTP*-Mn2+ structure is ionization-mediated (two H-bonds) rather than tautomerization-mediated (three H-bonds). Although the possibility of the enolization-mediated H-bond formation between them still exists based on the observed 2.9 Å distance between O6 of dG and O4 of dTTP*, a strong H-bond between N3 of dTTP* and the negatively charged N1 of the templating dG could override the unfavorable repulsive interaction between O4 of dTTP* and O6 of dG in the active site of polβ (Figure 1). Alternatively, the structure could result from a slow protonation at the O6 position of the deprotonated dG (paired with dTTP*) in the crystal environment.
A comparison of the dG•dTTP*-Mg2+ and the dG•dTTP*-Mn2+ ternary complex structures suggests that a large-scale conformational change may occur, albeit slowly, during the misincorporation of dTTP opposite dG by polβ (Figure 3D and E). During the conformational transition to the closed conformation, several conformational changes occur in the active site (Figure 3D). Asp256 is coordinated to the catalytic metal ion; the catalytic metal ion shifts 1.9 Å away from the position found in the open conformation toward the position found in the closed conformation; the incoming dTTP moves ∼3 Å toward the primer terminus base; O4 of dTTP disengages from N1 of dG; Asn279 engages with O2 of dTTP; the incoming dTTP forms the Watson–Crick-like base pair with dG and the α-helix N moves ∼6–9 Å toward the nascent base pair.
A comparison of the polβ-dG•dTTP*-Mn2+ and the published polβ-dC•dATP*-Mn2+ ternary complex structures shows significant conformational differences between the two mismatched complexes (Figure 3G). The dC•dATP*-Mn2+ complex adopts an ‘intermediate’ protein conformation and a staggered base pair conformation, whereas the dG•dTTP*-Mn2+ complex adopts a closed protein conformation and a coplanar base pair conformation, suggesting that polβ responds differently to different mismatches and adopts different ground-state conformations.
Structure of polβ incorporating a dCTP analogue opposite dA in the presence of Mg2+
To elucidate the mechanism of polβ-induced formation of dA•dCTP mismatch, we solved a ternary structure of polβ incorporating a nonhydrolyzable dCMPNPP (hereafter dCTP*) opposite the templating dA in the presence of the active-site Mg2+. The polβ-dA•dCTP*-Mg2+ structure was refined to 2.5 Å resolution.
The conformations of the dA•dCTP*-Mg2+ and the published dC•dATP*-Mn2+ complexes show several structural differences (RMSD = 0.913 Å) (Figure 4A and F). The most pronounced difference is found in the orientation of the N-terminal lyase domain (Figure 4B): the lyase domain of the dA•dCTP*-Mg2+ complex adopts an open conformation, while the dC•dATP*-Mn2+ complex forms an orientation that is similar to that of the closed conformation. In addition, the two structures show substantially different H-bonding networks in the nascent base pair binding pocket (Figure 4C and F). In the dC•dATP*-Mn2+ structure, the templating dC does not form any H-bonds with the incoming dATP*, and the dC•dATP* base pair is not H-bonded to Tyr271 and Asn279. In the dA•dCTP*-Mg2+ ternary structure, N6 of dA engages in an H-bonding interaction with N3 of dCTP* (Figure 4C). In addition, Tyr271 and Asn279 are H-bonded to N1 of dA and O2 of dCTP*. The distance between N6 of the templating dA and N4 of the dCTP* is 3.1 Å, suggesting that they interact via an unusual mechanism, such as the interaction between the N–H (N6 of dA or N4 of dCTP*) and the π-electrons of NH2 (N4 of dCTP* or N6 of dA). Last, the 3′-OH of the primer terminus is coordinated to the catalytic metal ion (2.4 Å) in the dA•dCTP*-Mg2+ complex, but is not coordinated to the dC•dATP*-Mn2+ complex (4.4 Å).
Figure 4.

Ternary structures of polβ with dA•dCTP* mismatch (PDB ID 4PHA and 4PHD). (A) Overall structure of the active site of the polβ-dA•dCTP*-Mg2+ complex. (B) Comparison of the lyase domains of the dA•dCTP*-Mg2+ (white) and dC•dATP*-Mn2+ (blue) structures. (C) Close-up view of the dA•dCTP*-Mg2+ structure. Key H-bonding interactions are indicated as dotted lines. (D) Comparison of the dA•dCTP*-Mg2+ (blue) and dG•dTTP*-Mg2+ (green) structures. (E) Comparison of the dA•dCTP*-Mg2+ (blue) and dG•dTTP*-Mn2+ (pale cyan) structures. (F) Comparison of the dA•dCTP*-Mg2+ (blue) and dC•dATP*-Mn2+ (yellow) structures. (G) Close-up view of the active site of the dA•dCTP*-Mn2+ structure. A 2Fo−Fc map contoured at 1σ around dCTP* and the templating dA. Key H-bonding interactions are indicated as dotted lines. (H) Comparison of the dA•dCTP*-Mg2+ (blue) and the dA•dCTP*-Mn2+ (magenta) structures.
The dA•dCTP*-Mg2+ structure is also different from the dG•dTTP*-Mg2+ structure (RMSD = 0.838 Å) (Figure 4D). In particular, the dA•dCTP*-Mg2+ structure adopts a staggered base pair conformation and an ‘intermediate’ α-helix N conformation, where the α-helix N shifts ∼5 Å toward the nascent base pair from the position observed in the dG•dTTP*-Mg2+ complex. This change in the position of the α-helix N appears to result in a slight shift of the catalytic metal ion toward Asp256 to coordinate directly, but weakly, with it (3.3 Å, Figure 4C). The dA•dCTP*-Mg2+ structure adopts a conformation that is closer to the closed conformation than the dG•dTTP*-Mg2+ structure. However, the incomplete coordination of the catalytic metal ion and the formation of the ‘intermediate’ protein conformation suggest that the dA•dCTP*-Mg2+ structure likely represents a ground-state conformation that is catalytically sub-optimal for nucleotidyl transfer.
Structure of polβ incorporating a dCTP analogue opposite dA in the presence of Mn2+
To gain structural insights into a pre-chemistry state of the dA•dCTP* complex, we determined a crystal structure of the polβ-dA•dCTP* complex in the presence of the active-site Mn2+. The resulting structure was refined to 2.2 Å resolution.
In contrast to the dG•dTTP* complexes that show a large-scale conformational change in the protein and DNA (RMSD = 1.297 Å), the dA•dCTP*-Mn2+ complex adopts a conformation that is essentially identical to that of the dA•dCTP*-Mg2+ complex (RMSD = 0.334 Å) (Figure 4C and G). The positions, orientations and H-bonding interactions of the templating dA, the incoming dCTP* and the minor groove interacting residues (Tyr271, Asn279 and Arg283) in the dA•dCTP*-Mn2+ complex are nearly indistinguishable from those observed in the dA•dCTP*-Mg2+ complex. The only notable difference is that Asp256, which is weakly coordinated to the catalytic metal ion in the dA•dCTP*-Mg2+ complex, is now fully coordinated (2.3 Å) to the catalytic metal ion, completing the coordination sphere (Figure 4G). However, completion of the catalytic metal ion coordination in the dA•dCTP* complexes does not induce the closed protein conformation. This finding suggests that polβ discourages the formation of the dA•dCTP mispair, as well as dC•dATP mispair, in the nascent base pair binding pocket by inducing a catalytically sub-optimal conformation, partially explaining the low insertion efficiency of polβ for this base pair (Table 1). The lack of a significant difference between the dA•dCTP*-Mg2+/Mn2+ complex structures is consistent with a modest 5-fold change in mismatch insertion efficiency upon the metal ion substitution (Table 1).
The dA•dCTP*-Mn2+ structure and published dC•dATP*-Mn2+ structure are quite different (RMSD = 0.873 Å) in the orientation of the lyase domain (not shown), highlighting the effect of mismatched DNA sequence on polβ structure. The dA•dCTP*-Mn2+ complex shows the completed coordination around the catalytic metal ion, whereas the dC•dATP*-Mn2+ complex shows the lack of coordination of the 3′-OH to the catalytic metal ion (4.4 Å).
MD simulation studies
To gain insights into how polβ discriminates the matched versus the mismatched base pairs and how dG•dTTP evades this discrimination mechanism, we carried out MD simulations based on the open and the closed ternary complex structures, respectively. We also evaluated the effect of the catalytic metal ion binding on the stability of each protein conformation (open versus closed) using MD simulations either with a single metal (i.e. the nucleotide-binding Mg2+) or with double metal ions (i.e. the nucleotide-binding and catalytic Mg2+). Supplementary Table S1 lists the simulated systems and their notations.
RMSDs of the thumb domain, which contains the α-helix N, and their fluctuations are compared first. The results are presented in Figure 5. Among the closed conformation simulations, the dG•dTTP system without the catalytic Mg2+ (dG•dTTPclosed-1Mg2+) shows the largest RMSD relative to the reference closed conformation (i.e. the X-ray polβ-dG•dTTP*-Mn2+ ternary structure); the RMSD rises quickly (around 35 ns) in the MD simulation (Supplementary Figure S2A). The results, together with the large fluctuation of the base pair distance for this system (Supplementary Figure S3A), suggest that the inability to form a stable Watson–Crick base pair between dTTP and dG and the absence of the catalytic metal ion in the active site destabilize the closed conformation. On the other hand, the corresponding double Mg2+ system retains the wobble base pair between dTTP and dG, and shows a relatively small RMSD throughout the entire simulation (Supplementary Figures S2A and S3A; see Supplementary Figure S3B for the wobble base pair). In the present MD simulations, we assumed a conventional protonation state for each nucleotide base, in which dG is then either ionized or tautomerized to form Watson–Crick-like base pair in the closed conformation as suggested by the dG•dTTP*-Mn2+ crystal structure. Nevertheless, no substantial structural changes were found from other closed systems including the control binary complex in the MD simulations (Figure 5 and Supplementary Figure S2A). The results imply that the closed conformation exhibits a relatively small discriminating power between the matched dG•dCTP and mismatched dG•dTTP systems, partly because the dG•dTTP system can adopt a wobble or a Watson–Crick-like base pair.
Figure 5.

Average RMSDs of the thumb subdomain of polβ and their fluctuations. In the left panel, RMSDs are compared for the closed conformation simulations, and in the right panel, for the open conformation simulations. For each coordinate during 150 ns MD simulation, RMSDs are computed relative to the open (red) and to the closed (black) reference conformation, respectively, in which the polβ-dG•dTTP*-Mn2+ ternary structure (for the closed conformation, PDB ID 4PGX) and the polβ-dG•dTTP*-Mg2+ ternary structure (for the open conformation, PDB ID 4PGQ) are used as their corresponding reference structures. ‘Apo’ denotes the single-nulcleotide gapped binary complex with the templating dG.
For the open conformation systems, the double Mg2+ systems show consistently larger RMSDs than their corresponding single Mg2+ systems (Figure 5). Although the differences are within the error range, the large RMSD suggests that the catalytic Mg2+ increases conformational flexibility of polβ in the open conformation. Base pair stability is also different between the single and the double metal systems and between the matched dG•dCTP and mismatched dG•dTTP systems. In particular, the dG•dCTP base pair forms quickly (<1 ns) and remains throughout the rest of the simulation in the double Mg2+ system, but it forms significantly slowly (around 113 ns) in the single metal dG•dCTPopen system (Supplementary Figure S3A, middle). In the dG•dTTPopen systems, although the MD simulations began with the pseudo-propeller twist base pair conformation, they quickly transitioned to the wobble base pair at the beginning of the MD, but the wobble base pair is weak and thus presents a large fluctuation in base-pair distance (Supplementary Figure S3A).
Conformational change to the closed conformation
The MD simulations for the open and closed conformations together with the present crystal structures suggest that the formation of the Watson–Crick base pair and binding of the catalytic metal ion in the enzyme's active site facilitate the open-to-closed conformational activation. We examined this mechanism using MD simulation of an ‘intermediate’ conformation, where the incoming nucleotide is base-paired with the templating dG, and the catalytic metal ion is placed in the positions found in the closed conformation while the α-helix N is in an open conformation. In particular, we examined the effects of the displacement of the catalytic metal ion to the position observed in the closed conformation, subsequent completion of the coordination sphere around this metal ion and the formation of Watson–Crick base pair on the closure of protein structure. As expected, we observed a spontaneous closure of the thumb domain in this simulation (Figure 6). At the beginning of the MD simulation, the thumb domain fluctuates around the open conformation, as it does in the open dG•dCTP system. Then, the RMSD decreases at ∼45 ns and nearly reaches the RMSD values of the closed conformation simulations at ∼125 ns. Several characteristic interactions, including Tyr271 and Arg283, are not fully established in this conformation (Supplementary Figure S4). Nevertheless, the present MD simulations suggest that the catalytic metal ion coordination plays a crucial role in the formation of the closed conformation. These results are further supported by the principal component analysis that is presented in the Supporting Information (Supplementary Figure S5).
Figure 6.

In the lower panel, the RMSDs of the thumb subdomain relative to the closed conformations are shown. The RMSD for the dG•dCTPInt system shows that the structure approaches the state of the closed conformation around 125 ns. In the upper panel, the RMSDs relative to the open conformation are presented for the dG•dCTPInt (black) and dG•dCTPopen-2Mg2+ (green) systems, respectively, showing that the ‘intermediate’ system deviates from the open conformation.
DISCUSSION
The dG•dTTP* and dA•dCTP* structures, in conjunction with the published dC•dATP* structure, provide insights into the spontaneous replication error and the replication fidelity mechanisms of polβ. First, these studies suggest that the polβ-induced formation of dG•dTTP and dA•dCTP mismatches occur through distinct mechanisms (Figure 7). In particular, dG•dTTP mismatch occurs through a Watson–Crick-like geometry facilitated by ionization, whereas dA•dCTP mismatch seems to occur through a non-Watson–Crick geometry. In addition, the large conformational variations observed in the dG•dTTP and the dA•dCTP mismatched structures indicate that polβ discriminates these mismatches, as well as the matched versus mismatched base pairs, in both the ground and the pre-chemistry states. This two-stage mismatch discrimination mechanism highlights the roles of the minor groove interaction and geometric selection, as well as the ability to form Watson–Crick or Watson–Crick-like base pair, in increasing the replication fidelity of polβ. Each role is discussed below.
Figure 7.

Ground-state and ‘pre-chemistry-state’ structures of polβ ternary complexes. The Mg2+-bound (left panels) and the Mn2+-bound (right panels) structures most likely represent the ground-state and the ‘pre-chemistry-state’ structures of the polβ ternary complexes, respectively. (A) The dG•dTTP*-Mg2+ structure. Tyr271 and dTTP form two H-bonds with the templating dG. (B) The dG•dTTP*-Mn2+ structure. Substituting Mn2+ for Mg2+ triggers an open-to-closed conformational change of the protein and a staggered-to-Watson–Crick conformational change of the dG•dTTP base pair. (C) The dA•dCTP*-Mg2+ structure. dA•dCTP* forms a non-coplanar conformation. (D) The dA•dCTP*-Mn2+ structure. The Mn2+ substitution does not induce a conformational change of the protein and the base pair. (E) Published polβ structure with dG•dCTP* and the active-site Mg2+ (PDB ID 1BPY).
Discrimination against mismatches in the ground state
A ternary structure of polβ with the active-site Mg2+ most likely represents a close approximation of the ground-state conformation of the polβ complex. The large variations in the ground-state conformations of the dG•dTTP* and dA•dCTP* complexes and the published dG•dCTP* complex suggest that polβ discriminates against mismatches based on the ability to form a stable Watson–Crick base pair in the ground state (Figure 7A, C and E). The ground-state conformations of dG•dTTP* and dA•dCTP* base pairs of the mismatched complexes are different from those of base pairs observed in polβ structure with the correct insertion. Unlike the matched complex that forms a stable Watson–Crick base pair (Figure 7E), the complexes with base mismatches do not (Figure 7A and C). These differences appear to result in different ground-state protein conformations: i.e. the open conformation for dG•dTTP*, the ‘intermediate’ conformation for dA•dCTP* and the closed conformation for dG•dCTP*.
Discrimination against mismatches in the pre-chemistry state
The ‘pre-chemistry’ state conformations of the dG•dTTP* and the dA•dCTP* complexes (i.e. the mismatched ternary complex structures with Mn2+) are also significantly different, suggesting that polβ recognizes different mismatches via different recognition mechanisms (Figure 7B and D). Whereas the dG•dTTP* complex exhibits the open-to-closed conformational reorganization upon the Mn2+ substitution, the dA•dCTP* complex does not, and remains in the ‘intermediate’ conformation. In addition, whereas the dG•dTTP*-Mn2+ complex adopts a Watson–Crick-like base pair conformation, the dA•dCTP*-Mn2+ complex adopts a staggered base pair conformation. These structural differences suggest that the dA•dCTP mismatch complex is more resistant to the conformational change to the closed conformation than the dG•dTTP mismatch, likely due to the inability of forming a Watson–Crick-like dA•dCTP base pair in the polβ active site. The modest ∼2-fold difference in the relative efficiencies for the dG•dTTP and the dA•dCTP complexes (0.049 versus 0.024, Table 1) may result from the offset of the favorable conformations of the dA•dCTP and the dG•dTTP complexes in the ground and the pre-chemistry states, respectively.
Implication of a Watson–Crick-like dG•dTTP base pair in the closed conformation state
Many DNA polymerases, including the A-family DNA polymerase BF, the B-family DNA polymerase RB69 and the Y-family DNA polymerases Dpo4 and polη, allow wobble conformation for purine•pyrimidine (i.e. G•T and A•C) base pair mismatches (7–10). By contrast, the pre-chemistry-state structure of polβ complex with dG•dTTP suggests that the X-family DNA polymerase polβ favors a Watson–Crick-like conformation over a wobble conformation for purine•pyrimidine base pairs in the closed conformation state (Figure 7B). This suggests a key strategy employed by polβ to discriminate against base pair mismatches and increase replication fidelity. The dG•dTTP mismatch escapes this discrimination presumably via ionization of the wobble dG•dTTP to form the Watson–Crick-like conformation. However, the dA•dCTP mismatch cannot escape the discrimination and remains in the staggered conformation (Figure 7D). It is possible that the formation of the Watson–Crick-like dG•dTTP involves a transient formation of a wobble dG•dTTP base pair, which is then ionized to generate the Watson–Crick-like base pair. The failure to form a stable wobble dG•dTTP base pair in the closed conformation state may be due to the rigid geometric constraints imposed by polβ's active site. For example, the DNA in the active site of some DNA polymerases (e.g. BF, T7) (7) is in A-DNA with a widened minor groove, whereas the DNA in the polβ active site is in B-DNA (16,29). The structural similarity between the dG•dCTP*-Mg2+ complex and the dG•dTTP*-Mn2+ complex suggests that the ground-state discrimination of dG•dCTP and dG•dTTP base pairs contributes to the replication fidelity of polβ to a greater extent than the pre-chemistry-state discrimination of the base pairs.
The same geometric constraints can also be applied to the dA•dCTP base pair to form the closed conformation. However, since the energetic penalty to form the Watson–Crick-like base pair (Figure 1) is expected to be high for dA•dCTP (36), polβ fails to form the Watson–Crick-like dA•dCTP base pair in the binding pocket of the closed conformation with high probability. This is evident from the dA•dCTP*-Mn2+ structure showing that even with Mn2+, the enzyme is in the ‘intermediate’ conformation and not in the closed conformation with Watson–Crick-like base pair (Figure 7D). This differs from some other DNA polymerases that allow a wobble or a Watson–Crick-like dA•dCTP or dC•dATP base pair. For example, the B-family DNA polymerase RB69 forms wobble base pair for dA•dCTP and dC•dATP likely through protonated templating bases (8). The A-family DNA polymerase BF with dA•dCTP mismatch forms a wobble dA•dCTP and an ‘ajar’ protein conformation with Mg2+, and a Watson–Crick-like dA•dCTP and a closed protein conformation with Mn2+, respectively (7).
Minor groove edge recognition by DNA polymerase may affect the conformations of mismatched replicating base pairs
The difference in the minor groove interactions at the nascent base pair binding pocket of DNA polymerases may contribute to the formation of various base pair geometries for the mismatches. Many DNA polymerases recognize the minor groove edge of either incoming nucleotide (e.g. BF, Dpo4, T7, Taq) or templating base (e.g. polη), but DNA polymerase that recognizes both edges of the nascent base pair is rare. Polβ recognizes the minor groove edges of both templating base and incoming nucleotide, implying that polβ may be more sensitive to the abnormality of the minor groove edges of the replicating base pair. Indeed, whereas BF, RB69, Dpo4 and polη accommodate wobble dG•dTTP or dT•dGTP base pair geometry in the active site, polβ does not, suggesting that polβ discriminates between wobble and Watson–Crick geometries better than other DNA polymerases. For example, our dG•dTTP-Mg2+, dA•dCTP-Mg2+/Mn2+ and published dC•dATP-Mn2+ structures with non-coplanar base pair conformations do not show the minor groove edge recognition by Arg283, whereas the dG•dTTP-Mn2+ and dA•dUTP-Mg2+ structures with coplanar base pair conformation show the interaction. Polβ may have to rely on strict minor groove edge recognition and geometric selection at the insertion site to maximize replication fidelity, because it does not have an intrinsic proofreading function. Polβ may sense the difference in the geometries of the wobble dG•dTTP and dA•dCTP base pairs and the canonical Watson–Crick base pair through its minor groove interaction at the insertion site, thereby discriminating wobble base pair from canonical Watson–Crick base pair prior to nucleotide incorporation.
Protein dynamics controls the accessibility of the closed conformation
The present X-ray structures and MD simulation results make it possible to propose a detailed sequence of events leading to the closed conformation and the mismatch discrimination mechanism of polβ. In the case of the correct insertion, when an incoming nucleotide binds opposite the templating base, the nascent base pair quickly forms the Watson–Crick conformation (Supplementary Figure S3A), and then the coordination around the catalytic metal ion completes. Then, the protein adopts a closed conformation via the intrinsic protein motion as observed in the ‘intermediate’ conformation simulation and the principal component analysis (Supplementary Figure S5). In the case of a misinsertion, the nascent base pair cannot form a stable Watson–Crick conformation in the active site, so the subsequent events (e.g. the completion of the coordination sphere of the catalytic metal ion) cannot occur or are slowed down. The MD simulation results and kinetic measurements suggest that the dG•dTTP mismatch escapes this screening mechanism by the formation of a transient wobble base pair, followed by deprotonation of the templating dG to form the Watson–Crick-like base pair. Consequently, the dG•dTTP mismatch complex adopts the closed conformation, but with a low probability, and carries out the catalytic insertion.
CONCLUSION
We characterized structures and dynamics of DNA polymerase β (polβ) with mismatched base pairs. The results reveal that polβ responds differently to different mismatches. In particular, we found that the dG•dTTP base pair forms a Watson–Crick-like base pair in the closed conformation, whereas the dA•dCTP base pair does not, partly due to high energy to form the Watson–Crick-like base pair in the polβ active site. This suggests that polβ discriminates between dG•dTTP and dA•dCTP mismatches based on the ability to form the Watson–Crick-like base pair in the pre-chemistry state, in which the Watson–Crick-like dG•dTTP base pair presumably forms via the ionization of dG. These findings, together with the published X-ray structures with the correct insertion, suggest that polβ senses subtle differences in the geometries of the wobble dG•dTTP and dA•dCTP base pairs and the canonical Watson–Crick base pair, and allows only a Watson–Crick-like conformation for purine•pyrimidine mismatches in the closed conformation. Overall, our studies provide new insights into the spontaneous replication error and the replication fidelity mechanisms of polβ.
ACCESSION NUMBERS
PDB ID: 4PGQ, 4PGX, 4PHA and 4PHD.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Instrumentation and technical assistance for this work were provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and the Institute for Cellular and Molecular Biology at the University of Texas at Austin. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
FUNDING
The National Institutes of Health (ES23101 to S.L.); Umeå University (to K.N.). Funding for open access charge: The National Institutes of Health (ES23101 to S.L.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Halliday J.A., Glickman B.W. Mechanisms of spontaneous mutation in DNA repair-proficient Escherichia coli. Mutat. Res. 1991;250:55–71. doi: 10.1016/0027-5107(91)90162-h. [DOI] [PubMed] [Google Scholar]
- 2.Bacolla A., Cooper D.N., Vasquez K.M. Mechanisms of base substitution mutagenesis in cancer genomes. Genes. 2014;5:108–146. doi: 10.3390/genes5010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keohavong P., Thilly W.G. Fidelity of DNA polymerases in DNA amplification. Proc. Natl Acad. Sci. U.S.A. 1989;86:9253–9257. doi: 10.1073/pnas.86.23.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bebenek K., Joyce C.M., Fitzgerald M.P., Kunkel T.A. The fidelity of DNA synthesis catalyzed by derivatives of Escherichia coli DNA polymerase I. J. Biol. Chem. 1990;265:13878–13887. [PubMed] [Google Scholar]
- 5.Lee H.R., Johnson K.A. Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 2006;281:36236–36240. doi: 10.1074/jbc.M607964200. [DOI] [PubMed] [Google Scholar]
- 6.Osheroff W.P., Jung H.K., Beard W.A., Wilson S.H., Kunkel T.A. The Fidelity of DNA Polymerase β during Distributive and Processive DNA Synthesis. J. Biol. Chem. 1999;274:3642–3650. doi: 10.1074/jbc.274.6.3642. [DOI] [PubMed] [Google Scholar]
- 7.Wang W., Hellinga H.W., Beese L.S. Structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis. Proc. Natl Acad. Sci. U.S.A. 2011;108:17644–17648. doi: 10.1073/pnas.1114496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia S., Wang J., Konigsberg W.H. DNA mismatch synthesis complexes provide insights into base selectivity of a B family DNA polymerase. J. Am. Chem. Soc. 2013;135:193–202. doi: 10.1021/ja3079048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaisman A., Ling H., Woodgate R., Yang W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y., Gregory M.T., Biertümpfel C., Hua Y.-J., Hanaoka F., Yang W. Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc. Natl Acad. Sci. U.S.A. 2013;110:8146–8151. doi: 10.1073/pnas.1303126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bebenek K., Pedersen L.C., Kunkel T.A. Replication infidelity via a mismatch with Watson-Crick geometry. Proc. Natl Acad. Sci. U.S.A. 2011;108:1862–1867. doi: 10.1073/pnas.1012825108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osheroff W.P. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase beta. J. Biol. Chem. 1996;271:12141–12144. doi: 10.1074/jbc.271.21.12141. [DOI] [PubMed] [Google Scholar]
- 13.Servant L., Bieth A., Hayakawa H., Cazaux C., Hoffmann J.-S. Involvement of DNA polymerase beta in DNA replication and mutagenic consequences. J. Mol. Biol. 2002;315:1039–1047. doi: 10.1006/jmbi.2001.5307. [DOI] [PubMed] [Google Scholar]
- 14.Canitrot Y., Frechet M., Servant L., Cazaux C., Hoffmann J.S. Overexpression of DNA polymerase beta: a genomic instability enhancer process. FASEB J. 1999;13:1107–1111. doi: 10.1096/fasebj.13.9.1107. [DOI] [PubMed] [Google Scholar]
- 15.Batra V.K., Beard W.A., Shock D.D., Pedersen L.C., Wilson S.H. Structures of DNA polymerase beta with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol. Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawaya M.R., Prasad R., Wilson S.H., Kraut J., Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 17.Batra V.K., Beard W.A., Shock D.D., Krahh J., Pedersen L.C., Wilson S.H. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vagin A., Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 19.Pelletier H., Sawaya M.R., Wolfle W., Wilson S.H., Kraut J. Crystal structures of human DNA polymerase beta complexed with DNA: implications for catalytic mechanism, processivity, and fidelity. Biochemistry. 1996;35:12742–12761. doi: 10.1021/bi952955d. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis I.W., Leaver-Fay A., Chen V.B., Block J.N., Kapral G.J., Wang X., Murray L.W., Arendall W.B., Snoeyink J., Richardson J.S., et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foloppe N., MacKerell A.D., Jr All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 24.MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., et al. All-atom empirical potential for molecular modeling and dynamic studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 25.MacKerell A.D., Feig M., Brooks C.L. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 27.York D.M., Darden T.A., Pedersen L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: a comparison of the Ewald and truncated list methods. J. Chem. Phys. 1993;99:8345–8348. [Google Scholar]
- 28.Batra V.K., Perera L., Lin P., Shock D.D., Beard W.A., Pedersen L.C., Pedersen L.G., Wilson S.H. Amino acid substitution in the active site of DNA polymerase β explains the energy barrier of the nucleotidyl transfer reaction. J. Am. Chem. Soc. 2013;135:8078–8088. doi: 10.1021/ja403842j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beard W.A., Wilson S.H. Structure and mechanism of DNA polymerase β. Chem. Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 30.Lin P., Batra V.K., Pedersen L.C., Beard W.A., Wilson S.H., Pedersen L.G. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase. Proc. Natl Acad. Sci. U.S.A. 2008;105:5670–5674. doi: 10.1073/pnas.0801257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabor S., Richardson C.C. Effect of manganese ions on the incorporation of dideoxynucleotides by bacteriophage T7 DNA polymerase and Escherichia coli DNA polymerase I. Proc. Natl Acad. Sci. U.S.A. 1989;86:4076–4080. doi: 10.1073/pnas.86.11.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freudenthal B.D., Beard W.A., Shock D.D., Wilson S.H. Observing a DNA polymerase choose right from wrong. Cell. 2013;154:157–168. doi: 10.1016/j.cell.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koag M.C., Lee S. Metal-dependent conformational activation explains highly promutagenic replication across O6-methylguanine by human DNA polymerase β. J. Am. Chem. Soc. 2014;136:5709–5721. doi: 10.1021/ja500172d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H., Eritja R., Bloom L.B., Goodman M.F. Ionization of bromouracil and fluorouracil stimulates base mispairing frequencies with guanine. J. Biol. Chem. 1993;268:15935–15943. [PubMed] [Google Scholar]
- 35.Watson J.D., Crick F.H. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 36.Moser A., Range K., York D.M. Accurate proton affinity and gas-phase basicity values for molecules important in biocatalysis. J. Phys. Chem. B. 2010;114:13911–13921. doi: 10.1021/jp107450n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.